
Abstract
Due to the high mutation and recombination rates of the influenza virus, current clinically licensed influenza vaccines and anti-influenza drugs provide limited protection against the emerging influenza virus epidemic. Therefore, universal influenza vaccines with high efficacy are urgently needed to ensure human safety and health. Passive immunization of influenza broadly neutralizing antibodies may become an ideal option for controlling influenza infection. CR9114 isolated from the peripheral blood mononuclear cells of healthy donors is a broadly neutralizing monoclonal antibody that targets different types of influenza viruses. As the adenovirus vector is one of the most promising delivery vehicles, we employed the chimpanzee adenoviral vector, AdC68, to express CR9114 as a universal anti-influenza vaccine, termed AdC68-CR9114, and evaluated its antibody expression and its broad spectrum of prophylactic and therapeutic effects in animal models. Based on our findings, AdC68-CR9114-infected cell expressed the broadly neutralizing antibody at a high level in vitro and in vivo, exhibited biological functions, and protected mice from different types of influenza virus infection at different time points. The findings from this study shed light on a new strategy for controlling and preventing influenza infection.
Similar content being viewed by others

Introduction
The influenza virus continues to serve as a severe threat to human health. It causes serious disease and even death among susceptible populations, as well as direct and indirect economic losses. According to the World Health Organization (WHO), seasonal influenza virus causes approximately 5 million infections and 250,000–500,000 deaths worldwide every year1. The WHO published the 2019–2030 Global Influenza Strategy (GIS)2 with the objectives of reducing the burden of seasonal influenza, minimizing the risk of zoonotic influenza, and preparing for the next influenza pandemic in all countries. The GIS has stressed the importance of influenza vaccines, drugs, and their shortages, thereby highlighting the urgent need to diversify and optimize current production capabilities and technologies, and seek new vaccines that offer stronger, broader, more long-lasting protection, and can be more rapidly produced.
Traditional influenza vaccines have been well-studied. Further, they have had a long history of use; however, their efficacy is poor in individuals older than 65 years. The effectiveness of the influenza vaccine is approximately 52% during the seasons when the vaccine and circulating viruses are well-matched3. If a significant mutation occurs in the circulating influenza virus, the efficacy of the vaccine is reduced to 36% (https://www.cdc.gov/flu/vaccines-work/effectivenessqa.htm). In addition, traditional vaccine cannot protect humans from a novel outbreak of the influenza virus, such as the H7N9 that occurred in 20134.
With the progress of the monoclonal antibody (mAb) approach, an increasing number of antibodies has been passively used in the prevention and control of infectious diseases5. For example, the delivery of mAbs with potent neutralizing activity against a broad range of isolates is a promising approach for the prevention and treatment of HIV infection6. The passive transfer of the anti-HIV antibody to a patient was found to significantly reduce the HIV titer7. Palivizumab, a newly approved anti-RSV mAb, has been routinely used as prophylaxis against respiratory syncytial virus8. To achieve viral infection control, anti-rabies immunoglobulins are usually administered to patients exposed to the rabies virus9,10. However, human immunoglobulins are rare and expensive, while equine rabies-immune globulin continues to induce some side effects9. Thus, the development of broadly neutralizing antibodies against the rabies virus may be a superior alternative11. Recently, the anti-influenza broadly neutralizing antibodies found in infected humans have provided new insights into universal influenza vaccine development12. In fact, researchers have attempted several methods to construct hemagglutinin stalk-based vaccines to activate broadly neutralizing antibodies, with minor effects achieved13. However, passive immunization of broadly neutralizing antibodies might be the most effective countermeasure for influenza infection.
C17914 is the first isolated influenza broadly neutralizing antibody that can neutralize all H1 and H2 strains. In addition, it can cross-neutralize the H5, H6, and H9 strains15. Compared with other broadly neutralizing mAbs that cross-neutralize several different subtypes of influenza A virus, such as CR6261, F10, 12D1, CR8020, and FI615, CR911416 is one of the most broadly neutralizing antibodies that has been identified to date. CR9114 binds to a conserved epitope in the HA stem and protects against lethal challenge from influenza A or B viruses. As a result, some broadly neutralizing antibodies against influenza have been used in clinical trials. The administration of the anti-HA antibody (VIS410)17 and anti-M2e antibody (TCN-032)18 significantly reduced the influenza virus titer in subjects. VIS410, a broadly neutralizing antibody engineered to bind the influenza A virus, has completed Ph2a studies in experimentally infected subjects19. MedImmune’s MEDI885220 and Genentech’s MHAA4549A have progressed to trials for naturally infected subjects19. New advances in biotech are also paving the way for more cost-effective immunoprophylaxis using broadly neutralizing antibodies19.
Because of the instability and high cost of the generic immunoglobulins, their administration to most patients is impractical. Hence, using a viral vector to express antibodies would be an ideal alternative. Adenoviral vectors have been widely used in vaccine development owing to their broad cell tropism, high gene expression, high immunogenicity, and mature production technology21. This commonly used adenoviral vector is human serotype 5 (AdHu5), a common-cold virus circulating in humans with a seropositive rate of 40–60%22,23. To circumvent the downside of pre-existing immunity to AdHu5, rare serotypes of human adenovirus or those derived from other species, such as chimpanzee, have been developed and applied in vaccine development. Chimpanzee adenoviruses, which rarely circulate in humans, have been engineered to carry different antigens and demonstrate remarkable safety and immunogenicity in clinical studies24. In this study, a chimpanzee-origin adenovirus vector with E1/E3 deletion, AdC68, was selected to express the broadly influenza-neutralizing antibody, CR9114, as a universal influenza vaccine, termed AdC68–CR9114, to offer both prophylactic and therapeutic protection against influenza infection.
Results
Expression of CR9114 in vitro and in vivo
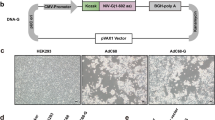
To assess the expression of CR9114 in vitro, Western blotting was performed. Different quantities of AdC68–CR9114 (108, 109, or 1010 virus particles (vp)) were used to infect HEK293 cells in a 6-well plate. AdC68–empty (1010 vp) was used as the negative control, while uninfected HEK293 cells were used as the blank controls. After 24 h, the cell supernatant was analyzed by reducing and nonreducing Western blotting with a horseradish peroxidase (HRP)-conjugated anti-human IgG antibody. As shown in Fig. 1a, the full-length antibody, CR9114, appeared at approximately 160 kDa in the nonreducing condition, but was resolved into the heavy chain (55 kD) and light chain (25 kD) under reducing conditions. For further validation, the CR9114 antibody was purified from the supernatant of infected HEK293 cells using a protein A column and subjected to reducing sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) with Coomassie Blue R-250 staining. The heavy and light chains are clearly presented in Fig. 1b. Sandwich ELISA was also performed to measure the concentration of CR9114 in the supernatant of infected HEK293 cells. In the cell supernatant, the concentration of the CR9114 antibody was up to 163 µg/ml at 24 h after 1010 vp AdC68–CR9114 infection. At 48 h, its concentration increased to 483 µg/ml (Fig. 1c). Therefore, AdC68–CR9114-infected cells efficiently expressed the CR9114 antibody in vitro.

To measure the CR9114 antibody level in vitro, different quantities of AdC68–CR9114 (1010, 109, or 108 virus particles (vp)) were used to infect the HEK293 cells in a 6-well plate. AdC68-gp (1010 vp) was used as the negative control, while the uninfected HEK293 cells were used as the blank controls. Supernatants were harvested at 24 and 48 h after infection. For the in vivo expression of the CR9114 antibody, mice were randomly divided into four groups (5 mice per group), and intranasally immunized with AdC68–CR9114 or AdC68–empty at 5 × 1010 vp/mouse in 30 μl of PBS, or intravenously administered AdC68–CR9114 or AdC68–empty at 5 × 1010 vp/mouse in 200 μl of PBS. All mice were subjected to bleeding every other day. a Western blot detection of the CR9114 antibody in the supernatant of infected HEK293 cells under nonreducing and reducing conditions. b The purified CR9114 antibody was boiled and subjected to SDS-PAGE with Coomassie Blue R-250 staining. c, d The concentrations of CR9114 in vitro and in vivo (five mice per group). Data in c, d are expressed as mean ± SD.
The serum samples of C57BL/6 mice immunized intravenously or intranasally with AdC68–CR9114 were collected every other day to determine the expression of CR9114 in vivo. The antibody was approximately 11,535 μg/ml at 24 h after intravenous immunization and increased as time progressed. Further, its concentration peaked on day 5 (21,284.7 µg/ml), and decreased to 8 μg/ml on day 13. Compared with the intravenously immunized group, the level of the CR9114 antibody in the intranasally immunized group was lower in the serum as it could not be detected at 24 h post immunization; however, it peaked on day 3 (7751.7 µg/ml) and declined to undetectable levels on day 7 post infection (Fig. 1d).
AdC68–CR9114 prophylactically or therapeutically protects mice from pH1N1 and influenza B virus
We used different immunization regimens to determine whether AdC68–CR9114 could prophylactically or therapeutically protect mice from the influenza infection. For the prophylactic experiments, C57BL/6 mice were intravenously administered 5 × 1010 vp AdC68–CR9114 virus and challenged with 10LD50 of the pH1N1 (A/California/7/2009) virus after 24 h. In addition, body weight and survival rate were monitored for 14 days. As shown in Fig. 2a, b, mice in the AdC68–CR9114 group rarely experienced any weight loss. In addition, they had a survival rate of 100%. Conversely, weight loss in the AdC68–empty group sharply decreased to 30% on day 11 post challenge; no mice were found to survive. Intranasal immunization was performed to evaluate the prophylactic efficacy of AdC68–CR9114 too. Briefly, C57BL/6 mice were intranasally treated with 5 × 1010 vp AdC68–CR9114 virus and challenged with 10LD50 pH1N1 virus after 24 h. As shown in Fig. 2c, d, mice in the control groups died on day 6 post challenge with the pH1N1 virus, whereas mice in the AdC68–CR9114 group showed limited weight loss and recovered with a survival rate of 100%. By comparing the intravenous with the intranasal regimen, the former was identified to be preferred for the prophylactic experiments.

For the prophylactic experiments, mice were divided into different groups and immunized with AdC68–CR9114 or AdC68–empty (5 × 1010 vp/mouse) via tail intravenous or intranasal immunization. Mice were challenged intranasally with 10LD50 of the pH1N1 influenza virus. Body weight and survival rate were monitored for 2 weeks. For the therapeutic experiments, mice were infected with 10LD50 of the pH1N1 virus, and intravenously or intranasally treated with 5 × 1010 vp AdC68–CR9114 or AdC68–empty virus at 6 h following the influenza challenge. a, b Weight loss and survival rate in the prophylactic group treated intravenously and challenged with pH1N1. c, d Weight loss and survival rate in the prophylactic group treated intranasally and challenged with pH1N1. e, f Weight loss and survival rate in the therapeutic groups treated intravenously with AdC68–CR9114 and challenged with pH1N1. g, h Weight loss and survival rate in the therapeutic group treated intranasally with AdC68–CR9114 and challenged with pH1N1. Five mice in each group. Data in a, c, e, g are expressed as mean ± SD.
For the therapeutic experiments, mice were first infected with 10LD50 pH1N1 virus, and intravenously or intranasally administered 5 × 1010 vp AdC68–CR9114 or AdC68–empty virus 6 h post influenza challenge. As shown in Fig. 2e, f, when intravenously injected, mice in the control group experienced serious weight loss and died on day 6 post challenge, and mice in the AdC68–CR9114 group had a sharp weight loss, with only 40% of mice surviving. In the intranasal immunization experiments, mice in the AdC68–CR9114 group did not experience weight loss and had a survival rate of 100% (Fig. 2g, h). Thus, we selected intranasal treatment for therapeutic use.
The prophylactic and therapeutic protection efficacy of AdC68–CR9114 against influenza B virus was evaluated. As shown in Fig. 3a–d, mice immunized intravenously for prophylactic purpose or treated intranasally with AdC68–CR9114 for therapeutic purpose survived from the influenza B (Yamagata lineage, B/phuket/3037/2013) challenge, whereas those in the control groups showed serious weight loss and mortality. Such findings demonstrate that AdC68–CR9114 protected mice from the influenza A and B infections, prophylactically and therapeutically.

Groups of mice were immunized with AdC68–CR9114 or AdC68–empty (5 × 1010 vp/mouse) via tail intravenous (for prophylactic purpose) or intranasal (for therapeutic purpose) immunization. Thereafter, they were challenged intranasally with the influenza B virus. Body weight and survival rate were monitored for 2 weeks. a, b Weight loss and survival rate in the prophylactic group treated intravenously and challenged with the influenza B virus. c, d Weight loss and survival rate in the therapeutic group treated intranasally and challenged with the influenza B virus. Five mice in each group. Data in a, c are expressed as mean ± SD.
AdC68–CR9114 prophylactically protects mice from the different subtypes of the influenza virus infection
To evaluate the potential of AdC68–CR9114 as a universal influenza vaccine for prophylactic use, we conducted a series of challenge studies with different subtypes of the influenza A viruses. Mice were intravenously injected with 5 × 1010 vp AdC68–CR9114 or 5 × 1010 vp AdC68–empty and challenged with 10LD50 of the different subtypes of influenza virus, covering PR8 (A/Puerto Rico/8/34), H3N1 (A/Archi/1/1968), H5N1 (A/environment/Hunan/6-69/2008), and H9N2 (A/chicken/Jiangsu/7/2002) after 24 h. The PR8 virus is the most widely used virus for influenza research, including influenza vaccine development25 and host-virus interaction26. H9N2 and H5N1 are two avian influenza viruses that belong to group 1, and H3N1 was selected to represent the group 2 virus. Body weight and the survival rate were monitored for 14 days. As shown in Fig. 4a–h, AdC68–CR9114 conferred full protection against all influenza viruses used in the study. However, no mice in the AdC68–empty group survived the challenge with any of the influenza virus strain.

Mice were intravenously administered 5 × 1010 vp AdC68–CR9114 or 5 × 1010 vp AdC68–empty, and 24 h post administration, they were challenged with 10LD50 of the different subtypes of influenza virus, PR8 (A/Puerto Rico/8/34), H3N1 (A/Archi/1/1968), H5N1 (A/environment/Hunan/6-69/2008), and H9N2 (A/chicken/Jiangsu/7/2002). Body weight and survival rate were monitored for 14 days. a, c, e, g Body weight loss after challenge with PR8, H3N1, H5N1, and H9N2, respectively. Data are expressed as mean ± SD. b, d, f, h Survival rate after challenge with PR8, H3N1, H5N1, and H9N2, respectively. Five mice in each group.
Viral loads and the pathological changes in the lungs after pH1N1 virus challenge
To further evaluate the protective effect of AdC68–CR9114 against the influenza virus, two groups of mice were intravenously administered 5 × 1010 vp AdC68–CR9114 or 5 × 1010 vp AdC68–empty and challenged with 10LD50 pH1N1 virus after 24 h. All mice were killed on the fifth day post challenge. Thereafter, sections of the lung tissues were fixed and stained with hematoxylin and eosin. Histological analysis of the lungs revealed that mice immunized with AdC68–CR9114 were close to the naive group, with no visible infiltration observed. However, mice in the AdC68–empty group developed severe perivascular and interstitial infiltrates, even necrosis, with an average pathogenic score of 4.6 (Fig. 5a, b). Viral loads were measured via RT-qPCR analysis and a TCID50 assay of the lung homogenates. As shown in Fig. 5c, d, the lung viral titers in the AdC68–CR9114 group were markedly lower than those in the AdC68–empty group.

a Five days after pH1N1 challenge, mice were sacrificed, and the lung sections were stained with hematoxylin and eosin. Arrows indicated the perivascular and interstitial infiltration of inflammatory cells and the lung consolidation. b Pathological scores of the lungs 5 days after challenge. c The viral titer in the lung tissues was determined by quantitative PCR. d Lung virus titer was determined by the TCID50 assay. Five mice in each group. ***p < 0.01. ns no significance (determined using one-way ANOVA).
Duration of the prophylactic and therapeutic protection induced by AdC68–CR9114
To determine the prophylactic duration of protection, groups of mice were intravenously administered 5 × 1010 vp AdC68–CR9114 and challenged with 10LD50 of the PR8 virus on days 1, 3, 7, 14, or 30. In addition, body weight and survival rate were monitored for 14 days. The purified CR9114 antibody and AdC68–empty were used as the positive and negative controls, respectively. As depicted in Fig. 6a, b, AdC68–CR9114 can afford full protection within 7 days post immunization and partial protection on day 14 post immunization. However, protection was not observed on day 30 post immunization.

For the assessment of the prophylactic protection, groups of C57BL/6 mice were treated with intravenous administration of 5 × 1010 vp AdC68–CR9114 and challenged with the 10LD50 PR8 virus at different time points. In addition, the mice were intraperitoneally treated with 200 μg of purified CR9114 antibody and then infected with 10LD50 PR8 virus at 24 h later. a Body weight change. b Survival rate. For the assessment of the therapeutic protection, groups of mice were infected with the 10LD50 PR8 virus and intranasally inoculated with AdC68–CR9114 at different time points or intraperitoneally administered 200 μg of purified CR9114 antibody at 24 or 48 h after infection. c Body weight change. d Survival rate. Five mice in each group. Data in a, c are expressed as mean ± SD.
To further assess the therapeutic timeline effect of AdC68–CR9114, groups of mice were infected with the 10LD50 PR8 virus and intranasally treated with AdC68–CR9114 at different time points. The purified CR9114 antibody inoculated intraperitoneally was used as the positive control. As shown in Fig. 6c, d, AdC68–CR9114 provided full protection from the PR8 virus at 6 h after challenge, partial protection at 24 h, and became invalid after 48 h. Therefore, it is best to treat influenza infection with AdC68–CR9114 within 12 h of its onset. However, the purified CR9114 antibody could confer full protection even at 48 h later (Fig. 6c, d).
Discussion
Vaccination is one of the most effective strategies against viral infection, including the influenza virus. The most commonly used influenza vaccine is a trivalent inactivated influenza vaccine whose composition is not fixed and can be adjusted according to the WHO’s forecasts. Besides the trivalent inactivated influenza vaccine, live attenuated influenza vaccine27 and subunit influenza vaccines are used in the clinic28. Sanofi has launched a tetravalent vaccine (VaxigripTetraTM) based on the trivalent vaccine administered to individuals ≥3 years old; this vaccine has been approved in Europe29. Nevertheless, the efficacy of the influenza vaccine may be significantly attenuated and can be invalid if a large mismatch exists between the vaccine and the new epidemic strain. Antiviral drugs and other treatments are a second intervention to combat influenza infection. Zanamivir and oseltamivir30, two neuraminidase inhibitors, have been licensed for clinical use in the treatment of influenza. Adamantane is another anti-influenza drug that targets the M2 protein and blocks the activity of the ion channel, which is essential for viral uncoating following entry into the host cell31. However, emergence of drug-resistant influenza virus has been detected32, which raises concerns regarding its widespread use. Broadly neutralizing antibodies against influenza would thus be an ideal option in influenza prevention and therapy.
Different strategies for expressing encoded mAbs have been applied in the prevention and control of influenza virus infections. Wilson et al.33 revealed that the adeno-associated virus 9 (AAV9) expressing a broadly neutralizing antibody, FI6, (AAV9.FI6) protected mice and ferrets from the heterotypic influenza virus infection when immunized intranasally. In a further study34, they found that intranasal immunization with AAV9.FI6 could protect old and immunodeficient mice from the influenza virus. Weiner et al.35 used DNA vectors to express two neutralizing antibodies targeting influenza A and B, respectively, and mixed the DNA vectors to protect mice from influenza A and B virus infection. Here, we developed a universal anti-influenza vaccine by using the chimpanzee adenoviral vector, AdC68, expressing CR9114, a broadly neutralizing antibody against influenza virus. AdC68 is a typical chimpanzee adenovirus with no pre-existing immunity in the majority of humans36. It has similar immunogenicity to that of AdHu5, and can elicit both humoral and cellular immune responses37.
Compared with the AAV vector, the adenoviral vector exerts a relatively short-term expression of the foreign gene of interest38. In this study, CR9114 was detected in the serum at 1 day after intravenous injection. Further, its level peaked on day 5 and was reduced to undetectable levels on day 13 post immunization. Hence, AdC68–CR9114 could prophylactically protect mice from influenza challenge for approximately 2 weeks. The influenza virus titer is known to reach its peak at 48 h after infection in human respiratory tracts. Thereafter, it slowly decreases until day 639. Thus, the duration of the neutralizing antibodies expressed by the adenovirus is sufficient to provide protection against influenza virus infection if this inoculation type is administered immediately following the first wave of the influenza outbreak. Based on the therapeutic application, all mice recovered from the influenza infection when intranasally administered AdC68–CR9114 within 6 h after the influenza infection. However, intraperitoneal injection with the purified CR9114 antibody can also be administered to mice suffering from influenza infection up to 48 h. Antiviral drugs are recommended for use within 48 h after infection, with earlier administration leading to better results40. Therefore, broadly neutralizing antibodies could be combined with anti-influenza drugs to treat drug-resistant influenza strains. After engineering a multimechanistic influenza B antibody, C12G6, Shen et al.41 found that it exhibited an addictive antiviral effect when co-administered with oseltamivir. In future studies, we will explore whether AdC68–CR9114 or purified CR9114 combined with antiviral drugs could synergistically combat influenza infection.
Regarding the antibody expression efficiency, the adenoviral vector is much stronger than the DNA35 and AVV vector33. The concentration of CR9114 expressed by the adenovirus in the serum reached 11,535 μg/ml at 24 h after intravenous immunization. However, 5 days after mice were administered 300 μg of the DNA plasmid, the serum antibody concentration could only increase to 10 μg/ml35. After the AVV9 vector was intranasally administered to mice, the concentrations of the antibody in the nose and lung were 0.5 and 2.0 μg/ml, respectively; however, its concentration in the serum was fairly low (approximately 120 ng/ml). According to the immunization methods employed in our study, 3 or 5 days were required to achieve the peak level of CR9114.
In this study, both intranasal immunization and intravenous injection provided 100% prophylactic protection. However, the concentrations of the neutralizing antibodies in the serum of the intravenously immunized mice were much higher and persisted much longer than in intranasally immunized mice. Thus, intravenous immunization was employed for preventive purposes. However, in the therapeutic experiments, the efficacy of nasal immunization was better than that of intravenous injection. We speculated the reason as the following: in the therapeutic experiment, mice were first infected with the influenza virus and then treated with the adenovirus-encoded antibody 6 h later. If the adenoviruses are administered intravenously, it may result in a toxic and potentially lethal reaction to the mice, and the toxicity of intravenously injected adenoviral vectors may be directly linked to the activation and destruction of Kupffer cells42. Although this phenomenon is transient, it may impair the first line of defense of the innate immune system and aggravate the symptoms of influenza infection in mice before the therapeutic antibody getting expressed and exerting its functions. If treated intranasally, adenoviruses were directly delivered to the respiratory tract and the lung; the amount of antibody produced locally might be higher than that of intravenous injection. Importantly, the innate immune system will not be hampered in this type of regimen. Our future efforts are thus focused on further exploiting the advantages of adenoviral vectors and improving the efficacy of this new anti-influenza vaccine.
Compared with the purified CR9114, AdC68–CR9114 was not as effective as purified antibody for therapeutic use in this study. The reason is that the antibody expression by the adenovirus requires time. Generally, antibody expressed by the adenovirus takes at least 24 h to be detected. However, influenza virus replicates quickly once it is seeded in the lung, and reaches its replicative peak 48 h after infection. If the antibody cannot appear in the early stage, it will be less effective. In this study, purified antibodies injected intraperitoneally could directly enter the circulation system to neutralize influenza virus effectively. Thus, the purified CR9114 even given at 48 h post influenza infection could confer full protection in mice, and AdC68–CR9114 was more effective when given within 6 h after influenza virus infection.
Altogether, we developed a universal influenza vaccine (AdC68–CR9114) based on a chimpanzee adenoviral vector that expresses influenza broadly neutralizing mAb. The recombinant adenovirus can express a high level of CR9114 in vitro and in vivo, exert biological functions, and cross-protect mice from different types of influenza virus infection at different time points. Therefore, the findings of this study could provide a landmark strategy for influenza prevention and control, and serve as an example for other infectious diseases.
Methods
Cells and viruses
Human embryonic kidney (HEK) 293 cells were bought from ATCC (USA), and Mardin–Darby canine kidney (MDCK) cells were bought from the Cell bank of Shanghai Institutes for Biological Science (Shanghai, China), and they were maintained in complete Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (Gibco, USA) and 2% penicillin and streptomycin (New Cell & Molecular Biotech, China). PR8 (A/Puerto Rico/8/34), pH1N1 (pandemic H1N1, A/California/7/2009), H3N1 (A/Archi/1/1968), H5N1 (A/environment/Hunan/6-69/2008), H9N2 (A/chicken/Jiangsu/7/2002), and influenza B (Yamagata lineage, B/phuket/3037/2013) were stored at −80 °C in our laboratory and propagated in chicken embryos for use in all experiments.
Construction of the recombinant adenovirus, AdC68–CR9114
E1-/E3-deleted pAdC68 vector is a chimpanzee-originated vector that was generated in our lab43. We obtained the sequences of the variable gene for the CR9114 light and heavy chain (GenBank: JX213639.1 and GenBank: JX213640.1) from the National Center for Biotechnology Information (NCBI) and combined them with the constant sequence of human IgG1 to obtain the full sequence of CR9114. CR9114 was subjected to codon optimization to improve its expression in human cells by Genewiz Biotech (Suzhou, China). Thereafter, it was cloned into a shuttle vector, pUC57–CASI, under the CASI promoter. CR9114 was then subcloned into an E1-/E3-deleted pAdC68 vector to obtain the recombinant adenoviral plasmid, pAdC68–CR9114. To rescue the recombinant adenovirus, pAdC68–CR9114 was linearized with Pac I and transfected into HEK293 cells using Lipofectamine 2000 (Invitrogen). The virus AdC68–CR9114 was propagated in HEK293 cells and purified by CsCl density gradient ultracentrifugation.
Antibody purification and identification
HEK293 cells were propagated to 30, 150-mm2 dishes and inoculated with AdC68–CR9114. The supernatants were collected, and the CR9114 antibody was purified using a Protein A column (GE Healthcare). The purified antibody was boiled and subjected to SDS-PAGE for Coomassie Blue R-250 staining.
Western blot
Different quantities of AdC68–CR9114 (1010, 109, or 108 virus particles (vp)) were used to infect HEK293 cells in a 6-well plate. AdC68–empty (1010 vp) was used as the negative control, while uninfected HEK293 cells were used as blank controls. After 24 h, the cell supernatant was analyzed by reducing and nonreducing Western blotting with a HRP-conjugated anti-human lgG (H&L) (Abcam, China, Cat. No. ab6759-HRP) at a dilution of 1:5000 to detect the expression of the CR9114 antibody. All blots derive from the same experiment and were processed in parallel.
ELISA
To measure the level of the CR9114 antibody in vitro, different quantities of AdC68–CR9114 (1010, 109, or 108 virus particles (vp)) were used to infect HEK293 cells in a 6-well plate. AdC68-gp (1010 vp) was used as the negative control, while uninfected HEK293 cells were used as blank controls. The supernatants were harvested at 24 and 48 h after infection. The supernatants and serum samples diluted in suitable proportions were used to determine the concentration of the CR9114 antibody using sandwich ELISA. The anti-human IgG Kappa (70 ng/well) (Southern Biotech, Cat. No. 2060-01) was coated on the ELISA plate at 4 °C overnight and blocked with 5% skim milk (200 μL/well) for 2 h at 37 °C. After blocking, the samples prepared above were added to the plate (100 μL/well) for 2 h at 37 °C. HRP-conjugated mouse anti-human IgG Fc (1:10,000, 100 μL/well) was added to each well and incubated for 1 h at 37 °C. This was followed by the washing steps and then the addition of the 3,3′,5,5′-Tetramethylbenzidine (TMB) substrate (New Cell & Molecular Biotech Co., Ltd., China) to observe the color reaction, which was stopped using a 2 M sulfuric acid (H2SO4) solution. Absorbance was measured at 450 nm using a microtiter plate reader (Thermo Scientific, Waltham, MA). To calculate the specific content of the expressed CR9114 antibody, a standard curve was generated according to the dosage of the purified CR9114 antibody.
Animal studies
All animal experiments were approved by the Institutional Animal Care and Use Committee and the Biosafety Committee of Institut Pasteur of Shanghai, while those related to the H5N1 virus were conducted in a biosafety level 3 laboratory following the standard operating protocols approved by the Institutional Biosafety Committee at the Shanghai Public Health Clinical Center, Fudan University. We have complied with all relevant ethical regulations for animal testing and research. Six- to eight-week-old female C57BL/6 mice were purchased from Beijing Vital River Laboratory (Beijing, China) and housed in the Biological Safety Level 2 laboratory of the Institute Pasteur of Shanghai.
For the in vivo expression of the CR9114 antibody, mice were randomly divided into four groups and intranasally immunized with AdC68–CR9114 or AdC68–empty at 5 × 1010 vp/mouse in 30 μl of PBS or intravenously administered AdC68–CR9114 or AdC68–empty at 5 × 1010 vp/mouse in 200 μl of PBS. All mice were subjected to bleeding every other day.
For the prophylactic protection assay, mice were divided into two groups and primarily immunized with AdC68–CR9114 or AdC68–empty (5 × 1010 vp/mouse, in 200 μl of PBS) through intravenous administration via the tail. After 24 h, mice were challenged intranasally with different groups of influenza A and B viruses, such as PR8, pH1N1, H9N2, H5N1, and H3N1 at doses of 10LD50 in 30 μl of PBS. Intranasal immunization was also implemented to evaluate the prophylactic efficacy of AdC68–CR9114. C57BL/6 mice were intranasally treated with 5 × 1010 vp of the AdC68–CR9114 virus and challenged with the 10LD50 pH1N1 virus and A/H3N1 virus after 24 h. To explore the prophylactic duration of AdC68–CR9114, mice were intravenously administered AdC68–CR9114 (5 × 1010 vp/mouse, in 200 μl of PBS) and challenged with 10LD50 PR8 in 30 μl of PBS after 1, 3, 7, 14, and 30 days. Mice intraperitoneally administered the purified CR9114 antibody (200 μg/mouse) or intravenously injected with the AdC68–empty (5 × 1010 vp/mouse, in 200 μl of PBS) served as the positive or negative controls.
For the therapeutic protection assay, mice were challenged with the influenza A virus of PR8 at a dose of 10LD50, and then treated intranasally or intravenously with AdC68–CR9114 or AdC68–empty (5 × 1010 vp/mouse, in 200 μl of PBS) at 6, 24, and 48 h, respectively, or treated by an intraperitoneal injection of purified CR9114 antibody (200 μg/mouse, in 1 ml of PBS) at 24 and 48 h, respectively. The body weights and survival rates of all mice were monitored daily for 14 days. The mouse was euthanized when a body weight loss greater than 30% of its pre-challenge weight was observed.
Viral loads in the lungs
Mice were euthanized on day 5 post challenge to dissect the lung tissues for histology and viral load detection. Briefly, a section of the weighted lung tissues was homogenized in the corresponding volume of DMEM containing 1% BSA and 1% antibiotics to obtain a 10% (w/v) suspension. A series of dilutions of this suspension was added to the monolayers of MDCK cells in 96-well plates, and the cells were cultured in an incubator at 37 °C. After 4 h, the virus titer was detected via a hemagglutination assay. A 50-μL volume of the supernatant from each well was added to a V-type 96-well plate and reacted with 50 μL 1% (v/v) of chicken RBCs (in PBS) for 15 min at room temperature. The virus titer was calculated using the Reed and Muench method44. Another section of the lung tissues was homogenized and resuspended in 1 mL of TRIzol reagent to extract total RNA for RT-qPCR analysis in order to measure and quantify the viral loads. Specifically, 1 μg of RNA from each sample was reverse-transcribed to cDNA according to the manufacturer’s protocol for the One-Step RT-PCR Kit (Roche). The viral loads in the lungs were measured via quantitative real-time transcription PCR. The primer pair for the mRNA of the influenza virus was F-5′ AAGACCAATCCTGTCACCTCTGA-3′, R-5′-CAAAGCGTCTACGCTGCAGTCC-3′, while that of the internal reference gene (GAPDH) was F-5′-CAATGTGTCCGTCGTGGATCT-3′, R-5′-GTCCTCAGTGTAGCCCAAGATG-3′. Data were analyzed using the 7900HT System SDS software (Applied Biosystems).
Histology
The third section of the murine lung tissues was fixed in 4% formaldehyde for 24 h at 4 °C and subjected to hematoxylin and eosin staining as previously described26. To assess the pathological changes in the lungs, the score was derived according to the following criteria: (1) no observable pathology, (2) perivascular infiltrates, (3) perivascular and interstitial infiltrates affecting <20% of the lobe section, (4) perivascular and interstitial infiltrates affecting 20–50% of the lobe section, and (5) perivascular and interstitial infiltrates affecting >50% of the lobe section.
Statistical analysis
All statistical analyses were performed using GraphPad Prism software version 7.0. The difference in the CR9114 antibody titer in vitro and the viral loads in the lungs among the groups were assessed by one-way analysis of variance (ANOVA). P values < 0.05 were regarded as statistically significant.
Reporting summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All data generated or analyzed in this study are available upon request from the authors.
References
Wu, X. et al. Progress of small molecular inhibitors in the development of anti-influenza virus agents. Theranostics 7, 826–845 (2017).
Global Influenza Strategy 2019–2030. https://apps.who.int/iris/bitstream/handle/10665/311184/9789241515320-eng.pdf?ua=1 (2019).
Darvishian, M., Bijlsma, M. J., Hak, E. & van den Heuvel, E. R. Effectiveness of seasonal influenza vaccine in community-dwelling elderly people: a meta-analysis of test-negative design case-control studies. Lancet Infect. Dis. 14, 1228–1239 (2014).
Jernigan, D. B. & Cox, N. J. H7N9: preparing for the unexpected in influenza. Annu Rev. Med. 66, 361–371 (2015).
Burton, D. R., Poignard, P., Stanfield, R. L. & Wilson, I. A. Broadly neutralizing antibodies present new prospects to counter highly antigenically diverse viruses. Science 337, 183–186 (2012).
Fuchs, S. P. & Desrosiers, R. C. Promise and problems associated with the use of recombinant AAV for the delivery of anti-HIV antibodies. Mol. Ther. Methods Clin. Dev. 3, 16068 (2016).
Stephenson, K. E. & Barouch, D. H. Broadly neutralizing antibodies for HIV eradication. Curr. HIV/AIDS Rep. 13, 31–37 (2016).
The IMpact-RSV Study GroupPalivizumab, a humanized respiratory syncytial virus monoclonal antibody, reduces hospitalization from respiratory syncytial virus infection in high-risk infants. Pediatrics 102, 531–537 (1998).
Wilde, H., Chomchey, P., Prakongsri, S., Puyaratabandhu, P. & Chutivongse, S. Adverse effects of equine rabies immune gobulin. Vaccine 7, 10–11 (1989).
Jones, P. D. Rabies vaccine failures. Lancet 1, 917–918 (1988).
De Benedictis, P. et al. Development of broad-spectrum human monoclonal antibodies for rabies post-exposure prophylaxis. EMBO Mol. Med. 8, 407–421 (2016).
Ekiert, D. C. et al. A highly conserved neutralizing epitope on group 2 influenza A viruses. Science 333, 843–850 (2011).
Krammer, F. & Palese, P. Influenza virus hemagglutinin stalk-based antibodies and vaccines. Curr. Opin. Virol. 3, 521–530 (2013).
Okuno, Y., Isegawa, Y., Sasao, F. & Ueda, S. A common neutralizing epitope conserved between the hemagglutinins of influenza A virus H1 and H2 strains. J. Virol. 67, 2552–2558 (1993).
Ekiert, D. C. & Wilson, I. A. Broadly neutralizing antibodies against influenza virus and prospects for universal therapies. Curr. Opin. Virol. 2, 134–141 (2012).
Dreyfus, C. et al. Highly conserved protective epitopes on influenza B viruses. Science 337, 1343–1348 (2012).
Wollacott, A. M. et al. Safety and upper respiratory pharmacokinetics of the hemagglutinin stalk-binding antibody VIS410 support treatment and prophylaxis based on population modeling of seasonal influenza A outbreaks. EBioMedicine 5, 147–155 (2016).
Ramos, E. L. et al. Efficacy and safety of treatment with an anti-m2e monoclonal antibody in experimental human influenza. J. Infect. Dis. 211, 1038–1044 (2015).
Corti, D. et al. Tackling influenza with broadly neutralizing antibodies. Curr. Opin. Virol. 24, 60–69 (2017).
Kallewaard, N. L. et al. Structure and function analysis of an antibody recognizing all influenza A subtypes. Cell 166, 596–608 (2016).
Tatsis, N. & Ertl, H. C. J. Adenoviruses as vaccine vectors. Mol. Ther. 10, 616–629 (2004).
Tatsis, N. et al. Adenovirus vector-induced immune responses in nonhuman primates: responses to prime boost regimens (vol 182, pg 6587, 2009). J. Immunol. 183, 7611–7611 (2009).
Douglas, J. T. Adenovirus-mediated gene delivery: an overview. Methods Mol. Biol. 246, 3–14 (2004).
Guo, J., Mondal, M. & Zhou, D. Development of novel vaccine vectors: chimpanzee adenoviral vectors. Hum. Vaccin Immunother. 14, 1679–1685 (2018).
Zhou, D. et al. A universal influenza A vaccine based on adenovirus expressing matrix-2 ectodomain and nucleoprotein protects mice from lethal challenge. Mol. Ther. 18, 2182–2189 (2010).
Song, Y. et al. Repeated low-dose influenza virus infection causes severe disease in mice: a model for vaccine evaluation. J. Virol. 89, 7841–7851 (2015).
Pebody, R., McMenamin, J. & Nohynek, H. Live attenuated influenza vaccine (LAIV): recent effectiveness results from the USA and implications for LAIV programmes elsewhere. Arch. Dis. Child. 103, 101–105 (2018).
Traynor, K. First recombinant flu vaccine approved. Am. J. Health Syst. Pharm. 70, 382 (2013).
Gresset-Bourgeois, V. et al. Quadrivalent inactivated influenza vaccine (VaxigripTetra). Expert Rev. Vaccines. 17, 1–11 (2018).
Shobugawa, Y. et al. Clinical effectiveness of neuraminidase inhibitors-oseltamivir, zanamivir, laninamivir, and peramivir-for treatment of influenza A(H3N2) and A(H1N1)pdm09 infection: an observational study in the 2010-2011 influenza season in Japan. J. Infect. Chemother. 18, 858–864 (2012).
Hay, A. J., Wolstenholme, A. J., Skehel, J. J. & Smith, M. H. The molecular-basis of the specific anti-influenza action of amantadine. Embo J. 4, 3021–3024 (1985).
Li, X. D. et al. Drug-resistant and genetic evolutionary analysis of influenza virus from patients during the 2013 and 2014 influenza season in Beijing. Microb. Drug Resist. 23, 253–260 (2017).
Limberis, M. P. et al. Intranasal antibody gene transfer in mice and ferrets elicits broad protection against pandemic influenza. Sci. Transl. Med. 5, 187ra172 (2013).
Adam, V. S. et al. Adeno-associated virus 9-mediated airway expression of antibody protects old and immunodeficient mice against influenza virus. Clin. Vaccin. Immunol. 21, 1528–1533 (2014).
Elliott, S. T. C. et al. DMAb inoculation of synthetic cross reactive antibodies protects against lethal influenza A and B infections. NPJ Vaccines 2, 18 (2017).
Zhang, S. et al. Seroprevalence of neutralizing antibodies to human adenoviruses type-5 and type-26 and chimpanzee adenovirus type-68 in healthy Chinese adults. J. Med. Virol. 85, 1077–1084 (2013).
Cohen, C. J. et al. Chimpanzee adenovirus CV-68 adapted as a gene delivery vector interacts with the coxsackievirus and adenovirus receptor. J. Gen. Virol. 83, 151–155 (2002).
Lai, C. M., Lai, Y. K. & Rakoczy, P. E. Adenovirus and adeno-associated virus vectors. DNA Cell Biol. 21, 895–913 (2002).
Taubenberger, J. K. & Morens, D. M. The pathology of influenza virus infections. Annu Rev. Pathol. 3, 499–522 (2008).
Leneva, I. A. et al. Virus susceptibility and clinical effectiveness of anti-influenza drugs during the 2010-2011 influenza season in Russia. Int J. Infect. Dis. 43, 77–84 (2016).
Shen, C. G. et al. A multimechanistic antibody targeting the receptor binding site potently cross-protects against influenza B viruses. Sci. Transl. Med. 9, eaam5752 (2017).
Schiedner, G. et al. A hemodynamic response to intravenous adenovirus vector particles is caused by systemic Kupffer cell-mediated activation of endothelial cells. Hum. Gene Ther. 14, 1631–1641 (2003).
Yang, Y., Chi, Y., Tang, X., Ertl, H. C. J. & Zhou, D. Rapid, efficient, and modular generation of adenoviral vectors via isothermal assembly. Mol. Biol. 113, 16.26.11–16.26.18 (2016).
Reed, L. J. & M., H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 27, 493–497 (1938).
Acknowledgements
This work was supported by the National Science and Technology Major Project (2018ZX10101004), National Natural Science Foundation of China (31870922 and 31670946), and Strategic Priority Research Program of the Chinese Academy of Sciences (XDB29040000) to D.Z., and partially supported by Postdoctoral Science Foundation of China (2018M632170) to X.W.
Author information
Authors and Affiliations
Contributions
X.W. and P.Z. contributed equally to this work. D.Z. conceived the idea and designed the experiments, and edited the paper. X.W. and P.Z. conducted most of the experiments, analyzed the data, and X.W. wrote the paper. M.W., K.Y., J.G., X.W., J.L., Z.F., G.W., and M.X. performed some of the experiments.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that the study was conducted in the absence of any commercial or financial relationships that could be construed as a potential competing interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, X., Zhou, P., Wu, M. et al. Adenovirus delivery of encoded monoclonal antibody protects against different types of influenza virus infection. npj Vaccines 5, 57 (2020). https://doi.org/10.1038/s41541-020-0206-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41541-020-0206-5
This article is cited by
-
Heterologous prime-boost immunizations with chimpanzee adenoviral vectors elicit potent and protective immunity against SARS-CoV-2 infection
Cell Discovery (2021)
-
Intranasal delivery of replicating mRNA encoding neutralizing antibody against SARS-CoV-2 infection in mice
Signal Transduction and Targeted Therapy (2021)
