
Abstract
Electrodeionization (EDI) is an electrically driven separations technology that employs ion-exchange membranes and resin particles. Deionization occurs under the influence of an applied electric field, facilitating continuous regeneration of the resins and supplementing ionic conductivity. While EDI is commercially used for ultrapure water production, material innovation is required for improving desalination performance and energy efficiency for treating alternative water supplies. This work reports a new class of ion-exchange resin-wafers (RWs) fabricated with ion-conductive binders that exhibit exceptional ionic conductivities—a 3–5-fold improvement over conventional RWs that contain a non-ionic polyethylene binder. Incorporation into an EDI stack (RW-EDI) resulted in an increased desalination rate and reduced energy expenditure compared to the conventional RWs. The water-splitting phenomenon was also investigated in the RW in an external experimental setup in this work. Overall, this work demonstrates that ohmic resistances can be substantially curtailed with ionomer binder RWs at dilute salt concentrations.
Similar content being viewed by others
Introduction
Electrochemical separations, which primarily consist of electrodialysis (ED), electrodeionization (EDI), and membrane capacitive deionization (MCDI/CDI),1 are a subset of technologies primarily used for deionization and other water treatment processes. These technologies offer distinct advantages for desalination over osmotic based technologies (e.g., reverse osmosis) in certain scenarios such as selective ionic sorption2,3,4,5 and deionization of liquid streams with relatively low dissolved ionic species concentrations (e.g., brackish water with less than 5000 mg L−1 6). Despite these advantages, both electrochemical and osmotic based separation technologies will continue to undergo materials research and process development because of the current stresses on global water resources, which are due to climate change and the increasing water, food, and energy demands associated with a growing population.
EDI is a commercial separation technology primarily deployed for ultrapure water production and remediation of industrial process waste streams.7,8 Its modular design and flexible operating parameters (e.g., adjustment of the cell’s operating voltage or current) make it uniquely versatile to carry-out a wide-range of ionic separations for various applications. EDI is similar to ED because both devices utilize the same basic structure consisting of two electrodes that are separated by a stack of alternating liquid compartments, which are partitioned by alternating cation and anion-exchange membranes. Application of an electric field drives the transport of ions towards their respective, oppositely-charged electrode. As a result, charged species are continuously removed from the diluate chambers and transferred into the adjacent concentrate chambers. EDI differs from ED because its traditional design features loosely packed cation and anion-exchange resin (CER and AER) particles in the diluate liquid chamber.9 These resins augment the ionic conductivity of dilute aqueous solutions in the diluate chamber of EDI. By lowering the ohmic resistances in the diluate compartment, the EDI stack is more thermodynamically efficient for removing ions in the more challenging dilute concentration regime.10
A drawback of conventional EDI is the utilization of loose resin beads that foster inconsistent process performance, stack leakage, and disruption of bulk liquid flow. In addition to these challenges, the loose particle bed in EDI requires routine maintenance.11,12,13 Over the past two decades, Argonne National Laboratory12 has addressed some of the challenges associated with EDI by substituting the packed compartment consisting of loose ion-exchange resin particles with a rigid, yet porous, ion-exchange resin wafer (RW) in which the ion-exchange resin particles are immobilized. The RW constitutes a mixture of CER and AER bound by polyethylene (PE)—a thermoplastic polymer. The ion-exchange resin beads supplement ionic conductivity and ion-exchange across the RW, while the PE binder keeps the resin beads stationary. Conventional RWs are about 20–35% porous14 and contain macropores that facilitate bulk liquid flow. Previous work has shown that RW-EDI provides significant advantages over conventional EDI in terms of the rate of removal of ions from liquids, energy efficiency, and process stability and consistency.6,12
To date, little material innovation has occurred for RW materials. The first generation of RW materials incorporated a latex based binder, but this was later replaced by PE, a thermoplastic, that exhibited better ionic separation and efficiencies and required shorter processing time for wafer manufacture.14,15 The ion-exchange resin bead chemistry has remained the same in RWs for desalination applications, and most activities related to manufacturing RW materials has focused on the composition of the RW (i.e., the binder content, porosigen amount, and the ion-exchange resin-particles’ content), and variation of the type of resin particles, which affects ion binding affinities and the ion-exchange capacity (IEC). The porosigen added to the RW during manufacturing serves as a sacrificial component that is leached in the final processing step to yield a porous material.13
Although the RW has a successful track record for augmenting the ionic conductivity of the diluate liquid streams and assisting in ion removal by ion-exchange,12 at the start of this work, it was posited that the presence of the non-conductive binder in the RW limits energy efficiency gains in EDI.14 The non-conductive binder obfuscates pathways for ion-exchange and ion transport between the solution and resin particles leading to larger ohmic drops that compromise EDI energy efficiency. Additionally, it was hypothesized that the non-conductive nature of the PE compromises the population of bipolar junction sites in the RW. These bipolar junctions are formed at the interface where CERs come into contact with AERs. The oppositely-charged tethered ionic moieties at this interface lead to an abrupt p-n type junction that dissociates water under an applied electric field.16,17,18,19,20 Under dilute conditions, the electric field drives water-splitting forming hydronium (H3O+) and hydroxide (OH−) ion carriers that enable electrical current flow in the unit, and these ions may undergo four fates: (i) they participate in ion-exchange with the resin-particles to regenerate the bed; (ii) they migrate to their respective ion-exchange membrane and are transferred into the concentrate stream; (iii) they recombine to form water; and (iv) the ions leave in the effluent stream, in different ratios, leading to a change in pH.21,22,23 Observations for water-splitting in the ion-exchange resin bed in EDI derive from measuring pH changes in the effluent stream and analyzing the device’s current efficiency.22 Water-splitting can also occur at the solution-ion-exchange membrane interface under the application of large voltages in ED resulting in pH changes of the effluent24 and with unequal ratios of fixed cations to fixed anions in the ion-exchange resin bed. From a theoretical perspective, ineffective water-splitting in the ion-exchange resins may hinder deionization and current efficiency of EDI. To recap, it is likely that a PE binder would not only derail the ionic conductivity of the RW but would also jeopardize the population of bipolar junction regions needed for water-splitting.
This paper reports the manufacture and performance of ionomer binder RWs with different configurations: (i) mixed resin with a cation-exchange ionomer (CEI) binder, (ii) anion-exchange resin (AER) only with CEI binder, (iii) mixed resin with an anion-exchange ionomer (AEI) binder, and (iv) cation-exchange resin (CER) only with AEI binder. The new ionomer binder RWs showed an impressive 3- to 5-fold improvement in ionic conductivity in the presence of dilute sodium chloride (NaCl) solution concentrations (≤500 mg L−1). The ionomer binder RWs provided a 25% faster separation in bench-scale EDI, and they also displayed a modest improvement in the energy efficiency (≈ 5%) when removing 99% NaCl from a 5000 mg L−1 NaCl aqueous solution. Finally, water-splitting in RWs was assessed in an external experimental setup. When compared to commercially available bipolar membranes, water-splitting in RWs was substantially less effective because of the absence of a water dissociation catalyst. Overall, the implications of this report highlight two salient points: (i) ionomer-based RWs offer the potential to reduce both the required capital equipment (e.g., size of EDI stack) and energy consumption for a particular electrochemical separation; and (ii) designing new RWs with effective bipolar junctions via the inclusion of a water dissociation catalyst will facilitate water-splitting and provide more thermodynamically efficient ionic separations in EDI desalination, especially for the dilute regime.16
Results and discussion
Figure 1a depicts the polymer binder chemical structures used to fabricate the four new RW materials. Figure 1b shows different configurations and pictures of the ionomer-based RWs. Two configurations of the ionomer binder RWs feature a single type of ion-exchange resin particles (e.g., CER only or AER only) bound by an oppositely-charged ionomer. It was envisaged that these two configurations maximize the number of bipolar junction points in the RW to improve the rate of water-splitting. This is because the mixed RWs with and without ionomer binder have a smaller probability that fixed cationic groups meet fixed anionic groups separated by a small gap on the nanoscale. Kohl and co-workers, using a simplified electrostatics model, report that the depletion width for bipolar junction regions is less than 2.5 nm.25 Hence, gaps between the fixed cationic and anionic groups much larger than 2.5 nm would be ineffective for splitting water. However, this simple model does not reflect a true bipolar junction in bipolar membranes as water dissociation catalysts are needed to effectively split water and these particles can be larger than 2.5 nm.16 The water-splitting performance of RWs will be discussed in more detail in subsequent sections.

a Chemical structures of ionomer polymer binders for RWs: (CEI: sodium sulfonate poly(ether ether ketone) (SPEEK) and AEI: quaternary benzyl n-methyl pyrrolidinium chloride poly(arylene ether sulfone)). b Cartoon representation and photographs of the four new RW ionomer binder materials. c Manufacturing scheme to prepare ionomer binder RWs.
The new manufacturing process used to fabricate ionomer binder RWs is depicted in Fig. 1c. The process commences with ionomer solution (14 weight% in N-methyl-2-pyrrolidone (NMP) solvent) mixed with the ion-exchange resin particles and NaCl as a porosigen. This mixture is poured into a stainless-steel mold that was first treated with a non-stick, cooking oil coating and positioned on a level surface in an oven. Then, the NMP solvent was evaporated overnight at 60 °C. The mold was closed with a stainless-steel top, and the enclosed mold with the ionomer binder RW was hot-pressed at 125 °C and 2 metric ton load for 2 h. Then, the ionomer binder RW was removed from the mold and immersed in 250 mL of deionized water to leach the NaCl leaving behind a porous RW. The new RW was rinsed with copious amounts water to remove residual NaCl and NMP solvent.
The selection of ionomer chemistries shown in Fig. 1a and the manufacturing scheme in Fig. 1c were inspired by known methods used to make membrane electrode assemblies for low temperature fuel cells.26,27 Both the AEI and CEI are linear polymers and are soluble in a variety of aprotic solvents including NMP, N,N-dimethylformamide, N,N-dimethylacetamide, and dimethylsulfoxide. Residual solvent present in the ionomer binders after the initial evaporation step makes them thermally processable for adhering the ionomer to the resin particles. However, the ionomer materials are not water soluble, which is an important requirement for use in RW-EDI. Other ionomer material chemistries, such as sodium sulfonate polystyrene and poly(vinyl benzyl pyridinium chloride-random-vinyl benzyl-4-fluorophenyethylamine), were assessed for fabricating ionomer binder RWs, but the mechanical quality of the RWs was poor upon removal from the mold, or the IEC of the ionomer was too high resulting in the RW falling apart in water (see Supplementary Fig. 1a to 1b). To make mechanically robust ionomer binder RWs, low IEC values of the poly(arylene ether) AEI and CEI (<1.5 mmol g−1) were needed. Typically, these ionomer binder chemistries are closer to 1.5–2 mmol g−1 when used as membranes and electrode binders in fuel cells. However, ionomer binders with these levels of IEC values caused unacceptable swelling of the RW when immersed in aqueous solutions. Hence, low IEC ionomer binders were important to making mechanically robust RWs in both dry and hydrated forms.
Figure 2a reports the ionic conductivity values of the RWs measured at different salt concentrations. Repeat measurements were performed for the mixed resin with PE binder and the mixed resin with CEI binder. The error bars are the absolute difference from the average. The ionic conductivity was measured in a flow-through mode device that encloses the RW in a cell and allows the salt solution to pass through continuously (see Supplementary Fig. 2a for the setup). Because the flow-through mode conductivity measurement is time consuming, high-throughput conductivity measurements were made with a 2-point static conductivity cell (see Supplementary Fig. 2b for the setup). Supplementary Fig. 2c provides the conductivity values for all RW variants (n = 3; standard error reported) and it is clear that the ionomer binders provide higher RW ionic conductivity over the non-conductive PE binder RW. Supplementary Fig. 2d compares the flow-through mode conductivity versus the static mode conductivity. Despite slight difference in absolute values, the trends for conductivity at different solution concentrations of NaCl are in agreement with those observed in the flow-through mode.

Ionic conductivity (κ) of resin wafers (a) at different NaCl concentrations. Error bars (n = 2; absolute difference from the average for the same sample—the error bars are very small) for mixed resin with CEI binder and mixed resin with PE binder. b Ionic conductivity normalized to ion-exchange capacity by weight (κwt*) at different salt concentrations.
Figure 2a and Supplementary Fig. 2c demonstrate that the ionic conductivity for each ionomer binder RW was higher across all salt concentrations when compared to the PE binder RW. Notably, the ionomer binder RWs composed of mixed resin with CEI and AEI binders showed the highest ionic conductivities across the NaCl concentrations. It is important to note that the ionic conductivity values of the NaCl solutions are also provided in Fig. 2a. The ionomer binder RW demonstrated that it can augment the spacer channel’s ionic conductivity up to 8 g L−1 NaCl solutions, while the PE binder RW only improves the ionic conductivity of NaCl solutions up to 3.5 g L−1. In other words, at 4 g L−1 NaCl solution concentration or greater in the spacer channel, the PE binder RW can no longer boost the ionic conductivity. These results emphasize the ionomer binder RWs’ versatility because they can supplement the ionic conductivity in RW-EDI’s diluate or concentrate compartments when the solution concentration is high as 8 g L−1 NaCl.
Figure 2b replots the ionic conductivity data normalized by the IEC of the RW on the basis of RW weight (meq g−1). Supplementary Fig. 2e reports the ionic conductivity data normalized by the IEC of the wafer on the basis of RW volume (meq mL−1). Table 1 reports the RWs’ IEC values per mass and per volume on a dry basis. These values account for fixed charge carrier contributions from both the binder (if applicable) and the ion-exchange resin particles. The normalized conductivity to IEC shown in Fig. 2b and Supplementary Fig. 2e yielded similar trends to those shown in Fig. 2a indicating that the ionomer binder RWs’ ionic conductivities were higher than the benchmark PE binder RW. Notably, a four-fold increase in normalized ionic conductivity was observed in the dilute salt concentration regime of <0.3 g L−1 when compared to PE binder RWs.
The presence of ionic groups in the binder provides more fixed charge carriers to supplement the ionic conductivity of the RW. The Nernst-Planck relationship indicates that ionic conductivity in electrolytes is a linear function of the fixed concentration of charge carriers7,28. Based on the theoretical relationship, it is plausible that the improvement in ionic conductivity might be solely attributed to the addition of fixed charge groups in the ionomer binder. However, the newly formulated RWs require less binder than the benchmark RW with PE binder, and normalizing the ionic conductivity of each RW to the RW’s IEC shows the concentration of fixed charge carriers alone cannot account for the increased ionic conductivity of the ionomer-based RWs. To better understand the ionic conductivity performance differences of the ionomer binder RWs versus PE binder RWs, electron microscopy was leveraged to inspect the RWs’ porous structure and binder distribution.
Figure 3a–e gives cross-sectional (left) and surface (right) SEM images of PE binder RW (benchmark) and ionomer binder RWs. The resin beads observed in each micrograph vary from 300 to 500 μm in size. Figure 3a corresponds to the PE binder RW, and the images show PE enveloping the surface of the ion-exchange resin particles with a relatively thick layer of PE and poor ion-exchange resin particle to ion-exchange resin particle contact. The large surface coverage with a thick PE binder hinders liquid solution contact with the ion-exchange resins, resulting in less ion-exchange and fewer pathways for ion transport from particle to particle. Figure 3b–e show distinctly different distributions of binder and particle confinement within the ionomer binder RWs when compared to the PE binder RW. From these images, the ionomer binder in each RW sample is thinner and more evenly distributed to provide better adhesion between the ion-exchange resin particles. Furthermore, the ionomer binder seems to cover particles’ surfaces less when compared to the PE binder RW. The ionomer binder RW structures also feature notably large, porous gaps that facilitate bulk liquid flow. This is important because the ionomer binder and exposed resin particles are capable of ion-exchange with the liquid solution.

SEM images of RWs (left: cross-section and right: surface) at 500 μm scale a mixed resin with PE binder. b mixed resin with CEI binder. c mixed resin with AEI binder. d AER with CEI binder. e CER with AEI binder.
In addition to Table 1 providing the IEC values of the RWs, the Table also gives the different RWs’ porosity values (i.e., free-liquid void space divided by wafer volume, Eq. 3). Two of the ionomer binder RWs, the mixed resin with CEI binder and CER with AEI binder, provided comparable porosity values to the benchmark PE binder RW (e.g., 23.8% and 24.3% versus 26.5%). The RWs consisting of mixed resin with AEI binder and the AER with CEI binder yielded lower porosity values—18.6% and 13.0%, respectively. It is important to mention that RW-EDI demonstrations were carried out with RWs composed of AER with CEI binder and mixed resin with CEI binder (these results are presented in the next section). The RW with AER and CEI binder had the smallest porosity value but still operated effectively in a RW-EDI bench-scale unit and with better performance than the benchmark PE binder RW. The RW-EDI with RWs consisting of mixed resin with a CEI binder exhibited similar performance to the separation run with RWs of AER with CEI binder. These results suggest that the RWs operate effectively in RW-EDI units with porosity values in the range 13–24%.
Electron microscopy and porosity measurements, in conjunction with RW-EDI demonstrations, revealed that the new ionomer binder RWs contained macropores and adequate porosity for bulk liquid flow. The first takeaway from these results substantiates the flexibility of the new manufacturing process to produce mechanically intact, conductive, and porous RWs. More importantly, the electron micrographs validate that the ionomer binder acts as a thinner adhesive between ion-exchange resin beads in the RW network when compared to PE binder. As an ion conducting adhesive, the ionomer binder enables facile ion transport from particle to particle in addition to greater uptake of ions from the liquid. These are key factors that govern the effectiveness of the delivery of ions to the membrane surfaces, which is necessary for the ultimate removal of ions from the diluate chamber.
The superior ionic conductivity of the ionomer binder RWs, in addition to their adequate porosity, motivated bench-scale RW-EDI studies to remove 99% NaCl (fed at 5000 mg L−1) from solution. This feed concentration was selected because it represents the upper end of the brackish water concentration regime with a model solution. The bench-scale unit featured four pairs of anion and cation-exchange membranes, and four diluate and concentrate compartments. Thus, the bench-scale EDI unit utilized four RWs for each EDI run with the different RW materials. The bench-scale RW-EDI was operated under constant voltage and with recirculation of the diluate and concentrate streams. Figure 4a reports the concentration of Cl− in the concentrate solution and the diluate solution versus time. The bench-scale RW-EDI unit was run continuously under recirculation until 99% removal of NaCl (measured as Cl−) occurred from the diluate solution. Both ionomer binder RWs, mixed resin with CEI binder and AER with CEI binder, resulted in a 25% faster removal of NaCl from the diluate solution when compared against the RW-EDI run with the benchmark wafer.

The EDI demonstrations were carried out in batch mode under recirculation until 99% of NaCl was removed from the diluate compartment. a Concentration of concentrate and diluate streams versus time. b Removal productivity of NaCl (i.e., ion removal flux) vs removal ratio. c Energy consumption for Cl− removal. Two EDI runs were performed with each RW material. The average result is given in each plot for each RW material, and the error bars represent the difference between the average and one of the collected data points for the same RW sample.
The faster removal of ions from the diluate stream with the ionomer binder RW was ascribed to the materials’ ability to promote a higher rate of ion removal flux over the range of NaCl concentrations in the diluate chamber. Figure 4b plots the removal productivity (the ion removal flux) from the diluate compartment versus the removal ratio (see Eq. 1). As the removal ratio increases, the amount of dissolved salt in the RW decreases resulting in greater ohmic resistance for the dilute chamber. The larger resistance hinders the ion removal flux from the diluate chamber, and thus, at constant applied voltage, the amount of electrical current that can be passed through the RW-EDI stack is lowered. Under the same operation conditions of applied voltages and feed flow rates, the removal productivity for the ionomer binder RWs as shown in Fig. 4b is 25% or more higher than the PE binder RW regardless of the removal ratio. The higher removal productivities are attributed to the ionomer binder RWs’ higher ionic conductivity values.
Figure 4c presents the energy use (kWh) per kg of Cl removed for the RW-EDI runs performed with different RWs along with their respective removal productivities attained at 99% removal. The ionomer RWs provided up to 4.3% reduction in energy consumption during the RW-EDI run in comparison to the benchmark RW. Although the ionomer binder RWs exhibited substantially better ionic conductivity and ion removal rates in RW-EDI, their improvement to energy consumption was marginal but still laudable. The energy consumption could be reduced to a greater extent by operating the RW-EDI unit using optimal conditions with a modulated applied electric field (i.e., constant current), cation/anion-exchange capacity ratio in RW and the feed flow rate. However, a constant current process would yield roughly the same time for clearing 99% of the NaCl from diluate stream (assuming similar charge efficiency values for each EDI run with the different materials). Each RW would likely require different optimal operating conditions for EDI, and in order to provide comparative data, the operation was not optimized in favor of any RW sample. Under optimal operating conditions (i.e., applied electric field, flow rate, etc.), RW-EDI has been reported to achieve <0.66 kWh/m3 for 90% removal of 5000 mg L−1 NaCl,12 whereas the desalination with a conventional RW in this study corresponded to an average energy consumption of approximately three times greater. From a process economics viewpoint, energy consumption for RW-EDI is directly related to the operating cost, whereas a RW-EDI unit capable of faster removal of ions from the dilute solution translates to lower capital costs (due to a smaller unit for deionization) and operational costs (primarily caused by using less ion-exchange membranes and RWs).12 Note that the pumping energy requirements for feed and concentrate streams are minimal in comparison to electrical energy consumption (see Supplementary table 2). The results in Fig. 4a–c demonstrate that ionomer binder RWs display faster ionic separations, while co-currently using less energy, making them extremely advantageous for RW-EDI processes.
Figure 5 shows the current-voltage (I–V) response (i.e., polarization curves) of RWs and bipolar membranes in a homemade 4-point cell used to assess water-splitting kinetics in bipolar membranes. The inset in Fig. 5 illustrates the low current response of RW materials and a bipolar membrane without a water dissociation catalyst. The bipolar membranes were assessed as a control against the RWs, and they include a commercial variant (Fumatech) and homemade variants with and without water dissociation catalysts. The homemade bipolar membranes consisted of a SPEEK cation-exchange membrane (CEM) adjoined to a quaternary benzyl n-methyl pyrrolidinium poly(2,6-dimethyl 1,4-phenylene oxide) (QAPPO) anion-exchange membrane (AEM). One homemade bipolar membrane contained a water dissociation catalyst (aluminum hydroxide (Al(OH)3) nanoparticles) and another did not contain a water dissociation catalyst. The RW materials assessed include the benchmark material (mixed resin with PE binder), CER with AEI binder, and the AER with CEI binder. Supplementary Fig. 3a-c shows the experimental setup and configuration of RWs, which was sandwiched between a CEM and an AEM, for testing. For the Fumatech and SPEEK-QAPPO bipolar membrane with a water dissociation catalyst, the onset potential drop for current flow was observed at 0.8 V indicating relatively facile water dissociation into H+ and OH− ion carriers in the bipolar junction of the membranes (note: the thermodynamic potential to split water in a bipolar junctions is 0.83 V18). However, the SPEEK-QAPPO bipolar membrane without a water dissociation catalyst and all RWs displayed onset potentials greater than 1 V and substantially lower current responses. These values indicate that the bipolar junctions within these materials have large overpotentials for water dissociation and a smaller population of bipolar junction regions (i.e., fewer sites to dissociate water which is needed for amplifying the current response).

The bipolar membranes tested include Fumatech (commercially available) and SPEEK-QAPPO with and without water dissociation catalysts. RWs evaluated include mixed ion-exchange resins with PE binder (benchmark material), CEI binder with AER, and an AEI binder with CER.
EDI processes can run continuously and without the need for chemicals for ion-exchange bed regeneration because oppositely-charged ion-exchange resin particles adjacent to each other and distributed throughout the bed can dissociate water into H+ and OH− charge carriers.21,22 While processing the diluate stream in EDI, very dilute conditions near the exit of the unit favor water dissociation to provide the ionic charge carriers and maintain EDI current flow. Once the H+ and OH− counterions are formed, a subset of them can exchange onto their oppositely-charged resins. The regenerated bed can then remove remaining salt ions from the interstitial solution through ion-exchange22 or by facilitating ion migration through the ion-exchange membranes. The reports for detecting water-splitting in mixed ion-exchange beds for EDI typically rely on monitoring the pH of the effluent stream and current efficiency of EDI under very dilute conditions. Herein, an external methodology was adopted in this report to assess water-splitting in RW materials. Because the ion-exchange particles are immobilized in RWs, these materials were conducive for assessment in a 4-point cell that is traditionally used for assessing water-splitting in bipolar membranes.16
Figure 5 shows water-splitting of AER with CEI binder RW that is 20-fold lower in current response at 2 V than bipolar membranes with a water dissociation catalyst (Fumatech) and 4-fold lower current response than a bipolar membrane without a water dissociation catalyst (SPEEK-QAPPO bipolar membrane without catalyst) at that same voltage. The previously described RWs fabricated with ionomer binder were hypothesized to facilitate better water-splitting over benchmark RWs by increasing the population of bipolar junction sites throughout the RW bed (see Supplementary Fig. 4). In order to test this hypothesis, RWs composed of a CEI binder with only AER and an AEI binder with CER were formulated. The current response for the RW with a CEI binder and AER was marginally better than the mixed RW with PE binder (see Supplementary Fig. 5), but it was significantly lower than the response observed with bipolar membranes featuring a water dissociation catalyst. In fact, it was more similar to a bipolar membrane without a water dissociation catalyst.17 The improved current response for the CEI binder AER RW over the benchmark mixed RW with PE binder was attributed to larger population of bipolar junctions throughout the RW sample. The Supplementary notes provides the basic continuum model to support this. The higher ionic conductivity and better water-splitting kinetics of the CEI binder AER RW over the benchmark material explained why this material demonstrated an almost 25% faster separation with a 4.3% reduction in energy usage for 99% removal. The insights attained from the results presented in Fig. 5 motivate our future studies to incorporate water dissociation catalysts in RWs and to maximize populations of bipolar junctions. Some examples include poly(acrylic acid), graphene oxide, or poly(vinyl pyridine) or metal oxides/hydroxide).29,30
Finally, it should be noted that pH changes were detected in the diluate stream under recirculation during RW-EDI runs (Supplementary Fig. 6) and the greatest pH change was observed for the RW composed of AER with CEI binder. The larger pH change observed during the run suggested that this RW material was most effective for water-splitting in EDI. This observation is in agreement with the water-splitting measurement using the external 4-pt measurement which indicated greater water splitting for the AER with CEI binder RW in comparison to the mixed resin with PE binder (Supplementary Fig. 5). However, it should be mentioned that unequal ratios of fixed anion-exchange and cation-exchange sites in the RW (see Supplementary table 1 for the amount of fixed anion-exchange and cation-exchange sites in RW samples) may also cause pH changes in the diluate chamber.31
Ionomer binder RWs were developed for EDI as a replacement for benchmark RWs, which are fabricated with a non-conductive PE binder. The ionomer binder RWs displayed superior ionic conductivity (3–5x improvement), while maintaining adequate porosity, resulting in faster removal of ions from aqueous streams with greater energy efficiency in RW-EDI demonstrations. Electron micrographs revealed that the ionomer binders were a thinner and better distributed adhesive throughout the RW bed to immobilize ion-exchange resin particles. The better binder distribution facilitated greater ion exchange between the liquid and resin particles and delivery of ions to the membranes. The water-splitting characteristics of these RWs were assessed in an external setup, and it was determined that all RW materials split water ×20 less effectively in comparison to bipolar membranes that contain water dissociation catalysts. Notably, the ionomer binder RWs’ ability to split water was on the same order of magnitude to bipolar membranes not featuring a water dissociation catalyst. These observations motivate future efforts to develop RW materials with water dissociation catalysts strategically placed in bipolar junction regions of RWs.
Methods
Materials
Commercially available cation-exchange resins (Purolite, PFC100E (IEC = 1.9 eq L−1), density = 1.27 g cm−3) and anion-exchanges resins (Purolite, PFA400 (IEC = 1.3 eq L−1), density = 1.07 g cm−3) were used in the resin wafer (RW) preparation. The cation-exchange resins consisted of sulfonated sodium polystyrene crosslinked with divinylbenzene. The anion-exchange resins were composed of quaternary benzyl trimethylammonium chloride polystyrene crosslinked with divinylbenzene. The polymer poly(ether ether ketone) (PEEK) was obtained from VICTREX. Udel® polysulfone pellets (PSf) were attained from Acros Organics, and the polymer had an average molecular weight of 60,000 g mol−1. Other chemicals, such as 97% sulfuric acid (H2SO4), sodium chloride (NaCl), 99.8% chloroform (CH3Cl), 95% paraformaldehyde, 98% chlorotrimethylsilane, 99% stannic chloride (SnCl4), 98% N-methyl pyrrolidine, 99% N-methyl pyrrolidone (NMP), 99.96% deuterated dimethylsulfoxide (d6-DMSO), and 99.6% deuterated chloroform (CDCl3), were attained from VWR or Sigma-Aldrich and used as is. Deionized water (DI H2O, 18.2 MΩ, <20 ppb TOC) was produced at the time of a particular synthesis or experiment with a Milli-Q Millipore Elix 10. Commercially available cation, anion, and bipolar membranes were used for the RW-EDI stack measurements (at Argonne National Laboratory) and were obtained from Ameridia (Neosepta CMX, AMX, and BP; ASTOM Corporation, Tokyo, Japan).
Poly(arylene ether) ionomer synthesis procedures
SPEEK ionomer binder: PEEK was sulfonated based on our previous work.1 In short, PEEK was dissolved in concentrated sulfuric acid (H2SO4) at room temperature. The degree of sulfonation (DS) in PEEK was monitored by assaying the reactor periodically throughout the reaction. After an aliquot was removed from the reactor, the sample was precipitated in DI H2O and rinsed excessively in DI H2O. After drying in a fume hood, the sample was dissolved in d6-DMSO and analyzed via 1H NMR (see Supplementary Fig. 7a and 7b for synthesis of SPEEK and 1H NMR spectrum with assignments). This procedure was repeated two or three times until the desired DS value of SPEEK was obtained. After the desired DS value was achieved, the remainder of the SPEEK in sulfuric acid batch was precipitated in excess DI H2O and rinsed excessively with DI H2O to neutralize excess H2SO4. The precipitated solid was collected by filtration and then dried in a fume hood overnight followed by vacuum drying at room temperature for 3 h. In this report, a DS of 0.4 was found suitable for making conductive but mechanically robust RWs. A too high of a DS value (i.e., >0.5) resulted in excess swelling of the RW. The ionomer solution used to prepare the resin wafer was prepared by dissolving SPEEK in NMP to make a 14 wt% solution.
QAPSf ionomer: Udel® PSF was chloromethylated following the procedure by Arges et al.32 In short, Udel® PSF was dissolved in CHCl3 at room temperature to prepare a 2 wt% solution in a round bottom flask with equipped with a magnetic stir bar. Paraformaldehyde and chlorotrimethylsilane (5:5:1 molar ratio to PSf repeat unit) was added to the flask. The flask was sealed with a rubber septum and the silicon oil bath containing the flask was heated to 55 °C. Then, SnCl4 (Lewis acid catalyst), a 2 wt% ratio to PSf added, was added slowly by syringe through the rubber septum. To monitor the degree of chloromethylation (DC) of PSf, samples were assayed from the flask over time. After withdrawing an aliquot from the flask, the chloromethylated polysulfone (CMPSf) solution was precipitated in methanol (5:1 volume ratio) and then vacuum filtered. The degree of chloromethylation (DC) of the batch obtained was 0.88 (see Supplementary Fig. 8a to 8c for QAPSf synthesis and 1H NMR spectra of CMPSF and QAPSf). Any CMPSf batch used above 0.9 DF yielded a QAPSf material that resulted in a swollen resin wafer with poor mechanical integrity.
To prepare the QAPSf solution used to make RWs, chloromethylated polysulfone in (NMP) was dissolved in NMP to make a 14 wt% solution. 1-methyl pyrrolidine was then added to the solution in the ratio of 2:1 to the amount of chloromethyl groups per repeat unit in CMPSf. The conversion of chloromethyl groups to quaternary benzyl N-methylpyrollidinum chloride groups was carried out for 24 h at 60 °C. The ionomer solution was then cooled to room temperature and stored until use in the manufacture of the RW.
Conventional resin-wafer (RW) synthesis
Synthesis of the conventional RW (i.e., Argonne’s benchmark material) is done by the addition of cation-exchange resins and anion-exchange resins in the ratio 1:1.3, to which polyethylene binder and sodium chloride are added to yield a mix ratio by mass of 2.0:1.0:0.5 of resins to binder to salt. The resulting mixture is packed to a resin mold. The typical dimensions of the mold for this work was 14 × 14 × 0.3 cm (other mold dimensions are possible). The mold is hot-pressed around 100–115 °C and with 2 metric tonnes of force for 30 min.
RW with cation-exchange ionomer (CEI) binder
A resin mixture is prepared by adding cation-exchange resins and anion-exchange resins in the ratio 1:1.3 and 13.8 wt% solution of SPEEK in NMP and sodium chloride in the mass ratio of 2.4:2:1 of resins to binder to salt. The resulting mixture is packed into a mold and dried in the oven at 60 °C for 12 h to remove residual solvent. It is then hot-pressed at 125 °C for 1.5 h with 2 metric tonnes of force. The same procedure was followed for the resin-wafer consisting of anion-exchange resin particles only with the CEI binder. Here, the mixture was prepared in the ratio of 2.4:2:1 of resins particles to CEI binder to salt.
RW with anion-exchange ionomer (AEI) binder
A resin mixture is prepared by adding cation-exchange resins and anion-exchange resins in the ratio 1:1.3 and 14 wt% solution of QAPSf in NMP and sodium chloride in the mass ratio of 2.4:2:1 of resins to binder to salt. The resulting mixture is packed into a mold and dried in the oven at 60 °C for 12 h to remove residual solvent. It is then hot-pressed at 150 ˚C for 1.5 h at 2 metric tonnes. The same procedure was followed for the resin-wafer only featuring cation-exchange resin particles with the AEI binder. This mixture is prepared with the ratio of 2.4:2:1 of resin particles to AEI binder to salt.
Ionic conductivity
Flow-through conductivity tests were performed using the 4-point probe method and an LCR meter (IM3533–01, Hioki USA, Cranbury, NJ). Wafers were cut into 1 × 1 in. squares, inserted into gaskets, and installed in custom-built flow-through cells consisting of stainless-steel electrodes as shown in Supplementary Fig. 2a. Five cells were arranged in series to perform measurement of five wafers simultaneously. Prior to each measurement, wafers were equilibrated in NaCl solution until steady state was achieved. Conductivity was measured as impedance using a frequency of 100 kHz and a voltage of 50 mV. The NaCl solution measurement was conducted using an identical apparatus and procedure excluding the wafer material. After all measurements were performed, the wafers were rinsed thoroughly with ultrapure water. Without removing the wafer from the test cell, duplicate measurements were performed for the mixed resin with CEI binder and mixed resin with PE binder at each NaCl concentration using freshly prepared NaCl solution. The electrical resistance across the water was continuously monitored with two electrodes during continuous flow until a steady resistance value was achieved. Equation 2 was used to determine the samples’ ionic conductivity under a flow-through setting.
Static conductivity tests were performed using a 2-point probe method (see Supplementary Fig. 2b) The cell consisted of platinum foil working electrodes adhered with silver epoxy onto stainless-steel collectors in a polytetrafluorethylene (PTFE) housing. A stainless-steel screw was used for making contact between the electrodes and the RW samples. The RWs were immersed and equilibrated in NaCl solution for 5 min (note: this caused the stainless-steel current collectors to also be immersed). Conductivity was measured using galvanostatic electrochemical impedance spectroscopy with 1 mA perturbation and in the frequency range of 100 kHz to 1 Hz. The high frequency resistance from the Nyquist plot was used in Eq. 2. The NaCl solution measurement was conducted using an identical apparatus and procedure excluding the wafer material.
where κ denotes the ionic conductivity of the wafer, t denotes the wafer thickness, A denotes the surface area, and R is the measured resistance value.
Porosity
A blue dextran solution (Sigma–Aldrich D5751) of 5 g L−1 concentration was prepared, and the concentration recorded as Cinitial was made by recording the UV-Vis absorbance of the solution at 620 nm and using a calibration curve that relates absorbance at 620 nm to concentration of blue dextran. The resin-wafer sample is then immersed into DI H2O to hydrate the ion-exchange materials. Then, the wafer was immersed in the 5 g L−1 blue dextran solution. After the immersion for a period of time, the excess blue dextran solution residing on the wafer surface was removed by blott drying using a Kimwipe. The adsorbed blue dextran from the wafer was purged from the resin-wafer using DI H2O, and the total volume of water used is recorded as Vfinal. The blue dextran concentration of the rinse solution was recorded as Cfinal by measuring the UV-Vis absorbance at 620 nm and using a calibration curve that relates absorbance at 620 nm to concentration of blue dextran. The free-liquid-void-space (FLVS), or also known as pore volume and the porosity (P), are estimated using the following Eqs. (3–4):
where w is the width, l is the length, and h is the height of the wafer.
IEC calculation
The theoretical IECs of the resin wafers were computed by averaging the IEC values of each of the components by weight and volume (see Eqs. 5 and 6).
where IECCER, IECAER, IECbinder denote the ion-exchange capacities of the cation-exchange, anion-exchange resins and binder, respectively, in milliequivalents per gram. mCER, mAER and mbinder, mTotal denote the weight of the cation-exchange, anion-exchange resins, binder and total weight of the wafer, respectively. xpolymer denotes the weight ratio of polymer in the ionomer binder used and VTotal denotes the total volume of the wafer, which is measured as the product of the wafer area and wafer thickness. \(\phi\) denotes the wafer porosity. Salt weight is not considered in these calculations as it is leached out by immersing the wafer in DI water.
SEM images
Cross-sectional and surface images of the resin wafer under were imaged under vacuum with a QuantaTM 3D Dual beam focused-ion beam scanning electron microscope instrument operated at 5 kV with field emission gun. The working distance ranged from 6 to 13 mm. For imaging the cross-section of resin-wafers, the samples had liquid nitrogen poured over them followed by cutting the sample immediately across the thickness. For the PE binder RWs only, a thin layer of 0.5 nm palladium-platinum was sputtered on the samples to enhance the contrast during imaging.
RW-EDI stack runs
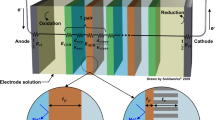
RW-EDI experiments were conducted using a homemade electrodialysis stack (see Supplementary Fig. 9) consisting of a stainless-steel cathode and dimensionally-stable anode (DSA). Ion-exchange membranes (active area = 14 mm2) were arranged in an alternating pattern as shown in Supplementary Fig. 9 to create feed/diluate compartments (~3.0 mm thick) containing resin wafers and concentrate compartments (~1.0 mm thick) for a total of four cell pairs. Experiments were conducted in batch mode using an initial concentration of 5 g L−1 NaCl for both the feed and concentrate solutions, a feed flow rate of 25 mL min−1, a concentrate flow rate of 50 mL min−1, and cell voltage of ~1 V/cell pair.
The energy consumption for salt removal in kWh per kg of salt removed during the RW-EDI demonstration was calculated by Eq. 7:
where V is the constant voltage applied per cell pair in V, I denotes the current in A, which is integrated over the charging time, t is the time, m denotes the mass of salt removed in kg, and 2.78 × 10–7 is the unit conversion factor from Joules to kWh.
Ion chromatography analysis
Chloride ion concentrations were measured with ion chromatography (882 Compact IC plus; Metrohm, Riverview, FL) equipped with chemical and CO2 suppression systems. Analyses were performed with Metrosep A Supp 5 150/4.0 analytical and guard columns, 3.2 mM Na2CO3/1.0 mM NaHCO3 as the eluent, a flow rate of 0.7 mL min−1, and 20 μL sample loop and injection volumes.
Water splitting in bipolar junctions
Water splitting in bipolar junction interfaces of the resin wafers and bipolar membranes was assessed using a home-built two compartment, 4-point electrochemical cell setup (see Supplementary Fig. 3). The cell consists of two Pt/Ir working electrode meshes, one in each compartment, and Ag/AgCl reference electrodes with Luggin capillaries intimately pressed against the membrane interfaces (in one instance a bipolar membrane and in another instance RW materials). The potential drop is measured across the RW bipolar membrane samples. The active area for the cell was 1.27 cm2, and the concentration of supporting electrolytes in each compartment was aqueous 0.5 M Na2SO4 electrolyte. Linear sweep voltammetry (LSV) was used to assess the current-voltage relationships of the samples in the 4-point cell. The LSV scan was carried out from 0.0 to 2.0 V at a 5 mV s−1 scan rate.
Data availability
All raw data used in this manuscript can be obtained for free by contacting the corresponding authors.
Change history
12 February 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41545-020-0066-6
References
Palakkal, V. M., Rubio, J. E., Lin, Y. J. & Arges, C. G. Low-resistant ion-exchange membranes for energy efficient membrane capacitive deionization. ACS Sustain. Chem. Eng. 6, 13778–13786 (2018).
Kim, Y.-J., Kim, J.-H. & Choi, J.-H. Selective removal of nitrate ions by controlling the applied current in membrane capacitive deionization (MCDI). J. Membr. Sci. 429, 52–57 (2013).
Lee, D.-H. et al. Selective lithium recovery from aqueous solution using a modified membrane capacitive deionization system. Hydrometallurgy 173, 283–288 (2017).
Tang, W., Kovalsky, P., Cao, B., He, D. & Waite, T. D. Fluoride removal from brackish groundwaters by constant current capacitive deionization (CDI). Environ. Sci. Technol. 50, 10570–10579 (2016).
Lado, J. J. et al. Removal of nitrate by asymmetric capacitive deionization. Sep. Purif. Technol. 183, 145–152 (2017).
Pan, S.-Y., Snyder, S. W., Ma, H.-W., Lin, Y. J. & Chiang, P.-C. Energy-efficient resin wafer electrodeionization for impaired water reclamation. J. Clean. Prod. 174, 1464–1474 (2018).
Strathmann, H. Ion-Exchange Membrane Separation Processes. (Elsevier, 2004).
Arar, Ö., Yüksel, Ü., Kabay, N. & Yüksel, M. Various applications of electrodeionization (EDI) method for water treatment—A short review. Desalination 342, 16–22 (2014).
Alvarado, L. & Chen, A. Electrodeionization: principles, strategies and applications. Electrochim. Acta 132, 583–597 (2014).
Mahmoud, A., Muhr, L., Grévillot, G., Valentin, G. & Lapicque, F. Ohmic drops in the ion-exchange bed of cationic electrodeionisation cells. J. Appl Electrochem 36, 277–285 (2006).
Zheng, X.-Y., Pan, S.-Y., Tseng, P.-C., Zheng, H.-L. & Chiang, P.-C. Optimization of resin wafer electrodeionization for brackish water desalination. Sep. Purif. Technol. 194, 346–354 (2018).
Pan, S.-Y., Snyder, S. W., Ma, H.-W., Lin, Y. J. & Chiang, P.-C. Development of a resin wafer electrodeionization process for impaired water desalination with high energy efficiency and productivity. ACS Sustain. Chem. Eng. 5, 2942–2948 (2017).
Ho, T., Kurup, A., Davis, T. & Hestekin, J. Wafer chemistry and properties for ion removal by wafer enhanced electrodeionization. Sep. Sci. Technol. 45, 433–446 (2010).
Lin, Y. J., Henry, M. P. & Snyder, S. W. Electronically and ionically conductive porous material and method for manufacture of resin wafers therefrom. US Patent, 7452920 (2008).
Arora, M. B., Hestekin, J. A., Lin, Y. J., Martin, E. J. S. & Snyder, S. W. Porous solid ion exchange wafer for immobilizing biomolecules. US Patent, 7306934 (2007).
Shen, C., Wycisk, R. & Pintauro, P. N. High performance electrospun bipolar membrane with a 3D junction. Energy Environ. Sci. 10, 1435–1442 (2017).
Yan, Z. et al. The balance of electric field and interfacial catalysis in promoting water dissociation in bipolar membranes. Energy Environ. Sci. 11, 2235–2245 (2018).
Unlü, M., Zhou, J. & Kohl, P. A. Hybrid polymer electrolyte fuel cells: alkaline electrodes with proton conducting membrane. Angew. Chem. Int. Ed. Engl. 49, 1299–1301 (2010).
Bauer, B., Gerner, F. J. & Strathmann, H. Development of bipolar membranes. Desalination 68, 279–292 (1988).
Strathmann, H., Rapp, H.-J., Bauer, B. & Bell, C. M. Theoretical and practical aspects of preparing bipolar membranes. Desalination 90, 303–323 (1993).
Meng, H., Peng, C., Song, S. & Deng, D. Electro-regeneration mechanism of ion-exchange resins in electrodeionization. Surf. Rev. Lett. 11, 599–605 (2004).
Lu, J., Wang, Y.-X. & Zhu, J. Numerical simulation of the electrodeionization (EDI) process accounting for water dissociation. Electrochim. Acta 55, 2673–2686 (2010).
Mani, K. N. Electrodialysis water splitting technology. J. Membr. Sci. 58, 117–138 (1991).
Nikonenko, V. V. et al. Desalination at overlimiting currents: state-of-the-art and perspectives. Desalination 342, 85–106 (2014).
Ünlü, M., Zhou, J. & Kohl, P. A. Hybrid anion and proton exchange membrane fuel cells. J. Phys. Chem. C. 113, 11416–11423 (2009).
Arges, C. G. et al. Quarternary ammonium and phosphonium based anion exchange membranes for alkaline fuel cells. ECS Trans. 33, 1903–1913 (2010).
Arges, C. G. & Zhang, L. Anion exchange membranes’ evolution toward high hydroxide ion conductivity and alkaline resiliency. ACS Appl. Energy Mater. 1, 2991–3012 (2018).
Sata, T. Ion Exchange Membranes. https://doi.org/10.1039/9781847551177 (2004).
Xu, T., Yang, W. & He, B. Water dissociation phenomena in a bipolar membrane. Sc. China Ser. B-Chem. 42, 589–598 (1999).
McDonald, M. B. & Freund, M. S. Graphene oxide as a water dissociation catalyst in the bipolar membrane interfacial layer. ACS Appl. Mater. Interfaces 6, 13790–13797 (2014).
Iurash, C. A., Nikonenko, V. V., Pismenskaya, N. D., Zabolotsky, V. I. & Volodina, E. I. Dependence of salt and water ion fluxes through ion-exchange membranes under electrodialysis on the ion-exchange bed composition. Desalination 124, 105–113 (1999).
Arges, G. C., Parrondo J., Johnson G., Nadhan A. & Ramani V. Assessing the influence of different cation chemistries on ionic conductivity and alkaline stability of anion exchange membranes. J. Mater. Chem. 22, 3733–3744 (2012).
Acknowledgements
The submitted manuscript has been created jointly by UChicago Argonne, LLC, Operator of Argonne National Laboratory (“Argonne”). Argonne, a US Department of Energy Office of Science laboratory, is operated under contract no. DE-AC02-06CH11357. Technical data collection of membrane separations was financially sponsored by the U.S. Department of Energy’s (DOE’s) Bioenergy Technologies Office (BETO). The setup for water-splitting measurements in bipolar membranes and RWs came from NSF Award #1703307 (PI Arges).
Author information
Authors and Affiliations
Contributions
V.M.P., L.V. (co-first author), C.G.A. and Y.J.L. designed research; V.M.P., L.V., Q.L. and S.K. performed research; V.M.P., L.V., C.G.A. and Y.J.L. analyzed data; and V.M.P., L.V., C.G.A. and Y.J.L. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Palakkal, V.M., Valentino, L., Lei, Q. et al. Advancing electrodeionization with conductive ionomer binders that immobilize ion-exchange resin particles into porous wafer substrates. npj Clean Water 3, 5 (2020). https://doi.org/10.1038/s41545-020-0052-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41545-020-0052-z
