
Abstract
The diagnosis of severe acute respiratory syndrome 2 (SARS-CoV-2) infection by quantitative PCR with reverse transcription (RT–qPCR) typically involves bulky instrumentation in centralized laboratories and an assay time of 1–2 h. Here, we show that SARS-CoV-2 RNA can be detected in 17 min via a portable device integrating reverse transcription, fast thermocycling (via plasmonic heating through magneto-plasmonic nanoparticles) and in situ fluorescence detection following magnetic clearance of the nanoparticles. The device correctly classified all nasopharyngeal, oropharyngeal and sputum samples from 75 patients with COVID-19 and 75 healthy controls, with good concordance in fluorescence intensity with standard RT–qPCR (Pearson coefficients > 0.7 for the N1, N2 and RPP30 genes). Fast, portable and automated nucleic acid detection should facilitate testing at the point of care.
Similar content being viewed by others

Main
Developing fast and reliable diagnostic tools is critical during the current coronavirus disease 2019 (COVID-19) pandemic, as diagnostic capacity at scale becomes critical to containing outbreaks and to reducing fatality rates1,2,3,4,5,6,7. In the absence of effective cures or vaccines for the disease, identifying as many infected individuals as possible (both symptomatic and asymptomatic) and then isolating them is the most effective way to prevent disease transmission5,6,8. Administering on-site and rapid tests is ideal for prompt patient triaging and contact tracing. Unfortunately, diagnostic tools with such capabilities are not readily available. Immunogenic lateral-flow assays are fast, small and cost effective, but are unsuitable for viral detection during the early disease phase9,10,11. Nucleic acid amplification tests based on PCR are the gold standard for COVID-19 confirmation, owing to their high analytical accuracy (~99%)12,13. A caveat, however, is that most PCR tests are carried out in centralized laboratories, which often incur logistic overheads (such as sample transfer and protection from degradation) and long turnaround times (2–3 d)14,15,16. The deployment of conventional PCR to point-of-care (POC) settings is also limited by lengthy assay times (~1–2 h) and the need for bulky instrumentation. New isothermal nucleic acid amplification technologies may shorten assay times, but they are less established than conventional PCR (because of supply chain issues and the need for clinical validation) and may have inferior accuracy17,18.
Rapid and portable thermocycling technology aims to bring nucleic acid amplification tests to POC settings. Thermoplasmonics is a promising method for fast heating19,20,21,22,23,24,25. It exploits the light-to-heat conversion mediated by plasmonic substrates26,27,28,29. This concept has been adopted for PCR thermocycling, in the form of a planar plasmonic substrate in a fluidic device20,30,31 or metallic nanoparticles in suspension22,32,33. Both methods have shown fast nucleic acid amplification, but challenges remain: nucleic acids after amplification are difficult to detect because plasmonic substrates would interfere with fluorescence reporters34,35. Improving the detection sensitivity often entails additional steps involving the use of external equipment (in particular, centrifugation (for particle removal), gel electrophoresis (for separate signal detection) or a microscope system) (Supplementary Table 1). These requirements would lengthen the total assay time, overshadowing the advantage of fast thermocycling, and would also complicate the workflow.
Here, we report the development of a fast one-pot PCR with reverse transcription (RT–PCR) technology for COVID-19 diagnostics. We named it nanoPCR. The system seamlessly integrates plasmonic thermocycling with fluorescent signal detection in a single device. A key concept is the use of dual-functional magneto-plasmonic nanoparticles (MPNs) for PCR applications. We noticed that most of the plasmonic effect is confined near the surface of plasmonic nanoparticles. By encasing a magnetic core with a plasmonic gold (Au) shell, we could thus achieve: (1) efficient plasmonic heating comparable to that by solid Au nanoparticles; and (2) rapid nanoparticle separation with external magnetic fields to perform in situ signal detection. Exploiting these advantages, we advanced a compact nanoPCR system that automatically executes reverse transcription, rapid PCR amplification and fluorescence detection with a single button press. The current prototype concurrently measures three samples within 17 min. The limit of detection is 3.2 gene copies per µl, which is comparable to those by benchtop PCR equipment. We further applied nanoPCR to test clinical samples (75 patients with COVID-19 and 75 controls). The nanoPCR device rapidly detected three gene targets (N1, N2 and RPP30) and achieved excellent diagnostic accuracy (>99%).
Results and discussion
We used nucleic acid targets recommended by the US Center for Disease Control and Prevention (Fig. 1a)36: N1 and N2 genes (for severe acute respiratory syndrome 2 (SARS-CoV-2) virus detection) and the human RPP30 gene (for human sample confirmation). Figure 1b shows the nanoPCR schematic for 17-min COVID-19 diagnostics (see Supplementary Video 1 for the whole workflow). The assay starts with extracting total RNAs from suspected patient samples (for example, nasopharyngeal/oropharyngeal swabs or sputum). Extracted RNA samples are then mounted inside a nanoPCR device, and the remaining assay steps are carried out automatically: (1) thermoplasmonic RT–PCR (11 min); (2) magnetic fluorescence switch (MFS) to remove MPNs from samples using an external magnetic field (3 min); and (3) fluorescence signal detection directly from tubes (<20 s). Overall, the nanoPCR offers practical advantages for on-site molecular diagnostics. The dual-functional MPNs enable both rapid nucleic acid amplification (via plasmonic heating) and the detection of fluorescent signal (via MPN clearing) in a one-pot assay format.

a, Target RNA regions for nanoPCR tests. The N1 and N2 genes are for SARS-CoV-2 detection, whereas the RPP30 gene serves as a control for human sample confirmation. ORF, open reading frame; S, spike; E, envelope; M, membrane; N, nucleocapsid. b, High-speed nanoPCR diagnostic flow for SARS-CoV-2 detection: (1) 3 min of RNA extraction using a disposable RNA preparation kit; (2) 11 min of RT–PCR by magneto-plasmonic thermocycling; and (3) 3 min of detection and diagnosis via MFS by application of an external magnetic field for MPN removal. c, A disposable RNA extraction kit with preloaded buffers and a simple plunge system. d, A compact nanoPCR instrument for POC application. The system houses a light source for plasmonic heating, a Ferris wheel for sample mounting, MFS magnets and optics for fluorescence detection. e, Circular array of the low-powered laser diodes and synchronized Ferris wheel for thermocycling of multiple samples. f, Syncing of laser illumination with Ferris wheel rotation to enable the processing of multiple samples without compromising the total assay time.
To facilitate sample preparation at POC settings, we designed a disposable RNA preparation kit (Fig. 1c and Supplementary Fig. 1). The kit has multiple chambers preloaded with RNA extraction reagents (see Methods for details) and a silica filter to capture RNA (that is, solid-phase extraction). After loading a test sample (20 µl), we sequentially actuated plungers to perform virus lysis, RNA capture and elution to PCR tubes. When tested with human coronavirus NL63, the kit achieved extraction yields comparable to those achieved by a conventional centrifugation method (Supplementary Fig. 2); however, the kit operation was equipment free and complete within 3 min.
We implemented a compact nanoPCR system, integrating key components for plasmonic RT–PCR. Specifically, the system had a light source for plasmonic heating, a rotating sample holder (Ferris wheel style), movable magnets and a fluorescent optical setup (Fig. 1d). A microcontroller coordinated the operation of individual modules: with a single button push, the system automatically executed plasmonic RT–PCR, MFS and fluorescence detection and displayed the results on an embedded screen (Supplementary Video 1). This self-contained system had a small form factor (15 × 15 × 18.5 cm3) and weighed ~3 kg (Fig. 1d).
As the plasmonic light source, we arranged weak-powered (80 mW) laser diodes in a circular array (Fig. 1e and Supplementary Fig. 3), which allowed for more even illumination on samples than using a single high-power (~1 W) laser. For the sample mounting, we adopted a Ferris wheel scheme to achieve higher throughput. The wheel rotation sequentially brought a sample under light illumination while other samples were air cooled. By syncing the wheel rotation and the illumination schedule (Fig. 1f), we could perform PCR reactions in multiple samples using an existing light source. The method thereby provided an economical way to scale up (see Supplementary Note 2 for details). The current POC prototype was designed for three samples. We also constructed a prototype that could process nine samples (Supplementary Fig. 4), which had the throughput of processing roughly 36 samples per hour.
The essential nanoPCR reagent was the dual-functional MPNs for plasmonic heating and magnetic clearing. The particle had a 16-nm magnetic core (Zn0.4Fe2.6O4) enclosed by a 12-nm-thick Au shell (Fig. 2a, left)37. Elemental mapping with energy dispersive X-ray spectroscopy (EDS) confirmed the MPNs’ core–shell nature (Fig. 2a, right). We further coated the particles with phosphine–sulfonate ligands, which stabilized MPNs by imparting negative surface charges. The hydrodynamic size of MPNs measured with dynamic light scattering (DLS) was ~50 nm without aggregation and they maintained excellent colloidal stability for 1 year without a change in size (Supplementary Fig. 5).

a, TEM images of MPNs with a core–shell (Zn0.4Fe2.6O4-Au) structure. Left: a 16-nm magnetic core was encased by a 12-nm Au shell. Right: elemental mapping of Au and Fe showed an area-specific distribution of the core and Au shell structure. b, Ultraviolet–visible cross-section absorption spectrum of an MPN measured with the integrating sphere. At a shell thickness of 12 nm, the highest absorbance was measured at 535 nm. c, Simulation of the electric field at 535 nm illumination. An intense electric field was confined to the Au surface. d, Linear profile of the maximum electric field enhancement factor (E/E0) (dotted line in c). e, Estimated magnetization curve of a single MPN, as measured using a vibrating sample magnetometer. MPNs are superparamagnetic at room temperature (20 °C). f, The clustered form of MPN was realized by dipolar interaction under magnetic field application (100 T m−1). g, Hydrodynamic sizes of MPNs before and after clustering, as measured by DLS. The size increased from 50 to 530 nm.
We characterized the MPNs’ optical properties, measuring plasmonic responses and simulating electric fields. With the 12-nm-thick Au shell, the MPNs showed plasmon resonance at a wavelength (λ) of 535 nm, which matched with the peak wavelength (532 nm) of the laser diode used in the system (Fig. 2b). The electric field map under plasmonic resonance, simulated by the boundary element method (see Methods), indicated that the field enhancement was particularly confined to both the inner and outer surfaces of the Au shell (Fig. 2c,d). In addition, the electric field enhancement (E/E0) and cross-section absorption (σabs) of a single MPN were similar to those of a solid 40-nm Au nanoparticle (Supplementary Fig. 6). These results supported that the core–shell structure could maintain the desired surface plasmonic properties while providing additional functionality through the core.
Magnetic measurements confirmed that the MPNs were superparamagnetic, with a magnetic moment of m = 7.52 × 10−19 A m2 per particle (Fig. 2e). Under the magnetic field generated by an external NdFeB magnet, MPNs clustered through dipole–dipole interactions, which increased the overall hydrodynamic size (from 50 to 530 nm) (Fig. 2f,g). The magnetic moment of clustered MPNs was estimated to be much higher (8.96 × 10−16 A m2)—approximately 1,200 times higher than a single MPN (see Supplementary Note 1). This feature enabled us to quickly clear MPNs from solution, facilitating the interference-free fluorescent detection.
We used MPNs as a volumetric heating source in the plasmonic RT–PCR. Shining light on the MPN-containing solution quickly heated the entire sample (Fig. 3a). The heating effect was dominant only when the incident wavelength matched to the MPNs’ plasmonic resonance (Fig. 3b), confirming the underlying thermoplasmonic mechanism. We further developed an analytical model to estimate the temperature increase in the MPN solution. Approximating a single MPN as a spherical heater, the temperature profile around the particle is given as ∆TMPN = (σabsI)/(4πRks), where ks is the thermal conductivity of the solution, I is the irradiance of the illumination and R is the particle radius38. Under our experimental conditions (σabs = 2.5 × 103 nm2, R = 20 nm, ks = 0.6 W m−1 K−1 for water and I = ~0.1 W mm−2), the overall temperature increase of the solution is estimated as ∆T = ∆TMPN(R/δ)N2/3, where N is the total number of MPNs and δ is the inter-particle distance39. With N particles suspended in volume V, δ = ~0.55(V/N)1/3, leading to ∆T = 2∆TMPN(N/V)V2/3. Therefore, a linear increase in ∆T with MPN concentration ([MPN]) is expected (Fig. 3c, dotted lines), which agrees well with the experimental observations (Fig. 3c, circles). The heating efficiency, however, deteriorated when [MPN] ≳ 3 × 1011 ml−1; this could be attributed to shifts in plasmonic resonance caused by inter-particle coupling40. To maximize the heating efficiency, we set [MPN] = 2.6 × 1011 ml−1 for the rest of the assay.

a, Thermal images of plasmonically heated MPN solution (light source = 1,000 mW at λ = 532 nm; solution volume = 10 µl; MPN concentration = 2.6 × 1011 particles per ml). The solution temperature changed from 25 to 90 °C within a few seconds upon laser illumination. b, Temperature profile of MPN solutions with different wavelengths of light illumination. The laser diode with a wavelength of 532 nm matched closely to the maximum absorption of MPN; thereby, faster heating was induced. c, Effect of MPN concentration on the temperature profile (dotted line: theoretical estimation; circle: measured values). The maximum temperature increase was observed at a concentration of 2.6 × 1011 particles per ml. At higher concentrations, the heating effect degenerated. d, High-speed plasmonic thermocycling profile of MPN solution (seven cycles per min; 58–90 °C). e, Single thermocycling of MPN solution (red dotted rectangle in d). The heating rate was 13.17 °C s−1 and the cooling rate was 4.94 °C s−1. The temperature deviations (right) were from five cycles at 58 (blue) and 90 °C (red). Data are mean ± s.e.m. The values were <1% compared with the target temperature. f, Schematic of the synchronized Ferris wheel system for multi-sample thermocycling. Sample rotation and laser illumination were synced to heat three samples. g, Temperature profiles of three samples on the wheel. Individual heating profiles were interleaved such that the total cycling time remained the same.
Applying MPN plasmonic heating, we could achieve rapid thermocycling (58 to 90 to 58 °C) at a rate of 8.91 s per cycle (Fig. 3d). The precision at target temperatures was excellent, with the coefficient of variations <1.64% (Fig. 3e). Note that we relied on the convective heat transfer to air for the sample cooling. The observed cooling time from 90 to 58 °C was ~6.5 s, which also agreed with the numerical modelling (Supplementary Fig. 7). Such fast cooling was possible mainly because the sample volume was small (10 µl). For COVID-19 detection, we tuned the Ferris wheel for three-sample measurement (Fig. 3f), with each sample targeting one of three genes for COVID-19 diagnostics (that is, N1, N2 and RPP30). By synching the wheel rotation with the heating cycle (2.43 s under light; 6.48 s with air cooling), we could continuously process three samples using a light source designed for a single PCR tube (Fig. 3g and Supplementary Fig. 8).
Next, we optimized the entire operation for SARS-CoV-2 gene detection (Fig. 4a). The process required maintaining a constant temperature for reverse transcription. We controlled the effective light power by modulating the on/off duty cycle, achieving the target temperature (42 °C) with small variations (<1 °C; Fig. 4a right). Testing with N1, N2 and RPP30 target genes, we confirmed that the 5-min reverse transcription produced sufficient numbers of complementary DNAs (see Methods and Supplementary Fig. 9). Following the reverse transcription reaction, we executed the PCR amplification (40 cycles per tube; 6 min). Gel electrophoresis on PCR products by nanoPCR (6 min) and by conventional benchtop instrument (2 h) showed matching bands (Fig. 4b), confirming comparable performance between two systems. With 5-min conventional PCR, no band was detected (Supplementary Fig. 10). We further assessed the purity of the PCR products by measuring ultraviolet absorbance ratios (A260/A230 and A260/A280), where A230, A260 and A280 are the ultraviolet absorbance at 230, 260 and 280 nm, respectively (Supplementary Fig. 11). The measured ratios were A260/A230 = ~2.2 and A260/A280 = ~1.8. A260/A230 was higher than A260/A280, indicating that nanoPCR produced high-quality PCR products.

a, Measured temperature profiles of a sample under nanoPCR. Left: 42 °C incubation for reverse transcription (5 min), followed by PCR amplification (40 cycles; 6 min), then signal detection steps (3 min). The whole RT–PCR process was complete within 11 min. Right: enlarged temperature profile during the isothermal reverse transcription phase. b, Gel electrophoresis images of PCR products from the 11-min nanoPCR and a 2-h conventional benchtop thermocycler, for N1, N2 and RPP30 genes. All of the bands were detected identically for both methods. NTC, non-target control. c, MFS for in situ detection of amplicons. Brightfield (BF) and fluorescence (FL) photographs of an assay mixture before and after 3 min of MFS application are shown. d, N1 gene fluorescence signal changes during MFS application (n = 3). 50% signal recovery was achieved at 3 min. e, Detection of N1, N2 and RPP30 genes before (−) and after (+) MFS application (n = 5 for each measurement). f, Evaluation of the LOD of nanoPCR with different amounts of target N1 RNA (n = 3). The detection threshold was set at three times the standard deviation of the signal from a blank. The LOD (dashed horizontal line) was 3.2 copies of gene per μl. g, Specificity evaluation for different strains of coronavirus (that is, N1 and N2 genes from SARS-CoV-2, SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV)) (n = 3 for each measurement). Data in d–g are mean ± s.d.
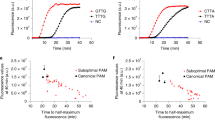
For fluorescence signal detection in situ, we applied MFS to clear MPNs in sample tubes. After completing the RT–PCR reaction, the nanoPCR system automatically moved a magnet array to the Ferris wheel (Fig. 4c, left; see Supplementary Video 1) and read out the fluorescence signal using an internal ultraviolet light-emitting diode (LED) and a photodiode (Supplementary Fig. 12). Under the field gradient (100 T m−1) created by the magnet, MPNs clustered and sedimented to the tube bottom (see Supplementary Note 1). We observed that MFS was indispensable for reliable measurements. Without MFS, amplicons’ signal from fluorescein amidites (FAMs) was barely detectable (Fig. 4c, right), presumably due to the spectral overlap between MPN absorption (λpeak = 535 nm) and FAM emission (λem = 517 nm). When tested with N1 amplicons, the fluorescence signal recovered to ~50% of its saturation level at 3 min after MFS (Fig. 4d and Supplementary Fig. 13). Applying this timing, we could reliably detect all three target genes (N1, N2 and RPP30; Fig. 4e). Of note, nanoPCR is inherently limited to end-point measurements, as the assay detects signal only after PCR reaction.
With operational parameters determined, we evaluated the analytical performance of nanoPCR, benchmarking it against conventional RT–PCR. To estimate the limit of detection (LOD), serially diluted samples containing N1, N2 or RPP30 target genes were prepared and subjected to the complete nanoPCR procedures (that is, reverse transcription, 40-cycle PCR and MFS). The LOD was calculated to be 3.2 copies per µl based on the standard formula (threshold = 3 × s.d. of a blank sample; Fig. 4f), which was comparable to that of conventional RT–PCR (2.0 copies per µl)41,42. Furthermore, nanoPCR-based N1 and N2 detection distinguished SARS-CoV-2 from other zoonotic coronaviruses, SARS-CoV and Middle East respiratory syndrome coronavirus, with negligible cross-reactivity (Fig. 4g).
To validate nanoPCR’s clinical applicability, we tested clinical samples (that is, nasopharyngeal/oropharyngeal swabs and sputum samples) from cohorts of patients with COVID-19 and control individuals without COVID-19 (see Supplementary Table 2 for patient information). COVID-19 infection status was independently confirmed by the Clinical Diagnostic Laboratory at Chonnam National University Hospital (Republic of Korea). We used the first 100 samples as a discovery cohort (50 patients with COVID-19 and 50 controls) and another 50 samples (25 patients with COVID-19 and 25 controls) as a validation cohort (Fig. 5a). Each patient sample was divided into two. One aliquot was processed using the nanoPCR and the other using a benchtop PCR system. Both assays used the same N1, N2 and RPP30 probes. As a positive reference, we also processed a control sample containing a known amount of reference RNA (synthetic N1 RNA).

a, Clinical study design. A total of 150 samples were analysed by nanoPCR and conventional RT–PCR. The first 100 samples were used as a discovery cohort and the remaining 50 were used as a validation cohort. b, Evaluation of analytical concordance between nanoPCR and RT–qPCR. The results were positively correlated (Pearson’s r values; rN1 = 0.87; rN2 = 0.78; rRPP30 = 0.70). The limit of quantification of both RT–qPCR and nanoPCR was plotted on each axis (broken lines parallel to the axes). Raw intensities from nanoPCR were used. c–e, Analysis of the discovery cohort. c, Control-normalized N1 and N2 signal levels (FN1 and FN2) for 50 patients with COVID-19 and 50 controls. d, F values were significantly higher in the patients with COVID-19 (+) than in controls (−) (****P < 0.0001; two-sided t-test; n = 100). e, ROC curves. The AUC was 1 for both N1 and N2. The F cut-off values for diagnostics were determined from the ROC curves: 0.15 (FN1) and 0.05 (FN2). f, The diagnostic accuracy was further confirmed with the validation cohort (n = 50). Cut-offs (dotted lines) from the discovery set were applied. As in d, F values were significantly higher in the patients with COVID-19 (+) than in controls (−) (****P < 0.0001; two-sided t-test). g, Waterfall distribution of FN1 and FN2 for all of the samples tested (n = 150).
Figure 5b shows the results from nanoPCR and quantitative PCR with reverse transcription (RT–qPCR). We compared raw fluorescence intensity I(nanoPCR) with –log2[Ct], where Ct is the cycle cut-off of RT–qPCR; these two quantities would be proportional to target gene concentrations. We observed a good concordance between these two methods. For each gene target, nanoPCR and RT–qPCR showed a strong positive correlation with the Pearson coefficient (r) values of 0.87 (N1), 0.78 (N2) and 0.70 (RPP30).
Next, we assessed the diagnostic accuracy of nanoPCR. As system-independent analytical measures, we defined FN1 and FN2 by normalizing the fluorescence intensities of the target genes (N1 and N2) with that of the positive reference (Fig. 5c). The FN1 and FN2 values were significantly higher (P < 0.0001 for both; two-sided t-test) in patients with COVID-19 than in controls (Fig. 5d). We further constructed receiver operating characteristic (ROC) curves for FN1 and FN2. The diagnostic accuracy was excellent, with an area under the curve of 1 (Fig. 5e) for both FN1 and FN2. From ROC curves, we determined optimal cut-offs, FTH_N1 = 0.15 for FN1 and FTH_N2 = 0.05 for FN2, that maximized the sum of sensitivity and specificity. When these cut-offs were applied to a separate validation set (25 patients with COVID-19 and 25 controls), nanoPCR maintained its high diagnostic power (Fig. 5f). Overall, rapid nanoPCR diagnostics correctly classified all of the clinical samples (n = 150) tested (Fig. 5g and Supplementary Fig. 14).
Overall, nanoPCR has the potential to decentralize COVID-19 molecular diagnosis. It has practical advantages: (1) the assay is based on well-established RT–PCR to produce more reliable and accurate (>99%) results than isothermal amplification tests17,18; and (2) nanoPCR considerably shortens the PCR reaction time to enable on-site diagnostics. With these advantages, nanoPCR could contribute to the decentralization of COVID-19 tests into mobile, ambulatory clinics, mitigating the logistic burden of sample transports. Our prototype system demonstrated such potential, meeting some of the criteria (for example, a sensitivity of >80%, a specificity of >97% and an assay time of <40 min) set by the World Healthcare Organization43. Also, MPN synthesis can be optimized for mass production where a single batch currently provides ~30,000 PCR reactions (~50 mg). For nanoPCR to be used in clinical settings, we envision further technical developments: (1) establishing MPN quality control and good manufacturing practice for mass production; (2) redesigning fluidic cartridges to minimize hands-on times in RNA extraction44; and (3) exploring new assay methods, such as heat-inactivated lysis45, to offer sample-in and answer-out tests. These efforts will expand nanoPCR’s utility in remote or resource-limited settings46,47. NanoPCR could also be applied for the prompt diagnosis of other infections, such as acquired immunodeficiency syndrome, tuberculosis and hepatitis48,49,50.
Methods
Synthesis of MPNs
The MPNs were synthesized as previously described37. Briefly, the magnetic core (M) was synthesized by nonhydrolytic thermal decomposition of Fe(iii) acetylacetonate and zinc chloride in oleic acid, oleylamine and trioctylamine at 330 °C. After washing the products with ethanol, 1 mg silica-coated magnetic core with an amine functional group (M@SiO2-NH2) was prepared by the sol-gel process of tetraethylorthosilicate and aminopropyltrimethoxysilane. The 2-nm colloidal Au nanoseeds were mixed with M@SiO2-NH2 to obtain an Au seed-coated magnetic core (M@Au = 2 nm) at room temperature for 5 h. To prepare the complete Au shell, the Au seeds were grown in 4.8 l of titrated Au precursors with 17.2 mg hydroxylamine hydrochloride (NH2OH) for 3 d. After separation with centrifugation and a magnetic column, typically the yield was higher than 65%, corresponding to 150 pmoles (~50 mg). The products were dispersed in 1 mg ml−1 bis(p-sulfonatophenyl)phenylphosphine dehydrate dipotassium salt solution for long-term storage.
Transmission electron microscopy (TEM) analysis and EDS mapping
TEM observations were made using an Atomic Resolution Analytical Electron Microscope (JEM-ARM200F; JEOL) at an acceleration voltage of 200 kV. MPNs were freshly prepared on a plasma-cleaned TEM grid (Ultra-Thin PELCO Grids for TEM; Ted Pella). Elemental analysis derived from EDS was performed under atomic resolution mapping followed by visualized indexing.
Plasmonic simulation of MPNs
Electrical fields of MPNs and Au nanoparticles were simulated using the MNPBEM Toolbox in MATLAB (MathWorks)50. The toolbox adopted the boundary element method approach39, solving Maxwell’s equations in metallic nanoparticles wherein homogeneous and isotropic dielectric functions are separated by abrupt interfaces. The simulation assumed that a particle was immersed in water. Wavelength-dependent dielectric functions for Au and magnetite were used.
Spectral analysis of MPNs
Diluted down to an optical density of 0.1 at λ = 535 nm, the aqueous solution of MPNs was subjected to ultraviolet light-aligned spectral analysis using an ultraviolet–visible spectrometer (V-760; Jasco) for absorbance analysis. To obtain scattering measurements for coefficient calculation, the solution was transferred into the integrating sphere (Quanta-phi; HORIBA Scientific) and analysed by photoluminescence spectroscopy (HORIBA Scientific).
Vibrating sample magnetometer measurements
An aqueous solution of MPNs was subject to measurement of magnetization using a vibrating sample magnetometer (VIBRATION 7407-S; Lake Shore Cryotronics). For mass magnetization, the mass of Fe, Zn and Au of each sample was determined using inductively coupled plasma mass spectrometry (7900 ICP-MS; Agilent Technologies).
DLS measurements
The hydrodynamic size of MPNs was measured using a DLS device (Nano ZS90; Malvern) with the following parameters: refractive index = 0.21 and absorption = 3.32.
Preparation of nanoPCR mix
NanoPCR mix (total volume = 10 μl) consisting of 0.2 μM specific primers (Bioneer; see Supplementary Table 3), 20 μM oligo dT, MPNs at an optical density of 4, 200 μM dNTPs (N0447S; New England Biolabs), 10 μM dithiothreitol, 2% dimethyl sulfoxide, 0.22 μM specific TaqMan probe (Integrated DNA Technologies) (or 1× SYBR Safe (Invitrogen) in the non-clinical study), 0.5 μl RNase Inhibitor (LS63; NIPPON Genetics EUROPE), 1 U FastGene Scriptase II (LS63-2; NIPPON Genetics EUROPE), 1 U Hot Start Taq DNA Polymerase (M0495; New England Biolabs), 0.4× Standard Taq Reaction Buffer (containing 10 mM Tris-HCl, 50 mM KCl and 1.5 mM MgCl2 (pH 8.3) at 1× concentration; New England Biolabs) and 0.2× Reverse Transcription Buffer (containing 10 mM Tris-HCl, 50 mM KCl and 0.1% Triton X-100 at 1× concentration (LS63; NIPPON Genetics EUROPE)) was prepared. A certain concentration of synthetic target RNA or viral RNA of clinical samples was applied to the mix, which was then subjected to RT–PCR in the nanoPCR device to automatically give a fluorescence result for the sample.
RNA preparation kit
We engineered a customized RNA-processing kit (20 × 68 × 83 mm3) using a three-dimensional (3D) printer (Form 3; Formlabs). Viral RNAs from collected biofluid samples could be purified via the kit’s silica gel membrane, where RNAs were bound, washed and eluted. Five individual chambers were used, one at a time, to purify the RNAs. The operational procedure of the kit was as follows: (1) transfer of the sample (20 µl) mixed with RNA shield (20 µl) onto the top of the membrane (15 s); (2) application of viral RNA buffer (400 µl guanidinium thiocyanate and acid phenol) for capsid degradation (15 s); (3) pre-washing and RNA immobilization with ion-exchange chromatography resin (1 ml; 2 min); (4) application of viral wash buffer (500 µl; containing ethanol) for debris washing (30 s); and (5) application of elution buffer (15 µl) and transfer to a tube preloaded with the customized nanoPCR reagents and primers (10 s). All of the reagents and buffers were from a commercial RNA extraction kit (R1034; Zymo Research) and were preloaded into the corresponding chambers before RNA processing. Due to the manufacturing shortcoming for mass production, the disposable RNA preparation kit was used sparingly (n = 5) with the sole aim of confirming its capability.
POC device configuration
A 3D-printed plastic housing (150 × 150 × 185 mm3) contained optoelectronic components and served as a dark chamber to exclude external light during the RT–PCR and fluorescence measurements. Three sample tubes (PCR-02-C; Corning) were installed separately onto the customized synchronized Ferris wheel. An array of 14 532-nm lasers (CK1872; CivilLaser) was installed for plasmonic heating, and a 310-nm ultraviolet LED (LED310W; Thorlabs) was added for fluorescence excitation. The tubes formed a trigonal planar arrangement to make an organized rotation with exact timing, and the bottom solution region of the tube in the reaction order was focused by the lasers (RT–PCR) or the ultraviolet LED (fluorescence detection). The lasers were arranged in a radial formation to enable a common focus on one reaction tube among the three tubes. The ultraviolet LED excited FAM dyes in the sample tube. The fluorescence signal was measured by a detector unit consisting of a bandpass filter (D560; Chroma) and a photodiode (S130C; Thorlabs). The incidence angle was about 55°, which prevented direct illumination onto the detector unit. A stepper motor (17HS3430; MotionKing) rotated the wheel at angles of 120.6°, 120.6° and 118.8°, and another stepper motor (28BYJ-48; Elegoo) managed the positioning of 52-N-grade neodymium magnets (10 × 10 × 10 mm3) for MFS. A 3D-printed component holder firmly set the locations of the lasers, the stepper motor and the LED to keep them precisely aligned. A microcontroller board (Arduino Uno R3; Arduino) managed the continuous and sequential operation of the elements, and pulse-width modulation was adopted to control the synchronized actions (Supplementary Fig. 8). The power supply of the nanoPCR device was managed by the PWR button while the nanoPCR procedure was initiated by pushing the PCR button. The fluorescence emission was measured using a sensor read by an optical power console (PM400; Thorlabs). A 12-V Li-ion battery pack supplied the whole system, excluding the console, for over 30 min. All custom-made parts were fabricated using two different 3D printers (3DWOX DP201 (Sindoh) and Form 3 (Formlabs)), considering the manageable resolution and product dimensions.
Clinical study
The nasopharyngeal and oropharyngeal specimens collected in the universal transport medium tubes (Asan Pharmaceutical) and sputum specimens were provided by the Chonnam National University Hospital laboratory (Institutional Review Board number CNUH-2020-106) and processed using an automated nucleic acid extraction system (AdvanSure E3 System; LG Chem) to extract viral RNAs. Nasopharyngeal or oropharyngeal swabs contained in universal transport medium were used as provided, whereas sputum samples were diluted with phosphate buffered saline followed by an extraction process. For consistency of the assay, and to avoid any instrumental deviation, one nanoPCR machine was used to assay all of the clinical samples. The results were then compared with the RT–qPCR results, which were also performed using one machine. The analytical measures (F) for diagnostics were defined as FN1 = IN1/IHK (for N1) and FN2 = IN2/IHK (for N2), where IN1 and IN2 are the raw intensity values for the N1 and N2 tests, respectively, and IHK is the fluorescence intensity of a positive control sample. ROC curves were generated using FN1 and FN2. From the discovery cohort, the optimal F cut-offs for COVID-19 diagnostics were determined by choosing values that maximized the sum of sensitivity and specificity: FTH_N1 = 0.152 (for N1) and FTH_N2 = 0.053 (for N2). The analyses were performed using R (version 4.0.2).
RT–qPCR of the clinical specimens
Viral RNA of the clinical samples was added to the RT–PCR mix composed of 0.4 μM specific primers (IDT), 200 μM dNTP (New England Biolabs), 0.1 μM TaqMan probe (IDT), 1.25 U Hot Start Taq DNA Polymerase (New England Biolabs), 1.25 U FastGene Scriptase (NIPPON Genetics EUROPE), 1× Standard Taq Reaction Buffer (New England Biolabs) and Reverse Transcription Buffer (NIPPON Genetics EUROPE). RT–qPCR was then carried out and monitored on a ViiA 7 Real-Time PCR System (Life Technologies) with the following protocol: 94 °C for 2 min, then 50 cycles of 92 °C for 8 s, 58 °C for 22 s and 72 °C for 40 s. The Ct value was automatically calculated using the system software and was correlated with the result of nanoPCR.
Gel electrophoresis
In 1× TBE (Tris/Borate/EDTA) running buffer, the PCR product solution was resolved on a 3% agarose gel containing 1× SYBR Safe (Invitrogen) as a staining dye at a constant voltage of 120 V for 40 min. A gel image was taken with a ChemiDoc MP Imaging system (Bio-Rad).
Statistical analysis
All of the data are presented as means ± s.d. from experiments repeated at least five times unless otherwise specified. Where appropriate, three times the standard deviation, added onto background values, was used as a threshold value for significance.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data that support the results of this study are available within the paper and its Supplementary Information files. The raw patient data are available from the authors on reasonable request, subject to approval from the Institutional Review Board of the Chonnam National University Hospital. Non-clinical data generated in this study, including source data and the data used to make the figures, are available in the Supplementary Information files.
Change history
17 December 2020
A Correction to this paper has been published: https://doi.org/10.1038/s41551-020-00676-8.
References
Winter, A. K. & Hegde, S. T. The important role of serology for COVID-19 control. Lancet Infect. Dis. 20, 758–759 (2020).
Cheng, M. P. et al. Diagnostic testing for severe acute respiratory syndrome-related coronavirus 2: a narrative review. Ann. Intern. Med. 172, 726–734 (2020).
Weissleder, R., Lee, H., Ko, J. & Pittet, M. J. COVID-19 diagnostics in context. Sci. Transl. Med. 12, eabc1931 (2020).
Wang, Y., Kang, H., Liu, X. & Tong, Z. Combination of RT‐qPCR testing and clinical features for diagnosis of COVID‐19 facilitates management of SARS‐CoV‐2 outbreak. J. Med. Virol. 92, 538–539 (2020).
Wang, C. J., Ng, C. Y. & Brook, R. H. Response to COVID-19 in Taiwan: big data analytics, new technology, and proactive testing. J. Am. Med. Assoc. 323, 1341–1342 (2020).
Salathé, M. et al. COVID-19 epidemic in Switzerland: on the importance of testing, contact tracing and isolation. Swiss Med. Wkly 150, w20225 (2020).
Kilic, T., Weissleder, R. & Lee, H. Molecular and immunological diagnostic tests of COVID-19: current status and challenges. iScience 23, 101406 (2020).
Ferretti, L. et al. Quantifying SARS-CoV-2 transmission suggests epidemic control with digital contact tracing. Science 368, eabb6936 (2020).
Krammer, F. & Simon, V. Serology assays to manage COVID-19. Science 368, 1060–1061 (2020).
Ainsworth, M. et al. Performance characteristics of five immunoassays for SARS-CoV-2: a head-to-head benchmark comparison. Lancet Infect. Dis. https://doi.org/10.1016/S1473-3099(20)30634-4 (2020).
Whitman, J. D. et al. Evaluation of SARS-CoV-2 serology assays reveals a range of test performance. Nat. Biotechnol. 38, 1174–1183 (2020).
Van Kasteren, P. B. et al. Comparison of commercial RT-PCR diagnostic kits for COVID-19. J. Clin. Virol. 128, 104412 (2020).
Jiang, F. et al. Review of the clinical characteristics of coronavirus disease 2019 (COVID-19). J. Gen. Intern. Med. 35, 1545–1549 (2020).
Carter, L. J. et al. Assay techniques and test development for COVID-19 diagnosis. ACS Cent. Sci. 6, 591–605 (2020).
Petralia, S. & Conoci, S. PCR technologies for point of care testing: progress and perspectives. ACS Sens. 2, 876–891 (2017).
Marx, V. PCR heads into the field. Nat. Methods 12, 393–397 (2015).
Mitchell, S. L. & George, K. S. Evaluation of the COVID19 ID NOW EUA assay. J. Clin. Virol. 128, 104429 (2020).
Park, G.-S. et al. Development of reverse transcription loop-mediated isothermal amplification (RT-LAMP) assays targeting SARS-CoV-2. J. Mol. Diagn. 22, 729–735 (2020).
You, M. et al. Ultrafast photonic PCR based on photothermal nanomaterials. Trends Biotechnol. 38, 637–649 (2020).
Son, J. H. et al. Ultrafast photonic PCR. Light Sci. Appl. 4, e280 (2015).
Gandolfi, M. et al. Ultrafast thermo-optical dynamics of plasmonic nanoparticles. J. Phys. Chem. C 122, 8655–8666 (2018).
Lee, J.-H. et al. Plasmonic photothermal gold bipyramid nanoreactors for ultrafast real-time bioassays. J. Am. Chem. Soc. 139, 8054–8057 (2017).
Donner, J. S., Morales-Dalmau, J., Alda, I., Marty, R. & Quidant, R. Fast and transparent adaptive lens based on plasmonic heating. ACS Photonics 2, 355–360 (2015).
Brolo, A. G. Plasmonics for future biosensors. Nat. Photonics 6, 709–713 (2012).
Kumar, A., Kim, S. & Nam, J.-M. Plasmonically engineered nanoprobes for biomedical applications. J. Am. Chem. Soc. 138, 14509–14525 (2016).
Brongersma, M. L., Halas, N. J. & Nordlander, P. Plasmon-induced hot carrier science and technology. Nat. Nanotechnol. 10, 25–34 (2015).
Ndukaife, J. C., Shalaev, V. M. & Boltasseva, A. Plasmonics—turning loss into gain. Science 351, 334–335 (2016).
Bae, K. et al. Flexible thin-film black gold membranes with ultrabroadband plasmonic nanofocusing for efficient solar vapour generation. Nat. Commun. 6, 10103 (2015).
Hogan, N. J. et al. Nanoparticles heat through light localization. Nano Lett. 14, 4640–4645 (2014).
Son, J. H. et al. Rapid optical cavity PCR. Adv. Healthc. Mater. 5, 167–174 (2016).
Lee, Y. et al. Nanoplasmonic on-chip PCR for rapid precision molecular diagnostics. ACS Appl. Mater. Interfaces 12, 12533–12540 (2020).
Vanzha, E. et al. Gold nanoparticle-assisted polymerase chain reaction: effects of surface ligands, nanoparticle shape and material. RSC Adv. 6, 110146–110154 (2016).
Kim, J., Kim, H., Park, J. H. & Jon, S. Gold nanorod-based photo-PCR system for one-step, rapid detection of bacteria. Nanotheranostics 1, 178–185 (2017).
Swierczewska, M., Lee, S. & Chen, X. The design and application of fluorophore–gold nanoparticle activatable probes. Phys. Chem. Chem. Phys. 13, 9929–9941 (2011).
Morozov, V. N., Kolyvanova, M. A., Dement’eva, O. V., Rudoy, V. M. & Kuzmin, V. A. Fluorescence superquenching of SYBR Green I in crowded DNA by gold nanoparticles. J. Lumin. 219, 116898 (2020).
Huang, C. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020).
Kim, J.-w et al. Single-cell mechanogenetics using monovalent magnetoplasmonic nanoparticles. Nat. Protoc. 12, 1871–1889 (2017).
Baffou, G. et al. Photoinduced heating of nanoparticle arrays. ACS Nano 7, 6478–6488 (2013).
De Abajo, F. G. & Howie, A. Retarded field calculation of electron energy loss in inhomogeneous dielectrics. Phys. Rev. B 65, 115418 (2002).
Jain, P. K. & El-Sayed, M. A. Plasmonic coupling in noble metal nanostructures. Chem. Phys. Lett. 487, 153–164 (2010).
Kralik, P. & Ricchi, M. A basic guide to real time PCR in microbial diagnostics: definitions, parameters, and everything. Front. Microbiol. 8, 108 (2017).
Forootan, A. et al. Methods to determine limit of detection and limit of quantification in quantitative real-time PCR (qPCR). Biomol. Detect. Quantif. 12, 1–6 (2017).
COVID-19 Target Product Profiles for Priority Diagnostics to Support Response to the COVID-19 Pandemic v.1.0 R&D Blueprint (World Health Organization, 2020); https://www.who.int/publications/m/item/covid-19-target-product-profiles-for-priority-diagnostics-to-support-response-to-the-covid-19-pandemic-v.0.1
Smyrlaki, I. et al. Massive and rapid COVID-19 testing is feasible by extraction-free SARS-CoV-2 RT-PCR. Nature Comm. 11, 4812 (2020).
Roda, A. et al. Smartphone-based biosensors: a critical review and perspectives. Trends Analyt. Chem. 79, 317–325 (2016).
Liu, D. et al. Trends in miniaturized biosensors for point-of-care testing. Trends Analyt. Chem. 122, 115701 (2020).
Van Schalkwyk, C., Maritz, J., Van Zyl, G. U., Preiser, W. & Welte, A. Pooled PCR testing of dried blood spots for infant HIV diagnosis is cost efficient and accurate. BMC Infect. Dis. 19, 136 (2019).
Devonshire, A. S. et al. The use of digital PCR to improve the application of quantitative molecular diagnostic methods for tuberculosis. BMC Infect. Dis. 16, 366 (2016).
Probert, W. S. & Hacker, J. K. New subgenotyping and consensus real-time reverse transcription-PCR assays for hepatitis A outbreak surveillance. J. Clin. Microbiol. 57, e00500–e00519 (2019).
Waxenegger, J., Trügler, A. & Hohenester, U. Plasmonics simulations with the MNPBEM toolbox: consideration of substrates and layer structures. Comput. Phys. Commun. 193, 138–150 (2015).
Acknowledgements
We thank S.-Y. Cheon for graphic designs and illustrations of figures and POC devices. This work was supported by the Institute for Basic Science (IBS-R026-D1). H.L. was supported in part by US NIH grants R01CA229777, R21DA049577 and U01CA233360, US DOD-W81XWH1910199 and DOD-W81XWH1910194 and the MGH Scholar Fund.
Author information
Authors and Affiliations
Contributions
J. Cheong, H.Y., J.-H.L., H.L. and J. Cheon conceived of and designed the project. J. Cheong and J.-U.L. synthesized the MPNs and performed the material characterizations. H.-J.C. provided the clinical sample. J. Cheong and C.Y.L. conducted the PCR experiments. J. Cheong and H.Y. worked on the nanoPCR design and manufacturing. J. Cheong, H.Y., J.-H.L., H.L. and J. Cheon wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Peer review information Nature Biomedical Engineering thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Supplementary information
Supplementary Information
Supplementary figures, tables and discussion.
Supplementary Video 1
Overall workflow of the nanoPCR device, showing the use of the RNA preparation kit followed by nanoPCR nucleic acid amplification, all within 17 min.
Supplementary Data 1
Raw data for each main figure.
Supplementary Code 1
Source code for the operation of the nanoPCR device.
Supplementary Code 2
Header file for the source code.
Rights and permissions
About this article

Cite this article
Cheong, J., Yu, H., Lee, C.Y. et al. Fast detection of SARS-CoV-2 RNA via the integration of plasmonic thermocycling and fluorescence detection in a portable device. Nat Biomed Eng 4, 1159–1167 (2020). https://doi.org/10.1038/s41551-020-00654-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41551-020-00654-0
This article is cited by
-
Rapid deep learning-assisted predictive diagnostics for point-of-care testing
Nature Communications (2024)
-
Rapid quantitative PCR equipment using photothermal conversion of Au nanoshell
Scientific Reports (2024)
-
Untethered Micro/Nanorobots for Remote Sensing: Toward Intelligent Platform
Nano-Micro Letters (2024)
-
Metallic nanoplatforms for COVID-19 diagnostics: versatile applications in the pandemic and post-pandemic era
Journal of Nanobiotechnology (2023)
-
Single-molecule RNA capture-assisted droplet digital loop-mediated isothermal amplification for ultrasensitive and rapid detection of infectious pathogens
Microsystems & Nanoengineering (2023)
