
Abstract
Rheumatoid arthritis (RA) is a complex autoimmune disease with an unknown aetiology. However, rheumatoid arthritis fibroblast-like synoviocytes (RA-FLS) play a significant role in initiating and perpetuating destructive joint inflammation by expressing immuno-modulating cytokines, adhesion molecules, and matrix remodelling enzymes. In addition, RA-FLS are primary drivers of inflammation, displaying high proliferative rates and an apoptosis-resistant phenotype. Thus, RA-FLS-directed therapies could become a complementary approach to immune-directed therapies by predicting the optimal conditions that would favour RA-FLS apoptosis, limit inflammation, slow the proliferation rate and minimise bone erosion and cartilage destruction. In this paper, we present a large-scale Boolean model for RA-FLS that consists of five submodels focusing on apoptosis, cell proliferation, matrix degradation, bone erosion and inflammation. The five-phenotype-specific submodels can be simulated independently or as a global model. In silico simulations and perturbations reproduced the expected biological behaviour of the system under defined initial conditions and input values. The model was then used to mimic the effect of mono or combined therapeutic treatments and predict novel targets and drug candidates through drug repurposing analysis.
Similar content being viewed by others


Introduction
Rheumatoid arthritis (RA) is a multifactorial disease that affects the articular joints of the human body and involves combinations of genetic factors and environmental triggers1,2,3. Cytokines such as tumour necrosis factor-alpha (TNF-α) and interleukin 6 (IL-6) are central players in RA, causing inflammation and tissue damage, whereas their inhibitors are considered among the main treatments for RA2,4,5. However, despite a growing number of such drugs, 40% of patients fail to respond to therapy adequately6,7. Recent studies have identified RA fibroblast-like synoviocytes (RA-FLS) as responsible for up to a quarter of the disease’s heritability8, attributing to these cells a causal role in disease pathogenesis. The primary roles of RA-FLS in RA are discussed below9,10. RA-FLS plays a central role in the pathogenesis of RA by activating the innate immune response. The immune response is maintained via the secretion of soluble molecules, such as proinflammatory cytokines IL-6, IL-1 and TNF, in response to environmental stimuli and interactions with other cells. Secretions of these molecules act as a positive feedback loop and eventually trigger the activation of RA-FLS into expressing the responses repeatedly9. Joint inflammation is the primary characteristic of RA. During joint inflammation in RA, FLS proliferate to form the pannus, which invades and destroys the cartilage. Major pathways involved in the perpetuation of inflammation are TNF, IL-6, IL-1 and IL-174,11.
The final stages of the disease involve the degradation of bone and cartilage due to chronic inflammation within the joint area. During chronic and sustained inflammation, RA-FLS secrete two groups of soluble molecules: (a) receptor activator of nuclear factor kappa-Β ligand (RANKL), a molecule that promotes osteoblasts differentiation to osteoclasts, cells that are responsible for bone degradation and bone resorption12,13 and (b) matrix metalloproteinases (MMPs), a group of matrix proteases responsible for the degradation and breakdown of numerous extracellular matrix components such as collagen, leading to the degradation of cartilage14,15. The synergistic activity of these factors (RANKL and MMPs) leads to the gradual degradation of bone and cartilage in the joint area, leading to stiffness, pain and eventually disability of movement. Lastly, RA-FLS aberrant proliferation contributes to pannus formation and joint destruction. RA-FLS are the critical cell types in the growth of pathological synovial tissue in RA, and inhibition of their proliferation is a potential antirheumatic therapy16. RA-FLS are shown to be apoptosis-resistant17. The intrinsic and extrinsic pathways regulate apoptosis. The intrinsic pathway is activated inside the cell following intracellular stress or injury, including mitochondrial proteins like BH3 interacting domain death agonist (BID), Bcl-2-associated X protein (BAX), B-cell lymphoma 2 protein (BCL2), and phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1), among others. The tumour protein P53 (TP53) transcription factor is a critical activator of the intrinsic pathway of apoptosis via activating mitochondrial proteins. TNF and FAS ligand (FASLG) mainly activate the extrinsic pathway. Ultimately, both pathways activate caspases, which initiate a proteolytic cascade leading to cellular death18.
The biological information regarding pathways implicated in the RA-FLS pathology has been assembled into a large-scale mechanistic network, the RA map19. While the RA map contains generic information about pathways implicated in RA that come from various sources, the RA-FLS is the dominant cell type. The map is a process description diagram20 and is part of the Disease Maps initiative21,22. This type of construct contains valuable disease-specific information encoded in human and machine-readable formats. Besides serving as templates for omic data visualisation, disease maps can also work as scaffolds for building mathematical models and sources of causal interactions23.
Dynamical modelling has been widely used to study and decipher complex biological processes that are otherwise hard to comprehend. Previous attempts to build computational models for RA have contributed a few kinetic models to study the role of proinflammatory and anti-inflammatory cytokines24 and bone erosion25, and also the behaviour of various cells, including RA-FLS in cartilage destruction in RA joints26. A hybrid mathematical modelling framework that describes pannus production in a tiny proximal interphalangeal (PIP) joint was also proposed27. Nevertheless, the need for kinetic parameters limits its use for large-scale molecular interaction networks. Lastly, a large-scale hybrid model covering signalling, gene regulation, and metabolism in RA-FLS was published28. The model focuses on the metabolic reprogramming of fibroblasts under hypoxic conditions in the arthritic joint and suggests a reverse Warburg effect as the origin of the observed metabolic switch.
Discrete logic-based qualitative modelling has been increasingly used to model large-scale networks for which kinetic data is scarce29,30. For example, in a Boolean model, each node can take only two values, 0 (FALSE) and 1 (TRUE). The next value of each variable is determined by a logical function (using the classical AND, OR, NOT operators) of the current values of its regulators (upstream nodes). The evolution of each node also depends on the updating scheme chosen. The synchronous scheme updates all variables in the model simultaneously; in the asynchronous scheme, the variables are updated individually in a non-synchronous manner31,32.
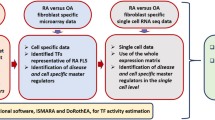
In this work, we use the state-of-the-art RA map19 and the tool CaSQ33 to build a large-scale, modular Boolean model of RA-FLS focused on inflammation, bone erosion, cartilage destruction, cell proliferation, and apoptosis. The model consists of five-phenotype-specific submodels (to be mentioned as modules later in the paper) that can be simulated individually and a five-phenotype global model (to be mentioned as the global model later in the paper). Systematic testing of different initial conditions using the modules and the global model showed that both the modules and the global model could reproduce small-scale experimental results from the literature. To search for meaningful steady states, we reduce calculations by propagating and eliminating fixed input components, as introduced in ref. 34 and implemented in the CoLoMoTo notebook35 in ref. 36. Input propagation consists of assigning fixed values to some of the model’s inputs and using Boolean algebra to simplify the rules of their downstream components. We also use a probabilistic framework to calculate phenotypic probabilities starting from predefined initial conditions. The RA-FLS model was used to study the effects of mono and combined therapy for RA and suggest potential targets that could enhance the desired phenotypic outcome. Furthermore, drug repurposing analysis identified possible drug candidates that have as targets the previously identified nodes. A new round of simulations was then performed to evaluate their impact on the cellular phenotype and, subsequently, on the arthritic joint (Fig. 1).

The RA map was converted into an executable Boolean model using the map-to-model framework described in ref. 33. Using single-cell omic datasets and literature studies, the RA generic model was subsequently enriched in RA-FLS-specific data. The RA-FLS model focuses on five phenotypes (apoptosis, cell proliferation, inflammation, matrix degradation, and bone erosion) characteristic of RA’s fibroblasts. Individual phenotype-specific submodels and a five-phenotype global model were created. Biological scenarios extracted from the literature were used to evaluate and validate the models’ behaviour leading to some modifications of the original models. The modified RA-FLS model was then used to test mono and combined RA therapies. Drug repurposing analysis and further drug combination simulations led to a panel of suggestions of drug combinations that are predicted to have a favourable outcome (apoptosis active, cell proliferation, inflammation, bone erosion and matrix degradation inactive).
Results
Enhancing the cell specificity of the initial network
RA-FLS is the most frequent cell type in the RA map, covering a total of 45%, followed by synovial tissue with 36%19. We used the updated fibroblast overlay list provided in refs. 37,38 to calculate the RA-FLS specificity of the global model. In this study, they used the RA fibroblast list provided in ref. 19 and updated it with the RNA-seq single-cell dataset available in the GEO database39 by performing Differential Expression Analysis (DEA) using BioTuring software and the Venice method. Using this list, out of 261 unique model components (proteins, genes, phenotypes), 194 are RA-FLS-specific. Other cell types primarily include synovial tissue with 29 components, peripheral blood mononuclear cells (PBMCs) with 25 components, along with the presence of other sources like blood, synovial fluid, T-helper (th1), macrophages and chondrocytes (Supplementary Table 1).
Five-phenotype-specific modules and global RA-FLS model
The tool bioLQM40 was used to extract the phenotype-specific modules and the global model using the SBML file produced by CaSQ33 using RA map (Supplementary Fig. 1) XML file as input (files available on GitLab). The size of the models in terms of nodes and reactions can be seen in Fig. 2a. One straightforward observation is that the size of the five-phenotype global model is not the sum of the subparts. Indeed, as seen in the Venn diagram (Fig. 2b), there is a core of 191 nodes shared by all modules, and only a few nodes are characteristic of each phenotype-specific module.

a Number of nodes of the phenotype-specific modules and the global model. b Venn diagram of all the five-phenotype-specific components. The core of 191 nodes is shared among all five modules, and only a few are characteristic of the corresponding phenotype-specific module.
Evaluating the RA-FLS model under different sets of input configurations using the input propagation method
The validation of each phenotype-specific module and the global model was performed by comparing the outcome of in silico analyses to RA-FLS-specific biological observations based on experimental evidence (Supplementary Table 2). For each module, we formulated prior knowledge based on small-scale experiments as observations and compared them with the results of the corresponding virtual experiment. Initial conditions were set to mimic the corresponding experimental settings. We used input propagation and identification of trap spaces to evaluate the phenotype-specific modules and the global model’s behaviour. First, we assessed the model’s behaviour by comparing the intended/ expected biological behaviour and the observed one after the process. In general, we faced some difficulties in assessing the models’ behaviour that can be summarised in the following categories: (a) not all components mentioned in the experimental settings included in the models, (b) experimental information not available for all components included in the models, (c) global behaviour consistent with experimental observations, but intermediate mechanisms only partially coherent. For some cases, further testing of the models revealed additional conditions needed to replicate the anticipated results. These additional “model conditions” that appeared to regulate the biological process are not experimentally proven (Supplementary Table 2). The perturbation experiments are also provided as knockout matrices for input conditions equal to 0 and equal to 1, respectively (Supplementary Tables 3 and 4).
Inflammation (Inflammation_phenotype in the models)
We formulated six biological scenarios to model Inflammation using the modules and the global RA-FLS model. TNF, one of the major pathways regulating Inflammation, was only able to activate the phenotype with the additional activation of the IKBA/NFKB/RELA complex. IKBA/NFKB/RELA complex is an input for activating the NFKB pathway, which is a key regulator of transcriptional responses to TNF41,42,43. IL-6 activation is a sufficient signal for activating Inflammation, as no additional inputs are needed to propagate the signal until the phenotype. Regarding IL-17, similar to TNF, the NFKB pathway is needed to activate the inflammation phenotype43,44,45.
Matrix degradation (Matrix_Degradation_phenotype in the models)
Cartilage destruction is one of the debilitating characteristics of RA. Activated RA-FLS have been shown to attach to the cartilage surface and release matrix-degrading enzymes. Matrix metalloproteinases (MMPs) play a pivotal role in cartilage destruction46. Our model reproduced the destructive role of two MMPs, MMP1 and MMP9, in activating the matrix degradation phenotype in both the phenotype-specific module and the global model.
Bone erosion (Bone_Erosion_phenotype in the models)
Bone erosion is another significant characteristic of RA. Synovitis, along with the production of proinflammatory mediators like Wnt (wingless-related MMTV integration site) and receptor activator of nuclear factor κB ligand (RANKL), results in the differentiation of bone-resorbing osteoclasts, thereby stimulating local bone resorption47. Secreted frizzled-related protein 5 (SFRP5), the primary upstream negative regulator of the WNT pathway when active, negatively regulates bone erosion48. Trap spaces showed that with both the module and the RA-FLS model, we could reproduce Wnt and RANKL (in the presence of SFRP5) biological behaviour49,50,51. Simulations of the models revealed a key role for SFRP5 in activating the bone erosion phenotype, as in its absence, the Wnt canonical pathway gets activated and further activates bone erosion.
Cell proliferation (Cell Growth/Survival/Proliferation_phenotype in the models)
Various growth factors like platelet-derived growth factor (PDGFA) and transforming growth factor beta 1 (TGFB1), and cytokines like TNF regulate the proliferation of RA-FLS52,53. Platelet-derived growth factor (PDGF) is an essential mitogen for fibroblasts, including RA-FLS54,55. PDGFA activated cell growth in both the phenotype-specific module and the global model. However, other pathways can still activate cell growth.
Apoptosis (Apoptosis_phenotype in the models)
The impaired apoptosis process of RA-FLS is responsible for synovial hyperplasia and joint destruction. Major pathways regulating apoptosis include extrinsic (FASLG and TNF) and intrinsic (mitochondrial pathway with BCL2 family proteins). Extrinsic pathways like FASLG and TNF contribute to the activation of apoptosis. The apoptosis module and the global model were able to reproduce this behaviour. AKT is another intracellular regulator of apoptosis (anti-apoptotic agent), which, when kept ON, protects RA-FLS against the apoptosis induced by FASL through inhibition of BID cleavage. However, this scenario was not reproducible, as discussed in the material and methods.
One of the reasons for the inconsistency was the presence of a direct inhibition (CAV1-negative regulator) to the apoptosis phenotype and the inferred logical formula that comprised OR gates between the activators and the absence of inhibitors. This direct inhibition reflects missing mechanistic details related to CAV1 regulating apoptosis. The RA map depicts this information using a negative influence directly at the phenotype glyph. While permitted by the CellDesigner tool56, the lack of mechanistic details leads to an inferred rule from the tool CaSQ that is not optimal, as the tool is designed to use reactions (SBGN-PD) and not influences (SBGN-AF) as input. Therefore, to reproduce the apoptotic-resistant nature of RA-FLS, we modified the logical formula changing the gates from OR to AND, keeping CAV1 as a dominant-negative regulator, in what we will denote the modified global model.
In our modified model, we need to have CAV1 set as active to act as a dominant inhibitor of apoptosis. This condition is justified and relevant from a biological point of view, as experiments have shown that MIR192—which acts as a CAV1 inhibitor in our model—suppresses cell proliferation and induces apoptosis in human RA-FLS by downregulating caveolin 1. Moreover, MIR192 appears to be downregulated in RA synovial tissues and restoring its expression restores the growth-suppressive effects on RA-FLS by targeting CAV157. Therefore, the model condition of having CAV1 always active implies the absence of its inhibitor, MIR192, in the RA settings.
All analyses were performed for both versions of the RA-FLS global model, unmodified and modified, as seen in Supplementary Table 2, to ensure that the formula change did not have any impact on the model’s behaviour regarding the other four phenotypes. All biological scenarios in Supplementary Table 2 were simplified and compiled into tables with the anticipated outcomes associated with Boolean values 0 (OFF) and 1 (ON). Value propagation analysis and trap space identification were applied, and heatmaps were used to visualise the results for both the global model and its modified version. We could not find experimental values for all the model inputs (88 inputs) in the RA condition. Therefore we opted to go for either 0 (no severe inflammation) or 1 (fully activated system, resembling more to an entirely inflammatory condition) for the remaining model inputs that were not part of the biological scenarios tested. In Supplementary Fig. 2, we can see the behaviour of the global and modified model when all inputs are zero. All the phenotypes exhibit the same behaviour except for apoptosis. In the modified model, an additional condition of MIR192, a regulator of the negative inhibitor of apoptosis set as active, was required to reproduce two relevant scenarios.
We further analysed the modified model with input conditions as 1 (Fig. 3a). Under these conditions, apoptosis remains OFF, and all other phenotypes remain ON, except for inflammation, which becomes inactive when IL-6 is OFF. Furthermore, when all inputs resemble an inflamed state in these phenotypic behaviours, many biological scenarios were still reproduced (as seen in Fig. 3b, represented with dark-grey colour). Lastly, some conflicts were observed between the expected and the displayed behaviour, represented in yellow. These conflicts were resolved with additional biological or model conditions in the next row.

Heatmap displaying the trap spaces for the tested biological scenarios in the modified global model (a) and the comparison between expected and obtained values shown with colour codes (b). The y axis shows all the tested scenarios’ names, as mentioned in Table 2, regarding all the five phenotypes as outcomes on the x axis with DEFAULT inputs set to one. †Represent scenarios where additional conditions were given as a known biological behaviour* or as model conditions**. Trap spaces colour codes: -1 (unfixed)  , 0 (OFF)
, 0 (OFF)  , 1 (ON)
, 1 (ON)  . Expectation graph colour codes: Score: expected value, obtained value; 1: Yes [OFF, OFF & ON, ON]
. Expectation graph colour codes: Score: expected value, obtained value; 1: Yes [OFF, OFF & ON, ON]  ; 0: No [ON, OFF & OFF, ON (conflict)]
; 0: No [ON, OFF & OFF, ON (conflict)]  , -1: Undefined
, -1: Undefined  .
.
In general, evaluating the models’ performance in regards to more mechanistic processes and intermediates that involved more complicated scenarios was not possible in several cases, as experimental information is currently missing, and we could not conclude on the coherence of these results. This is reflected in Fig. 3b (and also Supplementary Fig. 2B, D) for the cases represented with light grey colour. Nevertheless, these scenarios could serve as interesting hypotheses for further experimental testing.
Calculating continuous time phenotypic probabilities
The RA-FLS model, in both versions, was used to calculate phenotypic probabilities. We used the software MaBoSS to reproduce representative scenarios with different parameters (discussed in “Materials and methods”)58. One scenario per phenotype from those listed in Supplementary Table 2 was selected for simulations, and results showed that in all cases but apoptosis, the two model versions were able to reproduce the experimental observations. In brief, we performed the following simulations:
Inflammation
IL-6, one of the main pathways regulating inflammation, was set active (1), and the Inflammation phenotype was chosen as the output. As a result, the simulation confirmed the activation of inflammation (Fig. 4a).

a Simulation with IL6_Extracellular_space active and Inflammation as output. The Inflammation phenotype gets activated in the presence of IL-6. b Simulation with PDGFA active and Cell proliferation as output. The Cell proliferation phenotype gets activated in the presence of PDGFA. c Simulation with TNFSF11 (RANKL) inactive, SFRP5 active and Bone erosion as output. The Bone erosion gets deactivated in the presence of SFRP5 and the absence of TNFSF11. d Simulation with MMP1 inactive and Matrix degradation as output. Matrix degradation phenotype gets deactivated in the absence of MMP1. e Simulation with FASLG active, AKT2 active, BID inactive and Apoptosis as output (RA original global model). Apoptosis gets activated in the presence of FASLG, AKT2 and the absence of BID. f Simulation with FASLG active, AKT2 active, BID inactive and Apoptosis as output (RA modified global model). Apoptosis gets inactive in the presence of FASLG, AKT2 and the absence of BID due to the dominant-negative regulator CAV1.
Cell proliferation
When PDGFA was set as active (1), cell proliferation was activated (Fig. 4b).
Bone erosion
When TNFSF11 (RANKL) was set as inactive (0) and the negative regulator SFRP5 as active (1), simulations showed the deactivation of bone erosion (Fig. 4c).
Matrix degradation
MMP1 was set as inactive by mutating its value as 0 (because it is an internal component and not an input to the model, its value must be fixed so that it cannot be changed by any upstream regulation, leaving it at 0 for the duration of the simulation) with matrix degradation chosen as the output. The simulation resulted in the deactivation of the matrix degradation (Fig. 4d).
Apoptosis
FASGL and AKT2 were set as active (1) and BID inactive (0). In the unmodified RA-FLS global model, apoptosis was found to be active in this condition, while in the modified global model, apoptosis was found to be inactive due to the dominance of the CAV1-negative regulator (as seen in Figs. 4e and 5f).

a Trap spaces show that along with TP53 and MDM2, mitochondrial proteins like BAX and cytoplasmic protein CASP9 remain unfixed. b Simulation with CC shows TP53 and MDM2 oscillations while all inputs remain active. c Simulation with CC shows oscillatory behaviour of the mitochondrial proteins when all inputs remain active.
The results of the simulations are consistent with those from the trap space experiments for all scenarios tested.
Reproducing known oscillatory behaviour of the P53-MDM2 interactions using the RA-FLS model
P53 is known to contribute to oscillatory behaviour through the P53 -MDM2 interactions. In short, a negative feedback loop on p53 is produced when p53 stimulates Mdm2 transcription, which in turn targets p53 for destruction. These P53 and MDM2 feedback-loop oscillations have been validated by mathematical and experimental models59,60 in various cell types and conditions. Moreover, TP53 directly takes part in the intrinsic apoptosis process by interacting with the multidomain members of the Bcl-2 family, causing mitochondrial outer membrane permeabilisation61. We wanted to see if the modified RA-FLS model could reproduce this dynamic behaviour. First, we calculated trap spaces when all inputs were active. The trap spaces analysis showed that the values of the proteins BAX, CASP9, TP53 and MDM2 are unfixed, while those of proteins BAD and BID are fixed in an active state, and protein BCL2 is fixed in an inactive state (Fig. 5a). The modified RA-FLS model was further analysed to study if the unfixed proteins exhibited an oscillatory behaviour under these conditions. We used the web-based modelling platform Cell Collective62 and asynchronously simulated the model setting all inputs as active. As seen in Fig. 5b, the model can reproduce the oscillations between TP53 and MDM2 entities and in Fig. 5c, we observe the oscillations in the intrinsic mitochondrial pathway of apoptosis.
Reproducing mono and combined drug therapy in RA
Disease-modifying antirheumatic drugs (DMARDs), such as conventional synthetic (csDMARDs) (methotrexate, hydroxychloroquine), biologic (TNF-α inhibitors, IL-6 inhibitors), and targeted synthetic medications (tsDMARDs) (pan-JAK- and JAK1/2-inhibitors), have provided the most encouraging outcomes for the treatment of RA63,64. Many drugs targeting TNF and IL-6 are already established in clinical treatment65. While the symptoms of inflammation and pain could be relieved with the cocktails of these medications, 30–40% of the patients fail to fully respond to treatment and experience periods of disease remission and relapse66. In addition, the side effects caused by most medications and the financial cost limit their use after a specific dosage64,67. In Table 1, we provide a list of the mono and combined drug therapies used to treat RA. The RA-FLS model can mimic the effects of the drugs and predict the outcome of combined perturbations.
Analysis was performed with the modified version of the RA-FLS model, DEFAULT input conditions as 1, as they are closer to the inflamed conditions in the RA joint. In addition, we set CAV1 as 1 to reproduce the apoptosis-resistant phenotype of the cells. Results shown in Fig. 6 confirm that IL-6, targeted by Sarilumab, Tocilizumab and JAK, targeted by Tofacitinib, Baricitinib and Itacitinib, were able to downregulate inflammation. Under these conditions, all other phenotypes, except apoptosis, were found to be active and not impacted by the drug treatment, implying that their regulation is more complex and requires additional targeting. We also tested drug combinations that can be administered to RA patients. The combination of Methotrexate and Sarilumab was seen to deactivate inflammation successfully. Our results corroborate the experimental findings of clinical trials that demonstrated better efficacy of the sarilumab targeting IL-6, either as monotherapy versus adalimumab, which targets TNF or in combination with csDMARDs versus placebo and csDMARDs68. Indeed, in our modelling framework, all results with anti-TNF drugs were inadequate to entirely suppress the inflammation phenotype, suggesting that blocking TNF alone is insufficient to hamper the signal. Our results also follow the findings of ref. 69, which demonstrated that IL-6 suppression was more successful in downregulating key transcription factors leading to inflammation in patients treated with anti-TNF treatment.

Heatmap showing identified trap spaces regarding the five phenotypes while testing different drug targets with DEFAULT inputs conditions as 1 with the modified version of the RA-FLS model.
Identification of novel targets and candidate drugs via drug repurposing analysis
The main objective of our modelling study was to find conditions that can induce apoptosis and downregulate cell proliferation, inflammation, matrix degradation and bone erosion in RA-FLS. This included identifying targets and available drugs that could act upon those targets. To proceed, we decided to focus on the direct upstream regulators of each phenotype, study their role based on the logical formula that describes their effect on the phenotype, and search for the available drugs in clinical trials and in vivo and in vitro studies. The available drugs targeting the upstream regulators of each phenotype can be seen in Supplementary Table 5.
In our model, one of the core conditions to reproduce RA-FLS behaviour is setting CAV1 as always active (being a negative regulator of apoptosis), implying a downregulation of its inhibitor, MIR192, which is also an input. This condition is sufficient to reproduce the RA-FLS resistance to apoptosis along with the activation of cell proliferation, inflammation, bone erosion and matrix degradation. We kept the same default model conditions (all inputs active and MIR192 inactive) for the drug-targeting simulations using the modified RA-FLS model and the Cell Collective simulation platform62 (Supplementary Fig. 3).
According to the logical formula, apoptosis is activated when CAV1 is inhibited and at least one activator is present. Interestingly, the negative regulator of apoptosis, CAV1, can be targeted by third-generation bisphosphonates (BPs), as demonstrated in various studies, although not always RA-relevant. Incadronate inhibits CAV1 expression in PC3 prostate cells70. Furthermore, studies on human epithelial fibroblasts show the negative impact of BPs on the expression of genes essential for their growth and differentiation at medium-low therapeutic doses in the long term71,72. As a substitute for patients who did not respond to conventional medication or who did not have easy access to biologics treatment, BPs have also been tested in RA patients. More specifically, scientists studied intravenous pamidronate administration for refractory rheumatoid arthritis. Results showed that pamidronate infusions were beneficial for most patients, but the alleviation of symptoms did not last for more than six months73. More recently, Zoledronic acid (ZA) was tested in combination with Methotrexate (MTX) in 66 RA patients for its efficacy in inhibiting RA disease activity. The combined treatment of ZA and MTX effectively reduced disease activity, fracture risk and bone pain in patients with RA-derived secondary osteoporosis74. A clinical trial involving 28 patients to assess the effects of ZA on patients with early-stage RA and low disease activity was concluded in 2018, but the results are not yet available (https://clinicaltrials.gov/ct2/show/NCT02123264). ZA has also been used to induce cell apoptosis in human and murine osteoclast precursors and mature osteoclast-like cells by triggering ROS- and GSK-3β-mediated Mcl-1 downregulation75. According to our model, BPs could also alleviate RA symptoms and inflammation, as they could inhibit CAV1 and activate apoptosis in RA-FLS. The inhibitory effects of ZA on tumour-related growth factor IL-6 in hormone-resistant prostate cancer cell lines have also been studied. The study showed the inhibitory effect of ZA on IL-6 secretion, which in turn resulted in increased apoptosis76. Keeping the default model conditions, we performed the simulations targeting CAV1 with Zoledronic acid. The results showed that when CAV1 is inhibited, apoptosis will become activated (Supplementary Fig. 4).
Inflammation in RA is one of the most characteristic symptoms. The inflammation phenotype in the RA-FLS model has thirteen direct upstream regulators. Among them, we find TNF, IL-6, IL17A, IL1B and NFKB, which represent common RA targets, and chemokines, such as CXCL8, CXCL9, CXCL10 and CXC11, two interferon proteins IFNB1, IFNA1, and lastly IRF1, IRF5 and IRF7. From all the direct upstream regulators, IRF1, 5 and 7 seem to control the fate of phenotype and are all regulated upstream by the IL-6 pathway. We performed simulations under the default conditions targeting IL-6, resulting in the complete inactivation of inflammation (Supplementary Fig. 5).
Bone erosion is directly dependent on the activation of eight upstream regulators. Besides TNF, IL17A and IL1B, known targets of RA therapy, we also find IL7, TNFRSF11, FOS, JUN and NFATC1 to exert direct control over the phenotype. We were able to find drugs that inhibit all five of them, with one having been in clinical trials for patients with multiple sclerosis (MS), but based on misrepresentation of preclinical data (GSK2618960 for IL7R). Results showed that simultaneous blocking of IL7 and AP-1 (JUN and FOS) leads to the inactivation of bone erosion under the default model conditions (Supplementary Fig. 6).
Matrix degradation in the RA-FLS model seems to be controlled exclusively by zinc-dependent endopeptidases, namely four MMPs and a disintegrin and metalloproteinase with thrombospondin motif (ADAMTS) member, ADAMTS4. Among these proteins, MMP3 is the one that exerts complete control over the phenotype for the given default conditions, and targeting only MMP3 was sufficient to inhibit matrix degradation (Supplementary Fig. 7).
Regarding cell proliferation, eight direct upstream regulators can affect the phenotype, namely TNF, NFKB, P38, BCL2, RPS6KB1, ADAMTS9, CREB1 and YWHAQ. TNF and NFKB are both usual targets of RA therapy. Regarding NFKB, Bortezomib is a proteasome inhibitor that has been shown to downregulate its pathway in cancer cells77. Bortezomib was also shown to attenuate murine collagen-induced arthritis78 and was recently found to improve the joint manifestations of rheumatoid arthritis in three patients79. P38 was once considered a promising target for developing new anti-inflammatory drugs to treat RA and other inflammatory diseases. However, the results in clinical trials were disappointing80. We identified drugs for two more upstream regulators, namely BCL2 and CREB1, and drugs that could target FOXO1, the upstream transcription factor of YWHAQ. In default conditions, among the eight regulators, CREB1 and YWHAQ were shown to inhibit completely the cell proliferation phenotype when targeted (Supplementary Fig. 8).
To recapitulate, the RA-FLS model was used to predict the best combinations for activating apoptosis and inhibiting inflammation, cell proliferation, bone erosion and matrix degradation. According to the model, the desired effect can be achieved by blocking CAV1, IL-6, IL7 and AP-1 (JUN and FOS), CREB1 and YWHAQ, and MMP3. For blocking CAV1, we have three potential drugs, Pamidronate, Incadronate and Zoledronic Acid (ZA), with the latter having been tested for RA (Supplementary Table 5). Regarding IL-6, Tocilizumab and Sarilumab are common biologics administered to RA patients (Table 1). IL7 activity can be targeted with GSK2618960, a monoclonal antibody against IL7R (Supplementary Table 5). Concerning AP-1, we identified Acitretin and T-5224. Acitretin, a retinoid, has been tested in clinical trials for various diseases. While Acitretin acts by binding and activating all nuclear subtypes of retinoid X receptors and retinoic acid receptors, it has been suggested that retinoic acid and its receptors regulate the differentiation and proliferation of epidermal keratinocytes by acting as an antagonist of activating protein-1 (AP-1). In psoriasis, it was also used in a small cohort of patients in combination with Etanercept (anti-TNF) to improve response to therapy, with encouraging results. T-5224, a selective inhibitor of c-Fos/AP-1, has been tested in a murine model of acute kidney injury (Supplementary Table 5). For CREB1, we could only identify one inhibitor, 666-15, while for YWHAQ, we identified AS1842856, which can target and inhibit FOXO1, an upstream regulator of YWHAQ. However, both inhibitors have only been tested in mice studies. Lastly, for MMP3, a variety of broad-spectrum MMP inhibitors are available. Some have already been tested in RA patients (like Cipemastat or Apratastat) but failed to produce significant results. Most drugs targeting MMPs, in general, and MMP3, in particular, have been tested primarily on cancer disease (Supplementary Table 5).
Discussion
In this work, we focused mainly on the mechanisms regulating inflammation, matrix degradation, bone erosion, apoptosis, and cell proliferation, in RA-FLS. Our framework presents the experimental and clinical data and model results in an approachable manner which links various software applications and data. To build our large-scale model, we used the state-of-the-art RA map19 and the map-to-model framework proposed in ref. 33. Five-phenotype-specific modules and a global model were extracted and further enriched using RA-FLS-specific literature and single-cell data. The inferred models were further analysed using a variety of software, such as GINsim81, bioLQM40, MaBoSS82 and the CoLoMoTo notebook35. To our knowledge, the RA-FLS model is the first large-scale model that describes these cells’ signalling and gene regulation mechanisms in disease-specific settings.
Rheumatoid arthritis fibroblasts, are cells that possess complex traits, undergo phenotypic transformations and contribute to the sustained inflammation of the joints and the disease perpetuation. A simple phenomenological model cannot grasp the way multiple molecular pathways act in coordination to regulate this transformation. The debilitating symptoms of the disease include more than one phenotypic outcome, and one cannot claim to study sufficiently the mechanisms responsible for these outcomes by focusing only on a single subgroup of them. Especially so, when the aim is to propose novel targets, and probable repurposed drugs and combinations of them. While the challenge and the complexity are not negligible, Boolean formalism with its qualitative nature, can be used to efficiently scale up simulations, especially for converging signalling networks.
To evaluate the model’s behaviour, we mined experimental evidence based on cell-specific small-scale studies (knockouts and knock-ins) from the literature. We used value propagation34,36, trap spaces computation83,84, asynchronous simulations62,85 and continuous time Boolean stochastic modelling82 to analyse the models’ behaviour and evaluate the results. The employed methodologies not only reduced the amount of time needed to evaluate the behaviour of our large-scale models but also provided the means to comprehend intricate outcomes under specific circumstances.
The RA-FLS model has successfully reproduced various experimental observations impacting the five phenotypes of interest. However, inconsistencies were also identified. For example, while working on apoptosis, we observed the inability of the apoptosis phenotype to be inactive in the presence of a negative regulator. It highlighted the insufficient knowledge related to the biological processes involved in regulating the phenotype and the limitations of the automated model inference. To leverage the missing information, we modified the logical formula making the inhibitor dominant, a choice also justified by experimental evidence. The modified RA-FLS model reproduces many biological scenarios based on experimental evidence, at least in the input–output scale and for most of the phenotypes tested.
Furthermore, it successfully reproduces known oscillatory behaviour regarding TP53 and MDM2 along with the mitochondrial pathway proteins. The modules and the global model helped to understand the input and output (phenotype) relationships in a rather extensive network, along with the understanding of the regulatory processes at different biological scales. Considering it as a first step for understanding the RA-FLS behaviour in different sets of initial conditions, the model contributes toward a better understanding of the mechanisms that drive cell proliferation, inflammation, and resistance to apoptosis, bone erosion, and matrix degradation.
We used the RA-FLS model to perform mono and combined drug therapy simulations using drugs and combinations already administered to RA patients to understand better each drug’s different mechanisms of action and the combined effects on the cellular phenotypic outcomes. With all inputs active and mimicking the inflamed joint, IL-6 produces a stronger signal for inflammation, as TNF and IL-17 require the additional activation of NFkB to regulate inflammation. In addition, while different DMARDs have distinct modes of action, they interfere primarily with the main pathways of the inflammation cascade86. The activation of cell proliferation during drug testing and drug combinations shows that additional growth factors and signalling pathways are involved in controlling this process. As PDGFA is the only growth factor targeted in this instance, it seems insufficient to exert substantial control over the phenotype. Regarding bone erosion, the WNT non-canonical pathway signalling protein RAC12 (targeted by Methotrexate and Azathioprine) exerts partial control over the phenotype. However, bone erosion can also be triggered by other routes like RANKL (TNFSF11) and the WNT canonical pathway, thus keeping bone erosion active.
The next step was to identify possible targets that could enhance the desired effect of the treatment. Our goal was to achieve activation of apoptosis, suppression of inflammation, bone erosion, matrix degradation and cell proliferation for RA-FLS. To do so, we identified the direct regulators of the five different phenotypes and used drug repurposing analysis to identify drug candidates for these targets. More specifically, we used the logical formulae of the Boolean model to identify the direct regulators of each phenotype that could change the phenotypic state if targeted. We used several databases and dedicated sites to infer information about drugs and small molecules that target the action of these regulators. Some of these drugs had already been tested in RA or were administered in the context of other diseases. We had to tag discontinued drugs due to side effects, even when administered in a different disease context. For some drugs, information on toxicity or trial reports was limited. For example, CTS-1027, an MMP inhibitor, had been in phase II clinical trials by Conatus Pharmaceuticals (licensed from Roche) to treat Hepatitis C Virus (HCV) infection. However, this study was discontinued due to abnormalities and adverse events in a subset of clinical trial participants. This compound, originally discovered by Roche, showed positive results for osteoarthritis treatment, but no development report is available for this study (https://www.pharmacodia.com/yaodu/html/v1/chemicals/8651920f3ba2e5f8bcd3e58ba0b48584.html). Similarly, Marimastat, a broad MMP inhibitor, was discontinued in phase II trials, as it demonstrated drug toxicity linked to musculoskeletal pain and stiffness initially involving small peripheral joints of the hands87,88.
While our framework can be used to identify potential drugs and drug targets, we cannot assess toxicity or drug dosage but suggest new potentially interesting drug combinations to be tested experimentally. In Table 2, we recapitulate the identified drugs for the highlighted targets. Combining them could help activate apoptosis and downregulate inflammation, matrix degradation, bone erosion and cell proliferation in RA-FLS.
Our analyses demonstrated that besides input–output relationships, it is also necessary to understand and elucidate the underlying mechanisms that control the different phenotypes. Unit testing, wherein collections of tests specify the anticipated behaviour connected with distinct system modules, could be applied to verify local behaviour (see refs. 34,36). This could be of particular interest for understanding the complexity of the apoptosis mechanism in relation to the extrinsic and intrinsic (mitochondrial) pathways. Moreover, the resulting inconsistencies between prior knowledge and model behaviour could serve as a helpful indicator for re-evaluating the inferred logical formulae. The availability of more quality datasets could help to analyse the models under different sets of initial parameters and study their relation to the phenotypic outcomes. Furthermore, topological analysis of the model’s structural characteristics could reveal hubs and nodes with high centrality measures89, which could guide the combination of in silico knockouts (KOs) and knock-ins (KIs) for simulating the models.
Our approach supports using systems modelling in preclinical and clinical settings to provide insights into the possible outcomes of drug combinations. Further optimisation of the RA-FLS model using patient-specific omic data integrated with other clinical parameters could help towards personalised treatments.
Materials and methods
Generation of RA-FLS phenotype-specific modules and the global model
Analysing complex biological networks often requires a workflow that includes several different tools with different specifications and requires many additional steps to harmonise the whole procedure. The CoLoMoTo Interactive Notebook provides an integrated environment to execute, share, and reproduce analyses of qualitative models of biological networks35. The framework is available as a Docker image on Linux, MacOS and Microsoft Windows with the tools already installed and ready to be run with a Jupyter web interface executing the codes and visualising the results. The resulting notebook files can be shared and re-executed in the same environment. After installing the docker and accessing the latest colomoto-docker-V 2022-10-01 image, the notebook interface with Python modules for different tools was available to execute the analysis.
A global, comprehensive and fully annotated RA-specific map was published recently19,90. This map features components and interactions implicated in RA coming from various cell types. The RA-map CellDesigner XML file was first imported into the notebook using BioLQM and then converted into an executable RA-map model using CaSQ (CellDesigner as SBML-Qual), a tool for automated inference of large-scale, parameter-free Boolean models from molecular interaction maps based on network topology and semantics33. The executable RA model, in standard SBML-qual format91, can be further simulated and analysed using different modelling tools supporting the SBML-qual format. First, the RA model was sanitised using bioLQM to replace the model node Ids with node names. Then, the model was further used to extract five-phenotype-specific modules, apoptosis, cell proliferation, inflammation, matrix degradation, bone erosion, and a global model. The modules were extracted by selecting the phenotype and extracting its upstream part of the model through the connected components. Within this framework, we developed Python notebooks to validate RA-FLS-specific biological scenarios, drug testing and drug combination predictions.
Model annotation and use of standards
Besides building and analysing large-scale biological models, one of the main challenges in systems modelling is the use of standards and proper annotation of such models to enhance a model’s reusability92,93. The models obtained in our pipeline are in a standard format (SBML-Qual) and thoroughly annotated using the MIRIAM scheme94, including PubMed IDs and pathway identifiers to annotate components and reactions. However, these elements are maintained and visualised only when the models are imported into the Cell Collective platform62 and lost when the models are further analysed with our CoLoMoTo notebooks due to the sanitisation process.
Literature survey to find RA-FLS-specific biological scenarios
As our modelling efforts are based on RA-FLS, we did exhaustive literature mining to find biological scenarios to be tested for each phenotype with the individual modules and the global model. As a result, we formulated biological scenarios to be tested for each phenotype from the literature-5 for inflammation, 5 for bone erosion, 4 for cell proliferation, 2 for matrix degradation, and 4 for apoptosis phenotype (for more details, see Supplementary Table 2).
Identification of trap spaces
We used terminal trap spaces to evaluate the asymptotic behaviour of our RA-FLS model. A trap space (also known as a stable motif or symbolic steady state) is a subspace from which the system cannot escape83,84. In particular, all possible successors of every state of a trap space belong to the same subspace. A trap space is terminal if it does not contain any smaller trap space. Terminal trap spaces accurately represent the Boolean model’s asymptotic behaviour and provide a good approximation of all attractors in practice.
Trap spaces can be efficiently identified using constraint-solving techniques without performing the simulation. Here we used the implementation provided by the bioLQM software for the identification of terminal trap spaces in reduced models obtained after input propagation, as explained below.
Propagation of fixed components
Value propagation works on the propagation of assigned constant values to the corresponding downstream nodes. First, the cellular context is defined by assigning constant values to some model components, followed by a model reduction technique34. Then, for each fixed (constant) node, the corresponding value is inserted into the logical rule associated with each target node. If the rule can then be simplified to a constant, this new fixed value is further propagated into the logical rules of downstream nodes. This process is iterated until no further simplification can be made on the logical rules of the model36. Value propagation decreases the complexity of the model while preserving all attractors and terminal trap spaces.
Continuous time stochastic simulations
In this type of simulation, continuous time phenotypic probabilities are being calculated. The probabilities associated with each phenotype represent the number of stochastic simulations leading to each phenotype from predefined initial conditions (based on experimental evidence that mimic the disease state).
MaBoSS is a C + + software for simulating continuous/discrete-time Markov processes applied to Boolean networks58,82. MaBoSS uses a specific language for associating transition rates with each node. Given some initial conditions, MaBoSS applies Monte-Carlo kinetic algorithm to the network to produce time trajectories and thus can associate probabilities with asymptotic solutions. The MaBoSS simulations were used to recreate at least one biological situation from each phenotype in the form of time-dependent (max time 40) trajectories using chosen parameters, more details of which could be found in the provided Python notebooks.
Simulations using the web-based platform Cell Collective
We also used the modelling platform Cell Collective to simulate the global models62. Models in Cell Collective can be imported via the SBML-qual standard (CaSQ-produced SBML-qual model file was imported) or built from scratch. The Cell Collective SBML-qual import is compatible with model annotations and network layout. Furthermore, references kept in CellDesigner’s XML file’s MIRIAM section can be retrieved and seen in the Cell Collective platform.
Modified RA model
While validating the biological scenarios in the original model, we observed that while Apoptosis should have been OFF, the phenotypic node was always active. The inability of the phenotype to turn OFF was further examined by studying the regulatory rules, which indicated a lack of mechanistic information on the biological process. CAV1 was part of the OR rule for the apoptosis regulators, as can be seen below:
!CAV1_rna | CASP3_phosphorylated | CASP8 | TNFRSF10A_rna | TNFRSF10B_rna where `|` represents a disjunction and `!` is a negation.
The Boolean rule was modified appropriately to add CAV1 as a dominant regulator:
!CAV1_rna&(CASP3_phosphorylated | CASP8 | TNFRSF10A_rna | TNFRSF10B_rna, where `&` represents a conjunction.
This rule change generated a modified global model where apoptosis remains OFF in the presence of a dominant-negative regulator corresponding to an actual RA condition where apoptosis remains deficient.
Five-phenotype-specific individual modules (apoptosis, cell proliferation, inflammation, matrix degradation and bone erosion) and the global model were then extracted from the modified RA model. Finally, all the biological scenarios were again tested with the modified individual modules and the five phenotypes global model and compared with the unmodified modules and model results.
Drug-targeting (single, multiple targets) and drug combination analysis
We identified RA treatments that are administered regularly to RA patients and target different molecules, some of which are also RA-FLS specific (Table 2). We used both global models to simulate and analyse drug effects on the five selected phenotypes—Apoptosis, Cell proliferation, Inflammation, Matrix degradation and Bone erosion. We analysed the effect on the phenotypes for each drug by targeting its single and multiple targets in the global models. We also analysed the models by combining multiple drugs and their targets.
Drug repurposing
We checked the logical formulae of all five phenotypes and extracted their upstream first-level regulators, as shown in Supplementary Table 5. For each regulator, we searched drugs with all clinical phases - launched, preclinical, phase 1, phase 2, and phase 3. For the latter, besides bibliographic search, the following sites were also used: https://clue.io/repurposing-app95; http://db.idrblab.net/ttd/96; https://go.drugbank.com/97. The results of this effort can be seen in Table 2, where the asterisk * denotes drugs already used in RA. We also thoroughly studied the mechanism of action of all the drugs (wherever information was available). We checked the default behaviour of the RA-FLS modified model concerning the five phenotypes by keeping all the inputs active and MIR192 inactive. MIR192 is input and needs to be OFF to activate the negative regulator of apoptosis CAV1, thus contributing to the state of RA pathology.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All data are available in the GitLab repository of the lab: https://gitlab.com/genhotel/rheumatoid-arthritis-large-scale-computational-modeling/-/tree/main.
References
McInnes, I. B. & Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 365, 2205–2219 (2011).
McInnes, I. B. & Schett, G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 7, 429–442 (2007).
Ostrowska, M., Maśliński, W., Prochorec-Sobieszek, M., Nieciecki, M. & Sudoł-Szopińska, I. Cartilage and bone damage in rheumatoid arthritis. Reumatologia 56, 111–120 (2018).
Noack, M. & Miossec, P. Selected cytokine pathways in rheumatoid arthritis. Semin. Immunopathol. 39, 365–383 (2017).
Alunno, A., Carubbi, F., Giacomelli, R. & Gerli, R. Cytokines in the pathogenesis of rheumatoid arthritis: new players and therapeutic targets. BMC Rheumatol. 1, 3 (2017).
Wijbrandts, C. A. & Tak, P. P. Prediction of response to targeted treatment in rheumatoid arthritis. Mayo Clin. Proc. 92, 1129–1143 (2017).
Rein, P. & Mueller, R. B. Treatment with biologicals in rheumatoid arthritis: an overview. Rheumatol. Ther. 4, 247–261 (2017).
Ge, X. et al. Functional genomics atlas of synovial fibroblasts defining rheumatoid arthritis heritability. Genome Biol. 22, 247 (2021).
Bartok, B. & Firestein, G. S. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol. Rev. 233, 233–255 (2010).
Turner, J. D. & Filer, A. The role of the synovial fibroblast in rheumatoid arthritis pathogenesis. Curr. Opin. Rheumatol. 27, 175–182 (2015).
Choy, E. H. & Panayi, G. S. Cytokine pathways and joint inflammation in rheumatoid arthritis. N. Engl. J. Med. 344, 907–916 (2001).
Boyle, W. J., Simonet, W. S. & Lacey, D. L. Osteoclast differentiation and activation. Nature 423, 337–342 (2003).
Sato, K. & Takayanagi, H. Osteoclasts, rheumatoid arthritis, and osteoimmunology. Curr. Opin. Rheumatol. 18, 419–426 (2006).
Burrage, P. S., Mix, K. S. & Brinckerhoff, C. E. Matrix metalloproteinases: role in arthritis. Front. Biosci. 11, 529–543 (2006).
Yoshihara, Y. & Yamada, H. [Matrix metalloproteinases and cartilage matrix degradation in rheumatoid arthritis]. Clin. Calcium 17, 500–508 (2007).
Sandler, C. et al. Imatinib mesylate inhibits platelet derived growth factor stimulated proliferation of rheumatoid synovial fibroblasts. Biochem. Biophys. Res. Commun. 347, 31–35 (2006).
Zhao, J., Jiang, P., Guo, S., Schrodi, S. J. & He, D. Apoptosis, autophagy, netosis, necroptosis, and pyroptosis mediated programmed cell death as targets for innovative therapy in rheumatoid arthritis. Front. Immunol. 12, 809806 (2021).
Peter, M. E. Programmed cell death: apoptosis meets necrosis. Nature 471, 310–312 (2011).
Singh, V. et al. RA-map: building a state-of-the-art interactive knowledge base for rheumatoid arthritis. Database 2020, baaa017 (2020).
Rougny, A. et al. Systems biology graphical notation: process description language level 1 version 2.0. J. Integr. Bioinform. 16, 20190022 (2019).
Mazein, A. et al. Systems medicine disease maps: community-driven comprehensive representation of disease mechanisms. NPJ Syst. Biol. Appl. 4, 21 (2018).
Ostaszewski, M. et al. Community-driven roadmap for integrated disease maps. Brief. Bioinforma. 20, 659–670 (2019).
Touré, V., Flobak, Å., Niarakis, A., Vercruysse, S. & Kuiper, M. The status of causality in biological databases: data resources and data retrieval possibilities to support logical modeling. Brief. Bioinformatics 22, bbaa390 (2021).
Baker, M., Denman-Johnson, S., Brook, B. S., Gaywood, I. & Owen, M. R. Mathematical modelling of cytokine-mediated inflammation in rheumatoid arthritis. Math. Med. Biol. 30, 311–337 (2013).
Macfarlane, F. R., Chaplain, M. A. J. & Eftimie, R. Quantitative predictive modelling approaches to understanding rheumatoid arthritis: a brief review. Cells 9, 74 (2019).
Moise, N. & Friedman, A. Rheumatoid arthritis - a mathematical model. J. Theor. Biol. 461, 17–33 (2019).
Macfarlane, F. R., Chaplain, M. A. J. & Eftimie, R. Modelling rheumatoid arthritis: a hybrid modelling framework to describe pannus formation in a small joint. ImmunoInformatics 6, 100014 (2022).
Aghakhani, S., Soliman, S. & Niarakis, A. Metabolic reprogramming in rheumatoid arthritis synovial fibroblasts: a hybrid modeling approach. PLoS Comput. Biol. 18, e1010408 (2022).
Hemedan, A. A., Niarakis, A., Schneider, R. & Ostaszewski, M. Boolean modelling as a logic-based dynamic approach in systems medicine. Comput. Struct. Biotechnol. J. 20, 3161–3172 (2022).
Hall, B. A. & Niarakis, A. Data integration in logic-based models of biological mechanisms. Curr Opin. Syst. Biol. 100386. https://doi.org/10.1016/j.coisb.2021.100386 (2021).
Niarakis, A. & Helikar, T. A practical guide to mechanistic systems modeling in biology using a logic-based approach. Brief. Bioinformatics 22, bbaa236 (2021).
Abou-Jaoudé, W. et al. Logical modeling and dynamical analysis of cellular networks. Front. Genet. 7, 94 (2016).
Aghamiri, S. S. et al. Automated inference of Boolean models from molecular interaction maps using CaSQ. Bioinformatics 36, 4473–4482 (2020).
Saadatpour, A., Albert, Ré., Reluga, T. C. A reduction method for Boolean network models proven to conserve attractors. SIAM Stud. Appl Math. 12, 1997–2011 (2013).
Naldi, A. et al. The Colomoto interactive notebook: accessible and reproducible computational analyses for qualitative biological networks. Front. Physiol. 9, 680 (2018).
Hernandez, C., Thomas-Chollier, M., Naldi, A. & Thieffry, D. Computational verification of large logical models-application to the prediction of T cell response to checkpoint inhibitors. Front. Physiol. 11, 558606 (2020).
Zerrouk, N., Aghakhani, S., Singh, V., Augé, F. & Niarakis, A. A mechanistic cellular atlas of the rheumatic joint. Front. Syst. Biol. 2, 925791 (2022).
Zerrouk, N., Miagoux, Q., Dispot, A., Elati, M. & Niarakis, A. Identification of putative master regulators in rheumatoid arthritis synovial fibroblasts using gene expression data and network inference. Sci. Rep. 10, 16236 (2020).
Mizoguchi, F. et al. Functionally distinct disease-associated fibroblast subsets in rheumatoid arthritis. Nat. Commun. 9, 789 (2018).
Naldi, A. Biolqm: a Java toolkit for the manipulation and conversion of logical qualitative models of biological networks. Front. Physiol. 9, 1605 (2018).
Matsuno, H. et al. The role of TNF-alpha in the pathogenesis of inflammation and joint destruction in rheumatoid arthritis (RA): a study using a human RA/SCID mouse chimera. Rheumatology 41, 329–337 (2002).
Hayden, M. S. & Ghosh, S. Regulation of NF-κB by TNF family cytokines. Semin. Immunol. 26, 253–266 (2014).
Liu, T., Zhang, L., Joo, D. & Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2, 17023 (2017).
Sønder, S. U. et al. IL-17-induced NF-kappaB activation via CIKS/Act1: physiologic significance and signaling mechanisms. J. Biol. Chem. 286, 12881–12890 (2011).
Hata, K. et al. IL-17 stimulates inflammatory responses via NF-kappaB and MAP kinase pathways in human colonic myofibroblasts. Am. J. Physiol. Gastrointest. Liver Physiol. 282, G1035–G1044 (2002).
van der Laan, W. H. et al. Cartilage degradation and invasion by rheumatoid synovial fibroblasts is inhibited by gene transfer of TIMP-1 and TIMP-3. Gene Ther. 10, 234–242 (2003).
Cici, D., Corrado, A., Rotondo, C. & Cantatore, F. P. Wnt signaling and biological therapy in rheumatoid arthritis and spondyloarthritis. Int. J. Mol. Sci. 20, 5552 (2019).
Chen, H. et al. Bone marrow sFRP5 level is negatively associated with bone formation markers. Osteoporos. Int. 28, 1305–1311 (2017).
Pettit, A. R. et al. TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am. J. Pathol. 159, 1689–1699 (2001).
Kwon, Y.-J., Lee, S.-W., Park, Y.-B., Lee, S.-K. & Park, M.-C. Secreted frizzled-related protein 5 suppresses inflammatory response in rheumatoid arthritis fibroblast-like synoviocytes through down-regulation of c-Jun N-terminal kinase. Rheumatology 53, 1704–1711 (2014).
MacLauchlan, S. et al. Genetic deficiency of Wnt5a diminishes disease severity in a murine model of rheumatoid arthritis. Arthritis Res. Ther. 19, 166 (2017).
Rosengren, S., Corr, M. & Boyle, D. L. Platelet-derived growth factor and transforming growth factor beta synergistically potentiate inflammatory mediator synthesis by fibroblast-like synoviocytes. Arthritis Res. Ther. 12, R65 (2010).
Zhang, K.-S. et al. Effects of tumor necrosis factor alpha on the expression of programmed cell death factor 5 in arthritis. Orthop. Surg. 11, 698–704 (2019).
Thornton, S. C. et al. Identification of the major fibroblast growth factors released spontaneously in inflammatory arthritis as platelet derived growth factor and tumour necrosis factor-alpha. Clin. Exp. Immunol. 86, 79–86 (1991).
Remmers, E. F., Sano, H. & Wilder, R. L. Platelet-derived growth factors and heparin-binding (fibroblast) growth factors in the synovial tissue pathology of rheumatoid arthritis. Semin. Arthritis Rheum. 21, 191–199 (1991).
Funahashi, A., Morohashi, M., Matsuoka, Y., Jouraku, A. & Kitano, H. Celldesigner: a graphical biological network editor and workbench interfacing simulator. in Introduction to Systems Biology (ed. Choi, S.) 422–434 (Humana Press, 2007).
Li, S., Jin, Z. & Lu, X. MicroRNA-192 suppresses cell proliferation and induces apoptosis in human rheumatoid arthritis fibroblast-like synoviocytes by downregulating caveolin 1. Mol. Cell. Biochem. 432, 123–130 (2017).
Stoll, G. et al. MaBoSS 2.0: an environment for stochastic Boolean modeling. Bioinformatics 33, 2226–2228 (2017).
Lev Bar-Or, R. et al. Generation of oscillations by the p53-Mdm2 feedback loop: a theoretical and experimental study. Proc. Natl Acad. Sci. USA 97, 11250–11255 (2000).
Lahav, G. Oscillations by the p53-Mdm2 feedback loop. Adv. Exp. Med. Biol. 641, 28–38 (2008).
Vaseva, A. V. & Moll, U. M. The mitochondrial p53 pathway. Biochim. Biophys. Acta 1787, 414–420 (2009).
Helikar, T. et al. The Cell Collective: toward an open and collaborative approach to systems biology. BMC Syst. Biol. 6, 96 (2012).
Radu, A.-F. & Bungau, S. G. Management of rheumatoid arthritis: an overview. Cells 10, 2857 (2021).
Lin, Y.-J., Anzaghe, M. & Schülke, S. Update on the pathomechanism, diagnosis, and treatment options for rheumatoid arthritis. Cells 9, 880 (2020).
Bullock, J. et al. Rheumatoid arthritis: a brief overview of the treatment. Med. Princ. Pract. 27, 501–507 (2018).
Haschka, J. et al. Relapse rates in patients with rheumatoid arthritis in stable remission tapering or stopping antirheumatic therapy: interim results from the prospective randomised controlled RETRO study. Ann. Rheum. Dis. 75, 45–51 (2016).
Wang, W., Zhou, H. & Liu, L. Side effects of methotrexate therapy for rheumatoid arthritis: a systematic review. Eur. J. Med. Chem. 158, 502–516 (2018).
Genovese, M. C. et al. Efficacy and safety of sarilumab in combination with csDMARDs or as monotherapy in subpopulations of patients with moderately to severely active rheumatoid arthritis in three phase III randomized, controlled studies. Arthritis Res. Ther. 22, 139 (2020).
Miagoux, Q. et al. Inference of an integrative, executable network for rheumatoid arthritis combining data-driven machine learning approaches and a state-of-the-art mechanistic disease map. J. Pers. Med. 11, 785 (2021).
Iguchi, K., Matsunaga, S., Nakano, T., Usui, S. & Hirano, K. Inhibition of caveolin-1 expression by incadronate in PC-3 prostate cells. Anticancer Res. 26, 2977–2981 (2006).
Manzano-Moreno, F. J. et al. Impact of bisphosphonates on the proliferation and gene expression of human fibroblasts. Int. J. Med. Sci. 16, 1534–1540 (2019).
Jung, J. et al. Effects of an oral bisphosphonate and three intravenous bisphosphonates on several cell types in vitro. Clin. Oral. Investig. 22, 2527–2534 (2018).
Salesi, M., Mottaghi, P., Karimifar, M. & Farajzadegan, Z. Intravenous pamidronate for refractory rheumatoid arthritis. J. Res. Med. Sci. 17, 422–427 (2012).
Xie, J. et al. Zoledronic acid ameliorates the effects of secondary osteoporosis in rheumatoid arthritis patients. J. Orthop. Surg. Res. 14, 421 (2019).
Tai, T.-W. et al. Reactive oxygen species are required for zoledronic acid-induced apoptosis in osteoclast precursors and mature osteoclast-like cells. Sci. Rep. 7, 44245 (2017).
Asbagh, L. A., Uzunoglu, S. & Cal, C. Zoledronic acid effects interleukin-6 expression in hormone-independent prostate cancer cell lines. Int. Braz. J. Urol. 34, 355–363 (2008).
Sung, M.-H. et al. Dynamic effect of bortezomib on nuclear factor-kappaB activity and gene expression in tumor cells. Mol. Pharmacol. 74, 1215–1222 (2008).
Lee, S. W., Kim, J. H., Park, Y. B. & Lee, S. K. Bortezomib attenuates murine collagen-induced arthritis. Ann. Rheum. Dis. 68, 1761–1767 (2009).
Lassoued, S. et al. Bortezomib improved the joint manifestations of rheumatoid arthritis in three patients. Jt. Bone Spine 86, 381–382 (2019).
Canovas, B. & Nebreda, A. R. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 22, 346–366 (2021).
Chaouiya, C., Naldi, A. & Thieffry, D. Logical modelling of gene regulatory networks with GINsim. Methods Mol. Biol. 804, 463–479 (2012).
Stoll, G., Viara, E., Barillot, E. & Calzone, L. Continuous time Boolean modeling for biological signaling: application of Gillespie algorithm. BMC Syst. Biol. 6, 116 (2012).
Zañudo, J. G. T. & Albert, R. An effective network reduction approach to find the dynamical repertoire of discrete dynamic networks. Chaos 23, 025111 (2013).
Klarner, H., Bockmayr, A. & Siebert, H. Computing symbolic steady states of boolean networks. in Cellular Automata (eds Wąs, J. et al.) Vol. 8751, 561–570 (Springer International Publishing, 2014).
Helikar, T., Kowal, B. & Rogers, J. A. A cell simulator platform: the cell collective. Clin. Pharmacol. Ther. 93, 393–395 (2013).
Benjamin, O., Goyal, A. & Lappin, S. L. Disease modifying anti-rheumatic drugs (DMARD). In StatPearls (StatPearls Publishing, Treasure Island (FL), 2022).
Keystone, E. Treatments no longer in development for rheumatoid arthritis. Ann. Rheum. Dis. 61, ii43–ii45 (2002).
Rasmussen, H. S. & McCann, P. P. Matrix metalloproteinase inhibition as a novel anticancer strategy: a review with special focus on batimastat and marimastat. Pharmacol. Ther. 75, 69–75 (1997).
Durón, C., Pan, Y., Gutmann, D. H., Hardin, J. & Radunskaya, A. Variability of betweenness centrality and its effect on identifying essential genes. Bull. Math. Biol. 81, 3655–3673 (2019).
Singh, V. et al. Computational systems biology approach for the study of rheumatoid arthritis: from a molecular map to a dynamical model. Genomics Comput. Biol. 4, (2018).
Chaouiya, C. et al. SBML qualitative models: a model representation format and infrastructure to foster interactions between qualitative modelling formalisms and tools. BMC Syst. Biol. 7, 135 (2013).
Niarakis, A. et al. Addressing barriers in comprehensiveness, accessibility, reusability, interoperability and reproducibility of computational models in systems biology. Brief. Bioinforma. 23, bbac212 (2022).
Niarakis, A. et al. Setting the basis of best practices and standards for curation and annotation of logical models in biology-highlights of the [BC]2 2019 CoLoMoTo/SysMod Workshop. Brief. Bioinforma. 22, 1848–1859 (2021).
Le Novère, N. et al. Minimum information requested in the annotation of biochemical models (MIRIAM). Nat. Biotechnol. 23, 1509–1515 (2005).
Corsello, S. M. et al. The Drug Repurposing Hub: a next-generation drug library and information resource. Nat. Med. 23, 405–408 (2017).
Zhou, Y. et al. Therapeutic target database update 2022: facilitating drug discovery with enriched comparative data of targeted agents. Nucleic Acids Res. 50, D1398–D1407 (2022).
Wishart, D. S. et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 46, D1074–D1082 (2018).
Huang, J. et al. Promising therapeutic targets for treatment of rheumatoid arthritis. Front. Immunol. 12, 686155 (2021).
Rosengren, S., Corr, M., Firestein, G. S. & Boyle, D. L. The JAK inhibitor CP-690,550 (tofacitinib) inhibits TNF-induced chemokine expression in fibroblast-like synoviocytes: autocrine role of type I interferon. Ann. Rheum. Dis. 71, 440–447 (2012).
Li, G. et al. Celastrol inhibits lipopolysaccharide-stimulated rheumatoid fibroblast-like synoviocyte invasion through suppression of TLR4/NF-κB-mediated matrix metalloproteinase-9 expression. PLoS ONE 8, e68905 (2013).
Fang, Z. et al. High-throughput study of the effects of celastrol on activated fibroblast-like synoviocytes from patients with rheumatoid arthritis. Genes 8, 221 (2017).
Terabe, F. et al. Imatinib mesylate inhibited rat adjuvant arthritis and PDGF-dependent growth of synovial fibroblast via interference with the Akt signaling pathway. Mod. Rheumatol. 19, 522–529 (2009).
Bergström, B., Carlsten, H. & Ekwall, A.-K. H. Methotrexate inhibits effects of platelet-derived growth factor and interleukin-1β on rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Res. Ther. 20, 49 (2018).
Spurlock, C. F. et al. Methotrexate-mediated inhibition of nuclear factor κB activation by distinct pathways in T cells and fibroblast-like synoviocytes. Rheumatology 54, 178–187 (2015).
Mertens, M. & Singh, J. A. Anakinra for rheumatoid arthritis: a systematic review. J. Rheumatol. 36, 1118–1125 (2009).
Boyapati, A. et al. Sarilumab plus methotrexate suppresses circulating biomarkers of bone resorption and synovial damage in patients with rheumatoid arthritis and inadequate response to methotrexate: a biomarker study of MOBILITY. Arthritis Res. Ther. 18, 225 (2016).
Genovese, M. C. et al. Sarilumab plus methotrexate in patients with active rheumatoid arthritis and inadequate response to methotrexate: results of a phase III study. Arthritis Rheumatol. 67, 1424–1437 (2015).
Tugwell, P. et al. Combination therapy with cyclosporine and methotrexate in severe rheumatoid arthritis. The Methotrexate-Cyclosporine Combination Study Group. N. Engl. J. Med. 333, 137–141 (1995).
Fox, R. I. et al. Combined oral cyclosporin and methotrexate therapy in patients with rheumatoid arthritis elevates methotrexate levels and reduces 7-hydroxymethotrexate levels when compared with methotrexate alone. Rheumatology 42, 989–994 (2003).
Willkens, R. F. & Stablein, D. Combination treatment of rheumatoid arthritis using azathioprine and methotrexate: a 48 week controlled clinical trial. J. Rheumatol. Suppl. 44, 64–68 (1996).
Willkens, R. F., Sharp, J. T., Stablein, D., Marks, C. & Wortmann, R. Comparison of azathioprine, methotrexate, and the combination of the two in the treatment of rheumatoid arthritis. A forty-eight-week controlled clinical trial with radiologic outcome assessment. Arthritis Rheum. 38, 1799–1806 (1995).
Schapink, L., van den Ende, C. H. M., Gevers, L. A. H. A., van Ede, A. E. & den Broeder, A. A. The effects of methotrexate and hydroxychloroquine combination therapy vs methotrexate monotherapy in early rheumatoid arthritis patients. Rheumatology 58, 131–134 (2019).
Carmichael, S. J., Beal, J., Day, R. O. & Tett, S. E. Combination therapy with methotrexate and hydroxychloroquine for rheumatoid arthritis increases exposure to methotrexate. J. Rheumatol. 29, 2077–2083 (2002).
Inui, K. & Koike, T. Combination therapy with biologic agents in rheumatic diseases: current and future prospects. Ther. Adv. Musculoskelet. Dis. 8, 192–202 (2016).
Boleto, G., Kanagaratnam, L., Dramé, M. & Salmon, J.-H. Safety of combination therapy with two bDMARDs in patients with rheumatoid arthritis: A systematic review and meta-analysis. Semin. Arthritis Rheum. 49, 35–42 (2019).
Korhonen, R. & Moilanen, E. Abatacept, a novel CD80/86-CD28 T cell co-stimulation modulator, in the treatment of rheumatoid arthritis. Basic Clin. Pharmacol. Toxicol. 104, 276–284 (2009).
Author information
Authors and Affiliations
Contributions
A.Ni. designed the study, supervised, performed simulation experiments, and analysed results. V.S. did literature search, contributed to notebooks’ development, calculated trap spaces, performed simulation experiments, and analysed the results. A.Na. designed and developed notebooks for trap spaces and input propagation and performed verification experiments. S.S. advised on methodology, supervised and analysed results. A.Ni. and V.S. prepared figures and wrote the first draft of the manuscript. All authors contributed text and read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Singh, V., Naldi, A., Soliman, S. et al. A large-scale Boolean model of the rheumatoid arthritis fibroblast-like synoviocytes predicts drug synergies in the arthritic joint. npj Syst Biol Appl 9, 33 (2023). https://doi.org/10.1038/s41540-023-00294-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41540-023-00294-5
