
Abstract
Influenza A virus, with the limited coding capacity of 10–14 proteins, requires the host cellular machinery for many aspects of its life cycle. Knowledge of these host cell requirements not only reveals molecular pathways exploited by the virus or triggered by the immune system, but also provides further targets for antiviral drug development. To uncover novel pathways and key targets of influenza infection, we assembled a large amount of data from 12 cell-based gene-expression studies of influenza infection for an integrative network analysis. We systematically identified differentially expressed genes and gene co-expression networks induced by influenza infection. We revealed the dedicator of cytokinesis 5 (DOCK5) played potentially an important role for influenza virus replication. CRISPR/Cas9 knockout of DOCK5 reduced influenza virus replication, indicating that DOCK5 is a key regulator for the viral life cycle. DOCK5’s targets determined by the DOCK5 knockout experiments strongly validated the predicted gene signatures and networks. This study systematically uncovered and validated fundamental patterns of molecular responses, intrinsic structures of gene co-regulation, and novel key targets in influenza virus infection.
Similar content being viewed by others


Introduction
The influenza A virus (IAV), a member of the Orthomyxoviridae family, is the causal agent of an acute respiratory tract infection suffered annually by 5–20% of the human population. IAV can cause high mortality in humans, with 250,000–500,000 deaths per year worldwide.1 In pandemic years, influenza infection can lead to even higher mortality rates, as seen in the most extreme case with the 1918 Spanish influenza pandemic.2 Of particular concern is the threat of emerging highly pathogenic avian influenza viruses such as H5N1 and H7N9, which—although not easily transmissible human-to-human—have an unusually high death rate. Current treatments are focused on vaccines and drugs that target viral proteins. However, both of these approaches have limitations as vaccines require annual development to match the antigenic strains circulating, while viral proteins have an impressive capacity to evolve resistance against anti-viral agents.3 With the expression of 14 functional proteins for viral replication and virulence, the repertoire of gene products on the pathogen side is limited. The viral life cycle and the replication of the IAV are dependent on hijacking host-cell biological processes to facilitate entry, replication, assembly, and budding. The recognition that a suite of mammalian host proteins is required for IAV infection and replication presents additional targeting strategies that may be less prone to deflections by the quickly mutating viral genome.
IAV entry is a dynamic process that is comprised of six different steps:4 (i) attachment to the target cell, (ii) internalization into cellular compartments, (iii) endosomal trafficking to the perinuclear region, (iv) fusion of viral and endosomal membranes, (v) uncoating, and (vi) import of the viral genome into the nucleus. After nuclear import three more steps are required: genome replication/transcription and translation; vRNP transport from the nucleus to the cytoplasm; and virus assembly and release.5
Influenza infection activates a number of host defense pathways, including the innate and adaptive immune responses, the induction of cytokines, and activation of apoptosis.6 The detection of viral particles (in particular nuclear acids) by toll-like receptors (TLR7) of the MyD88, NF-kB pathway,7,8 as well as cytosolic proteins such as RIG-I (DDX58)9,10 of the MDAF/MAVS pathway and their trigger of interferon expression via the activation of transcription factors including IRF3 and IRF7, have been well studied. Unfortunately, much less is known about downstream host defense factors and signaling pathways.
Large-scale genome-wide studies of viral host factors and corresponding cellular networks have been conducted since 2008. Because RNAi-based screening technology was not well established in mammalian cells at that time, Drosophila was tested as an experimental platform to characterize host–virus interactions during influenza infections.11 With the development of mammalian RNAi-based screening, a comprehensive analysis of mammalian host cell functions in influenza virus replication became feasible.12,13,14 Known protein–protein interactions were used and superimposed with the RNAi screening data to construct functional host–pathogen interaction networks relevant for the influenza life cycle. A different tactic was then employed15 to combine yeast two-hybrid analyses, genome-wide transcriptional gene expression profiling, and an RNAi screening. More recent approaches revisited transcriptomic data but used weighted gene co-expression network analysis (WGCNA) to construct a host-influenza regulatory network.16
In this study, we employed an integrative network-based approach to identify host response co-expression networks in influenza A virus infection. Integration of differential gene expression, data driven correlation, and co-expression networks enabled reconstruction of novel signaling maps underlying influenza infection and host response. We predicted and validated DOCK5 as a key driver and potential host factor that is important for influenza virus infection.
Results
Twelve cell-based time-series gene expression data sets from ten published studies were assembled (Table 1) to identify key processes and key regulators in influenza infections. Cell lines, such as human alveolar basal epithelial cells (A549) and cultured human airway epithelial cells (Calu-3), as well as primary human bronchial epithelial cells (HBEC) were used to capture the overall response of the primary target tissues after influenza infection. The influenza A viruses used for infection include strains of subtypes H1N1 (including the resurrected strain of the 1918 pandemic), H5N1, H5N2, and H9N2. The cell-based experimental platforms include time-series data up to 72 h post-infection but the majority cover time post-infection to 24 h. Thus this study focused primarily on the initial steps of the influenza infection and the host innate immune response.
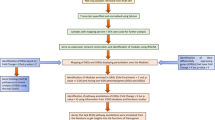
These data sets were processed separately by an integrative network analysis approach (Fig. 1), which was primarily based on weighted gene co-expression networks.17,18 We first identified differentially expressed genes in each data set that showed significant change (significant response genes, SRG) or trend (Jonckheere Trend Genes, JTG) during influenza infection. Individual sets of differentially expressed genes (DEG) were further assembled to consensus sets. Weighted co-expression network analysis was applied to each data set to identify modules of highly co-regulated genes that were further analyzed to derive consensus modules through a clustering analysis of the module–module similarity matrix (see details in the Methods section). The consensus modules were prioritized by their enrichment for both SRGs and JTGs, and the key regulators (hub genes) in each module were determined by network connectivity. A top key regulator (DOCK5) of the top ranked module involved in viral replication was validated through CRISPR-Cas9-based knockout experiments. The impact of DOCK5 knockout on the replication of the virus in A549 cells was determined by titration of the culture supernatants using a TCID50 assay. Molecular responses to DOCK5 knockout, i.e., DOCK5’s target genes, were determined by RNA sequencing of the DOCK5-knockout cell lines and were used to validate the predicted gene networks and differentially expressed gene signatures.

Overview of the proposed integrative network-based approach to influenza infection. a Twelve publically available gene expression data sets for studying influenza infection were curated. b ANOVA was used to identify significant response genes (SRG) that were differentially expressed by taking into account the time-series. c Genes showing significant up-regulation or down-regulation along the time-series were identified by the Jonckheere trend analysis and they were called Jonckheere trend genes (JTG). d Weighted co-expression network analysis was applied to each data set to identify modules comprised of highly co-regulated genes. Consensus modules were further determined by the clustering analysis of the similarities between all the modules from the 12 data sets. e The consensus modules were rank-ordered by their enrichment for both SRGs and JTGs. f Key regulators of the top-ranked module were determined by network connectivity. Key driver centered un-weighted co-expression networks were then constructed by correlation analysis of the individual data sets. g Key regulator (DOCK5) predicted by the network analysis was validated through CRISPR-Cas9-based knockout experiments. h The impact of DOCK5 knockout on the replication of the virus in A549 cells was determined by titration of the culture supernatants using a TCID50 assay. i Molecular response to DOCK5 knockout, i.e., DOCK5’s target genes, was determined by RNA sequencing data from the CRISPR-Cas9-based knockout experiments. j The predicted gene networks and differentially expressed gene signatures were validated by their enrichment of DOCK5 target genes identified through the validation experiments
Gene set enrichment analysis with well-established gene sets from gene ontology (GO)19 and MSigDB20 were used to assess biological functions. Influenza-specific processes were evaluated by enrichment calculations using published gene sets including the influenza host factors,5,21 the inflammasome,22 the interferon stimulated genes (ISGs)23, and the known host defense factors from InnateDB.24 All p values reported in the text are corrected for multiple testing unless otherwise specified. These gene sets, along with a number of abbreviations used throughout the manuscript, are described in Table 2.
Differential expression analysis uncovers genes responding to influenza infection
Given the time series information across the different data sets, we were particularly interested in the temporal response of expressed genes. For this purpose, ANOVA was used to identify differentially expressed genes across the time series (SRGs; see Supplementary Information for details). In addition, a complementary non-parametric Jonckheere trend analysis25 was used to identify significant up-regulated or down-regulated genes across all measured time points (JTGs) using a threshold of 0.05 for the corrected p-values.
The conservation of SRGs and JTGs across the 12 data sets was then evaluated. Although the data sets include a variety of different cell lines infected with different influenza virus subtypes and strains, the resulting SRG signatures significantly overlap with each other based on the Fisher’s exact test (FET) (p-values range between 1.37e−11 and 0). As shown in Fig. S1, there are three groups without explicit preference of virus strain or cells, including (i) a large group of H1N1, H5N1, and H5N2 virus strains infecting A549 and Calu-3 cells; (ii) the H1N1/1918 strain with the original (gn37_8) and modified NS1 protein (gn37_7); and (iii) H9N2 avian influenza in A549 cells (gn31H9), and H1N1/PR8 in HBEC (gn19).
We identified 2898 SRGs, including 416 up-regulated JTGs (JTGsup), 1526 down-regulated JTGs (JTGsdown), and 956 non-JTGs, in at least 6 of the 12 studies at a false discovery rate of 5%, while 1462 SRGs (146 JTGsup, 711 JTGsdown, and 605 non-JTGs) were common to at least seven studies (Fig. 2a). Enriched pathways were characterized by gene otology (GO) categories and MSigDB gene signatures (Fig. 2b; Table S1). As expected, the up-regulated genes are involved in immune system response, in particular type 1 interferon signaling (FET p = 1.09e−6, 3.9-fold). Well conserved are the host defense pathways comprised of MX1, IFITM, IRF7, and OAS, which are known ISGs that control IAV infections,26 among other interferon-induced genes. The down-regulated genes are associated with small molecule/lipid metabolic processes and localization. The heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) is the highest ranked gene significantly expressed in all 12 data sets and predominantly down-regulated (in 9/12 data sets). It is a member of a family of ubiquitously expressed hnRNPs, which are RNA-binding proteins associated with pre-mRNAs in the nucleus and that influence pre-mRNA processing, as well as other aspects of mRNA metabolism and transport. HnRNPA1 is one of the most abundant core proteins of hnRNP complexes; it plays a key role in the regulation of alternative splicing.27 Together with splicing factor 2 (SF2), it regulates alternative splicing of interferon regulator factor-3 (IRF3).28 Mediated by transportin 1 (TNPO1),29 hnRNPA1 shuttles between the nucleus and the cytosol.30 Other highly ranked members of the hnRNP family involve the related hnRNPD and the synaptotagmin binding cytoplasmic RNA interacting protein (SYNCRIP), both significantly expressed in 11 data sets. Although SYNCRIP is not known to be involved in influenza infection, it is a host factor involved in hepatitis C virus RNA replication,31 and required by the HCV IRES for translation-competent 48S complex formation.32 SYNCRIP has previously been reported to be associated with immune functions.33 Other SRGs, potentially responsible for host defense, include the CD59 molecule (a cell surface glycoprotein that is involved in lymphocyte signal transduction), C–C motif chemokine ligand 5 (CCL5) and MALT paracaspase that may play a role in NFκB activation. A list of the best DEGs together with their consensus trend are shown in Table S2. Due to the diverse cell lines and virus strains used, even well conserved SRGs such as hnRNPA1, which is an SRG in all 12 data sets, show diverse gene expression with up-regulation in 2 and down-regulation in 9 data sets. Similarly, host defense genes experience diverse regulation depending on infected tissue and viral strain. For example, IRF7 is up-regulated in 10/12 data sets, MX1 is up-regulated in 9/12 data sets, and RIG-I (DDX58) is up-regulated in 7/12 and down-regulated in 3/12 data sets.

Conservation of the differentially expressed gene sets including ANOVA-based SRGs and Jonckheere trend analysis-based JTGs, across multiple data sets. SRGs(n) and JTGs(n) refer to the SRGs and JTGs shared by n data sets (n = 1, 2, …, n), respectively. a The number of SRGs(n) (black line), the number of up-regulated JTGs(n) (red line), and the number of down-regulated JTGs(n) (blue line). b A Venn diagram of the overlap between SRGs(7), and the up-regulated and down-regulated JTGs(7). Biological functions refer to the pathways enriched in the corresponding sets and the numbers in parentheses indicate the corrected FET p-values
Co-expression network analysis reveals intrinsic gene–gene co-regulation structures underlying influenza infection
To understand how the genes interact with each other during influenza infection, we performed WGCNA of the 12 data sets.17,34,35 The 12 weighted co-expression networks consist of a total of 1191 modules with 9–8529 members (Fig. S2). We further identified 282 consensus modules (CMs) of sizes between 20 and 2500 through an average link-based hierarchical clustering method using the Jaccard similarity measure for modules (see Supplemental Information). These consensus modules were ranked by the significance of the enrichment for the previously identified DEG signatures (see Methods and Table 3). The most important CM (turquoise) captures common system responses against influenza infections enriched for GO biological processes, such as translational elongation/termination (FET p = 4.7e−5, 4.8-fold) and viral reproduction (FET p = 4.0e−7, 2.3-fold). Enrichment analysis using the canonical pathway collection from MSigDB20 identifies similar virus response-related pathways, such as peptide chain elongation (FET p = 3.6e−5, 4.4-fold) and influenza life cycle (FET p = 2.5e−5, 3.6-fold) enriched in the top ranked consensus module. This turquoise CM is also most enriched for the known influenza targets. Other CMs are involved in more specific processes during the influenza infection process. Figure S3 shows the detailed information about the 100 top-ranked CMs through a circular heatmap representation of the enrichment for the DEG signatures, GO/MSigDB functions as well as influenza, inflammation, and innate immunity gene sets (Table 3).
Meta-analysis of connectivity in the co-expression networks of the 12 data sets, by summing log 10 (network connectivity) obtained from the weighted co-expression networks, identified MDM2 (MDM2 proto-oncogene) and DOCK5 (dedicator of cytokinesis 5) as the top two most connected genes among the SRGs (see Table S3). Both DOCK5 and MDM2 are differentially expressed in 9 of the 12 data sets.
While the functions of MDM2 have been well studied (it was mentioned by 7312 papers in PubMed), very little is known about DOCK5’s function in general (only 35 papers about DOCK5 in PubMed). P53 modulation by MDM2 during viral infection has been reported in the literature. MDM2 and p53 polymorphisms were show to be associated with the development of hepatocellular carcinoma in patients with chronic hepatitis B virus infection.36 Furthermore, the MDM2-dependent inhibition of p53 is required for Epstein-Barr virus B-cell growth transformation and infected cell survival.37 With respect to IAV infection, the accumulation of p53 in IAV infected cells is due to the stabilization of p53 associated with compromised MDM2-mediated ubiquitination of p53.38 Jonckheere trend analysis reveals that MDM2 is up-regulated in 5 data sets and down-regulated in 4, whereas DOCK5 is up-regulated in 1 data set and down-regulated in 5 data sets (Fig. 3). In comparison, typical host defense genes, such as chemokine CCL5, interferon α inducible protein 27 (IFI27), OAS2, and IRF7 are up-regulated in the majority of the data sets. In contrast, known host factors, such as NXF1 (up-regulated and down-regulated in 3 data sets and 1 data set, respectively), COPA (up-regualted and down-regulated in 2 and 5 data sets, respectively), or SF3B1 (up-regulated and down-regulated in 2 data sets and 1 data set, respectively) show similar diverse expression across the data sets (Table S3). Thus, a simple expression analysis is insufficient to decipher the functional role of host factors during the influenza life cycle.

Expression profiles of DOCK5 in 12 data sets. The time-series responses of DOCK5 in the 12 data sets are shown together with the adjusted p-value after ANOVA time series analysis and significant trend after Jonckheere trend analysis. DOCK5 can be observed to be significantly up-regulated in 1 and down-regulated in 5 data sets - “Significant” is defined to be statistically significant after Jonckheere test and requiring an overall fold change of 1.2 or greater between measurements at time t = 0 day and the last time-point in the time-series
A DOCK5-centered network captures key biological processes in influenza infection
As WGCNA does not generate actual networks, we explicitly constructed DOCK5-centered unweighted co-expression networks for comparison with DOCK5 knockout (DOCK5-ko) signatures. To do this, we first identified the genes significantly (FDR < 0.05) correlated with DOCK5 in each of our 12 assembled data sets, and then defined the consensus correlations conserved across at least 7 of the data sets (Table S4). The genes significantly correlated with DOCK5 in at least n data sets are termed DOCK5-correlated consensus gene set, DOCK5-CCGS(n), where n = 2, 3, …, 12. Table S4 shows the sizes of these DOCK5-CCGS with corresponding genes listed in Table S5. Eight genes are correlated with DOCK5 in 11 data sets, including abhydrolase domain containing 2 (ABHD2), acetyl-CoA acyltransferase 2 (ACAA2), CD47 molecule, DEAD-box helicase 17 (DDX17), karyopherin subunit alpha 4 (KPNA4), tumor suppressor protein neurofibromin 2 (NF2), RuvB like AAA ATPase 1 (RUVBL1), and the trans-golgi network protein 2 (TGOLN2). ABHD2 was shown to be important in Hepatitis B virus propagation.39 DDX17 was shown to promote production of HIV-1 particles,40 facilitate viral RNA synthesis in H5N1 infection of human cells,41 and regulate alternative splicing.42 KPNA4 is an importin and docks proteins with NLS signals to the nuclear pore complex. NF2 coordinates collective migration of epithelial cells.43 RUVBL1 regulates the Fanconi anaemia core complex.44
To understand the functions of DOCK5 in influenza infection and defense, we performed a comprehensive functional analysis of the DOCK5-centered network conserved in a majority of the data sets, i.e., DOCK5-CCGS(7), through enrichment tests of known pathways and relevant gene signatures, as well as the previously identified differentially expressed gene sets including SRGs(7) and JTGs(7). We distinguished JTGs(7) with up-trends, i.e., JTGsup(7), from those with down-trends (JTGsdown). We considered the following gene signatures in addition to functional GO and MSigDB genes sets: (i) two sets of influenza host factors derived from siRNA data5,21 (sets i a and i b, respectively), (ii) a set of “inflammasome” genes,22 (iii) a set of host defense genes relevant for innate immunity,24 and (iv) a set of interferon-stimulated genes.23 DOCK5-CCGS(7) is enriched for a number of pathways including the ER-nucleus signaling, response to ER stress, RNA localization, Golgi vesicle transport, viral process, modulation by virus of host morphology, RNA splicing, and cellular protein metabolic process (Table S6). The 985 genes shared by DOCK5-CCGS(7) and SRGs(7) (FET p < 1e−320, 4.3-fold) are involved in processes required for both the viral life cycle and antiviral host-defense. DOCK5-CCGS(7) and JTGsup(7) share 226 genes (FET p = 6.2e−76, 3.5-fold) that are associated with interferon signaling and host response to virus, while the 762 genes shared by DOCK5-CCGS(7) and JTGdown(7) (FET p = 1.1e−239, 3.2-fold) are involved in lipid and fatty acid metabolism (Tables S7 and S8).
Known influenza host factors are enriched in DOCK5-CCGS(7) (FET p = 0.035, 1.6-fold) (Table S9). The inflammasome signature is significantly enriched in DOCK5-CCGS(7) (FET p = 3.6e-5, 1.2-fold), SRGs(7) (FET p = 1.2e−8, 1.4-fold) and JTGsdown(7) (FET p = 5.4e−7, 1.4-fold) (Table S10). The innate immune system-related genes curated by ImmuneDB are significantly enriched in DOCK5-CCGS(7) (FET p = 9.3e−10, 1.4-fold), SRGs (FET p = 5.6e−12, 1.7-fold), JTGsup(7) (FET p = 2.7e−15, 2.6-fold), but not in JTGdown(7), indicating the typical activation of innate immune response. All non-empty intersections between DOCK5-CCGS(7), SRGs(7), and JTGsup(7) are also enriched for the ImmuneDB genes (Table S11). However, the down-regulated innate immune system genes from the intersection of DOCK5-CCGS(7) and JTGsdown(7) (FET p = 0.05, 1.4-fold change) indicate potential modulation of the immune system by DOCK5 (see next section). DOCK5-CCGS(7) and SRGs(7) are significantly enriched for interferon stimulated genes (ISGs) with FET p = 2.3e−20 (3.0-fold) and 5.3e−20 (2.2-fold), respectively (Table S12), suggesting interferon stimulation in the DOCK5-centered network and the overall response. The analysis above demonstrates that the DOCK5-CCGS(7) network captures many aspects of the immune response.
DOCK5 is a potential host factor for influenza infection
To validate the functional role of DOCK5 during influenza infection, we knocked out DOCK5 in human lung epithelial A549 cell lines using the CRISPR/Cas9 genome editing system.45 Virus replication in these knockout cell lines was compared to that in the wild type parental A549 cell lines. Four DOCK5-ko A549 clones (DOCK5-c20, c25, c28, and c41) were selected and compared to the wild type A549 (A549-wt) for their capacity to support the replication of influenza virus. The cells were infected with an H1N1 virus (A/Puerto Rico/8/1934) or an H3N2 virus (A/New York/238/2005) and the viral titers at days 1, 2, and 3 post infection were determined using a TCID50 assay. The influenza viruses replicated to significantly lower titers in DOCK5-ko cells than in the parental A549 cell line (Fig. 4a, b). At day 1, IAV replication in the DOCK5-ko cells was statistically indistinguishable from replication in DOCK5-wt. At day 2, the absence of functional DOCK5 protein resulted in a 20-fold decrease (or 1.30 log10 reduction) of the H1N1 viral titer (8.12–363.08 fold decrease corresponding to 0.91–2.56 log10 reduction in the 4 DOCK5-ko cell lines compared to A549-wt) and an 8.51 fold decrease (or 0.93 log10 reduction) of the H3N2 viral titer (3.16–40.73-fold decrease corresponding to 0.50–1.61 log10 reduction in the 4 DOCK5-ko cell lines compared to A549-wt). At day 3, the effect of DOCK5 knockout was even more pronounced: the H1N1 viral titer was reduced by 436.51-fold or 2.64 log10 reduction (81.28–8709.64-fold decrease corresponding to 1.91–3.94 log10 reduction in the 4 DOCK5-ko cell lines) and the H3N2 viral titer decreased by 52.48-fold or 1.72 log10 (7.41–87.10-fold decrease corresponding to 0.87–1.94 log10 reduction in the 4 DOCK5-ko cell lines compared to DOCK5-wt). A hierarchical linear model (hLM) was employed using both the time past infection and wt/clone information as parameters. All 4 DOCK5-ko clones showed significantly different time series responses compared to the wild type cells (hLM p-values of range between 5.3e−7 and 5.7e−3; Table S13). Similar to some known host factors, DOCK5 moderately compromises influenza replication when using siRNA depletion (Fig. 5). The compromised replication of both the H1N1 virus (Fig. 4a) and the H3N2 virus (Fig. 4b) in the absence of functional DOCK5 protein indicates that DOCK5 is a potential host factor involved in the life cycle of influenza virus.

Effect of DOCK5 knockout on the replication of influenza virus in A549 cells. H1N1 virus (a) and H3N2 (b) virus were inoculated at MOI of 0.0005 TCID50/cell and 0.005 TCID50/cell, respectively, at day 0. The replication of the virus in the wild type and DOCK5 knockout A549 cells was determined by titration of the culture supernatants over 3 days using a TCID50 assay. Error bars represent standard deviation (SD). c Intersections among six selected DOCK5-ko DEG signatures are shown. The matrix of solid and empty circles at the bottom illustrates the “presence” (solid green) or “absence” (empty) of the gene sets in each intersection. The numbers to the right of the matrix are set sizes. The colored bars on the top of the matrix represent the intersection sizes with the color intensity showing the p-value significance. The intersections are ordered by p-values of the super exact test. The corresponding function for each gene set was also shown at the bottom and on the right hand-side of the matrix

Distribution of influenza host factors after siRNA screens. A set of 254 siRNA-based host-factors5,21 have been used to assess their effect on the influenza virus life cycle. Corresponding Z-scores have been scaled and combined. Host-factors with the lowest Z-scores, e.g., NXF1, induce most severe effect on viral replication after siRNA knock-down
Since the parental A549 cell line used for CRISPR/Cas9 knockout is composed of a heterogeneous population of cells, the clonal variability of the selected DOCK5-ko clones (DOCK5-c20, -c25, -c28, and -c41) may have resulted in the variable, though significantly reduced capacity of these cells to support the replication of influenza viruses (Fig. 4a, b). DOCK5-c28, whose cellular morphology and proliferation rate are closest to the A549-wt cells, was chosen as a representative clone to determine the effects of knockout perturbation on host RNA responses (Fig. 4c). Real-time qPCR was used to further quantify the expression level of some important genes in the wild type and DOCK5-c28 A549 cells, under infection conditions of Mock, H1N1, and H3N2.
The DOCK5-regulated transcriptome involves cytokinesis, vesicle trafficking, and splicing
The transcriptional program regulated by DOCK5 was characterized by sequencing mRNA from the cells of the representative clone DOCK5-c28 under different conditions as combinations of infection status (H1N1, H3N2, or Mock infection) and DOCK5 perturbation status (with or without DOCK5-ko). These data sets are referred to as the “validation data set”. Differential expression analysis was performed on the following “contrasts” using Bioconductor’s limma package: relative DOCK5-ko vs. wild type (the relative expression of a gene was the ratio of the gene’s expression in H1N1/H3N2 infection to that in the corresponding MOCK), and absolute DOCK5-ko vs. wild type (the absolute expression values in H1N1, H3N2, and MOCK infections were used). A fold change cutoff of 1.2 and an FDR threshold of 5% were employed to identify the corresponding DEG signatures.
Without influenza infection (i.e., MOCK infection), DOCK5 predominantly modulates cell migration, as indicated by the significant enrichment of cell receptor signaling (FET p = 1.3e−8, 2.4-fold), cell communication (4.9e−4, 1.3-fold), cell migration (FET p = 6.1e−3, 1.9-fold), extracellular matrix receptor interaction (FET p = 8.6e−3, 2.8-fold), and integrin pathways (FET p = 2.5e−2, 3.8-fold) in the DEG signature in the MOCK DOCK5-ko cells in comparison with the MOCK wildtype cells (Tables S14 and S15). Similarly, enriched pathways can be observed in the case of infection (Tables S16–S18). During infection, pathways that are up-regulated by the knockout of DOCK5 involve carbohydrate metabolism (FET p = 3.3e−4, 1.5-fold), in particular aminoglycan processes (FET p = 2.4e−3, 2.2-fold), lysosome functions (FET p = 5.8e−3, 2.1-fold), and phospholipid metabolism (FET p = 2.0e−2, 1.9-fold). Whereas down-regulated pathways in DOCK5-ko conditions compared to DOCK5-wt include cell adhesion (FET p = 1.0e−6, 1.6-fold), extracellular matrix receptor interaction (FET p = 9.4e−6, 2.8-fold), and integrin pathways (FET p = 3.0e−3, 2.5-fold). After removing genes responding to MOCK infection from the DOCK5-ko DEG signatures in the case of infection, we performed enrichment tests of functional pathways. DOCK5 knockout induces downregulation of cell adhesion (FET p = 2.7e−13, 1.7-fold), GPCR ligand binding (FET p = 6.1e−9, 2.2-fold), GPCR signaling (FET p = 5.2e−7, 1.9-fold), and extracellular matrix receptor interaction (FET p = 2.8e−6, 2.7-fold). However, no pathways were enriched in the case of upregulated genes by DOCK5-ko (Tables S19 and S20).
Figure 4c shows the highly significant overlap among these DEG signatures of DOCK5-ko (Tables S21 and S22 for details). The genes up-regulated by DOCK5-ko in both H1N1 and H3N2 infections are enriched for enzyme linked receptor protein signaling, cell communication, cell adhesion, and cell migration pathways; whereas the down-regulated genes are involved in single-multicellular organism process, generic transcription, and integrin pathways. Given the highly significant overlap between these DEG signatures under the H1N1 and H3N2 infections, we focus on the signature observed under H3N2 infection although results for all DEG signatures are presented in the supplementary documents. The 2863 up-regulated genes induced by DOCK5-ko under the H3N2 infection are termed sgDOCK5-DEG+, while the 4512 down-regulated genes are called sgDOCK5-DEG.
The striking conservation of the expression of hnRNPs and SYNCRIP across 11 studies suggests that splicing is a significant process during influenza infection. To further investigate this process, we evaluated differential exon splicing and exon usage based on the RNAseq data from the samples in the DOCK5-ko experiments. The number of genes that displayed differential exon usage ranges between 13 and 2851, depending on the specific scenario (Table S23). Among all the different scenarios, the cysteinyl-tRNA synthetase 2 (CARS), glucosyltransferase ALG5, and non-imprinted in Prader-Willi/Angelman Syndrome 1 (NIPA1) showed the most significant different exon usage. There are 137 exon variants in a collagen gene COL7A1, 118 exon variants in high density lipoprotein binding protein (HDLBP), and 112 exon variants in microtubule-actin crosslinking factor 1 (MACF1). COL7A1 is responsible for anchoring epithelial cells to the stroma, HDLBP is known to support cell proliferation, and MACF1 couples the microtubule network to cellular functions. Despite a small snapshot, these three top-ranked genes do suggest a role for DOCK5 in cytokinesis. In the MOCK infection, genes and corresponding pathways relevant for splicing are subject to differential splicing/exon usage (Table S24). H3N2 infection induces differential exon usage in pathways responsible for cellular macromolecular metabolic processes, i.e., protein modification/protein phosphorylation/signaling as well as regulation of apoptosis (Tables S25 and S26). A weaker, but nevertheless significant, differential exon usage can be observed during H1N1 infection with respect to viral process pathways and pathways responsible for virus-induced modulation of host morphology and physiology (not shown).
Validation of the DOCK5-centered network
We validated the previously identified consensus modules and DOCK5-centered networks (DOCK5-CCGS(n)) by the DEG signatures identified from the DOCK5 knockout experiments (sgDOCK5-DEGS) to achieve an in-depth understanding of the biological functions of DOCK5 (Fig. 6). This analysis was done for the DEG signatures from 15 configurations (Fig. S5 and S6; Table S27) a detailed discussion was included in the Supplemental Information. Among 282 consensus modules, 5 are significantly enriched for the genes up-regulated or down-regulated by DOCK5 during MOCK infection, 27 for the genes upregulated by DOCK5 knockout during H1N1 infection, 31 for the genes upregulated by DOCK5 knockout during H3N2 infection, 7 for the genes down-regulated by DOCK5 knockout during H1N1 infection, and 16 for the genes down-regulated by DOCK5 knockout during H3N2 infection. Eleven of the top 13 modules in Table 3 are significantly enriched for sgDOCK5-DEGS. These modules have different functions, such as viral reproduction, single organism cellular process, and organelle organization, indicating that DOCK5 regulates a diversity of biological processes during IAV infection.

Validation of DOCK5 centered gene co-expression networks (DOCK5-CCGS) by the gene signatures (sgDOCK5-DEGS) identified from the DOCK5 knockout experiments. a The enrichment of the DOCK5 centered network for the differentially expressed gene sets induced by the knockout of DOCK5 under H1N1, H3N2, and MOCK infections. The x-axis shows the DOCK5-centered co-expression network DOCK5-CCGS(n) conserved in at least n data sets, n = 2,3,…,12. The y-axis indicates the—log10 of corrected FET p-values. Boxed labels show corresponding fold-changes. b A subnetwork of DOCK5-CCGS(7), i.e., the genes correlated with DOCK5 in at least 7 data sets shows the genes shared by DOCK5-CCGS(7), DOCK5’s knockout signature (sgDOCK5-DEG+) and the union of the consensus differential expression gene signature SRGs(7) and the consensus down-regulated gene signature JTGsdown(7). The selected nodes are up-regulated by DOCK5 in either the H1N1 or H3N2 infection. The left and right color sectors in each node indicate whether it was differentially expressed in H1N1 infection (blue for significant differential expression and grey for non-significant one) and H3N2 infection (cyan for significant differential expression and grey for non-significant one), respectively
Highly significant overlap among DOCK5-CCGS(7), SRGs(7), JTGs(7), and sgDOCK5-DEG+ by the Super Exact Test (SET)46 strongly validated our predicted centered network, as shown in Fig. 7 (see also Fig. S4 and S7). The intersection of DOCK5-CCGS(7), SRGs(7), and sgDOCK5-DEG+ includes 24 interferon-stimulated genes, i.e. ISGs (Table S28). Among the 24 genes, only ISG20, IL6, and STAT1 are predominantly up-regulated across the data sets while all other genes are either up-regulated or down-regulated in comparable numbers of data sets. ISG20, an antiviral ribonuclease required for viral replication,47 shows a large difference in expression between the DOCK5-wt cells and DOCK5-ko cells. ISG20 was up-regulated by over 8-fold 2 days post-infection. In the DOCK5-ko cells, the expression of ISG20 increased to over 20-fold 2 days after post-infection.

Intersections amongst DOCK5-CCGS(7), SRGs(7), JTGsup(7), JTGsdown(7) and sgDOCK5-DEG+ by Super Exact Test. The bar chart plot shows the combinations of the five gene sets with non-empty intersections. The matrix of solid and empty circles at the bottom illustrates the “presence” (solid green) or “absence” (empty) of the gene sets in each intersection. The numbers to the right of the matrix are set sizes. The colored bars on the top of the matrix represent the intersection sizes with the color intensity showing the p-value significance. The intersections are ordered by p-values of the super exact test
Although DOCK5-CCGS(7) is not significantly enriched for sgDOCK5-DEG− (i.e., genes that are downregulated in DOCK5-ko compared to DOCK5-wt under the H3N2 virus infection), the genes in their intersection are involved in regulation of transcription and phosphate metabolic processes. Some of the most conserved up-regulated genes are in the overlap between DOCK5-CCGS(7) and sgDOCK5-DEG- and they include the vesicle associated membrane protein 5 (VAMP5), as well as cytokines. VAMP5 is a member of the VAMP/synaptobrevin family and the SNARE superfamily, and may participate in vesicle trafficking events potentially (indirectly) controlled by DOCK5. VAMP5 is significantly upregulated in DOCK5-wt cells. Under DOCK5-ko conditions, upregulation of VAMP5 is significantly reduced. Furthermore, VAMP5 is a curated target of the JUN transcription factor. Thus, VAMP5 expression could be potentially modulated by JUN, alternatively under direct control of DOCK5. The observed restriction of up-regulation in the DOCK5-ko cells compared to the wild type may indicate a decline in host defense response. As a functional DOCK5 is absent in DOCK5-ko, so are the DOCK5-dependent processes during influenza infection. Therefore expression of the predominately host defense genes (such as CXCL11, IRF7, or JUN) may no longer be required at a high transcriptional level. According to the ENCODE data, JUN, as a transcription factor, could potentially regulate DOCK5.48 In the RNAseq experiments, JUN itself is up-regulated in DOCK5-wt and not in the DOCK5-ko cells. Data from the library of integrated network-based cellular signatures (LINCS, see Supplemental Information for details) indicates a feedback between JUN and DOCK5, with JUN down-regulating DOCK5 as transcription factor and DOCK5 (indirectly) activating JUN.
The overlap between DOCK5-CCGS(7) and the sgDOCK5-DEGs consists of genes such as TGOLN2. As discussed above, TGOLN2 is significantly correlated with DOCK5 in 11 out of 12 data sets. The genes correlated with TGOLN2 within the DOCK5 neighborhood include golgins such as GOLGA2, GOLGA4, GOLGB1, GLG1, and GOLPH3. Proteomic analysis shows that GLG1 binds to the influenza virus PB1-F2 protein49 though the consensus siRNA screening result indicates it does not function as a host factor during the influenza virus life cycle.50 The intersection of DOCK5-CCGS(7) and the sgDOCK5-DEG+ also includes AP1S1, which is part of the clathrin coat assembly complex linking clathrin to receptors in coated vesicles. These vesicles are involved in endocytosis and Golgi processing. The DOCK5 network neighbors also include a member of the lysosomal ATPases (ATP6V1A) and COPG2, one of the 7 subunits of the coatomer 1 vesicular transport complex (COPI). The lysosomal (or vacuolar) ATPase complex was suspected to be required for influenza vesicle entry.14 A similar assessment has been made for COPI. Presumably, DOCK5 utilizes COPG2 as an entry point for COPI modulation.
Another process potentially modulated by DOCK5, is mRNA processing, in particular splicing. Prominent members of the spliceosome include hnRNPA1, hnRNPD, and SYNCRIP that are all commonly expressed as SRGs in almost all data sets, as discussed above. These 3 genes are correlated with DOCK5 in at least 8 data sets. SYNCRIP is a host factor that was shown to be involved in hepatitis C virus RNA replication,31 and required by the HCV IRES for translation-competent 48S complex formation.32 It was also previously found to be associated with immune functions.33 Other genes in the intersection between DOCK5-CCGS(7) and sgDOCK5-DEG+ are the Influenza virus NS1 binding protein (NS1BP, IVNS1ABP, or hnRNP-I) and splicing factors 3 (SF3A1, SF3A3, and SF3B1). NS1BP is a required viral host factor relevant for pre-mRNA processing, mRNA metabolism, and transport.5 It is also a key mediator of IAV gene expression, in particular viral RNA splicing.51 Furthermore, NS1BP modulates tumor suppressor, and potential host defense gene sirtuin 3 (SIRT3), which is down-regulated during infection in 9/12 data sets (with no significant directional change in expression in the remaining 3 data sets). The exosome component 6 (EXOSC6) is another member in the overlap that is differentially expressed in 9/12 data sets and significantly correlated with DOCK5 in 10/12 data sets. The exosome controls alternative splicing by mediating the gene expression and assembly of the spliceosome complex.52 EXOSC6, hnRNPs, NS1BP, and the splicing factors form a tight sub-network within DOCK5-CCGS(7). Nuclear pore proteins assemble an additional class of genes that are required for splicing/mRNA processing, as they are responsible for the transport of vRNPs into the nucleus. Our data show that NUP35, NUP50, NUP93, NUP133, and NUP210 are correlated with DOCK5.
The potential overall effect of DOCK5-ko on general cellular functions such as splicing has been evaluated by analyzing functional processes of the corresponding DEGs between the relative expression in DOCK5-wt vs. DOCK5-ko during H1N1/H3N2 infections, and the relative expression in DOCK5-wt vs. DOCK5-ko during MOCK infections (Tables S29 and S30). Disregarding directional DEG responses, functional processes involve multicellular organismal processes, extracellular matrix and structure organization, cell adhesion, and cell–cell signaling.
Pathways that are up-regulated in the DOCK5-wt scenario, i.e., pathways that we have identified to require DOCK5 functionality, involve extracellular matrix receptor interaction, GPCR ligand binding, and unfolded protein response. Whereas, pathways that are down-regulated, i.e., activated pathways after DOCK5-ko, are cellular glucuronidation, fibroblast apoptotic process, and cellular response to xenobiotic stimulus. These enriched functions may indicate a potential breakdown of cellular integrity due to lack of DOCK5 functionality. Although such a breakdown of cellular integrity can also be caused by viral infection and induced cell death, we are confident that such a scenario and the potential influence on the identified role of DOCK5 can be excluded. First, validation experiments and data sets with intermediate to high multiplicity of infections (MOIs) were used, the latter well controlled due to the consensus approach. Second, the identified effects between DOCK5 and target genes are correlation-based and the causal role of DOCK5 in this process has been identified in combination with the DOCK5-ko experiments, as described. A simple difference in proliferation/cell death would not display such a causal pattern with DOCK5 as center. Furthermore, pathways relevant for apoptotic processes have been observed in DOCK5-CCGS(7) as discussed above, but not in overall significantly expressed genes, such as SRGs(7) or JTGs(7) (see Table S7). Also, apoptotic pathways were only identified as being significantly expressed between DOCK5-wt and DOCK5-ko scenarios (Tables S29 and S30). Thus, the causal role of DOCK5 in these processes can be established and potential effects caused by virus-induced cell death ruled out.
Given the information on differential exon splicing/usage from the DOCK5-ko RNAseq data, genes and processes that show differential splicing effects induced by DOCK5 were further investigated. We specifically evaluated the overlap between DOCK5-induced differentially spliced genes and the DOCK5 network neighborhood (Table S31). DOCK5-CCGS(7) is most significantly enriched for the gene set of differential exon usage after H3N2 infection (FET p = 1.43e−119, 2.0-fold) and their overlap is associated with protein modification, signal transduction, regulation of localization, adherence junction, and regulation of the SMAD2/3/4 transcriptional activity (Tables S32 and S33). In particular, the overlap is enriched for influenza virus host factors,5 indicating that DOCK5 may be required for the regulation of mRNA processing and splicing of genes relevant for the influenza life cycle. We further explored if and how gene splicing affected by influenza infection differs from gene splicing potentially induced by DOCK5-modulated splicing processes. Differential splicing between influenza-infected and mock-infected cells were compared with or without the perturbation of DOCK5. DOCK5-CCGS(7) is enriched for the differentially spliced gene signatures (WT: H3N2 vs Mock, FET p = 1.1e−24, 2.2-fold change; DOCK5-ko: H3N2 vs. Mock, FET p = 5.8e−9, 2.0-fold change), the intersections are not enriched for influenza host factor signatures or the inflammasome. Furthermore, none of the original differentially spliced gene sets show any significant overlap with influenza host factor genes or inflammasome genes. Therefore, DOCK5 is likely to be required for proper splicing, in particular of host genes relevant for the influenza life cycle.
In order to verify the role of DOCK5 in modulating key cellular processes predicted by the network analysis and confirmed by the RNAseq data, we further measured mRNA levels of highly responding genes in DOCK5-CCGS(7) using real-time quantitative PCR (RT-qPCR). The genes were selected based on the transcriptome sequencing data (requiring strong transcriptional response, i.e., significant difference between DOCK5-wt and DOCK5-ko cells) and network analyses. These genes include upstream transcription factor JUN that may influence DOCK5 expression and two potential downstream targets of DOCK5 (ISG20 and VAMP5) representing examples of DOCK5-mediated processes such as immune system quenching (ISG20/JUN) and vesicle transport (VAMP5). Their expression profiles are similar to the DOCK5-c28 transcriptome data, but significantly different from that in the wild type A549 cells (Fig. 8). For ISG20, its upregulated expression during both H1N1 and H3N2 influenza infection in mRNA level was significantly enhanced in DOCK5-ko cells compared to DOCK5-wt cells (Fig. 8a, b). At day 1, neither ISG20 nor JUN showed significant difference between DOCK5-wt and DOCK5-ko cells during the infections but VAMP5 was already significantly down-regulated by DOCK5-ko (H1N1: p = 0.0056, 0.1-fold change; H3N2: p = 0.0020, 0.19-fold change). At day 2, ISG20 was significantly up-regulated by DOCK5-ko (H1N1: p = 0.03, 1.8-fold change; H3N2: p = 0.0013, 2.5-fold change; Fig. 8a, b) while JUN and VAMP5 were down-regulated by DOCK5-ko during IAV infection (Fig. 8c–f). Specifically, at day 2, JUN expression was down by about 70% (H1N1: p = 0.045, 0.35-fold change; H3N2: p = 0.076, 0.27-fold change) and VAMP5 was down by about 90% (H1N1: p = 0.0038, 0.087-fold change; H3N2: p = 0.0046, 0.10-fold change) in DOCK5-ko cells compared to DOCK5-wt cells.

Validation of the changes of expression of DOCK5 key targets under different scenarios as combinations of the DOCK5 perturbations (DOCK5-wt and DOCK5-ko) and virus infections (H1N1 and H3N2 infections) by RT-qPCR at day 1 and day 2 post infection. The expression of ISG20 (a, b), JUN (c, d) and VAMP5 (e, f) was measured by RT-qPCR during H1N1 (a, c, e) and H3N2 (b, d, f) infections. Difference in expression of each gene between DOCK5-wildtype (WT) and DOCK5-ko (DOCK5) during either H1N1 or H3N2 infection was assessed by t-test. Significant differences were indicated by *(p < 0.05), **(p < 0.01) and ***(p < 0.005)
We have further evaluated the potential effect of influenza proteins on DOCK5 and identified cellular functions relevant for the influenza life cycle. For this purpose, we used experimentally obtained protein–protein interaction data of influenza protein interactions with host factors. Durmuş and Ülgen assembled pathogen–host interactions of 11 DNA virus families and 15 RNA virus families, including influenza.53 We extracted the influenza A specific network consisting of 11 influenza proteins and 1621 host factors. Significant overlap between human host factors interacting with 9 viral proteins and 231 targets in DOCK5-CCGS(7) were observed (Fig. S8 and Table S34). Among the most significantly enriched overlap, with fold enrichment of 2 or higher, are human host factors interacting with influenza proteins HA, M1, M2, NA, and NP. Although primary functions of the corresponding intersections are according to the processes modulated by the specific viral protein (e.g., the M1 protein and early phase of viral life cycle function, or ribonucleoprotein complex assembly and RNA transport with respect to NP), a majority of the functions involve mRNA processing and splicing (Tables S35 and S36). Other functions involve viral process, the proteasome, and protein localization and transport. The latter processes are indicated by host factors interacting with the viral M2 and NS1 proteins. Thus, these findings validate the significant impact of DOCK5 on the genes discussed and corresponding pathways.
Discussion
In this study, we systematically analyzed a large amount of gene expression data from 12 molecular studies of influenza infection covering MOIs between 0.1 and 5 (median 2.5, see Table 1). We first identified differentially expressed genes using ANOVA and then derived robust consensus DEG signatures across multiple studies. Many of those DEGs have been known to play essential roles during influenza infections. By employing non-parametric Jonckheere trend analysis we identified significant up-regulated and down-regulated genes required for viral replication or activated as a host defense. Type I interferon signaling and the up-regulation of interferon stimulated genes or ISGs, such as MX1, IFITM, IRF7, and OAS, among other ISGs, are well conserved across all 12 studies. On the other hand, down-regulated genes are associated with small molecule/lipid metabolic processes and localization. The heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) is the highest ranked gene significantly expressed in all 12 data sets and predominantly down-regulated. Together with splicing factor 2 (SF2), it regulates alternative splicing of interferon regulator factor-3 (IRF3).28 Mediated by transportin 1 (TNPO1),29 hnRNPA1 shuttles between the nucleus and the cytosol.30 Other highly ranked members of the hnRNP family involve the related hnRNPD and the synaptotagmin binding cytoplasmic RNA interacting protein (SYNCRIP), both significantly expressed in 11 data sets.
To further understand the co-regulation among the genes in response to influenza infection, we performed gene co-expression analysis to identify 1191 modules from 12 studies, which were further used to derive 282 consensus co-expression modules. These consensus modules have functions from viral reproduction to RIG-I signaling and Golgi associated vesicle biogenesis. We formally rank-ordered the 282 consensus modules by their enrichment for the ANOA and Jonckheere Trend analysis-based DEG signatures derived from the individual studies. Two members of the top-ranked module, MDM2 and DOCK5, are most connected across all 12 co-expression networks and they were predicted to be the top drivers of the gene networks and potential host factors for influenza infection. We sought to comprehensively examine the role of DOCK5 during influenza infection. We explicitly constructed DOCK5-centered networks, which capture many known processes and host factors for influenza infection, including the ER-nucleus signaling, response to ER stress, RNA localization, Golgi vesicle transport, viral process, modulation by virus of host morphology and RNA splicing, as well as the cellular protein metabolic process. We validated experimentally DOCK5 and its co-regulated networks. DOCK5 was knocked out in human lung epithelial A549 cell lines and virus replication was compared to that in the wild type parental A549 cell lines. The influenza viruses replicated to significantly lower titers in DOCK5-ko cells than in the parental A549 cell line indicating impairment of viral replication without functional DOCK5. We also characterized the transcriptional program regulated by DOCK5 by sequencing mRNA from infected and uninfected wild type and DOCK5-ko cells. Our network approach, combined with knockout data and comparative analysis between different genetic and infection scenarios, validates the causal role—be direct or indirect—of DOCK5 in these processes. Three genes, ISG20, JUN, and VAMP5, were selected and their expression re-validated by RT-qPCR. The co-regulatory network was validated by the DEG signatures identified from the DOCK5 knockout experiments to achieve an in-depth understanding of the biological functions of DOCK5. Our results demonstrate that DOCK5 is a host factor that is potentially required for viral replication in cell culture. We further demonstrated by our coexpression network analysis approach that DOCK5 not only modulates processes that are important for the IAV life cycle but also potentially subverts the host defense response by directly compromising key defense genes, or by indirectly affecting cellular factors required for host defense.
In particular, we have identified three key processes in the centered network: (i) vesicle transport, (ii) pre-mRNA processing, and (iii) host defense. Figure 9 shows an overview of cellular processes, in particular vesicle trafficking and splicing, controlled by DOCK5. Cellular transport and viral trafficking are essential processes during the viral life cycle and the data suggest that DOCK5 could modulate influenza virus trafficking within the cell. DOCK5 also potentially influences gene regulation of trafficking-relevant genes. The trans-Golgi network protein 2 (TGOLN2), golgins, and the Golgi phosphoprotein 3 display highly conserved interactions between each other as well as with DOCK5 (Fig. 9 rhs/”Budding”). Additional vesicle trafficking-associated genes, such as VAMP5, are highly up-regulated during IAV infection. VAMP5 may function together with TGOLN2 in the trans-Golgi complex (Fig. 9 rhs/”Budding”) though VAMP5 does not mediate vesicle fusion with plasma membrane t-SNAREs.54 Loss of DOCK5 significantly decreases VAMP5 expression, indicating the importance of DOCK5 for these cellular processes.

DOCK5 is a key regulator for splicing and transport. Virus entry and initial transport to the nucleus is shown with regulatory links to the spliceosome, intracellular trafficking, and control of budding. In particular, CD81 potentially controls both entry/initial transport as well as budding. DOCK5 causally influences the V-type ATPases and CLINT1 with respect to transport, as well as NS1 BP, potentially for splicing. As a RAC specific GEF, DOCK5 transduces signals for cytoskeleton rearrangement as a host defense response against IAV infection. DOCK5 also down-regulates splicing by directly modulating hnRNPA1, NS1 BP, and SYNCRIP as well as SF3A1/SF3B1 and DHX36, in addition to down-regulation of NUP93 and NUP210. Blue colored nodes are down-regulated by DOCK5. Green nodes are in the DOCK5-CCGS(7) neighborhood but not causally controlled by DOCK5. Closely spaced nodes indicate protein–protein interaction, black solid lines are material transport, blue dashed lines indicate DOCK5 induced gene regulation, solid red lines denote induced functions based on published studies (see text)
Other genes, such as a member of the adaptor protein complex 1 (AP1S2), which mediates the recruitment of clathrin to the Golgi complex, as well as the V-type ATPases, relevant for acidification and fusion of the cellular compartments, seem to be transcriptionally controlled by DOCK5. Together with COPG2, another cellular player for early processes during the influenza life cycle (i.e., uncoating and fusion, which is mediated by viral hemagglutinin, HA), as well as budding, DOCK5 potentially modulates these first steps of IAV entry via AP1S1, ATP6V1A, and COPG2 (Fig. 9 lhs/”Entry”). DOCK5 may function in a regulatory feedback loop between COPI and NS1 BP. Another potential regulatory dependency was observed between transcription factor JUN and DOCK5. According to LINCS data, JUN down-regulates DOCK5. Conversely, JUN expression is significantly reduced under DOCK5-ko conditions, indicating an (indirect) activation by DOCK5 and the potential existence of a feedback loop.
The spliceosome, which is responsible for mRNA processing and NS1BP in particular, is another target that is most likely modulated by DOCK5. NS1BP binds to the predominantly cytosolic SYNCRIP, which itself is a member of the heterogeneous nuclear ribonucleoproteins. Other members of this complex, which are all controlled by DOCK5, are hnRNFA1, hnRNPA3, hnRNPD, hnRNPR, and hnRNPU, together with the splicing factor 3 components SF3A1, SF3A2, and SF3B1 (Fig. 9 center/”Spliceosome”). DOCK5 seems to be required for the proper function of the splicing machinery, by potentially mediating transport of splicing factors (via KPNA4 and TNPO1), such as hnRNPA1 between the cytoplasm and the nucleus, or by directly modulating alternative splicing responsible genes, such as EXOSC6 and DDX17. Required influenza host factors, such as splicing genes (PTBP1 and SF3A1, nuclear pore protein NUP98) as well as trafficking genes, are differentially spliced by DOCK5 induction. Compared to the modulation of mRNA processing and splicing induced by NS1, DOCK5 seems to be required for splicing of genes relevant for the influenza life cycle.
Although not the predominant function, DOCK5 may be responsible for inhibiting specific host defense mechanisms to promote viral replication. We were able to identify 2 host defense processes that were up-regulated by DOCK5—the interferon-induced gene ISG20 and cytokine NAMPT.
Availability of data from different cell lines and IAV strains, with different multiplicity of infection scenarios, allowed us to use a consensus approach at all levels of our multi-scale analysis, including capturing a consensus environment. This facilitated the discovery of universal processes that are essential during IAV infection. Thus, overall evidence indicates that DOCK5 plays an important role in the gene-regulatory networks that potentially modulate host processes required for influenza infection by regulating intra-cellular trafficking and splicing, as well as subverting host defenses.
By combining an integrative network approach, a state-of-the-art gene knockout technique, and RNAseq data, this study uncovered and validated fundamental patterns of molecular responses, intrinsic structures of gene co-regulation, and novel key targets in influenza virus infection. Our findings pave a way for further functional investigations to identify novel therapeutic targets against influenza infection.
Methods
A brief description of key methods and sample description is provided below, whereas complete details are discussed in the supplement.
Modulation of virus growth and validation
To abolish the expression of functional DOCK5 proteins in A549 cells, the CRISPR/Cas9 genome editing system was used to introduce frameshift deletions into the DOCK5 coding region of A549 cells. Briefly, CRISPR sgRNA was designed using the CRISPR Design tool45 and two pairs of oligo nucleotides (pair 1: 5′-CACCGTATGGCCCACGCGGACAATC-3′ and 5′-AAACGATTGTCCGCGTGGGCCATAC-3′; pair 2: 5′-CACCGGATAAATCGGAGCGAGCATT-3′ and 5′-AAACAATGCTCGCTCCGATTTATCC-3′) were selected, synthesized, individually annealed, and ligated into the pSpCas9(BB)-2A-Puro (PX459) V2.0 vector, following established protocols.45 The resultant plasmids pBZ321A6 and pBZ322A5 were transfected into human lung epithelial A549 cells. After puromycin selection, cells were cloned and sequenced to identify the ones with desired frameshift deletions. Four DOCK5-ko A549 clones (DOCK5-c20, c25, c28, and c41) were expanded by culture and compared to the wild type A549 (A549-WT) for their capacity to support the replication of influenza A virus strains.
To compare the replication kinetics of the influenza A virus in A549-wt and A549-DOCK5-ko cells, cells were seeded into 12-well plates to approximately 80% confluency (~2 × 105 cells/well) at the time of infection. A/Puerto Rico/8/1934 (H1N1) and A/New York/238/2005 (H3N2) viruses were inoculated into the cells at MOI of 0.0005 (100 TCID50/well) and MOI of 0.005 (1000 TCID50/well), respectively. The supernatants were collected at days 1, 2, and 3 post-infection and titrated by TCID50 assay using MDCK cells. The inoculum was also back-titrated and the titer was used to represent the day 0 titer (Fig. 4). The medium for culture of the wild type and knockout A549 cells was F12K supplemented with 10% FBS and the medium for virus infection was MEM supplemented with 1% anti–anti (Thermo Fisher Scientific), 0.15% BSA fraction V (Thermo Fisher Scientific), and 1 μg/ml tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-trypsin (Worthington, Lakewood, NJ). Three biological replicates were tested at each time point for each genotype, resulting in a total of 24 samples per each of the two viruses. A power analysis based on Cohen’s effect size estimate55 indicates that our experimental design will allow a ~80% power to detect an effect size of larger than 0.6 in viral production rate difference between DOCK5 KO and wild-type.
A representative A549-DOCK5-ko clone, DOCK5-c28, was selected to determine the effects of DOCK5 knockout on global gene expression upon influenza virus infection. A549-WT and A549-DOCK5-c28 cells were each infected at MOI of 0.5 TCID50/cell of the PR8 (H1N1) virus, the A/New York/238/2015 (H3N2) virus, or Mock infected. Cells were harvested at 1 and 2 days post infection; total cellular RNA was extracted, and genomic DNA removed by DNase treatment. The frameshift deletion in the DOCK5-c28 clone compared to the DOCK5-wt is shown in Fig. S9.
Analysis of RNA sequencing data
Single-ended RNA-seq data was generated using the Illumina HiSeq 2500 platform. The sequencing reads were aligned to the human hg19 genome using star aligner (version 2.5.0b). Following read alignment, featureCounts56 was used to quantify gene expression at the gene and exon levels based on Ensembl gene model GRCh37.70. Genes with at least 1 count per million (CPM) reads in at least one sample were considered expressed, otherwise absent and hence discarded. The gene level read counts data was normalized,57 multi-dimensional scaling, and cluster analysis were performed to check for potential sample outliers.
Quantification of gene expression (RT-qPCR)
Wild type A549 and DOCK5-ko clones (c20, c25, c28, and c41) were infected as described above, and extracted RNA was used as a template for RT-qPCR. The RT-qPCR was performed using the TaqMan® RNA-to-Ct™ 1-Step Kit and the TaqMan® Gene Expression Assays (Thermo Fisher Scientific, Inc.). The assay IDs are Hs01103582_s1 for JUN, Hs01105383_g1 for VAMP5, Hs00158122_m1 for ISG20, Hs00353740 and Hs99999905_m1 for NR3C1, Hs00227848_m1 for DOCK5, and Hs00213893_m1 for WBSCR22. GAPDH was used as an internal control for the quantification because its level is unchanged upon virus infection. The 2−ΔΔCT method was used to analyze the relative changes in gene expression.58
Data sets and sample processing
We compiled from GEO 8 microarray expression profiles (Table 1) of human cell cultures infected with different IAV subtypes and strains yielding 12 distinct data sets. We subjected the expression data to log2 transformation and quantile normalization.
Identification of differentially expressed genes
We used three distinct methods to identify differentially expressed genes. The t-test was used to identify differentially expressed genes (DEGs) between case and control. We used one-way ANOVA to determine significantly responding genes (SRGs) depending on time post infection as a single independent factor. Multi-factorial analysis of temporal expression data between wildtype and DOCK5-ko genotypes were performed by a hierarchical linear model (hLM) after Limma.59 Significantly up-regulated and down-regulated genes depending on time post infection were identified by the non-parametric Jonckheere trend analysis.25
Gene co-expression network analysis
We performed weighted gene co-expression network analysis (WGCNA) to identify 1191 modules of highly co-expressed genes from the assembled 12 data sets. Using the Jaccard-Needham dissimilarity measure for assessing module–module similarity and the hierarchical clustering analysis, the 1191 modules were further grouped into 282 clusters, i.e., consensus modules.
Co-expression modules and consensus modules (CMs)
We performed WGCNA17,18 to identify 1191 modules of highly co-regulated genes from the assembled 12 data sets. We further identified consensus modules conserved across multiple data sets based on the hierarchical clustering analysis of using the Jaccard-Needham dissimilarity matrix for the 1191 modules.
The total relevance of each consensus module to influenza infection was calculated by summarizing the enrichment of the DEG signatures: \(G_j = \mathop {\prod }\nolimits_i g_{ji}\), where, \(g_{ji}\) is the relevance of a consensus j to a signature i. \(g_{ji}\) is defined as \(\left( {{\rm max}_j\left( {r_{ji}} \right) + 1 - r_{ji}} \right)/\mathop {\sum }\nolimits_j r_{ji}\), where \(r_{ji}\) is the ranking order of the significance level of the overlap between the consensus module j and the signature i.
Enrichment analysis and internal verification of data
To functionally annotate gene signatures and gene modules identified in this study, we performed enrichment analysis of the established pathways and signatures including the gene ontology (GO) categories and MSigDB, and the subject area specific gene sets including influenza host factors, inflammasome, interferome, and InnateDB.
Data availability statement
Analytic results are available in a large number of supplementary tables. All raw RNA-sequencing data (single FASTQ files) as well as the processed CPM matrix from this study have been deposited into the Gene Expression Omnibus (GEO) under Accession Number GSE104168.
Code availability
The R code and the R package WINA for the coexpression network analysis are available at doi:10.7303/syn7221264.2.
References
WHO. Fact sheet Number 211 http://www.who.int/mediacentre/factsheets/fs211/en/ (2009).
Marcotte, E. M., Xenarios, I. & Eisenberg, D. Mining literature for protein–protein interactions. Bioinformatics 17, 359–363 (2001).
Bright, R. A. et al. Incidence of adamantane resistance among influenza A (H3N2) viruses isolated worldwide from 1994 to 2005: a cause for concern. Lancet 366, 1175–1181 (2005).
Edinger, T. O., Pohl, M. O. & Stertz, S. Entry of influenza A virus: host factors and antiviral targets. J. Gen. Virol. 95, 263–277 (2014).
Watanabe, T., Watanabe, S. & Kawaoka, Y. Cellular networks involved in the influenza virus life cycle. Cell Host Microbe 7, 427–439 (2010).
Barber, G. N. Host defense, viruses and apoptosis. Cell Death Differ. 8, 113–126 (2001).
Lund, J. M. et al. Recognition of single-stranded RNA viruses by toll-like receptor 7. Proc. Natl Acad. Sci. USA 101, 5598–5603 (2004).
Kawai, T. & Akira, S. Signaling to NF-kappaB by toll-like receptors. Trends Mol. Med. 13, 460–469 (2007).
Yoneyama, M. et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5, 730–737 (2004).
Rehwinkel, J. et al. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 140, 397–408 (2010).
Hao, L. et al. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature 454, 890–893 (2008).
Brass, A. L. et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139, 1243–1254 (2009).
Karlas, A. et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 463, 818–822 (2010).
Konig, R. et al. Human host factors required for influenza virus replication. Nature 463, 813–817 (2010).
Shapira, S. D. et al. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell 139, 1255–1267 (2009).
Li, C. et al. Host regulatory network response to infection with highly pathogenic H5N1 avian influenza virus. J. Virol. 85, 10955–10967 (2011).
Zhang, B. et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153, 14 (2013).
Zhang, B. & Horvath, S. A. general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 4, Article17. (2005).
Gene Ontology, C. Gene ontology consortium: going forward. Nucleic Acids Res. 43, D1049–D1056 (2015).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Ward, S. E. et al. Host modulators of H1N1 cytopathogenicity. PLoS One 7, e39284 (2012).
Wang, I. M. et al. Systems analysis of eleven rodent disease models reveals an inflammatome signature and key drivers. Mol. Syst. Biol. 8, 594 (2012).
Rusinova, I. et al. Interferomev2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. 41, D1040–D1046 (2013).
Breuer, K. et al. InnateDB: systems biology of innate immunity and beyond-recent updates and continuing curation. Nucleic Acids Res. 41, D1228–D1233 (2013).
Jonckheere, A. R. A distribution-free k-sample test against ordered alternatives. Biometrika 41, 133–145 (1954).
Iwasaki, A. & Pillai, P. S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 14, 315–328 (2014).
Matlin, A. J., Clark, F. & Smith, C. W. Understanding alternative splicing: towards a cellular code. Nat. Rev. Mol. Cell Biol. 6, 386–398 (2005).
Guo, R. et al. HnRNP A1/A2 and SF2/ASF regulate alternative splicing of interferon regulatory factor-3 and affect immunomodulatory functions in human non-small cell lung cancer cells. PLoS One 8, e62729 (2013).
Fridell, R. A., Truant, R., Thorne, L., Benson, R. E. & Cullen, B. R. Nuclear import of hnRNP A1 is mediated by a novel cellular cofactor related to karyopherin-beta. J. Cell Sci. 110, 1325–1331 (1997).
Gustin, K. E. & Sarnow, P. Effects of poliovirus infection on nucleo-cytoplasmic trafficking and nuclear pore complex composition. Embo J. 20, 240–249 (2001).
Liu, H. M. et al. SYNCRIP (synaptotagmin-binding, cytoplasmic RNA-interacting protein) is a host factor involved in hepatitis C virus RNA replication. Virology 386, 249–256 (2009).
Park, S. M. et al. Translation-competent 48S complex formation on HCV IRES requires the RNA-binding protein NSAP1. Nucleic Acids Res. 39, 7791–7802 (2011).
Pichlmair, A. et al. Viral immune modulators perturb the human molecular network by common and unique strategies. Nature 487, 486–490 (2012).
Wang, I. M. et al. Systems analysis of eleven rodent disease models reveals an inflammatome signature and key drivers. Mol. Syst. Biol. 8, 594 (2012).
Zhu, J. et al. Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat. Genet. 40, 854–861 (2008).
Yoon, Y. J. et al. MDM2 and p53 polymorphisms are associated with the development of hepatocellular carcinoma in patients with chronic hepatitis B virus infection. Carcinogenesis 29, 1192–1196 (2008).
Forte, E. & Luftig, M. A. MDM2-dependent inhibition of p53 is required for Epstein-Barr virus B-cell growth transformation and infected-cell survival. J. Virol. 83, 2491–2499 (2009).
Wang, X. et al. Stabilization of p53 in influenza A virus-infected cells is associated with compromised MDM2-mediated ubiquitination of p53. J. Biol. Chem. 287, 18366–18375 (2012).
Ding, X., Yang, J. & Wang, S. Antisense oligonucleotides targeting abhydrolase domain containing 2 block human hepatitis B virus propagation. Oligonucleotides 21, 77–84 (2011).
Lorgeoux, R. P., Pan, Q., Le Duff, Y. & Liang, C. DDX17 promotes the production of infectious HIV-1 particles through modulating viral RNA packaging and translation frameshift. Virology 443, 384–392 (2013).
Bortz, E. et al. Host-and strain-specific regulation of influenza virus polymerase activity by interacting cellular proteins. MBio 2, e00151–00111 (2011).
Honig, A., Auboeuf, D., Parker, M. M., O’Malley, B. W. & Berget, S. M. Regulation of alternative splicing by the ATP-dependent DEAD-box RNA helicase p72. Mol. Cell. Biol. 22, 5698–5707 (2002).
Das, T. et al. A molecular mechanotransduction pathway regulates collective migration of epithelial cells. Nat. Cell Biol. 17, 276–287 (2015).
Rajendra, E., Garaycoechea, J. I., Patel, K. J. & Passmore, L. A. Abundance of the Fanconi anaemia core complex is regulated by the RuvBL1 and RuvBL2 AAA+ATPases. Nucleic Acids Res. 42, 13736–13748 (2014).
Ran, F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013).
Wang, M., Zhao, Y. & Zhang, B. Efficient test and visualization of multi-set intersections. Sci. Rep. 5, 16923 (2015).
Espert, L. et al. ISG20, a new interferon-induced RNase specific for single-stranded RNA, defines an alternative antiviral pathway against RNA genomic viruses. J. Biol. Chem. 278, 16151–16158 (2003).
Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Kroeker, A. L., Ezzati, P., Halayko, A. J. & Coombs, K. M. Response of primary human airway epithelial cells to influenza infection: a quantitative proteomic study. J. Proteome Res. 11, 4132–4146 (2012).
Tripathi, S. et al. Meta- and orthogonal integration of influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe 18, 723–735 (2015).
Tsai, P. L. et al. Cellular RNA binding proteins NS1-BP and hnRNP K regulate influenza A virus RNA splicing. PLoS Pathog. 9, e1003460 (2013).
Zhang, L. et al. The exosome controls alternative splicing by mediating the gene expression and assembly of the spliceosome complex. Sci. Rep. 5, 13403 (2015).
Durmus, S. & Ulgen, K. O. Comparative interactomics for virus–human protein–protein interactions: DNA viruses versus RNA viruses. FEBS Open Bio. 7, 96–107 (2017).
Hasan, N., Corbin, D. & Hu, C. Fusogenic pairings of vesicle-associated membrane proteins (VAMPs) and plasma membrane t-SNAREs–VAMP5 as the exception. PLoS One 5, e14238 (2010).
Cohen, J. Statistical power analysis for the behavioral sciences. (Lawrence Erlbaum Associates, 1977).
Liao, Y., Smyth, G. K. & Shi, W. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Livak, K. & Schmittgen, T. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)). Method 25, 402–408 (2001).
Smyth, G. K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, Article 3. (2004).
Acknowledgements
This work was supported by the grants U01AI111598 from NIH/National Institute of Allergy and Infectious Diseases (NIAID, http://www.niaid.nih.gov/ ), U01AG046170 from the NIH/National Institute on Aging (NIA, http://www.nia.nih.gov/), RF1AG054014 from NIH/NIA, U01AG052411 from NIH/NIA and R01CA163772 from NIH/National Cancer Institute (NCI, http://www.cancer.gov/). U01AG046170 is a component of the AMP-AD Target Discovery and Preclinical Validation Project. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
B.Z. perceived the concept and designed the study. E.S. and E.G. contributed to study design. C.F. curated data and performed statistical and network analyses. M.W. and W.S. implemented key statistical tools. E.G. designed and oversaw the validation experiments. T.C., G.M. and B.Z. performed validation experiments. C.F., B.Z., M.W., M.S., T.C., G.M., B.Zhou and E.G. drafted the manuscript. All the authors read, edited and approved of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Forst, C.V., Zhou, B., Wang, M. et al. Integrative gene network analysis identifies key signatures, intrinsic networks and host factors for influenza virus A infections. npj Syst Biol Appl 3, 35 (2017). https://doi.org/10.1038/s41540-017-0036-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41540-017-0036-x
