
Abstract
Due to the rampant rise in obesity and diabetes, consumers are desperately seeking for ways to reduce their sugar intake, but to date there are no options that are both accessible and without sacrifice of palatability. One of the most promising new ingredients in the food system as a non-nutritive sugar substitute with near perfect palatability is D-psicose. D-psicose is currently produced using an in vitro enzymatic isomerization of D-fructose, resulting in low yield and purity, and therefore requiring substantial downstream processing to obtain a high purity product. This has made adoption of D-psicose into products limited and results in significantly higher per unit costs, reducing accessibility to those most in need. Here, we found that Escherichia coli natively possesses a thermodynamically favorable pathway to produce D-psicose from D-glucose through a series of phosphorylation-epimerization-dephosphorylation steps. To increase carbon flux towards D-psicose production, we introduced a series of genetic modifications to pathway enzymes, central carbon metabolism, and competing metabolic pathways. In an attempt to maximize both cellular viability and D-psicose production, we implemented methods for the dynamic regulation of key genes including clustered regularly interspaced short palindromic repeats inhibition (CRISPRi) and stationary-phase promoters. The engineered strains achieved complete consumption of D-glucose and production of D-psicose, at a titer of 15.3 g L-1, productivity of 2 g L-1 h-1, and yield of 62% under test tube conditions. These results demonstrate the viability of whole-cell catalysis as a sustainable alternative to in vitro enzymatic synthesis for the accessible production of D-psicose.
Similar content being viewed by others

Introduction
The deadly rise in sedentary lifestyle and access to calorie-dense foods has tripled global obesity rates from 1975 to 20161. Almost 40% of adults world-wide are considered overweight, a major risk factor associated with cardiovascular disease, diabetes, musculoskeletal disorders, and some cancers1. Within the United States alone, nearly 50% of adults are predicted to have obesity by 2030, with almost 25% of adults having severe obesity2. In the U.S., increases in severe obesity is predicted to disproportionally affect women, Black non-Hispanic adults, and adults with household income <$50,000, which could lead to further socioeconomic disparities as a result of obesity-associated medical complications, chronic disease, lower life expectancy, and weight stigma2. One critical factor in combating this issue is in the development of new tools and cost-effective ingredients that enable the food industry to play a role in providing nutritionally balanced products. One of the most immediate impacts that can be made is through the substitution of sucrose and high-fructose corn syrup with zero and low-calorie sugar substitutes. As such, the market for sugar substitutes is expected to reach 20.6 billion USD by 20253. However, this cannot be done in a manner that sacrifices enjoyment or cost for the customer. One of the most promising approaches to thread this delicate needle appears to be through the use of rare sugars.
Rare sugars are monosaccharides uncommonly found in nature, which differ slightly in structure from common sugars such as glucose and fructose. Many rare sugars are non-nutritive and possess potential health benefits such as anti-inflammatory, anti-viral, and tumor-suppressing capabilities, making them intriguing targets for pharmaceuticals and as sugar substitutes in food items4. They are being studied as sustainable alternatives to herbicides, fungicides, insecticides, and plant growth and immune system modulators4. One such rare sugar, D-psicose, is a Generally Recognized as Safe, zero-calorie, C3 epimer of D-fructose, with 70% the sweetness of sucrose5. Marketed to consumers as “allulose”, D-psicose has a desirable flavor profile along with favorable browning, hygroscopic, and solubility properties. Additionally, studies have correlated consumption of D-psicose to antihyperglycemic, antihyperlipidemic, antiparasitic, and antioxidant health benefits4. As with most rare sugars, a limitation to the study of D-psicose and its implementation in industry is the lack of economic, large-scale production. D-Psicose is naturally found in fruits, wheat, and syrups, but only in minute concentrations6. D-Psicose, along with other rare sugars, could be extracted from their natural sources, but their low abundance would lead to costly purification and the waste of consumable biomass.
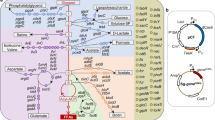
Current commercial production processes of D-psicose utilize a combination of D-tagatose-3-epimerase (DTEase) and D-psicose-3-epimerase (DPEase) to catalyze the conversion from fructose7,8,9. Both enzymes operate under high temperature and alkaline pH to epimerize the C3 carbon of D-fructose to form D-psicose8. Attempts to bolster production have focused on engineering these enzymes for improved catalytic efficiency, thermostability, and ability to perform at lower pHs and temperatures4,10. Despite concerted efforts to improve DPEases and DTEases, this method inherently suffers from limited yield due to a lack of thermodynamic driving force, and therefore has not achieved conversion yields greater than 50%8,10. The epimerization of D-fructose to D-psicose is reversible and has a predicted ΔG’° of +5 kJ mol-1, making it thermodynamically unfavorable and more likely to favor D-fructose at equilibrium (Fig. 1a)11. Low conversion results in a mixed solution of D-fructose and D-psicose, making isolation and purification by simulated moving bed chromatography or anion exchange resin a major challenge10,12. Methods that achieved conversions greater than 50% relied on the inclusion of toxic or expensive cofactors10.

a The current industrial method for D-psicose production leads to limited yield (~50%) due to a positive ΔG’°. b In the proposed biosynthetic pathway of D-psicose, the dephosphorylation step thermodynamically drives production forwards due to a large negative ΔG’m under cellular reactant concentrations of 1 mM. c The proposed pathway for the biosynthetic production of D-psicose in E. coli. Deleted steps are in blue. Overexpressed steps are in red. PTS, the phosphotransferase system; AlsE, D-allulose 6-phosphate 3-epimerase, HxpB hexitol phosphatase B.
To overcome this barrier, phosphorylation and dephosphorylation can be used to provide thermodynamic incentive and a driving force towards D-psicose production (Fig. 1b). Carrying a predicted ΔG’m of −31.1 kJ mol-1 at typical physiological concentrations of 1 mM, the dephosphorylation of D-psicose-6-phosphate (P6P) to D-psicose is a highly favorable reaction11. Dephosphorylation has been utilized as a thermodynamic sink for an in vitro enzyme system to produce D-psicose from the polysaccharide maltodextrin13. This system required five core and four auxiliary enzymes operating at elevated temperatures and resulted in a mixed solution of D-glucose, D-fructose, and D-psicose. Although successful at generating some D-psicose, this system suffered from the same limitations as other in vitro systems including enzymes which only operate at high temperatures, the need to isolate and purify pathway enzymes, and reliance on phosphorylating agents or polysaccharides to provide energy for phosphorylation. Additionally, D-fructose was generated as a side product and unable to be reassimilated back into the production pathway, which would lead to increased separation and purification cost in an industrial setting. As such, phosphorylation/dephosphorylation is more suited for whole-cell catalysis, where the enzymatic machinery and cofactors are readily available and side products such as D-fructose can be reused by the cells. In whole-cell catalysis, living cells play the role of chemical factory by assembling enzymes for chemical production, providing cofactors for enzymes to operate, and excreting products for simplified purification, all under environmentally friendly production conditions and temperatures14,15,16,17. Feedstocks for living cells can consist of sugars from agricultural waste, reducing competition with commercial food production. The technology and infastructure required for whole-cell catalysis and fermentation is established on an industrial scale, including for the model organism, Escherichia coli. Like most organisms, E. coli utilizes phosphorylation/dephophorylation of sugars as part of sugar consumption and central carbon metabolism18. In an attempt to overcome the inherent limitations of in vitro biosynthesis, a biosynthetic pathway using heterologous enzymes was constructed in E. coli to produce D-psicose from D-fructose19. However, cultures produced a minimal amount of D-psicose (~1.6 g L-1) and suffered from poor growth, even when supplemented with glycerol as an additional carbon source. Most recently, the supplementation of ATP and sodium hexametaphosphate was shown to improve D-psicose production from D-glucose in E. coli20. However, the supplementation of ATP and sodium hexametaphosphate drastically increases production cost, and the system did not consume all glucose and produced D-fructose as a side product, which would increase the cost of downstream processing. As demonstrated by the studies listed above, in vivo D-psicose production has great potential, but further research into enzyme selection, carbon flux regulation, feedstock optimization, and balancing energetics are required to achieve industrially relevant levels of production. Through our research, we attempt to address all four of these subjects.
In this study, we discovered that E. coli is capable of producing D-psicose from D-glucose without the expression of exogenous genes (Fig. 1b). Within E. coli, carbon metabolism begins with the phosphotransferase system (PTS), where D-glucose is simultaneously phosphorylated and transferred across the cell membrane18. Alternatively, glucose can be transported across the cell membrane by galactose proton symporter GalP, after which it is phosphorylated by glucokinase Glk21,22. D-glucose-6-phosphate (G6P) is then isomerized to D-fructose-6-phosphate (F6P), which while typically directed into glycolysis, we diverted towards D-psicose production using static and dynamic carbon flux regulation techniques18. F6P can be epimerized to P6P using the native D-allulose-6-phosphate 3-epimerase (AlsE), which is then dephosphorylated to D-psicose using the phosphatase hexitol-phosphatase B (HxpB) and excreted from the cell. By utilizing E. coli’s native genes, we successfully generated D-psicose from D-glucose at yields greater than 50%, without the need for heterologous enzyme expression.
We further improved the capacity of E. coli to produce D-psicose by additionally expressing alsE and hxpB and removing or regulating competing metabolic pathways, including the pentose phosphate pathway (PPP), glycogen biosynthesis, glycolysis, the D-allose degradation pathway, and the D-mannose degradation pathway. D-glucose import was supplemented by additionally expressing the native galactose proton symporter gene, galP, and the glucokinase gene, glk. To further increase production while maintaining cellular viability, we explored multiple strategies for dynamic gene regulation and carbon flux partitioning. During the growth phase, cells need more energy to rigorously grow, and genes related to glycolysis should be expressed. When the cells enter the stationary phase and are not actively growing, carbon flux can be diverted from glycolysis to D-psicose production. To balance carbon partitioning depending on growth phase, we explored two strategies to dynamically regulate the expression of key metabolic and D-psicose production genes: inducer-free stationary-phase promoters23,24,25 and clustered regularly interspaced short palindromic repeats interference (CRISPRi)26,27.
Results and discussion
Design of a thermodynamically favorable D-psicose production pathway
Currently, the primary method for production of D-psicose involves the in vitro enzymatic isomerization of D-fructose to D-psicose, a thermodynamically unfavorable process that results in incomplete product formation (~50%) and requires expensive purification (Fig. 1a)10. To overcome the thermodynamic limitations of this method, we proposed a pathway driven by the phosphorylation and dephosphorylation of sugars, a process whose required cofactors and enzymes are readily available within living organisms (Fig. 1b, c). Therefore, we chose the model organism E. coli to host the D-psicose biosynthetic pathway.
The proposed pathway begins with the assimilation of D-glucose into E. coli via the PTS18, which converts D-glucose to G6P (Fig. 1c). Alternatively, D-glucose can be assimilated by the galactose proton symporter GalP21, after which it is phosphorylated to G6P by glucokinase Glk22 (Fig. 1c). G6P is then isomerized to F6P via glucose 6-phosphate isomerase (Gpi)18. Here, the proposed pathway diverges from native carbon metabolism. Many enzymes are promiscuous, utilizing a variety of substrates. As such, we theorized E. coli may natively possess enzymes capable of producing D-psicose given the right conditions and alterations to sugar metabolism. F6P could be epimerized to P6P, which could then be dephosphorylated in a final, thermodynamically favorable step to D-psicose (Fig. 1c). We theorized that placing this favorable reaction at the end of our biosynthetic pathway drives flux through the production pathway by way of restoring equilibrium28,29. As P6P is favorably dephosphorylated to D-psicose and excreted from the cell, equilibrium is restored by generating more P6P from F6P.
Assessing E. coli’s native D-psicose production capabilities
To assess E. coli’s native capability of producing D-psicose, we tested production in E. coli MG1655 and MG1655-derived strain AL3601, which carries the gene encoding for T7 RNA polymerase30, in M9P media (Supplementary Table 1, Methods). Without any genetic manipulations, neither strain produced detectable levels of D-psicose (Fig. 2a).

Cells were grown in M9P media with 10 g L-1 glucose at 37 °C to OD600 ~ 0.4, then grown at 30 ˚C for 24 h. At OD600 ~ 0.4, 1 mM IPTG was added (a, b, e, f). a D-psicose production in MG1655 and AL3601 (Supplementary Table 1) with and without the deletion of pfkA and/or alsE. b Various sugar phosphatases with AlsE were tested for D-psicose in AL3601. c The AlphaFold predicted structure of HxpB with P6P (199 Å3) located in the active site pocket (429 Å3), with volumes calculated using MoloVol and CAVER, respectively66,67. d ASP173 interacts with the magnesium ion, which in turn positions the phosphate in P6P for nucleophilic attack by ASP15. Residues GLU22, TRP25, LEU52, SER117 are predicted to form hydrogen bonds (shown as dotted yellow lines) with the hydroxyl groups and position the P6P for hydrolysis. e The operon of alsE and hxpB was expressed under PT7 and PLlacO1 in AL3601 and AL1050 (Supplementary Table 1) respectively. ΔOD600 indicates the difference in OD600 at 0 h and 24 h. f Comparison of the effect of gene deletions on the production of D-psicose. Error bars indicate s.d. (n = 3 biological replicates).
We hypothesized that carbon flux must be purposely directed towards D-psicose production by accumulating the upstream metabolite, F6P (Fig. 1c). The major metabolic pathway competing for F6P is glycolysis, as F6P is preferentially converted to D-fructose 1,6-bisphosphate by phosphofructokinase A and B (PfkA and PfkB, Fig. 1c). To build up F6P pools within the cell, the gene encoding PfkA, which accounts for about 90% of phosphofructokinase activity31,32, was deleted in MG1655 and AL3601, generating AL4058 and AL3694 respectively (Supplementary Table 1). ∆pfkA strains produced D-psicose at 0.24 g L−1 in AL4058 and 0.15 g L−1 in AL3694, indicating that E. coli contains the enzymes necessary for producing D-psicose, most likely from F6P (Fig. 2a). D-psicose production was not detected in cultures grown in M9P media without glucose.
Elucidating enzymes involved in D-psicose production
We identified D-allulose 6-phosphate 3-epimerase (AlsE) as a potential candidate for the conversion of F6P to P6P33. AlsE assimilates D-psicose into central carbon metabolism by converting P6P to F6P. Under high concentrations of F6P, AlsE has shown reverse activity and is capable of converting F6P to P6P33. To confirm whether AlsE is involved in the production of D-psicose, alsE was deleted in AL3694 and AL4058, resulting in strains AL4063 and AL4082 (Supplementary Table 1). Neither strain produced detectible levels of D-psicose, suggesting that AlsE is the epimerase involved in the production of D-psicose (Fig. 2a).
E. coli has a number of phosphatase enzymes with potential activity towards P6P34. We chose candidate phosphatases with a wide range of activity towards various sugar substrates and tested hexitol phosphatase B (HxpB), sugar phosphatase YbiV, sugar phosphatase YidA, hexitol phosphatase A (HxpA), α-D-glucose-1-phosphate phosphatase YihX, and phosphosugar phosphatase YigL34. To test each phosphatase for the conversion of P6P to D-psicose, genes for each phosphatase were individually expressed from PT735, along with alsE on an expression plasmid (Supplementary Table 2). The plasmids were introduced to AL3601 (pfkA+). After 24 h, the strain harboring pAL1946 (PT7:alsE-hxpB) produced the most D-psicose, at 0.55 g L−1, suggesting that HxpB is an excellent phosphatase candidate for D-psicose production (Fig. 2b). The strain harboring pAL1947 (PT7:alsE-ybiV) or pAL2351 (PT7:alsE-yidA) produced 0.21 g L−1 and 0.20 g L−1 of D-psicose respectively, while cultures containing pAL2348 (PT7:alsE-hxpA), pAL2352 (PT7:alsE-yihX), and pAL2349 (PT7:alsE-yigL) did not generate detectable D-psicose (Fig. 2b).
Identification of critical P6P binding motifs
A combination of AlphaFold36 and the Rosetta Molecular Suite37 were utilized to evaluate predicted binding modes between each of the six phosphatases and P6P. Phosphatases with activity towards P6P (HxpB, YbiV, and YidA, Fig. 2b) were predicted to form at least two internal hydrogen bonds and one additional hydrogen bond with the terminal hydroxyl group of P6P (Fig. 2c, d and Supplementary Fig. 1). Conversely, phosphatases without activity towards P6P (HxpA, YihX, and YigL, Fig. 2b), were predicted to not form hydrogen bonds with the terminal hydroxyl group of P6P (Supplementary Fig. 1). Phosphatase HxpB, which produced the highest D-psicose titer (Fig. 2b), was predicted to form hydrogen bonds between active site residues and the four hydroxyl groups of P6P (Fig. 2d). These predictions show that a minimum of three hydrogen bonds, including a hydrogen bond to the terminal hydroxyl group, is essential for binding P6P in a catalytically competent orientation. An example of the input files used, including constraints, RosettaScripts XML, and ligand params can be found in the Supplementary Information.
Comparing expression system for D-psicose production
In AL3601, the T7 RNA polymerase expression system, which includes the T7 RNA polymerase (RNAP) gene under an IPTG-inducible PlacUV5 promoter30, was utilized. However, we observed decreased growth when IPTG was added to induce expression of T7 RNAP (Fig. 2e). Thus, an alternative expression system in which the operon of alsE and hxpB was expressed from the IPTG-inducible promoter PLlacO1 was tested38. Under induced conditions (with IPTG), the strain with PLlacO1:alsE-hxpB produced D-psicose similarly to the strain with PT7:alsE-hxpB (Fig. 2e). Importantly, the strain with PLlacO1:alsE-hxpB showed better repression under uninduced conditions (without IPTG) and did not have a growth burden under induced conditions compared to the strain with PT7:alsE-hxpB (Fig. 2e). Thus, the PLlacO1 expression system was used for further studies.
Increasing D-psicose production by removing competing pathways
E. coli relies on two primary glycolytic pathways to metabolize glucose: the PPP and glycolysis (also known as the Embden Meyerhof Parnas pathway)31. The branching point between D-psicose production and the PPP occurs when glucose-6-phosphate dehydrogenase (Zwf) converts G6P to 6-phospho-D-glucono 1,5-lactone (Fig. 1c)39. The branching point between D-psicose production and glycolysis occurs when one of two phosphofructokinases, PfkA or PfkB, converts F6P to D-fructose 1,6-bisphosphate (Fig. 1c)31. We established that the deletion of pfkA leads to the development of D-psicose production in comparison to our unmodified base strains (Fig. 2a).
In addition to the PPP and glycolysis, the allose degradation pathway has potential to divert carbon flux away from D-psicose production by reassimilating P6P back into central carbon metabolism40. The rpiB gene encodes for allose-6-phosphate isomerase (RpiB), which may be able to convert P6P to aldehydo-D-allose 6-phosphate (Fig. 1c).
To redirect carbon flux away from central carbon metabolism and towards D-psicose production, gene knockouts ∆pfkA, Δzwf, and ΔrpiB were constructed in AL1050, resulting in Strain 1 (Table 1). Those three deletions resulted in a fourfold increase in D-psicose production at 2.31 g L−1 of D-psicose (Fig. 2f).
Through knocking out pfkA, zwf, and rpiB, carbon flux was channeled into the D-psicose production pathway. In particular, knocking out pfkA and zwf should lead to an increase in F6P availability within cells31. These results reaffirmed our suspicions that F6P plays a key role as precursor to P6P, and that accumulation of F6P is necessary to drive carbon flux through the production pathway.
Identification of a side product
When analyzing samples from Strain 1, we observed a significant peak on the high-performance liquid chromatography (HPLC) chromatogram that did not align with D-glucose, D-fructose, or D-psicose standards, or any media component. Using gas chromatography–mass spectrometry (GC-MS) analysis, we found the retention time and mass spectrum of the unknown peak matched the retention time and spectrum of D-mannose (Supplementary Fig. 2).
Under standard conditions, the D-mannose pathway involves the assimilation of D-mannose 6-phosphate (M6P) into central carbon metabolism by the reversible isomerization of M6P to F6P via mannose-6-phosphate isomerase (ManA)41. An accumulation of F6P could cause this reaction to run in reverse, generating M6P and D-mannose as a result. To test the hypothesis that this side product was D-mannose, we deleted manA in Strain 1, resulting in Strain 2 (Table 1).
The deletion of manA resulted in a considerable decrease in D-mannose production. Strain 2 with IPTG generated only 0.69 g L−1 of D-mannose compared to Strain 1 with IPTG, which produced 2.49 g L−1 of D-mannose (Supplementary Fig. 3). Complementary to the reduction in D-mannose production, the production of D-psicose in Strain 2 was 1.5 times higher than in Strain 1 (Fig. 2f). While not fully eliminated, the unwanted production of D-mannose was greatly decreased by the deletion of manA. Subsequent production was therefore carried out in strains containing ΔpfkA, Δzwf, ΔrpiB, and ΔmanA.
Although the D-mannose pathway was eliminated, there may be other D-fructose epimer pathways continuing to compete for carbon flux. Eliminating the PPP and limiting glycolysis can cause a substantial increase in cellular F6P pools, which if not funneled efficiently into D-psicose biosynthesis, may be acted upon by other epimerases or isomerases. In short, increased F6P availability may allow enzymes not normally observed to engage with the substrate and generate other sugar products.
Utilization of a stationary phase promoter
Constructing production pathways within a microorganism requires careful carbon partitioning between essential metabolic processes and production, especially when working around central carbon metabolism42,43,44. Within our system, dynamically balancing carbon flux between glycolysis and the D-psicose pathway is critical for maximizing both cellular viability and D-psicose production.
The life cycle of an E. coli culture includes 5 distinct phases: lag, logarithmic, stationary, death, and long-term stationary phase23. The lag phase occurs when cells are inoculated into media and adjust their metabolic processes according to their new environment. Given favorable conditions, cells will rapidly grow and divide, entering the logarithmic phase. It is at this time that enzymes related to central carbon metabolism are most important, and the transcription of corresponding genes will be upregulated. Once the cells sense environmental stressors such as scarcity of media nutrients, their growth and division slows, and the culture enters the stationary phase. Here, culture density plateaus and genes related to stress response are expressed24,45. The transcription of these genes is regulated in part by the σ38-subunit of RNA polymerase, which recognizes the promoter region of a gene23.
We chose to utilize E. coli’s native gene regulatory system to balance carbon flux by affixing the D-psicose production genes, alsE and hxpB, downstream of a stationary phase-active promoter23,24,25. This should prevent the production pathway from competing with central carbon metabolism for carbon flux during the logarithmic phase of growth, a time when cells need carbon to rigorously grow and divide.
Four promoters previously shown to have activity during the stationary phase were chosen for testing using Green Fluorescent Protein as a reporter: PgadB, PcbpA2, PihfA4, and Pdps23,24,25. Each promoter was cloned upstream of sfgfp on an expression plasmid (Supplementary Table 2). The strongest promoter, PgadB, had an expression strength 100 times as strong as IPTG-induced promoter PLlacO1, in addition to having an expression time correlating to late logarithmic or early stationary phase (Supplementary Fig. 4a). The second strongest promoter, PcbpA2, closely followed the expression timing of induced PLlacO1, but with approximately two-thirds the expression strength (Supplementary Fig. 4b). PihfA4 and Pdps also followed the expression timing of PLlacO1, but with lower expression strength than PcbpA2. (Supplementary Fig. 4c).
Due to its strong expression during the stationary phase, PgadB was used to express the operon of alsE and hxpB (pAL2247, Supplementary Table 2). To evaluate the effects of the initial glucose concentration, we tested D-psicose production with 10, 20, and 40 g L−1. Under varying glucose concentrations (10, 20, and 40 g L−1) Strain 3 (PgadB:alsE-hxpB, Table 1) consistently produced greater titers of D-psicose and grew with a greater ΔOD600, which represents the difference in optical density at 600 nm (OD600) at 0 and 24 h, compared to Strain 2 (PLlacO1:alsE-hxpB, Fig. 3a). The highest D-psicose titers were achieved at a glucose concentration of 40 g L−1, which allowed Strain 3 to produce 6.92 g L−1 of D-psicose and grow with a ΔOD600 of 5.2, while Strain 2 produced 4.55 g L−1 and grew with a ΔOD600 of 4.0. The initial glucose concentration of 40 g L−1 was used for further studies.

a Cells were grown in M9P media with various concentrations of glucose to OD600 ~ 0.4 at 37 °C, then grown at 30 ˚C for 24 h. At OD600 ~ 0.4, 1 mM IPTG was added for the PLlacO1 constructs. The operon of alsE and hxpB was expressed under PLlacO1 (pAL2001) and PgadB (pAL2247) in AL3756 and AL3990 respectively (Strain 1 and 2, Table 1). b Strain 1 and 2 were grown in M9P media with 40 g L−1 glucose at 37 °C to OD600 of ~0 (no culturing at 37 °C), ~0.4, or ~1, then grown at 30 ˚C for 24 h. When the temperature was shifted to 30 ˚C, 1 mM IPTG was added for the PLlacO1 constructs. c The operon of galP and glk were expressed under PLlacO1 (pAL2264, Supplementary Table 2). Strain 4 (AL3990 with pAL2264 and pAL2247, Table 1) was grown in M9P media with 40 g L−1 glucose to OD600 ~ 1, then grown at 30 ˚C for 24 h. At OD600 ~ 1, 1 mM IPTG was added to induce PLlacO1:galP-glk. Specific titer (g−1 L−1 OD600−1) indicates titer per final OD600. d Comparison of the effect of gene deletions on the production of D-psicose. ptsG, ptsH, and/or pgm were deleted in Strain 4. D-psicose production was done as described in (c). Error bars indicate s.d. (n = 3 biological replicates).
Next, the impact of timing on the temperature shift from 37 °C to 30 °C was tested. Our preliminary test showed that 37 °C was suitable for cell growth while 30 °C was suitable for production. To determine the optimal timing of the shift from 37 °C to 30 °C, cultures were grown to an OD600 of ~0 (no culturing at 37 °C), ~0.4, or ~1 at 37 °C, then grown at 30 °C and induced as necessary. Across all strains, cultures moved to 30 °C at a later OD600 produced higher titers of D-psicose and grew with a greater ΔOD600 (Fig. 3b). When shifted at an OD600 ~ 1, Strain 3 produced the highest titers of D-psicose, at 9.13 g L−1, and grew with a ΔOD600 of 5.6. Strain 2 produced 5.42 g L−1 of D-psicose and grew with a ΔOD600 of 4.4.
We theorized that despite being a stronger promoter than PLlacO1, the expression timing of PgadB prevents D-psicose production enzymes from siphoning carbon away from central metabolism during a key period of growth. This allows for cultures to grow more robustly and produce higher titers of D-psicose. The use of endogenous, growth phase-associated promoters eliminates the need for costly chemical inducers and allows cultures to self-regulate pathway expression depending on growth and cellular viability. PgadB:alsE-hxpB (pAL2247, Supplementary Table 2) was thereafter used in production experiments.
Supplementing glucose import using GalP and Glk
Continuous glucose import, especially during the stationary phase of growth, is critical to the production of D-psicose. One consequence of limiting carbon flux through glycolysis by knocking out pfkA is the reduction in downstream metabolites, such as phosphoenolpyruvate (PEP)46. PEP is of particular concern, as it is utilized by the PTS to import and phosphorylate glucose. A reduction in PEP availability due to decreased flux through glycolysis may have an impact on the ability to assimilate glucose and produce D-psicose46,47. Additionally, it has been shown that increased G6P or F6P pools in a ΔpfkA mutant leads to degradation of ptsG mRNA that encodes for membrane receptor IICBGlc of the PTS complex48.
To enhance glucose import without the use of the PTS in the ΔpfkA background, we additionally expressed galP and glk from a plasmid. The galP gene encodes for the galactose proton symporter GalP21, which is capable of importing glucose. The glk gene encodes for glucokinase Glk22, which phosphorylates glucose to G6P (Fig. 1c). These genes were cloned downstream of PLlacO1, generating plasmid pAL2264 (Supplementary Table 2). Induction of the expression of galP-glk in Strain 4 (Strain 3 with PLlacO1:galP-glk, Table 1) substantially increased D-psicose production and allowed the strain to produce 13.81 g L−1 with a specific titer of 3.2 g L−1 OD600−1 and yield of 55% (Fig. 3c). In comparison, Strain 4 without IPTG produced 8.67 g L−1 of D-psicose, with a specific titer of 1.3 g L−1 OD600−1 and a yield of 49%. Yield was calculated based on the possibility that for every 1 mol of D-glucose consumed, 1 mol of D-psicose could be produced, leading to a theoretical maximum yield of 100%. Interestingly, expression of galP and glk imposed a growth burden that resulted in lower ΔOD600 (Fig. 3c), potentially due to overexpression of membrane proteins competing for membrane translocation machinery49.
Manipulating glucose utilization and metabolism
It has been shown that the PTS utilizes ~50% of PEP to transport and phosphorylate glucose50. To further balance cellular PEP supply and promote glucose import using GalP-Glk, we attempted to incapacitate the PTS by knocking out genes ptsG, encoding for membrane receptor IICBGlc, and ptsH, encoding for phosphocarrier protein HPr, in AL3990 (Supplementary Table 1)46.
Another source of glucose siphoning away from the D-psicose pathway is through glycogen biosynthesis. Glycogen is stored for usage during times of starvation, which for our purpose is unnecessary. The deletion of pgm, which encodes for phosphoglucomutase Pgm, prevents E. coli from producing glycogen51. As such, pgm was deleted in the production strains (Supplementary Table 1).
While Strain 4 with ΔptsG appeared detrimental to psicose production (ΔptsG, ΔptsG ΔptsH and ΔptsG Δpgm), Strain 4 with ΔptsH has no effect (Fig. 3d). Δpgm was beneficial for production, particularly when considering specific titers (Fig. 3d). Strain 5 (Strain 4 with Δpgm, Table 1) produced 14.66 g L−1 of D-psicose, with a specific titer of 5.5 g L−1 OD600−1 and yield of 58% (Fig. 3d).
Inhibiting glycogen biosynthesis by knocking out pgm removes a source of carbon to central metabolism which would normally be used to help cells grow under periods of starvation52. This combined effect of impaired growth and enhanced production lead to an increase in specific titer going from Strain 4 to Strain 5 (Fig. 3d).
Although some studies have shown the elimination of the PTS through gene knockouts helps restore intracellular PEP balance and rescue growth46, we found knocking out ptsG or ptsH to be deleterious or neutral to D-psicose production. Phosphorylated and dephosphorylated forms of IICBGlc and HPr take part in signaling cascades related not only to carbon metabolism, but also global gene expression through expression of RNA polymerase sigma subunits, including the aforementioned σ38-subunit and logarithmic phase-associated σ70-subunit24,45,46,53,54. Given the use of PgadB to express alsE and hxpB, eliminating portions of the PTS may reduce the expression of the D-psicose production pathway genes.
Dynamic regulation of glycolysis using CRISPRi
While the deletion of pfkA successfully redirected carbon flux towards D-psicose production, glycolysis remains active through PfkB. Completely shutting down glycolysis by deleting pfkB did not allow cells to grow under our culture conditions, so we attempted to dynamically limit pfkB expression to times it was essential. Cells require carbon flux through glycolysis when building biomass during the logarithmic phase of growth42,43,55. During the stationary phase, carbon flux can be reduced through glycolysis and redirected to D-psicose production. In order to dynamically regulate the expression of pfkB, we implemented a CRISPRi system26,27 targeting pfkB on the genome.
The CRISPRi system utilizes an inactivated Cas9, dCas9, which when recruited by a single guide RNA scaffold (sgRNA) can precisely target and block transcription initiation by RNA polymerase26. The dcas9 gene was cloned under an anhydrotetracycline (aTc)-inducible promoter, Ptet56, along with a constitutively expressed sgRNA sequence targeting a gene of interest. To confirm functionality of the CRISPRi system and determine where the sgRNA should target in order to achieve the greatest inhibition of expression, three different sgRNA sequences were designed to repress the expression of sfgfp under PLlacO1 (Supplementary Fig. 5). The sgRNAs targeted the upstream, middle, and downstream sequence of PLlacO1 (Supplementary Fig. 5a). We found the sgRNA targeting the middle of PLlacO1 lead to greatest reduction in fluorescence. Successive sgRNA targeting the promoter region of pfkB was designed with homology to the middle of the promoter sequence.
Further exploration of the CRISPRi system involved expressing dcas9 and sgRNA from either the same or separate plasmids (Supplementary Fig. 6). In the single-plasmid system, Ptet:dcas9 and a constitutively expressed sgRNA sequence targeting the promoter region of pfkB (Supplementary Fig. 6a) or no targeting sequence were cloned onto the same plasmid (Supplementary Table 2). For the separate-plasmid system, Ptet:dcas9 was cloned onto one plasmid, and the constitutively expressed sgRNA sequence targeting the promoter region of pfkB or no targeting sequence were cloned onto a different plasmid (Supplementary Table 2). Each CRISPRi system was introduced into AL4186 (Supplementary Table 1), and growth was measured 24 h after induction with 100 ng mL−1 aTc. The separate-plasmid system caused a greater inhibition of growth, most likely because the sgRNA was expressed from a high copy number plasmid, rather than a low copy number plasmid as was the case in the single-plasmid system. When production was tested using the separate-plasmid CRISPRi system in Strain AL3990 with pAL2247 (Supplementary Table 1 and 2), very little D-psicose was produced, regardless of aTc induction. As such, the single-plasmid CRISPRi system was used for production.
The single-plasmid CRISPRi system was installed in Strain 5, generating Strain 6 (empty guide) and Strain 7 (sgRNA targeting pfkB, Table 1). When tested for D-psicose production, Strain 7 with CRISPRi showed reduced cell growth and D-psicose titer, resulting in higher specific titer in comparison to Strains 5 and 6 (Fig. 4a). Strain 7 with aTc produced 11.40 g L−1 of D-psicose with a specific titer of 3.6 g L−1 OD600−1 and yield of 62%. Strain 7 without aTc produced 13.65 g L−1 of D-psicose with a specific titer of 3.8 g L−1 OD600−1 and yield of 60%.

a CRISPRi was used to knock down pfkB. The sgRNA targeting the promoter region of pfkB or without targeting sequence was expressed from a constitutive promoter. dcas9 was expressed from the aTc-inducible Ptet. Strain 5, 6, and 7 (Table 1) were grown in M9P media with 40 g L−1 glucose at 37 °C to OD600 ~ 1, after which 1 mM IPTG and 100 ng mL−1 aTc were added and cells were grown at 30 ˚C for 24 h. b High cell density D-psicose production. Strain 7 cultures was grown in M9P media with 40 g L−1 glucose at 37 °C until an OD600 of ~1, after which they were induced with 1 mM IPTG and 100 ng mL−1 aTc and grown for another 30 min. Cultures were then spun down and resuspended in M9P media with 40 g L−1 glucose, 1 mM IPTG, and 100 ng mL−1 aTC to an OD600 of ~10 and grown at 30 ˚C for 8 h. Error bars indicate s.d. (n = 3 biological replicates). c HPLC chromatogram of the high cell density experiment with Strain 7 cultured for 24 h. The elution of D-glucose occurs at ~3.36 min, and the elution of D-psicose occurs at ~5.45 min.
D-psicose production under high culture density conditions
The rate of D-glucose consumption and D-psicose production of Strain 7 (Table 1) was monitored for 10 h at media glucose concentrations of 3, 5, and 10 g L−1 (Supplementary Fig. 7). We found the rate of D-glucose consumption and D-psicose production to be similar across cultures, independent of media glucose concentrations. Cultures fed 3 g L −1 of glucose consumed all glucose over the course of the experiment.
To decouple growth and production, and minimize limitations on production by glucose availability, we cultured Strain 7 under high cell density conditions in an excess of available D-glucose over a shorter period of time (Fig. 4b). Strain 7 (Table 1) was grown to an OD600 of ~1 before being induced with 100 ng mL−1 aTc and 1 mM IPTG and grown for a further 30 min. Cells were then pelleted and resuspended to an OD600 of ~10 with M9P containing 40 g L−1 glucose, 100 ng mL−1 aTc, and 1 mM IPTG. Samples were taken and analyzed at 0, 4, and 8 h. Over the course of 8 h, Strain 7 produced 15.3 g L−1 of D-psicose with a specific titer of 1.4 g L−1 OD600−1, yield of 43%, and productivity of 1.9 g L−1 h−1 (Fig. 4b). Although D-psicose titers produced at 0–4 h and 4–8 h were similar, at 7.3 and 8.0 g L−1, the yield at 4–8 h (53%) was higher than that at 0–4 h (35%). The productivity at 4–8 h was 2.0 g L−1 h−1. Higher yield may be the result of cells responding to their high-density culture conditions, as our production system relies on the activation of stationary phase promoter PgadB. Overall, the system achieved industrially relevant production with high yield (>0.5 gproduct gsubstrate-1) and high productivity (>1 g L−1 h−1) comparable to bioethanol57, despite being cultured in unoptimized test tube conditions.
Strain 7 was able to consume all media glucose at 24 h (Fig. 4c). Full consumption of substrate is a desirable property of microbial production, as it is costly to separate D-psicose from mixtures of glucose and/or fructose. Depleting glucose from the media allows for easier extraction and purification of D-psicose in an industrial setting.
Conclusion
Within this study, we applied whole-cell catalysis as a strategy for producing the industrially relevant, rare sugar D-psicose. Living cells possess the capability of assembling stereo/regioselective enzymes, providing necessary cofactors, and secreting products for easy purification, all under environmentally friendly production conditions14,15,16,17. Whole-cell catalysis technology and infrastructure is already established industrially, and the model organism E. coli can be fed feedstocks that do not compete with commercial food production.
The elimination of competing pathways and additional expression of native E. coli genes (alsE, hxpB, galP, and glk) along with the use of static and dynamic gene regulation strategies lead to a strain capable of producing D-psicose using a thermodynamically favorable biosynthetic pathway from a readily available feedstock. The highest titer of D-psicose produced was 15.3 g L−1 with a specific titer of 2.0 g L−1 OD600−1 under test tube conditions. The highest yield achieved was 62%, surpassing current industrial standards. Furthermore, the strain’s ability to consume all D-glucose present in media greatly simplifies downstream purification requirements.
Overall, the engineered strain represents a critical step in producing D-psicose and other rare sugars in an efficient, cost-effective manner. The ability to produce rare sugars in bulk will help address rising global obesity rates by providing low-calorie sugar alternatives for ultra-processed foods1,2,4. Increased production of rare sugars will also grant access to sustainable pesticides for the agricultural industry, and medicinally relevant monosaccharides for the pharmaceutical industry4. The strategy developed in this study has the potential to drive transformative changes in our ability to produce, measure, and control the human-food relationship, developing a world where easily attainable metabolic health leads to happier, healthier, longer lives for everyone.
Methods
Reagents
All enzymes involved in the molecular cloning experiments were purchased from New England Biolabs (NEB). All synthetic oligonucleotides were synthesized by Integrated DNA Technologies. Sanger Sequencing was provided by Genewiz. D-Psicose and D-mannose were purchased from Sigma-Aldrich. D-Glucose was purchased from Fisher Scientific.
Strains and plasmids
All strains and plasmids used in this study are listed in Supplementary Table 1 and 2, respectively. All oligonucleotides are listed in Supplementary Table 3. Plasmids for D-psicose production were constructed using sequence and ligation independent cloning58. The constructed plasmids were verified via sequencing. A guide to the construction of plasmids used in this study is detailed in Supplementary Table 4.
Genome modifications such as gene deletion and gene insertion were constructed using CRISPR-Cas9-mediated homologous recombination59. Linear DNA repair fragments for gene deletions and insertions were constructed by amplifying genomic or plasmid DNA via PCR assembly60. Plasmids encoding sgRNA for CRISPR-Cas9-mediated homologous recombination were constructed using Q5 site-directed mutagenesis (New England Biolabs) using pTargetF plasmid (Addgene #62226) as a template. All genomic modifications were verified via sequencing. A guide to the CRISPR-Cas9-mediated gene modifications used in this study is detailed in Supplementary Table 5.
Predictive modeling
The Rosetta Molecular Suite was used to dock the ligand, psicose-6-phosphate, into the phosphatases: HxpA, HxpB, YigL, YidA, YihX, YbiV37. P6P AM1-BCC partial charges were assigned using the Antechamber suite from AMBER61. For the initial protein structures, we used the AlphaFold Protein Structure Database36,62. The AlphaFold models were then prepared for docking by using Rosetta Relax63. The ligand was then placed in the active site. Rosetta GALigand Dock was then used to dock P6P conformers into each phosphatase64. Distance and angle constraints were integrated to maintain the D-psicose and magnesium ion in catalytically competent geometry for hydrolysis. A total of 2000 simulations were run for each phosphatase, and the top 10 best scoring outputs sorted by constraint score, protein-ligand interface energy, and total system energy score were selected for analysis.
Culturing media
Overnight cultures were grown at 37 °C in 3 mL of Luria-Bertani (LB) media with appropriate antibiotics. Antibiotic concentrations were as follows: spectinomycin (50 μg mL−1), ampicillin (200 μg mLSLIC1), kanamycin (50 μg mLSLIC1), gentamycin (3.75 μg mL−1). M9 minimal media consists of 33.7 mM Na2HPO4, 22 mM KH2PO4, 8.6 mM NaCl, 9.4 mM NH4Cl, 2 mM MgSO4, 0.1 mM CaCl2, A5 trace metals mix (2.86 mg L−1 H3BO3, 1.81 mg L−1 MnCl2⋅4H2O, 0.079 mg L−1 CuSO4⋅5H2O, 49.4 μg L−1 Co(NO3)2⋅6H2O), varying concentrations of glucose, and appropriate antibiotics. M9P media for the psicose production consists of M9 minimal media supplemented with 5 g L−1 of yeast extract and appropriate antibiotics. No D-psicose production was detected when cultures were grown in M9P media without glucose. Inducer concentrations are as follows: isopropyl-β-D-1-thiogalactopyranoside (IPTG) (1 mM), anhydrotetracycline (aTc) (100 ng mL−1). OD600 was measured with a Synergy H1 hybrid plate reader (BioTek Instruments, Inc.).
Fluorescence assays
Overnight cultures were inoculated at 1% into 300 μL of LB media in a 96-well black-walled fluorescence assay plate. Cells were grown at 37 °C, 250 rpm, until OD600 ~ 0.4. Cultures were then induced with IPTG if necessary and grown at 37 °C, 250 rpm, for 24 h. Fluorescence was measured at an excitation wavelength of 485 nm and emission wavelength of 510 nm with a Synergy H1 hybrid plate reader (BioTek Instruments, Inc.).
D-Psicose production
For regular cell density production experiments, overnight cultures were inoculated at 1% into 3 mL of M9P media. Cells were grown at 37 °C until the OD600 described, then induced with IPTG and aTc if necessary, and grown at 30 °C for 24 h.
For high cell density production experiments in M9P media, overnight cultures were inoculated at 2% into 50 mL of M9P media. Cells were grown at 37 °C until OD600 ~ 1. Cultures were then induced with IPTG and aTc if necessary and grown for a further 30 min. Cultures were centrifuged at 5000 g for 15 min and resuspended in M9P media with IPTG and aTc if necessary to a target OD600. Cultures were grown at 30 °C for 24 h.
HPLC analysis
Analysis of D-psicose, D-glucose, and D-mannose concentrations was performed using HPLC (Shimadzu) equipped with a refractive index detector (RID) 10 A and Rezex™ RCU-USP sugar alcohol column (Phenomenex). Mobile phase was comprised of 100% MilliQ water. Samples were run with an injection volume of 1 μL at a flow rate of 0.5 mL min−1 for 7 min, with the column oven at 83 °C and RID cell temperature of 40 °C.
To prepare samples for HPLC analysis, 300 μL of culture was centrifuged at 17,000 g for 5 min. Supernatants were applied to a 0.2 μm PVDF hydrophilic membrane 96 well filter plate and centrifuged at 17,000 g for 2 min into a polystyrene 96 well.
GC-MS analysis
GC-MS analysis was performed by the UC Davis West Coast Metabolomics Center. Chemical standards (D-psicose, D-mannose, D-glucose, D-galactose, D-erythrose, D-tagatose, and D-threose) were purchased from Sigma Aldrich.
To analyze via GC-MS, 4 μL of spun-down culture supernatant were dried down and derivatized by adding 10 μL of 40 mg mL−1 methoxyamine hydrochloride (Sigma-Aldrich) in pyridine (Sigma-Aldrich) and shaking at 30 °C for 1.5 h. Subsequently, 90 μL of N –methyl-N-(trimethylsilyl)-trifluoroacetamide (MSTFA) (Sigma-Aldrich) was added with a standard mixture of 13 fatty-acid methyl esters (FAMEs) (Sigma-Aldrich) as retention index markers and shaken at 80°C for 30 min. Samples were immediately transferred to crimp top vials and injected onto each GC − MS instrument. A LECO Pegasus IV TOF MS was coupled to an Agilent 7890 GC system installed with a Restek RTX-5Sil MS column (29.70 m length, 0.25 mm i.d, 0.25 μM df, 95% dimethyl/ 5% diphenyl polysiloxane film) with an additional 10 m guard column. 1 μL of the derivatized sample was injected into the GC in splitless mode at an injection temperature of 275 °C and a constant flow of 1 mL min−1. The initial oven temperature was held at 50 °C for 1 min and ramped at a rate of 20 °C min−1 to 330 °C that was maintained for 5 min for a total run time of 20 min. The mass spectrometer was used under electron ionization mode at +70 eV. Mass spectra were acquired from 85 to 500 m/z at a scan rate of 17 Hz and 250 °C source temperature. Binbase was used for metabolite annotation and reporting65.
Data availability
The datasets generated in this study are available from the corresponding author on request. The E. coli strains and the plasmids used in this study are available upon request.
References
Blüher, M. Obesity: global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 15, 288–298 (2019).
Ward, Z. J. et al. Projected U.S. State-Level prevalence of adult obesity and severe obesity. N. Engl. J. Med. 381, 2440–2450 (2019).
MarketandMarket. Sugar Substitutes Market by Type (High Fructose Syrup, High-Intensity Sweetener, Low-Intensity Sweetener), Composition, Application (Beverages, Food Products, and Health & Personal Care Products), and Region - Global Forecast to 2025. (2020).
Mijailovic, N., Nesler, A., Perazzolli, M., Aït Barka, E. & Aziz, A. Rare sugars: recent advances and their potential role in sustainable crop protection. Molecules 26, 1720 (2021).
FDA. GRAS Notice (GRN) No. 693. Encycl. Toxicol. 417–420 (2017).
Oshima, H., Kimura, I. & Izumori, K. Psicose contents in various food products and its origin. Food Sci. Technol. Res. 12, 137–143 (2006).
Itoh, H. et al. Purification and characterization of D -Tagatose 3-Epimerase from Pseudomonas sp. ST-24. Biosci. Biotechnol. Biochem. 58, 2168–2171 (1994).
Jiang, S. et al. Review on D-Allulose: in vivo metabolism, catalytic mechanism, engineering strain construction, bio-production technology. Front. Bioeng. Biotechnol. 8, 26 (2020).
Armetta, J. et al. Biosensor-based enzyme engineering approach applied to psicose biosynthesis. Synth. Biol. 4, ysz028 (2019).
Hu, M., Li, M., Jiang, B. & Zhang, T. Bioproduction of D-allulose: properties, applications, purification, and future perspectives. Compr. Rev. Food Sci. Food Saf. 20, 6012–6026 (2021).
Noor, E. et al. An integrated open framework for thermodynamics of reactions that combines accuracy and coverage. Bioinformatics 28, 2037 (2012).
Liu, J. J. et al. Overcoming the thermodynamic equilibrium of an isomerization reaction through oxidoreductive reactions for biotransformation. Nat. Commun. 10, 1–8 (2019).
Li, Y., Shi, T., Han, P. & You, C. Thermodynamics-driven production of value-added d -Allulose from inexpensive starch by an in vitro enzymatic synthetic biosystem. ACS Catal. 11, 5088–5099 (2021).
Nielsen, J. et al. Engineering synergy in biotechnology. Nat. Chem. Biol. 10, 319–322 (2014).
Rabinovitch-Deere, C. A., Oliver, J. W. K., Rodriguez, G. M. & Atsumi, S. Synthetic biology and metabolic engineering approaches to produce biofuels. Chem. Rev. 113, 4611–4632 (2013).
Nielsen, J. & Keasling, J. D. Engineering cellular metabolism. Cell 164, 1185–1197 (2016).
Liao, J. C., Mi, L., Pontrelli, S. & Luo, S. Fuelling the future: microbial engineering for the production of sustainable biofuels. Nat. Rev. Microbiol. 14, 288–304 (2016).
Fraenkel, D. G. & Neidhardt, F. C. Glycolysis. in Escherichia coli and Salmonella: cellular and molecular biology 189–198 (ASM Press, 1996).
Guo, Q. et al. Metabolically engineered Escherichia coli for Conversion of D-Fructose to D-Allulose via phosphorylation-dephosphorylation. Front. Bioeng. Biotechnol. 10, 947469 (2022).
Guo, Y. et al. Irreversible biosynthesis of D-allulose from D-glucose in Escherichia coli through fine-tuning of carbon flux and cofactor regeneration engineering. J. Sci. Food Agric. 103, 5481–5489 (2023).
Henderson, P. J. F., Giddens, R. A., & Jones Mortimer, M. C. Transport of galactose, glucose and their molecular analogues by Escherichia coli K12. Biochem. J. 162, 309–320 (1977).
Meyer, D., Schneider-Fresenius, C., Horlacher, R., Peist, R. & Boos, W. Molecular characterization of glucokinase from Escherichia coli K-12. J. Bacteriol. 179, 1298–1306 (1997).
Pletnev, P., Osterman, I., Sergiev, P., Bogdanov, A. & Dontsova, O. Survival guide: Escherichia coli in the stationary phase. Acta Nat. 7, 22–33 (2015).
Weber, H., Polen, T., Heuveling, J., Wendisch, V. F. & Hengge, R. Genome-wide analysis of the general stress response network in Escherichia coli: sigmaS-dependent genes, promoters, and sigma factor selectivity. J. Bacteriol. 187, 1591–1603 (2005).
Shimada, T. et al. Classification and strength measurement of stationary-phase promoters by use of a newly developed promoter cloning vector. J. Bacteriol. 186, 7112–7122 (2004).
Qi, L. S. et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 (2013).
Ge, C. et al. Redesigning regulatory components of quorum-sensing system for diverse metabolic control. Nat. Commun. 13, 1–12 (2022).
Robinson, P. K. Enzymes: principles and biotechnological applications. Essays Biochem. 59, 1–41 (2015).
Xiao, Q., Niu, J., Liu, H., Liu, Y. & Zhou, X. High Conversion of d-Fructose into d-Allulose by Enzymes Coupling with an ATP Regeneration System. Mol. Biotechnol. 61, 432–441 (2019).
Siebenlist, U., Simpson, R. B. & Gilbert, W. E. coli RNA polymerase interacts homologously with two different promoters. Cell 20, 269–281 (1980).
Hollinshead, W. D. et al. Examining Escherichia coli glycolytic pathways, catabolite repression, and metabolite channeling using Δpfk mutants. Biotechnol. Biofuels 9, 1–13 (2016).
Hellinga, H. W. & Evans, P. R. Nucleotide sequence and high‐level expression of the major Escherichia coli phosphofructokinase. Eur. J. Biochem. 149, 363–373 (1985).
Chan, K. K., Fedorov, A. A., Fedorov, E. V., Almo, S. C. & Gerlt, J. A. Structural basis for substrate specificity in phosphate binding (β/α)8-barrels: D-allulose 6-phosphate 3-epimerase from Escherichia coli K-12. Biochemistry 47, 9608–9617 (2008).
Kuznetsova, E. et al. Genome-wide analysis of substrate specificities of the Escherichia coli haloacid dehalogenase-like phosphatase family. J. Biol. Chem. 281, 36149–36161 (2006).
Studier, F. W. & Moffatt, B. A. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189, 113–130 (1986).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Leman, J. K. et al. Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nat. Methods 17, 665–680 (2020).
Lutz, R. & Bujard, H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 25, 1203–1210 (1997).
Fraenkel, D. G. Selection of Escherichia coli mutants lacking glucose-6-phosphate dehydrogenase or gluconate-6-phosphate dehydrogenase. J. Bacteriol. 95, 1267–1271 (1968).
Roos, A. K., Mariano, S., Kowalinski, E., Salmon, L. & Mowbray, S. L. d-Ribose-5-Phosphate Isomerase B from Escherichia coli is also a functional d-Allose-6-Phosphate isomerase, while the mycobacterium tuberculosis enzyme is not. J. Mol. Biol. 382, 667–679 (2008).
Gao, H., Chen, Y. & Leary, J. A. Kinetic measurements of phosphoglucose isomerase and phosphomannose isomerase by direct analysis of phosphorylated aldose-ketose isomers using tandem mass spectrometry. Int. J. Mass Spectrom. 240, 291–299 (2005).
Tan, S. Z. & Prather, K. L. Dynamic pathway regulation: recent advances and methods of construction. Curr. Opin. Chem. Biol. 41, 28–35 (2017).
Li, Y. et al. Fine tuning the glycolytic flux ratio of EP-bifido pathway for mevalonate production by enhancing glucose-6-phosphate dehydrogenase (Zwf) and CRISPRi suppressing 6-phosphofructose kinase (PfkA) in Escherichia coli. Microb. Cell Fact. 20, 1–12 (2021).
Gupta, A., Reizman, I. M. B., Reisch, C. R. & Prather, K. L. J. Dynamic regulation of metabolic flux in engineered bacteria using a pathway-independent quorum-sensing circuit. Nat. Biotechnol. 35, 273–279 (2017).
Ueguchi, C., Misonou, N. & Mizuno, T. Negative control of rpoS expression by phosphoenolpyruvate: carbohydrate phosphotransferase system in Escherichia coli. J. Bacteriol. 183, 520–527 (2001).
Gosset, G. Improvement of Escherichia coli production strains by modification of the phosphoenolpyruvate:sugar phosphotransferase system. Microb. Cell Factories 4, 14 (2005).
Brockman, I. M. & Prather, K. L. J. Dynamic knockdown of E. coli central metabolism for redirecting fluxes of primary metabolites. Physiol. Behav. 176, 100–106 (2016).
Morita, T., El-Kazzaz, W., Tanaka, Y., Inada, T. & Aiba, H. Accumulation of glucose 6-phosphate or fructose 6-phosphate is responsible for destabilization of glucose transporter mRNA in Escherichia coli. J. Biol. Chem. 278, 15608–15614 (2003).
Jensen, H. M., Eng, T., Chubukov, V., Herbert, R. A. & Mukhopadhyay, A. Improving membrane protein expression and function using genomic edits. Sci. Rep. 7, 1–14 (2017).
Holms, W. H. The central metabolic pathways of Escherichia coli: relationship between flux and control at a branch point, efficiency of conversion to biomass, and excretion of acetate. Curr. Top. Cell. Regul. 28, 69–105 (1986).
Eydallin, G. et al. Genome-wide screening of genes affecting glycogen metabolism in Escherichia coli K-12. FEBS Lett. 581, 2947–2953 (2007).
Preiss, J. Bacterial glycogen synthesis and its regulation. Annu Rev. Microbiol 38, 419–58 (1984).
Park, Y. H., Lee, C. R., Choe, M. & Seok, Y. J. HPr antagonizes the anti-σ70 activity of Rsd in Escherichia coli. Proc. Natl. Acad. Sci. USA 110, 21142–21147 (2013).
Görke, B. & Stülke, J. Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat. Rev. Microbiol. 6, 613–624 (2008).
Yang, Y. et al. Sensor-regulator and RNAi based bifunctional dynamic control network for engineered microbial synthesis. Nat. Commun. 9, 1–10 (2018).
Gossen, M. et al. Transcriptional activation by tetracyclines in mammalian cells. Science 268, 1766–1769 (1995).
Mohd Azhar, S. H. et al. Yeasts in sustainable bioethanol production: a review. Biochem. Biophys. Rep. 10, 52–61 (2017).
Li, M. Z. & Elledge, S. J. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat. Methods 4, 251–256 (2007).
Jiang, Y. et al. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl. Environ. Microbiol. 81, 2506–2514 (2015).
Xiong, A. S. et al. A simple, rapid, high-fidelity and cost-effective PCR-based two-step DNA synthesis method for long gene sequences. Nucleic Acids Res. 32, e98 (2004).
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A. & Case, D. A. Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 (2004).
Varadi, M. et al. AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 (2022).
Tyka, M. D. et al. Alternate states of proteins revealed by detailed energy landscape mapping. J. Mol. Biol. 405, 607–618 (2011).
Park, H., Zhou, G., Baek, M., Baker, D. & Dimaio, F. Force field optimization guided by small molecule crystal lattice data enables consistent sub-angstrom protein-ligand docking. J. Chem. Theory Comput. 17, 2000–2010 (2021).
Lai, Z. et al. Identifying epimetabolites by integrating metabolome databases withmass spectrometry cheminformatics. Nat. Methods 15, 53 (2018).
Stourac, J. et al. Caver Web 1.0: identification of tunnels and channels in proteins and analysis of ligand transport. Nucleic Acids Res. 47, W414–W422 (2019).
Maglic, J. B. & Lavendomme, R. MoloVol: an easy-to-use program for analyzing cavities, volumes and surface areas of chemical structures. J. Appl. Crystallogr. 55, 1033–1044 (2022).
Acknowledgements
This work was supported by Mars, Incorporated.
Author information
Authors and Affiliations
Contributions
J.E.T., D.S.K.P., A.Z., T.C., J.D., J.S. and S.A. designed research; J.E.T., D.S.K.P., A.Z., J.N.G., A.B.J.L. and E.P.R. performed the experiments; J.E.T., D.S.K.P., A.Z., E.P.R., O.F., J.B.S., and S.A. analyzed data; J.E.T., D.S.K.P, E.P.R., J.B.S. and S.A. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare the following financial interests which may be considered as potential competing interests: J.E.T., D.S.K.P., A.Z., J.N.G., A.A., T.A.C., A.B.J.L., J.B.S. and S.A. are inventors on the patent application related to this study.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Taylor, J.E., Palur, D.S.K., Zhang, A. et al. Awakening the natural capability of psicose production in Escherichia coli. npj Sci Food 7, 54 (2023). https://doi.org/10.1038/s41538-023-00231-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41538-023-00231-0
