
Abstract
The development of effective solid adsorbents is necessary to remove organic pollutants from wastewater in response to the global water crisis. The present study investigates the adsorption behavior of dye pollutants on a 1, 3, 6, 8-tetrakis (4formylphenyl) pyrene (TFPPy) for the self-assembly of an innovative three-dimensional (3D) covalent organic frameworks (COFs). Molecular dynamics (MD) and the well-tempered metadynamics simulation were used to evaluate the ability of COFs to adsorb seven types of dye pollutants (DPs). These include methyl orange (MO), gentian violet (GV), brilliant green (BG), tolonium chloride (TC), methylene blue (MB), anionic congo red (CR), and rhodamine B (Rh B). To examine this process, several descriptors are calculated from the simulation trajectories, including interaction energies, root-mean-square deviation, radial distribution function, solvent-accessible surface area, mean square displacement, and the number of hydrogen bonds to figure out the process. After analyzing the data, it was found that the MO/COFs system displayed exceptional stability. It was confirmed by a significant value of −448.54 KJmol−1 for total interaction energy. The density profile of dye molecules in the MO and Rh B-COFs systems exhibits the highest peaks at approximately 325.79, and 26.75 kg/m³, respectively. The free energy values for the DPs/COFs complexes at their global minima reached about, −512.127, −435.603, −381.244, −300.313, and −283.230 kJmol-1 for MO/GV/BG/TC/ and Rh B with the COFs, respectively. The nature of the design of TFFPy-COF’s unique, ultrafast kinetics, high adsorption capacity, and good reusability endow them with great potential for removing dyes in an aqueous environment.
Similar content being viewed by others


Introduction
Organic pollutants in water, especially persistent organic pollutants (POPs)1, have adverse effects on all living organisms due to their high toxicity2, long-term persistence3, high solubility4, and resistant to natural degradation3. One of these pollutants are dyes which are used in industries5 such as dyeing5, paper6, printing7, plastic8, leather9, and pharmaceutical industries10. Dumping the wastes of these industries in freshwater has harmful ecotoxicological effects and causes respiratory problems11, skin irritation12, itching, lung disease11 and increases the risk of cancer in humans13.
Colors consist of two components: color agent and color enhancer14. The dyeing agent imparts color to the water, while the color enhancer increases the solubility of the coloring agent and allows it to adhere to surfaces15. Many dyes are very stable to light and temperature, which slows down their degradation16. This long-term degradation cycle can make the natural degradation of paints challenging over time. Organic dyes such as methyl orange (MO)17, gentian violet (GV)18, cationic brilliant green (BG)19, tolonium chloride (TC)20, methylene blue (MB)21, anionic congo red (CR)22, and rhodamine B (Rh B)23, are common pollutants which widely used in industrials. These dyes are classified as biohazardous substances due to their high toxicity24, long-term persistence25 in the environment, mutation in DNA26, and carcinogenicity27. Among the mentioned dyes, methyl orange and Congo red have N = N groups in their structure that are known as azo dyes28,29,30, methylene blue is a thiazine dye31, and rhodamine B is a fluorescent dye32. Bright green color is used in various fields such as medicine and veterinary medicine33, but it shows high toxicity, especially in contact with skin and eyes, ingestion, and inhalation34. Gentian violet is a synthetic organic compound35 that is used in various industries like textiles36, cosmetics, and medicine. The high pollution of these colors has caused serious concerns for the environment and living organisms. Therefore, it is necessary to prevent their spread and remove them from polluted waters with effective methods37. Various strategies have been investigated for the removal of water pollutants, including adsorption38, membrane processes39, and photocatalytic degradation40. Among the mentioned methods, the adsorption of pollutants using covalent organic frameworks (COFs) has attracted much attention41,42,43,44. In 2005, Yaghi45 and his colleagues discovered COFs that belong to the category of crystalline porous materials and consist of lightweight elements linked by covalent bonds. These frameworks can take on a two-dimensional (2D) or three-dimensional (3D) structure46,47. 3D COFs featuring spatially periodic networks offer notable advantages compared to their 2D counterparts. These advantages include interconnected channels and more extensive exposure to active sites48. Due to their unique properties like the well-defined structure49, tunable pore sizes for the encapsulation of a diverse range of guest molecules50, excellent physical and chemical stability51, ease of functionalization52, thermal stability53, extensive internal surface areas for separation54, and catalytic processes55, show great potential as high-performance membranes for capturing and separating dye pollutants. In recent years, there has been extensive research into the realm of 3D COFs, uncovering their wide-ranging applications across diverse fields48. These include adsorption56, gas storage57, separations58, heterogeneous catalysis58, conductivity59, fluorescence60, chemical sensing61, and battery technology62. The capacity to meticulously engineer COFs with varying pore sizes and enhanced specific surface areas during both design and production underscores their potential. So, their inherent hydrophilic properties make them promising for desalination and extracting contaminants such as pharmaceuticals, dyes, pesticides, and pollutants. Despite the promising potential inherent in structural diversity and high-yielding synthesis, COFs have primarily been studied for their capacity to adsorb dyes from aqueous media48,63,64,65.
The unique characteristics of COFs arise from their layered structure, where covalent bonds connect repetitive units within each layer, and van der Waals interactions play a crucial role in binding these layers together66. Moreover, Attributes like surface charge and hydrophilicity significantly influence the performance of COFs. Ensuring the stability of COF materials when exposed to water is crucial for their effective utilization in water treatment39,67,68. Moreover, it is essential to explore various environmental factors, including reaction time and temperature69, to comprehensively understand the removal mechanism70. The adsorption phenomenon hinges on the interaction between the adsorbent and adsorption, making it crucial to delve into mechanistic details. Understanding these intricacies holds significant importance, as it plays a pivotal role in shaping the foundation for the development of future COFs materials. Therefore, various challenging factors such as designing COFs for aquatic applications present67 (e.g., COF stability in acidic71), modifying COFs’ pore size69, pollutant concentrations72, neutral73, basic environments74, material safety75, expensive building blocks55, recyclability and reusability76, and limited adsorption; thus, researchers should attempt to resolve all of these main challenges and limitations77. Up to now, only a few papers about the application of COF-based materials in environmental have been used and their adsorption mechanism for eliminating contaminants. For instance, Ghahari et al.41,78 designed a 2D covalent organic framework to explore its capacity for capturing and removing environmental pollutants. Also, polyimide-based covalent-organic frameworks through the condensation reaction of tris (4-aminophenyl) amine (TAPA) with pyromellitic dianhydride (PMDA) is synthesized and evaluated their practical application for the removal of MO and Rh B from industrial wastewater by ref. 79 Their results showed that the COFs are good candidates to serve as an efficient adsorbent for the removal of MB with high selectivity in the post-treatment of the dyeing process. Konavarapu et al.80 synthesized three triazene-based imine-functionalized covalent organic frameworks (COFs 1–3) for the removal of organic dye pollutants. Their result showed that the COFs can used as a promising adsorbent for removing anionic and cationic organic dye pollutants from aqueous solutions. In another work, Huo et al.81 prepared the COF with O-linked flexible 2, 4, 6-tris (p-formyl phenoxy)-1, 3, 5-triazine for removing methylene blue from wastewater. Their results illustrate that COF has highly adsorption capacity for stability in aqueous and functions as a robust adsorbent. In a similar vein, Razavi et al.42 investigated the Cu-BTC-metal-organic framework (MOF) to evaluate the interaction energy of dye pollutants with -NH2-MOF, -OH-MOF, and -NO2-MOF to remove Rh B pollutants in the wastewater. From these examples, it can be drawn out that COFs are an ideal platform for dye capture and pore structure modification can efficiently increase dye sorption capacity.
Herein, molecular dynamics (MD) and well-tempered metadynamics simulations are employed for investigating the adsorption affinity of a 3D COF TFPPy in removing seven different dyes contaminants (methyl orange, tolonium chloride, rhodamine b, methylene blue, brilliant green, Gentian violet cat, and congo red) in aquatic ecosystems. Furthermore, the well-tempered metadynamics is applied to analyze the free energy surface (FES) to adsorb pollutants on COF’s structure. Meanwhile, detailed analyses showed that MO/COFs and then GV/COFs systems exhibit the highest free energy, interaction energy, and adsorption affinity towards covalent organic frameworks. Hydrogen bond formation and van der Waals (vdW) forces were identified as the primary adsorption mechanisms. Therefore, the intriguing observation of the high selectivity exhibited by these 3D COFs toward dyes prompted us to delve into the underlying factors contributing to these exceptional performance characteristics.
Result and discussion
MD simulation
In this study, to investigate the adsorption capacity of COFs substrate toward seven different dye pollutants (MO, GV, BG, TC, MB, CR, and Rh B), MD simulations and a series of analyses on their outputs are performed. The analyses can provide detailed insight into the energy of interacting molecules, the spontaneity of the process, the distribution of dye molecules around the adsorbent, and the stability of the seven systems (Table 1): (1) COFs-MO; (2) COFs-GV; (3) COFs-BG; (4) COFs-TC; (5) COFs-MB; (6) COFs-CR and; (7) COFs-Rh B that have been designed and investigated.
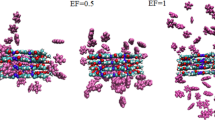
Our results reveal that an increasing number of dye pollutant molecules are adsorbed on the adsorbent surface during the simulation time (see Fig. 1 and Supplementary Fig. 1).

Water molecules are not shown for clarity. a MO/COFs (b) GV/COFs (c) CR/COFs (d) Rh B/COFs. Water molecules are not shown for clarity.
A close inspection of the presented snapshots (Fig. 1) displays that almost all contaminant molecules are adsorbed in the inner cavity or outside the COFs surface. These results reveal that the dye molecules have a strong affinity towards the cavity of the COFs materials and its surface which leads to a strong interaction between the dye pollutants (DPs) and the three adjacent COFs layers. In the overall design of an effective water treatment system, it is crucial to consider both the availability of active sites and the size of the nanopores on the surface. In this regard, the distance between identical monomers in the COFs material is approximately 5.4 Å and 12.90 Å. The pore size of COFs is a crucial factor influencing the adsorption of dye molecules (and dyes sizes are about MB 12.4 Å, GV 12.1 Å, BG 11.6 Å, RH B 12.9 Å, TC 6.4 Å, BG 8.8 Å, MO 12.1 Å, CR 22.1 Å). Therefore, the studied three-dimensional COFs has a high adsorption capacity for pollutants, which can be attributed to their large surface area. To accurately assess the adsorption capacity of the selected COFs, further quantitative evaluations are necessary, which are elaborated upon below.
Interaction energies
To gain a better understanding of the DPs adsorption mechanism on COFs surfaces, the interaction energies are studied. Figure 2 shows the Lennard-Jones (L-J), electrostatic, and total energies for the adsorption process. The comparison among the total energy values of the investigated systems shows that the most interaction energy belongs to the MO/COFs and GV/COFs systems with values of −448.54 and −435.40, respectively.

The interaction energy between dye molecules and COFs.
It’s noteworthy to highlight that MO and GV dyes exhibit the highest affinity for interacting with COFs, underscoring that the adsorption of MO/GV dyes onto the adsorbent surface constrains their mobility. MO, characterized by a common azo-anionic group (-N = N-) linked to two substituted aromatic rings and the neutral form (R-SO3H), shares a distinctive structure82. Additionally, GV, a triphenylmethane dye with a robust aromatic molecular structure, features benzaldehyde or the azo group (R–N = N–R′)83. Consequently, the adsorption of GV/MO fosters OH-π interactions between benzene rings or active sites (–CHO group) on the COFs adsorbent surface. Notably, hydrogen bonding mechanisms, specifically X–H⋯π (X = C, N, O; π = aromatic system) interactions, and the formation of π − π stacking interactions between the benzene ring of MO/GV and COFs enhance molecular rigidity, impeding their movement. It is noteworthy that, the outcomes substantiate that the adsorbent surface is well-suited for the adsorption of these dye molecules, with a robust electrostatic interaction formed between the adsorbent and MO/GV molecules. A close inspection of Fig. 2 indicates that the total interaction energy between COF and DPs have negative values in all studied systems, and they have the following order;
MO/COFs> GV/COFs> BG-COFs> TC-COFs> MB-COFs> CR-COFs> Rh B-COFs. Our findings reveal that the interactions gradually increase, and more dye molecules are adsorbed on the substrate with the passage of time. A detailed examination of the electrostatic and L-J energies shows that the L-J energy plays a crucial role in the interaction and total energy of COFs/dyes systems (see Fig. 2).
As shown in Fig. 2, the MO/COFs and GV/COFs systems had the most average number of adsorbed dye pollutants molecules at the end of the simulation. While the lowest average number of adsorbed drug molecules is related to the Rh B/COFs system. These findings are consistent with the depicted snapshots in Fig. 1.
Stability of systems
By examining the time-dependent root mean square deviation (RMSD) between the DPs and the COF, the stability of the investigated systems can be assessed. The RMSD diagrams demonstrate that all studied systems reached equilibrium after 100 ns (see Fig. 3). As depicted in Fig. 3, the RMSD curves of all systems display regular geometric fluctuations and the morphology of COFs remained stable throughout total simulation time, for all systems. It is noteworthy that the DPs cross a short path of 20 Å to reach a substrate with a large contact surface area and eventually adsorbed on the surface or diffuse within the cavity of the substrate. This feature enhances the stability and removal speed of pollutants in aquatic environments. Accordingly, it can be expected that this 3D COFs acts as an appropriate candidate for DPs removal.

a Mo/COFs; b GV/COFs; c BG/COFs; d TC/COFs; e MB/COFs; f CR/COFs; g Rh B/COFs systems, respectively.
Dispersion of molecules in water
The interaction between molecules and their environment, including solvents, depends on the level of exposure to the solvent. In molecular dynamics simulations using solvent-accessible surface area (SASA) analysis, it is possible to investigate the contact area between different molecules, including MO/GV/BG/TC/MB/CR and Rh B DPs/COFs and the aqueous medium. SASA is an acronym for solvent accessible surface area, which is the surface area of a molecule that is available for interaction with a solvent. In this study, to determine contact areas between DPs and the solvent, SASA analysis is carried out that evaluates the capacity of COFs to offer suitable areas for DPs removal. It is expected that when DPs are less dispersed in water and more adsorbed on COFs, the average SASA value decreases. Upon careful examination of Supplementary Figure 2, it is evident that the hydrophobic SASA of all studied systems decreases over time throughout the simulation. The cause of this phenomenon is due to the rise in π-π interactions between DPs and the aromatic rings of the substrate. This results in a large build-up of DPs around the structure of COFs, leading to a reduction in the area of dye molecules that are accessible to the solvent.
The obtained results show the following trend for the accessible surface of the investigated systems with solvents: MO/COFs< GV/COFs< BGN/COFs< TC/COFs< MB/COFs< CR/COFs< Rh B/COFs.
The average hydrophobic solvent-accessible surface area values for MO/GV/BG/TC/MB/CR and Rh B-COFs are 32.402 nm2, 41.701 nm2, 53.51 nm2, 66.3 nm2, 73.39 nm2, 81.22 nm2, and 106.60 nm2, respectively. The least SASA value belongs to MO/COFs (32.402 nm2), and the highest SASA values observed for the Rh B-COFs (106.60 nm2), in comparison to the other dyes, confirm that the Rh B-COFs system has a larger surface area available for interaction with water molecules. A good agreement was also found between our experimental prediction and theoretical and experimental data by Wang, Khedri, Gao, Pande, and Adeleke et al.84,85,86,87,88.
Hydrogen bonds formation potential
The number of hydrogen bonds (n-HB) for all systems has been indicated in Fig. 4. It is clear that after the adsorption of DPs on the surface of COFs, the number of hydrogen bonds between the DPs and the surface of COFs increases. The active sites of the adsorbent are involved in the formation of hydrogen bonding with the dyes (MO, GV, BG, TC, etc.). According to both theoretical and experimental41,42,73,74 evidence, the presence of these HBs can increase substrate adsorption capacity and system stability. The active sites of the adsorbent are involved in the formation of hydrogen bonding with the dyes. Inspection of Fig. 4 shows the following order for the n-HB between COFs structure and Dyes: MO/COFs> GV/COFs > BGN/COFs> TC/COFs> MB/COFs> CR/COFs> Rh B/COFs. Therefore, the MO-COFs and Rh B-COFs systems have the most and least average number of hydrogen bonds, respectively. Notably, fewer hydrogen bonds in the COFs/Rh B system can be due to more formation of HB between the dye molecules and water. So, it can be concluded that the hydrogen bonding between DPs and the COFs surface can be the driving force for the adsorption of DPs on the surface of the COFs structure. However, our results show that van der Waals interactions have a more significant role than electrostatic interactions in the adsorptiontof the mentioned dye89,90,91,92,93,94,95.

Against the simulation time between COFs and Dye molecules.
Mean square displacement (MSD)
Obtained results can be better presented through the mean squared displacement (MSD) calculations to gain the diffusion coefficient of organic dye pollutants, which can be calculated based on Eq. (1)
Where N is the number of particles to be averaged, xn (t0) is the referenced position of each particle and xn (t) is the specific position of a particle at time.
Furthermore, the self-diffusion coefficient (Di) can be calculated using Einstein’s relation (Eq. 2).
Where d/dt represents the slope of the MSD curve.
As seen in Fig. 5, the MSD curve for the MO/COFs system has a lower slope and less self-diffusion coefficient than the other dye/COFs systems. It’s important to mention that the lower slope in the MSD curves indicates that the adsorption of dye molecules on the adsorbent surface limits the mobility of the dye molecules. This fact can be related to the strong interaction of dye molecules with the active sites of COFs, which is caused by the formation of hydrogen bonds between the dimethyl amino phenyl azo group (C-N = N-C) and the substrate, as well as van der Waals forces, particularly π-π interactions. Also, less Di value of MO molecule in the MO/COFs system can be due to its lower molecular weight, which makes it migrate faster towards the COFs surface. Our findings show that the slope of the MSD curve for the Rh B molecule is more than the other DPs, which is probably due to more mass and strong repulsive interactions between Rh B and COFs. The values of the diffusion coefficient for systems with the most adsorption (MO/COFs) and the minimum adsorption (Rh b/COFs) on the substrate are equal to 0.0099 × 1e-5 and 0.0128 × 1e-5 cm2/s, respectively. These outcomes are confirmed by the RDF diagrams in Fig. 6, which demonstrate the possibility of the presence of Rh b molecules at a further distance from the adsorbent than in other systems. Likewise, the RDF diagram for MO/COFs, which is the most stable complex, shows that the probability of finding MO molecules at the closest distance to the adsorbent is higher than the other systems.

Time evolutions of MSD for the different dyes in the simulated COFs systems.

A between pollutants dye molecules and COFs. a Mo/COFs; b GV/COFs; c BG/COFs; d TC/COFs; e MB/COFs; f CR/COFs; g Rh B/COFs systems, respectively.
Pair distribution functions of Pollutants
In addition, molecular interactions can be examined through the radial distribution functions (RDF) of adsorbate molecules located at a distance ‘r’ from the adsorbent slabs. In this analysis, we investigate the distribution of dye molecules around the COFs structure using the RDF. The RDF, is studied using the following equation:
Where: g(r) represents the radial distribution function, which quantifies the probability of locating a dye molecule at a distance ‘r’ from a reference substrate. The n (r) denotes the number density of molecules at a specific distance ‘r’ from the reference molecule. Δr corresponds to the width of the shell at distance ‘r’, and ρ signifies the total number density of molecules in the investigated system.
Figure 6 presents the RDF analysis for the studied systems, providing critical information about the interaction between dye molecules and substrate. The height of the RDF peaks in the systems follows this order of MO/COFs > GV/COFs > BG/COFs > MB/COFs > CR/COFs > Rh B/COFs.
Intermolecular forces between dye molecules and COF are an influential factor in RDF analysis. The results of RDF analysis show that, after equilibrium, most of the DPs are absorbed. As seen in Fig. 6, the strongest RDF peak is observed for the MO/COFs system, which is located at a distance of 0.48 nm from the surface of the COF. This fact confirms that the MO dye molecule has the most interaction with the COFs surface, which is due to the interaction between the aromatic rings of COFs with the terminal parts and the aromatic rings of MO molecule.
Interestingly, the highest peak of function g(r) for the GV molecule is observed almost 0.29 nm further away compared to the MO molecule. It may be due to the bigger size and more steric effect of the GV molecule, which caused the repulsive force to be stronger. Furthermore, a careful examination of Fig. 6 reveals that as the interaction energy decreases, the intensity of the RDF peaks appears at a further distance. These observations correlate well with the other obtained results in this investigation41,42,96,97.
Density profile
The density profile reflects the probability of water’s presence at a specific location. Considering the density profile (ρ) is essential due to the variation in water concentration among the different systems (where ρ signifies the density of water molecules). However, we have calculated the density profile of dye molecules for MO/GV/BG/TC/MB/CR and Rh B/COFs systems in the z-plane of the structure. The density distribution of dye molecules across the substrate is illustrated in Fig. 7. As depicted in Fig. 7, three distinct oscillating density peaks can be observed for each system, suggesting the presence of three-layer structures within the COFs that confine water molecules. The distribution of water molecules near the substrate exhibits a high degree of symmetry, characterized by multiple peaks that reflect the influence of the COFs structure on the adsorption of DPs. Furthermore, the presence of three peaks within a short range signifies the existence of a local hydrogen bonding structure. Specifically, the first hydrogen bond peak is observed at approximately 0.5 nm for MO/GV systems, while for other designs, it occurs at 1.5 nm, as anticipated.

For dye molecules in the studied systems. a MO-GV, and BG-COFs and (b) TC-MB-CR, and Rh B-COFs systems.
The overall shape of the density profiles is almost similar, but a significant difference in the height of the peaks has been observed, so that the highest peak in MO/GV/BG/TC/MB/CR and Rh B-COFs systems is approximately 325.79, 171.972, 135.106, 133.936, 107.186, 65.71 and 26.75 kg/m3, respectively.
The obtained results show that the MO-COFs system has the highest peak height compared to other systems, which can be attributed to the high tendency of MO molecules to form hydrogen bonds and interact with the substrate. It is noteworthy that the highest peak in the density profile may result from a decrease in the number of water molecules surrounding the substrate’s surface, where the highest concentration of hydrogen bonds and π-π stacking interactions between dye molecules and the structure are present.
Metadynamics
The well-tempered metadynamics simulation is a method that accelerates rare events and estimates the free energy surface (FES) to describe complex molecular systems. In this study, the Parrinello and Laio method, which calculates the FES as a function of distances between the centers of masses (COMs) of two components, is utilized. Metadynamics simulations are used to investigate the free-energy surface for the interaction of dye molecules with COFs surface in MO/GV/BG/TC and Rh B/COFs systems. Figure 8 illustrates the FES as a function of the distance between the COM of the dye molecules and the COFs substrate. As the DPs move toward the COFs, the adsorption processes during the metadynamics simulations for FES have become more negative. These systems encounter energetic boundaries and local minima as they approach their respective global minima. The free energy values for the COFs/PPMs complexes at their global minima are approximately −512.127, −435.603, −381.244, −300.313, and −283.230 kJmol−1 in the MO/COFs, GV/COFs, and BG/ COFs, TC COFs, and Rh B COFs systems, respectively. The free energy results are in complete agreement with the interaction energies presented in Table 2. Additionally, the MO/GV/TC and Rh B dye molecules experience a relative energy barrier of approximately 249.59, 285.51, 1 39.2, and 135.4 kJ mol−1, respectively, when they reach a distance of ~ d = 3.23 nm, 3.97 nm, 2.33 nm, and 1.18 nm at the water-substrate interface. Once this barrier is crossed, the free energy value decreases as the dye molecules approach the outside and inner of the COF’s cavities, which leads to the attainment of the most stable state. The investigated of the free energy surface between COFs and pollutants with the recent computational findings41,42,78,98,99, indicate that this 3D COFs is a good candidate for the removal of pollutants.

For transferring dye molecules (MO/GV/BG/TC and Rh B) towards the COFS materials. a MO/COFs; (b) GV/COFs; (c) BG/COFs; (d) TC/COFs; (e) Rh B/COFs.
In summary, we have demonstrated the successful application of porous COFs structure with accessible cavities to adsorb colored pollutants in water and study their host-guest interactions.
The COF’s negative surface charge makes them attractive for adsorbing dye molecules, allowing us to remove dyes from aqueous mixtures. Molecular dynamics and metadynamics simulations are employed to investigate the adsorption of dye molecules onto or into the covalent organic framework in an aqueous environment. Our results reveal that the dye molecules spontaneously adsorbed on the substrate in all investigated systems. It is worth noting that the adsorption of DPs onto the COFs in MO/COFs is higher compared to other systems. Analysis of interaction energies highlights the crucial role of Lennard-Jones interactions in the adsorption of dye molecule contaminants on the COFs. The total interaction energy values for the examined systems indicate that the MO/COFs and GV/COFs systems exhibit the highest interaction energies, with values of −448.54 and −435.40, respectively. The RMSD diagrams indicate that all the investigated systems achieved equilibrium by the 100 ns mark.
The results from the other analyses, including the number of hydrogen bonds, SASA, mean-squared displacement, and radial distribution function, support the findings from the interaction energy analysis.
Specifically, the MO/COFs system exhibited the highest number of H-B. Moreover, The MO/COFs have the lowest SASA value of 32.402 nm2, while the Rh B-COFs have the highest at 106.60 nm2. This indicates that the Rh B-COFs system has a larger surface area than the other dyes, enabling it to interact with water molecules more efficiently. The MO/COFs system had the lowest diffusion coefficient approximately D = 0.0099 × 10−5 cm2/s. The highest RDF peak for the MO molecule at the closest of the surface is located at a distance of 0.48 nm. Notably, the highest peak in the density profile can be attributed to a reduction in the number of water molecules surrounding the substrate’s surface, particularly in the region where the highest concentration of hydrogen bonds and π-π stacking interactions between dye molecules and the structure occurs. The FES analysis of the studied systems reveals that achieving a global minimum requires DPs to surmount an energy barrier. Our metadynamics simulations, which aim to determine the adsorption behavior, demonstrate that the FES’s global minimum depends on the influences of the active sites on the COF’s surface. The obtained results from free energy are in agreement with the interaction energies. Overall, our results confirm that COFs are a good candidate for the adsorption of DPS.
Methods
MD simulations section
The MD simulation is applied to investigate the potential of COFs as a promising candidate for the elimination of dye-contaminated wastewater. It is worth mentioning that the initial structure of the COFs is taken from the X-ray data, which is reported by Dalapati et al.100, Jin et al.87, Wang et al.86, Yusuf et al.85, Chen et al.83, and Gao et al.’s work82. Dalapati et al.100. explored the chemical stability of the COF across various organic solvents, including hexane, Tetrahydrofuran, MeOH, aqueous HCl (1 M), and NaOH (1 M) solutions, at 25 °C for 24 h. Their findings revealed that the COF samples remained intact under these conditions, with no observable decomposition. Furthermore, they highlighted the preservation of high crystallinity in the COF samples. This exceptional chemical stability in diverse solvents is uncommon, underscoring the robust chemical nature of the monomer linker101. Herein, we utilized the 3D structure based on 1, 3, 6, 8-tetrakis (4 formyl phenyl) pyrene (Fiure1, TFPPy), which is a self-assembly and hydrophilic covalent organic framework to remove dye pollutants from water. The organic monomers form covalent interactions and are arranged through non-covalent bonds like aromatic stacking, van der Waals forces, and hydrogen bonding in the COF structure. As a result, the COF structure exhibits enduring high crystallinity and porosity. These non-covalent bonds between layers can significantly impact bulk properties such as surface area, porosity, electrical conductivity, crystallinity, and stability in the COF, contributing to its overall characteristics101.
In this study, the self-assembly COF system based on TFPPy with three layers is designed for removing dye pollutant molecules including MO, GV, BG, TC, MB, CR, and Rh B in water. In the designed simulation systems, DPs are located around the adjacent COF layers (Fig. 9); this position permits them to interact with four COF layers simultaneously, maximizing the COFs/DPs interaction strength. The schematic representation of the COF and its monomer is shown in Fig. 10. The DPs are initially randomly positioned at least 20 Å away from the framework surface. We observed spontaneous association for dyes within 100 ns of simulation time. A representative frame taken from the MD simulations of DPs is displayed in Fig. 9. The structure of the 3D COFs is designed with three layers using the Gauss View software102. MD simulations are conducted using the GROMACS software package (version 2019.2)103 with the CHARMM force field104. The simulation box has dimensions of 9 × 9 × 10 nm³ and contains COFs at the center, along with 6 dye molecules. The system is dissolved in a 6140 TIP3P water model105, mimicking the neutral medium. Periodic boundary conditions are applied to prevent interactions between components and neighboring cells. The particle mesh Ewald method is employed to manage non-bonded electrostatic and Lennard-Jones interactions within a 1.4 nm cutoff. The temperature and pressure are controlled at 310 K and 1 bar, respectively, utilizing the Nose-Hoover thermostat106 and the Parrinello-Rahman barostat107. The linear constraint solver algorithm108 is employed to maintain the bond lengths at their equilibrium values. Initially, the simulation system is relaxed through energy minimization using the steepest descent algorithm109 to reduce unfavorable interactions. Subsequently, MD simulations were performed for 100 ns using a time step of 1.5 fs. The initial configuration of the systems under investigation is depicted in Fig. 9. The Visual Molecular Dynamics (VMD) package110 is utilized for visualizing the simulated results. It is important to note that, in this study, the impact of different dye molecules on the adsorptions by COFs is considered.

For (COFs/dyes, such as methyl orange (MO), gentian violet (GV), cationic brilliant green (BG), Tolonium chloride (TC), methylene blue (MB), anionic Congo red (CR), and rhodamine B (Rh B)). Color code Carbon: cyan, Hydrogen: gray, Sulfur: yellow, and Nitrogen: blue.

Schematic representation of COFs, COF and TFPPY. Color code: C, cyan; O, red; N, blue; and H, white.
Metadynamics simulations
The well-tempered metadynamics simulation technique111, proposed by Parrinello et al.112 is utilized to identify the free energy surface (FES) with respect to the collective variables (CVs). The metadynamics simulations are performed using the PLUMED plug-in version 2.5.2113, which is integrated into the Gromacs 2019.04 package. In the well-tempered metadynamics algorithm, an initial Gaussian height of 1.0 kJ mol−1 and a width of 0.25 Å are employed. A bias factor of 15 is used, and deposition occurs every 500-time steps. It is important to note that the metadynamics simulations are conducted for 100 ns across all investigated systems. Overall, the metadynamics simulations presented in this study provide a reliable approximation of the free energy landscape for COFs, with the systems ranked as MO > GV > BG > TC > MB > CR > Rh B.
Data availability
Authors can confirm that all relevant data are included in the article and/or its supplementary information files.
References
Naghdi, S. et al. Recent advances in application of metal-organic frameworks (MOFs) as adsorbent and catalyst in removal of persistent organic pollutants (POPs). J. Hazard. Mater. 442, 130127 (2023).
Nagar, N., Saxena, H., Pathak, A., Mishra, A. & Poluri, K. M. A review on structural mechanisms of protein-persistent organic pollutant (POP) interactions. Chemosphere 332, 138877 (2023).
Najam, L. & Alam, T. Occurrence, distribution, and fate of emerging persistent organic pollutants (POPs) in the environment. In Emerging contaminants and plants: interactions, adaptations and remediation technologies 135–161 (Springer, 2023).
Nizzetto, L., Grung, M. & Nøklebye, E. Transport of POPs from source to sea. In Managing Persistent Organic Pollutants in India: Case Studies on Vapi and Surat, Gujarat 63–78 (Springer, 2023).
Aslam, A. A., Irshad, A., Nazir, M. S. & Atif, M. A review on covalent organic frameworks as adsorbents for organic pollutants. J. Clean. Prod. 400, 136737 (2023).
Sriram, G. et al. Recent trends in the application of metal-organic frameworks (MOFs) for the removal of toxic dyes and their removal mechanism-a review. Sustain. Mater. Technol. 31, e00378 (2022).
Castillo-Suárez, L. A., Sierra-Sánchez, A. G., Linares-Hernández, I., & Martínez-Miranda, V. A critical review of textile industry wastewater: green technologies for the removal of indigo dyes. Int. J. Environ. Sci. Technol. 27, 1–38 (2023).
Tubić, A. et al. Sorption potential of microplastics for azo-and phthalocyanine printing dyes. Dye. Pigment. 209, 110884 (2023).
Adeyemo, A. A., Adeoye, I. O. & Bello, O. S. Metal organic frameworks as adsorbents for dye adsorption: overview, prospects and future challenges. Toxicol. Environ. Chem. 94, 1846–1863 (2012).
Adegoke, K. A., Agboola, O. S., Ogunmodede, J., Araoye, A. O. & Bello, O. S. Metal-organic frameworks as adsorbents for sequestering organic pollutants from wastewater. Mater. Chem. Phys. 253, 123246 (2020).
Dey, S., Tripathy, B., Kumar, M. S. & Das, A. P. Ecotoxicological consequences of manganese mining pollutants and their biological remediation. Environ. Chem. Ecotoxicol. 5, 55–61 (2023).
Khan, W. U., Ahmed, S., Dhoble, Y. & Madhav, S. A critical review of hazardous waste generation from textile industries and associated ecological impacts. J. Indian Chem. Soc. 100, 100829 (2022).
Jamshaid, M., Khan, A. A., Ahmed, K. & Saleem, M. Heavy metal in drinking water its effect on human health and its treatment techniques-a review. Int. J. Biosci. 12, 223–240 (2018).
Ramesh, M. & Muthuraman, A. Flavoring and coloring agents: Health risks and potential problems. In Natural and artificial flavoring agents and food dyes 1–28 (Elsevier, 2018).
Chequer, F. M. D. et al. Textile dyes: dyeing process and environmental impact. Eco Friendly Text. Dye. Finish. 6, 151–176 (2013).
Chithambararaj, A., Sanjini, N. S., Velmathi, S. & Bose, A. C. Preparation of h-MoO 3 and $α$-MoO 3 nanocrystals: comparative study on photocatalytic degradation of methylene blue under visible light irradiation. Phys. Chem. Chem. Phys. 15, 14761–14769 (2013).
Serban, G. V. et al. Removal efficiency and adsorption kinetics of methyl orange from wastewater by commercial activated carbon. Sustainability 15, 12939 (2023).
AitAli, S. et al. Application of central composite design and response surface methodology for the study of extraction of gentian violet dye in aqueous solution by polystyrene membrane modified with oleic acid. Environ. Prog. Sustain. Energy. 42, e14200 (2023)
Gul, S. et al. Removal of brilliant green dye from water using ficus benghalensis tree leaves as an efficient biosorbent. Materials (Basel) 16, 521 (2023).
Abedi, M. H., Ahmadmoazzam, M. & Jaafarzadeh, N. Removal of cationic tolonium chloride dye using Fe3O4 nanoparticles modified with sodium dodecyl sulfate. Chem. Biochem. Eng. Q. 32, 205–213 (2018).
Houas, A. et al. Photocatalytic degradation pathway of methylene blue in water. Appl. Catal. B Environ. 31, 145–157 (2001).
Chen, H. & Zhao, J. Adsorption study for removal of Congo red anionic dye using organo-attapulgite. Adsorption 15, 381–389 (2009).
Ren, J. et al. Magnetic Core@ Shell Fe3O4@ Polypyrrole@ sodium dodecyl sulfate composite for enhanced selective removal of dyestuffs and heavy metal ions from complex wastewater. Langmuir 39, 10098–10111 (2023).
Ulson, S. M. et al. Removal of COD and color from hydrolyzed textile azo dye by combined ozonation and biological treatment. J. Hazard. Mater. 179, 35–42 (2010).
Kishor, R. et al. Ecotoxicological and health concerns of persistent coloring pollutants of textile industry wastewater and treatment approaches for environmental safety. J. Environ. Chem. Eng. 9, 105012 (2021).
Roy, U. et al. Dye removal using microbial biosorbents. Green Adsorbents Pollut. Remov. Innov. Mater. 253–280 (2018).
Mathur, N., Bhatnagar, P. & Sharma, P. Review of the mutagenicity of textile dye products. Univers. J. Environ. Res. Technol. 2, (2012).
Hsueh, C.-C. & Chen, B.-Y. Comparative study on reaction selectivity of azo dye decolorization by Pseudomonas luteola. J. Hazard. Mater. 141, 842–849 (2007).
Jia, Y. et al. Performances and mechanism of methyl orange and Congo red adsorbed on the magnetic ion-exchange resin. J. Chem. Eng. Data 65, 725–736 (2020).
Ismail, M. et al. Green synthesis of zerovalent copper nanoparticles for efficient reduction of toxic azo dyes congo red and methyl orange. Green. Process. Synth. 8, 135–143 (2019).
Movsesyan, Z. O., Antonyan, A. P. & Vardevanyan, P. O. Binding mechanism of methylene blue with single-stranded synthetic polynucleotides poly (rA) and poly (rU). J. Appl. Spectrosc. 90, 742–746 (2023).
Han, W. et al. Construction of multilayered boron nitride nanosheets for improving optical performance and stability of rhodamine B. Dye. Pigment. 215, 111244 (2023).
Ahmad, A., Jawaid, M., Ibrahim, M. N. M., Yaqoob, A. A. & Alshammari, M. B. Nanohybrid Materials for Treatment of Textiles Dyes (Springer Nature, 2023).
Jia, Z. et al. QD-based fluorescent nanosensors: production methods, optoelectronic properties, and recent food applications. Compr. Rev. Food Sci. Food Saf. 22, 4644–4669 (2023).
Bhuyan, A. & Ahmaruzzaman, M. Novel Zn (II)-imidazolate polymorph MOF loaded with Mo-QD clusters for solar light-driven H2O2-assisted synergistic photosensitized degradation of gentian violet. J. Alloy. Compd. 950, 169747 (2023).
Sathiyavimal, S. et al. Facile synthesis and characterization of hydroxyapatite from fish bones: Photocatalytic degradation of industrial dyes (crystal violet and Congo red). Prog. Org. Coat. 148, 105890 (2020).
Kordbacheh, F. & Heidari, G. Water pollutants and approaches for their removal. Mater. Chem. Horiz. 2, 139–153 (2023).
Ahmadian, M., Derakhshankhah, H. & Jaymand, M. Recent advances in adsorption of environmental pollutants using metal–organic frameworks-based hydrogels. Int. J. Biol. Macromol. 231, 123333 (2023).
Liu, X. et al. Recent progress of COFs membranes: design, synthesis and application in water treatment. Eco-Environ. Health (2023).
Palas, B. Nanohybrid-based catalysts for degradation of dyes from aqueous solution. In Nanohybrid Materials for Treatment of Textiles Dyes 343–379 (Springer, 2023).
Ghahari, A., Raissi, H., Pasban, S. & Farzad, F. Proposing two-dimensional covalent organic frameworks material for the capture of phenol molecules from wastewaters. Npj Clean Water 5, 28 (2022).
Razavi, L., Raissi, H., Hashemzadeh, H. & Farzad, F. Strategy to improve Cu-BTC metal-organic frameworks performance in removal of Rhodamine B: MD and WT-MtD simulations assessment. NPJ Clean Water 5, 47 (2022).
Gendy, E. A., Oyekunle, D. T., Ifthikar, J., Jawad, A. & Chen, Z. A review on the adsorption mechanism of different organic contaminants by covalent organic framework (COF) from the aquatic environment. Environ. Sci. Pollut. Res. 29, 32566–32593 (2022).
da Silva, V. D. et al. Covalent Organic Frameworks as promising materials for the removal of metal and organic pollutants from water. Mater. Today Sustain. 21, 100279 (2022).
Cote, A. P. et al. Porous, crystalline, covalent organic frameworks. Science 310, 1166–1170 (2005).
Gui, B. et al. Crystallization of dimensional isomers in covalent organic frameworks. J. Am. Chem. Soc. 145, 11276–11281 (2023).
Zhu, D. et al. Three-dimensional covalent organic frameworks with pto and mhq-z topologies based on Tri-and tetratopic linkers. Nat. Commun. 14, 2865 (2023).
Guan, X., Chen, F., Qiu, S. & Fang, Q. Three-dimensional covalent organic frameworks: from synthesis to applications. Angew. Chem. Int. Ed. 62, e202213203 (2023).
Tan, K. T. et al. Covalent organic frameworks. Nat. Rev. Methods Prim. 3, 1 (2023).
Zhang, Y. et al. Harnessing self-repairing and crystallization processes for effective enzyme encapsulation in covalent organic frameworks. J. Am. Chem. Soc. 145, 13469–13475 (2023).
Qiu, J. et al. Porous covalent organic framework based hydrogen-bond nanotrap for the precise recognition and separation of gold. Angew. Chem. 135, e202300459 (2023).
Solanki, S., Prakash Nair, P., Saxena, R. & Singh, R. Recent advances in metal-organic-framework-based composites for efficient sequestration of organophosphorus pesticides (OPPs). ChemistrySelect 8, e202203450 (2023).
Huang, L. et al. Monolithic covalent organic frameworks with hierarchical architecture: attractive platform for contaminant remediation. Chem. Mater. 35, 2661–2682 (2023).
Fan, H. et al. Covalent organic framework–covalent organic framework bilayer membranes for highly selective gas separation. J. Am. Chem. Soc. 140, 10094–10098 (2018).
Cheng, H.-Y. & Wang, T. Covalent organic frameworks in catalytic organic synthesis. Adv. Synth. Catal. 363, 144–193 (2021).
Guo, X. et al. Colyliform crystalline 2D covalent organic frameworks (COFs) with quasi-3D topologies for rapid I2 adsorption. Angew. Chem. Int. Ed. 59, 22697–22705 (2020).
Wu, M.-X. & Yang, Y.-W. Applications of covalent organic frameworks (COFs): From gas storage and separation to drug delivery. Chin. Chem. Lett. 28, 1135–1143 (2017).
Zhi, Y., Wang, Z., Zhang, H.-L. & Zhang, Q. Recent progress in metal-free covalent organic frameworks as heterogeneous catalysts. Small 16, 2001070 (2020).
Bian, G., Yin, J. & Zhu, J. Recent advances on conductive 2D covalent organic frameworks. Small 17, 2006043 (2021).
Ding, H. et al. An AIEgen-based 3D covalent organic framework for white light-emitting diodes. Nat. Commun. 9, 5234 (2018).
Li, W., Ren, P., Zhou, Y., Feng, J. & Ma, Z. Europium (III) functionalized 3D covalent organic framework for quinones adsorption and sensing investigation. J. Hazard. Mater. 388, 121740 (2020).
Wei, C. et al. Covalent organic frameworks and their derivatives for better metal anodes in rechargeable batteries. ACS Nano 15, 12741–12767 (2021).
Zhong, X., Lu, Z., Liang, W., Guo, X., & Hu, B. (2020). The fabrication of 3D hierarchical flower-like δ-MnO 2@ COF nanocomposites for the efficient and ultra-fast removal of UO 2 2+ ions from aqueous solution. Environmental Science: Nano. 7, 3303–3317.
Yu, S. et al. Recent advances in metal-organic framework membranes for water treatment: a review. Sci. Total Environ. 800, 149662 (2021).
Yao, L. et al. Bismuth oxychloride-based materials for the removal of organic pollutants in wastewater. Chemosphere 273, 128576 (2021).
Tang, J., Su, C. & Shao, Z. Covalent organic framework (COF)-based hybrids for electrocatalysis: recent advances and perspectives. Small Methods 5, 2100945 (2021).
Wang, J. & Zhuang, S. Covalent organic frameworks (COFs) for environmental applications. Coord. Chem. Rev. 400, 213046 (2019).
Wu, J. et al. Porous polymers as multifunctional material platforms toward task-specific applications. Adv. Mater. 31, 1802922 (2019).
He, G., Zhang, R. & Jiang, Z. Engineering covalent organic framework membranes. Acc. Mater. Res. 2, 630–643 (2021).
Tang, H. et al. Recent advances in nanoscale zero-valent iron-based materials: characteristics, environmental remediation and challenges. J. Clean. Prod. 319, 128641 (2021).
Wu, C., Wang, X., Zhu, T., Li, P. & Xia, S. Covalent organic frameworks embedded membrane via acetic-acid-catalyzed interfacial polymerization for dyes separation: enhanced permeability and selectivity. Chemosphere 261, 127580 (2020).
Niu, L. et al. One-Step mechanochemical preparation of magnetic covalent organic framework for the degradation of organic pollutants by heterogeneous and homogeneous Fenton-like synergistic reaction. Sep. Purif. Technol. 294, 121145 (2022).
Li, Z., He, T., Gong, Y. & Jiang, D. Covalent organic frameworks: pore design and interface engineering. Acc. Chem. Res. 53, 1672–1685 (2020).
Rasheed, T. Covalent organic frameworks as promising adsorbent paradigm for environmental pollutants from aqueous matrices: perspective and challenges. Sci. Total Environ. 833, 155279 (2022).
Sun, T., Xie, J., Guo, W., Li, D.-S. & Zhang, Q. Covalent–organic frameworks: advanced organic electrode materials for rechargeable batteries. Adv. Energy Mater. 10, 1904199 (2020).
Mallakpour, S., Azadi, E. & Hussain, C. M. Emerging new-generation hybrids based on covalent organic frameworks for industrial applications. N. J. Chem. 45, 7014–7046 (2021).
Wang, Z., Zhang, S., Chen, Y., Zhang, Z. & Ma, S. Covalent organic frameworks for separation applications. Chem. Soc. Rev. 49, 708–735 (2020).
Ghahari, A. & Raissi, H. Design of a hydroxy channel based on the selectivity of water permeation via ions exclusion. https://doi.org/10.1038/s41545-022-00210-0.
Hsu, Y.-C., Sil, M. C., Lin, C.-H. & Chen, C.-M. Modification of covalent organic framework by hydrolysis for efficient and selective removal of organic dye. Appl. Surf. Sci. 612, 155890 (2023).
Konavarapu, S. K. & Biradha, K. Luminescent triazene-based covalent organic frameworks functionalized with imine and azine: N2 and H2 sorption and efficient removal of organic dye pollutants. Cryst. Growth Des. 19, 362–368 (2018).
Huo, J., Luo, B. & Chen, Y. Crystalline covalent organic frameworks from triazine nodes as porous adsorbents for dye pollutants. ACS Omega 4, 22504–22513 (2019).
Pormazar, S. M. & Dalvand, A. Adsorption of Direct Red 23 dye from aqueous solution by means of modified montmorillonite nanoclay as a superadsorbent: Mechanism, kinetic and isotherm studies. Korean J. Chem. Eng. 37, 2192–2201 (2020).
Nayak, A. K. & Pal, A. Enhanced adsorption of gentian violet dye from water using lignocellulosic agricultural waste modified with di- And tri-carboxylic acids: Artificial intelligence modeling, practical comprehension, mechanistic and regeneration analyses. J. Environ. Chem. Eng. 9, 105578 (2021).
Pande, V. et al. Molecular docking and molecular dynamics simulation approaches for evaluation of laccase-mediated biodegradation of various industrial dyes. J. Biomol. Struct. Dyn. 40, 12461–12471 (2022).
Gao, Z., Hu, B., Wang, H., Wang, J. & Cheng, M. Computational insights into the sorption mechanism of environmental contaminants by carbon nanoparticles through molecular dynamics simulation and density functional theory. Phys. Chem. Chem. Phys. 22, 27308–27319 (2020).
Khedri, M. et al. Removal of phenazopyridine as a pharmacological contaminant using nanoporous metal/covalent-organic frameworks (MOF/COF) adsorbent. Appl. Mater. Today 25, 101196 (2021).
Wang, Y., Cui, X., Zhang, P., Wang, Y. & Lu, W. Synthesis of porphyrin porous organic polymers and their application of water pollution treatment: a review. Environ. Technol. Innov. 29, 102972 (2023).
Adeleke, V. T., Adeniyi, A. A. & Lokhat, D. Coagulation of organic pollutants by Moringa oleifera protein molecules: in silico approach. Environ. Sci. Water Res. Technol. 7, 1453–1464 (2021).
Uddin, M. J., Ampiaw, R. E. & Lee, W. Adsorptive removal of dyes from wastewater using a metal-organic framework: a review. Chemosphere 284, 131314 (2021).
Li, Y. et al. Highly stable $β$-ketoenamine-based covalent organic frameworks (COFs): synthesis and optoelectrical applications. Front. Optoelectron. 15, 38 (2022).
Gao, C. et al. Enhancing the iodine adsorption capacity of pyrene-based covalent organic frameworks by regulating the pore environment. Macromol. Rapid Commun. 44, 1–7 (2023).
Chen, Z. et al. Novel one-dimensional covalent organic framework as a H+ fluorescent sensor in acidic aqueous solution. ACS Appl. Mater. Interfaces 13, 1145–1151 (2021).
Yusuf, K. et al. Inverse gas chromatography demonstrates the crystallinity- dependent physicochemical properties of two-dimensional covalent organic framework stationary phases. https://doi.org/10.1021/acs.chemmater.2c03448 (2023).
Wang, Y. et al. Nanoscale 0D/1D Heterojunction of MAPbBr 3 /COF toward efficient LED-driven S–S coupling reactions. ACS Catal. 15493–15504 https://doi.org/10.1021/acscatal.3c03051 (2023).
Jin, E. et al. Designed synthesis of stable light-emitting two-dimensional sp2 carbon-conjugated covalent organic frameworks. Nat. Commun. 9, 1–10 (2018).
Wang, W., Jia, Y., Zhou, S. & Deng, S. Removal of typical PFAS from water by covalent organic frameworks with different pore sizes. J. Hazard. Mater. 460, 132522 (2023).
Mousavi, S. Z. et al. Elucidating the sorption mechanisms of environmental pollutants using molecular simulation. Ind. Eng. Chem. Res. 62, 3373–3393 (2023).
Pasban, S. & Raissi, H. PNIPAM/Hexakis as a thermosensitive drug delivery system for biomedical and pharmaceutical applications. Sci. Rep. 12, 14363 (2022).
Haghi, A., Raissi, H., Hashemzadeh, H. & Farzad, F. Designing a high-performance smart drug delivery system for the synergetic co-absorption of DOX and EGCG on ZIF-8. RSC Adv. 10, 44533–44544 (2020).
Dalapati, S., Jin, S., Gao, J., Xu, Y., Nagai, A., & Jiang, D. (2013). An azine-linked covalent organic framework. J. Am. Chem. Soc. 135, 17310–17313.
Dhanabalan, K. et al. Water splitting catalysts using non-metal covalent organic frameworks: a review. Int. J. Hydrogen. Energy https://doi.org/10.1016/j.ijhydene.2023.08.178 (2023).
Garc\’\ia-Valverde, M., Cordero, N. A. & de la Cal, E. S. GAUSSVIEW®AS A TOOL FOR LEARNING ORGANIC CHEMISTRY. In EDULEARN15 Proceedings 4366–4370 (2015).
Abraham, M. J. et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25 (2015).
Moss, F. R. et al. Platform: molecular dynamics I. https://doi.org/10.1016/j.bpj.2016.11.971 (2017).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Evans, D. J. & Holian, B. L. The nose–hoover thermostat. J. Chem. Phys. 83, 4069–4074 (1985).
Podio-Guidugli, P. On (Andersen–)Parrinello–Rahman molecular dynamics, the related metadynamics, and the use of the Cauchy–Born rule. J. Elast. 100, 145–153 (2010).
Hess, B., Bekker, H., Berendsen, H. J. C. & Fraaije, J. G. E. M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997).
Meza, J. C. Steepest descent. Wiley Interdiscip. Rev. Comput. Stat. 2, 719–722 (2010).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Dama, J. F., Parrinello, M. & Voth, G. A. Well-tempered metadynamics converges asymptotically. Phys. Rev. Lett. 112, 240602 (2014).
Barducci, A., Bussi, G. & Parrinello, M. Well-tempered metadynamics: a smoothly converging and tunable free-energy method. Phys. Rev. Lett. 100, 20603 (2008).
Bonomi, M. et al. PLUMED: a portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 180, 1961–1972 (2009).
Author information
Authors and Affiliations
Contributions
Afsaneh Ghahari: devised the computational protocol and prepared the model systems, performed all calculations, analyzed the data, writing-software, and wrote and edited the original and the revised manuscript, reviewing- editing, edited the original and the revised version of the manuscript. Farzaneh Farzad: supervision. writing-software, reviewing- editing, edited the original and the revised version of the manuscript. Reza Azadnejad: devised the computational protocol and prepared the model systems, performed all calculations, analyzed the data, and wrote and edited the original manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ghahari, A., Farzad, F. & Azadnejad, R. The strategy of three-dimensional Covalent Organic Frameworks to exclude dye contaminants in aqueous solutions. npj Clean Water 7, 27 (2024). https://doi.org/10.1038/s41545-024-00311-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41545-024-00311-y
