
Abstract
Osteoporosis is characterized by decreased bone mass, microarchitectural deterioration, and increased bone fragility. High-fat diet (HFD)-induced obesity also results in bone loss, which is associated with an imbalanced gut microbiome. However, whether HFD-induced obesity or HFD itself promotes osteoclastogenesis and consequent bone loss remains unclear. In this study, we developed HFD-induced obesity (HIO) and non-obesity (NO) mouse models to evaluate the effect of HFD on bone loss. NO mice were defined as body weight within 5% of higher or lower than that of chow diet fed mice after 10 weeks HFD feeding. NO was protected from HIO-induced bone loss by the RANKL /OPG system, with associated increases in the tibia tenacity, cortical bone mean density, bone volume of cancellous bone, and trabecular number. This led to increased bone strength and improved bone microstructure via the microbiome-short-chain fatty acids (SCFAs) regulation. Additionally, endogenous gut-SCFAs produced by the NO mice activated free fatty acid receptor 2 and inhibited histone deacetylases, resulting in the promotion of Treg cell proliferation in the HFD-fed NO mice; thereby, inhibiting osteoclastogenesis, which can be transplanted by fecal microbiome. Furthermore, T cells from NO mice retain differentiation of osteoclast precursors of RAW 264.7 macrophages ex vivo. Our data reveal that HFD is not a deleterious diet; however, the induction of obesity serves as a key trigger of bone loss that can be blocked by a NO mouse-specific gut microbiome.
Similar content being viewed by others

Introduction
Osteoporosis, characterized by decreased bone mass, microarchitectural deterioration and increased bone fragility, is the most prevalent chronic metabolic bone disease, subsequent increases in the risk of fracture1,2,3. Bone loss results from an imbalance in bone remodeling4,5,6,7. Although there are many preventive methods and treatments for bone loss, their application requires exertion by patients, who may limit their physical exercise, or includes unwanted side effects and drug costs, such as hormone therapies. These drawbacks decrease their utilization and increase the risk of bone loss. Therefore, diet intervention is considered the safest and most easily implemented treatment to improve bone health and reduce bone loss8,9,10,11,12.
Abnormal bone metabolism and resorption are influenced by many factors, such as genetics, hormones, age, and nutrition, among which dietary factors play an important role13,14,15. A high-fat diet (HFD) has become a common dietary pattern worldwide16,17,18. Recent studies suggest that an HFD not only leads to obesity but also induces metabolic abnormalities and absorption of bone, resulting in reduced bone mass and low bone strength, thus increasing the risk of spontaneous and traumatic bone injury19,20,21,22. This suggests that excess fat is detrimental to bone health23,24,25. However, while it has been considered a poor dietary choice, an HFD does not invariably lead to obesity26,27,28. Moreover, non-obese individuals with an HFD and their associated bone health have barely been studied, which raises the question as to whether HFD-induced obesity or an HFD alone leads to bone loss.
Gut microbiota, known as the second gene pool of the human body, plays a critical role in regulating host physiology, including bone remodeling in animal subjects and human patients29,30,31. Intestinal microbiomes mediate bone metabolism by regulating immune status, endocrine coordination, flora composition, and their generated metabolites32,33,34. Short-chain fatty acids (SCFAs), the main product of human bacterial fermentation, enhance body weight density and regulate osteoclastogenesis to prevent bone loss, which is an effective regulator of osteoclast (OC) metabolism and bone homeostasis35,36,37,38. Furthermore, SCFAs regulate the number and function of colonic regulatory T cells (Tregs) through inhibiting histone deacetylases (HDACs) and activating the G-protein-coupled receptor signaling cascade in mice39,40,41. Tregs have been shown to ameliorate arthritis and bone loss in autoimmune animal models and ovariectomized rats, respectively42,43,44. However, how they are involved in bone loss in HFD-induced obese mice is unclear.
In the present study, we show that HFD-induced obesity leads to gut dysbiosis and consequent bone loss. Furthermore, using biomechanical and structural bone parameters, we demonstrate that HFD-fed non-obese (NO) mice still build up healthy bones despite long-term HFD. These osteological improvements were mediated by gut microbiome-SCFA-regulation and were transferrable to obese mice through fecal microbiota transplantation (FMT). Mechanistically, we show that endogenous gut SCFA-mediated Tregs and HDACs are critical for maintaining bone health through the gut microbiome-bone axis.
Results
HFD-induced obesity, but not HFD alone, leads to bone loss
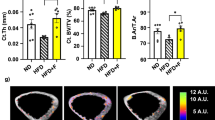
To address whether HFD alone or HFD-induced obesity contribute to the observed bone loss, HIO mice were defined as having body weight higher than 20% of chow diet (CD) mice, while NO mice were defined as body weight within 5% of the CD weight range after 10 weeks of HFD feeding. After that, NO and HIO mice were fed HFD for another 10 weeks. NO mice receiving an extended HFD had a stable weight, but significantly lower serum triglycerides (TG) and low-density lipoprotein (LDL) compared to HIO mice during these later 10 weeks (Fig. 1a–c and S1a–d) followed by reduced numbers and smaller sizes of fat vacuoles in hepatocytes and white adipose tissues (Fig. S1e–g). This was accompanied by a trend toward a significant increased Young’s modulus (Fig. 1d), suggesting preferred tenacity of the tibia, with no significant changes in maximum bending stress and maximum bending load (Fig. S1h, i) in a three-point bending test.

a Study design. Tibia were harvest after 20 weeks HFD intervene for HIO and NO mice on bone property analysis. b Body weight changes after chow diet (n = 14) and HFD (n = 81) fed during 70 days. c Body weight of HIO, NO, and CD mice (n = 7) after 10 weeks of HFD or chow diet, respectively. d Young’s modulus (n = 5). Tibia was analyzed by μ-CT (n = 4–6) to access (e) the mean/density of cortical bone, (f) generate representative cortical bone images, (g) three-dimensional reconstruction images of cancellous bone, (h) BV/TV of tibia trabecular, (i) trabecular number (Tb. N), (j) SIM of Trabecula and (k) trabecular separation/spacing (Tb. Sp). l Immunohistochemistry of OPG and RANKL expression (12×), length of the scale bar is 1000 μm. Data are presented as means ± standard errors of the means (± SEM). Results determined by one-way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001.
Biomechanical changes were also reflected at a structural level. HIO mice were significantly different from CD mice, while NO mice were similar. However, while the decrease in mean/density of cortical bone was enhanced in NO compared to HIO mice (Fig. 1e, f), it was not significantly different and there were no effects on cortical thickness (Ct.Th) (Fig. S1j). As an extension of cortical bone into cancellous bone, the quality of trabecular bone is closely related to its microstructure. The μ-CT and three-dimensional reconstruction images (Fig. 1g) showed that HIO had reduced bone volume to tissue volume (BV/TV) ratio of tibia trabecular, while NO had markedly elevated the value (Fig. 1h). A robust reduction in trabecular bone (Tb.N) (Fig. 1i) and structure model index (SMI) (Fig. 1j) and increases in trabecular separation (Tb.Sp) (Fig. 1k) were observed in HIO mice, whereas the NO group reversed these changes. Based on the above findings, we investigated whether osteoclastogenesis was involved. By immunostaining tibia, we determined substantially higher osteoprotegerin (OPG) and lower receptor activator of nuclear factor-κB ligand (RANKL) expression in NO mice, suggesting inhibition of OC differentiation compared with the HIO group (Fig. 1l). RANKL, as a type II transmembrane protein, is the only factor found to induce osteoclast differentiation and development by combined with receptor activator of nuclear factor-κB (RANK). Moreover, OPG and RANKL competitive combination with RANK to inhibit bone resorption and promote bone formation45. These data show that NO mice retained normal biomechanical and structural bone parameters despite a long-term HFD, unlike the HIO mice.
The gut microbiome determines the differences between obese and non-obese mice under HFD
Next, we investigate why the same HFD led to different levels of weight gain with entirely different bone qualities. Recent evidence suggests an interaction between gut microbiota and bone metabolism5. We, therefore, investigated whether microbiota compositions were altered with HFD in NO versus HIO mice. As shown by principal component analysis (PCA), the NO group was closer to the CD group than to the HIO group on PC1 which explained 59.77% of the total variation, indicating that the gut microbiota composition of NO was similar to the CD group (Fig. S2a).
Alpha analysis showed that the operational taxonomic unit (OTU) number and PD_Whole_tree index were increased in NO compared to HIO mice, indicating greater diversity of bacterial species (Fig. 2a, b). In addition, the Chao1 and ACE indices, which take into account species abundance, were increased in NO mice (Fig. 2c, d). In contrast to the HIO group, the ratio of Firmicutes to Bacteroidetes was markedly decreased in the NO group, suggesting that the change in the ratio was related to the development of obesity46 (Fig. 2e, f). Notably, bacterial differential abundance at the phylum level of Actinobacteria and Patescibacteria in NO were significantly lower compared to the HIO group, which were previously shown to be increased in obese patients and in the adipose tissue of obese people47,48, respectively (Fig. 2g, h). The family taxa heat map (Fig. 2i) revealed an HIO microbiota signature associated with an increase in families of Erysipelotrichaceae, Enterococcaceae, Peptostreptococcaceae and Corynebacteriaceae (Fig. S2b) which are negatively related with obesity49,50,51,52. HIO microbiota were also associated with increases in disease-associated families Saccharimonadaceae, Aerococcaceae, and Eggerthellaceae (Fig. S2c), which are related to genetic risk for autoimmunity, schizophrenia and depression, respectively53,54,55. NO microbial composition was, however, different than the HIO microbial composition, as well as CD one, with overall increases in SCFA-producing bacteria Akkemansiaceae, Prevotellaceae, Ruminococcaceae, Lachnospiraceae, Bacteroidaceae and Enterobacteriaceae56,57,58, and obesity negative-related bacteria Rikenellaceae and Muribaculaceae (Fig. S2d)26,59,60, suggesting that the NO gut environment favored the selective expansion of SCFA-producing or obesity negative-related bacterial taxa.

a–d OTU number, PD_Whole_tree, chao1 and ACE indices of alpha diversity. e Relative abundance of phyla. f Ratio of Firmicutes/Bacteroidetes. g, h Relative abundance of Acrtnobacteria and Patescibacteria. i Heat map representing differentially abundant taxa (genus with higher hierarchy family name) in CD, HIO and NO groups, which has been normalized to the range of −4 to 4. j Fecal SCFAs content composed of acetate, propionate, butyrate and isobutyrate, dimethylbutyric acid, n-valeric acid, isovaleric acid, and caproic acid. k Correlations between kinds of genus and eight types of SCFAs in CD, HIO and NO groups. l Volcano plot showing differentially expressed metabolites in feces from HIO and NO mice; red dots indicate upregulated in HIO group, blue dots indicate downregulated in HIO group. m Summary of metabolic pathways which include discriminating metabolites. Green rectangle—significant metabolites with an increased fold change in NO vs. HIO comparison. Red rectangle—significant metabolites with an decreased fold change in NO vs. HIO comparison. n = 5 per group for high-throughput sequencing of the 16S rDNA gene of fecal bacteria; n = 5–7 per group for Fecal metabolomics. Correlations were performed using Pearson test by “R” packet. Two-way ANOVA was used for SCFAs content analysis of (j). Data are presented as means ± SEM. One-way ANOVA was used to analyze significant differences in the rest of the data. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To confirm that changes in gut microbiota composition correlated with SCFAs, we analyzed the fecal SCFA content and its correlation with the genera identified. There was little difference between CD and NO mice, while as expected, HIO mice differed for most SCFAs. Strikingly, NO had increased acetate and butyrate content compared to that of HIO mice. NO mice also had increased propionate content but it was not significant (Fig. 2j). Furthermore, correlation analysis showed that the genera Alistipes, Bacteroides, Lachnospiraceae_NK4A136_group, prevotellaceae_UCG-001, Ruminiclostridium_9 and Ruminococcaceae_NK4A214_group were positively correlated with isovaleric acid, isobutyric acid or dimethybutyric acid (Fig. 2k). Consistently, the relative abundance of those genera was over-represented in NO mice (Fig. S2e). Significant correlations were also observed between Roseburia, Butyricicoccus and Ruminiclostridium_6 and butyrate or acetic acid content. NO mice exhibited lower relative abundance of Lachnospiraceae_UCG-006 (Fig. S2f) than HIO mice, which was negatively correlated with N-valeric acid, propionic acid, isobutyric acid, caproic acid, butyrate or acetic acid (Fig. 2k). These data suggest that NO mice resist HFD-induced obesity through increased SCFA-producing bacteria in their gut.
As critical regulators, gut microbiota should change other metabolites in addition to SCFA content. To reveal the metabolic phenotype related to the gut microbiome, which could potentially explain why the same HFD results in different body weights, we performed metabolomic profiling of the feces of HIO and NO mice. Through comparison of the two groups, 50 fecal metabolites were identified with significant peaks in negative and positive ionization modes (Fig. 2l and S2g, h). Analysis using the Kyoto Encyclopedia of Genes and Genomes (KEGG) showed that enrichment of metabolites in NO pathways primarily involved phenylalanine, tyrosine and tryptophan biosynthesis; phenylalanine metabolism; alanine, aspartate, and glutamate metabolism; and pyruvate metabolism as compared to HIO (Fig. S2i). In the integrated metabolic pathways ((Fig. 2m), tryptophan catabolic fluxes were enhanced in the HIO gut, as characterized by increased levels of 6-hydroxymelatonin (−1.65-fold), 3-methylindole pyruvate (−1.44-fold), and 3-indoleacetonitrile (−1.35-fold), and decreased levels of indolylmethylthiohydroximate (+1.45-fold), and 3-methyldioxyindole (+1.62-old) (Fig. S2j). Furthermore, increased levels of tyrosine metabolite of sinapine (−1.42 fold), 1,2-dehydroreticuline (−1.59 fold), phenol (−1.37 fold) and fumarate (−1.73 fold) were shown in HIO mouse feces (Fig. S2j). We also observed that all three purine metabolite levels including 5-hydroxyisourate (−1.55 fold), xanthine (−1.50-fold), and hypoxanthine (−1.46-fold) were reduced in NO mice (Fig. S2j), which was negatively correlated with the bone parameters of SMI, Tb.N, BV/TV, bone mean/density and Young’s modulus, but positively correlated with Tb.sp (Fig. S2k).
SCFA treatment ameliorates HFD obesity-induced bone loss
To confirm whether microbiota impacts the bone parameters through SCFA production, we first administrated a SCFA mix intragastrically to HIO mice. SCFAs improved the trabecular meshwork at the femoral head with relatively increased reticular formation and a neatly arranged and complete microstructure. In addition, cartilage collagen fibers also recovered from a disordered loose and dissolved arrangement to become more matured (Fig. 3a). These structural improvements were accompanied by reduced fragility of bones in SCFA-treated mice, as shown by the biomechanical measurements during the three-point test (Fig. 3b–d). The SCFA mix also elevated acetate and propionate levels in feces, which were mainly produced by the applicable gut microbiota (Fig. 3e). Correspondingly, SCFA gavage significantly enhanced free fatty acid receptor 2 (Ffar2) expression which can be activated by SCFA in the colon, small intestine, and spleen (Fig. 3f). To directly elucidate whether SCFAs were involved in the prevention of bone loss in NO mice through regulation of SCFA-producing gut microbiota, we transplanted the fecal microbiota (FMT) from NO mice into HIO mice (Fig. 3g), while maintaining the diet (HFD for HIO, NO, HIO+FMT and NO+FMT mice; chow for CD and CD+FMT control mice). Alpha-diversity analysis showed higher ACE, Chao1, and PD_Whole_tree indices in NO+FMT mice after the transplantation than those of the CD+FMT and HIO+FMT groups (Fig. S3a). Interestingly, alpha diversity was elevated after FMT in all groups compared to pretransplantation. At the family level, the heat map showed Eggerthellaceae, Enterococcaceae and Erysipelotrichaceae were more abundant in the HIO+FMT group than in the CD+FMT and NO+FMT groups (Fig. 3h and S3b); in addition, NO+FMT mice had increased the abundance of Akkemansiaceae, which was partly in accordance with the results obtained before the transplantation (Fig. 3h and S3c; Fig. S2b–d). Ffar2 mRNA expression in colon and lymph node tissue was significantly increased after NO mice fecal transplantation and showed positive correlation with Candidatus_Saccharimonas, Lactobacillus, Lachnospiraceae_UCG-006, Ruminococcaceae_UCG-014, and Ruminococcaceae_NK4A214_group; and negative correlation with Enterococcus, Blautia and Rikenella (Fig. 3i, j and S3d). Together, these data demonstrated that SCFAs produced by the gut microbiome were involved in inhibition of bone loss by NO.

a H&E staining of backbone and masson trichrome staining (100×), length of the scale bar is 500 μm. b Maximum bending stress. c Maximum bending load. d Young’s modulus (n = 3). e Fecal SCFAs content composed of acetate, propionate and butyrate (n = 5). f Relative Ffar2 mRNA expression in colon, small intestine, spleen, and lymph node (n = 5). g Study design. Five-week-old HFD-fed mice were colonized with feces from the indicated mice for 10 weeks. h Heat map of the selected most differentially abundant features at the family level. i Relative Ffar2 mRNA expression in colon and lymph (n = 4). j Correlations between two different locations of recipients and kinds of genus. Correlations were performed using Pearson test between two different locations of recipients and kinds of genus by “R” packet. k–o μ-CT was conducted to determine the bone parameters of BV, Tb. Th, Tb. N, Tb. Sp, and obtain 3D images (n = 3). p Relative mRNA expression of Rankl, Opg, Rankl/Opg, Trap and Ctsk (n = 3–4). Data are presented as means ± SEM. Results determined by one-way ANOVA for (b–d) and (k–n), and two-way ANOVA for (e, f, i, p). *p < 0.05, **p < 0.01, ****p < 0.0001.
To uncover whether the transplanted microbiota enhanced bone strength, we also performed a similar analysis for parameters of bone tissue. Fecal transplantation of NO microbiota significantly increased the values of BV, Tb.Th, and Tb. N, but decreased Tb.Sp values in HIO+FMT mice, which was consistent with the μ-CT bone imaging results, suggesting that any femur bone loss had been substantially alleviated (Fig. 3k–o). In contrast, the mean/density and BV/TV values remained unchanged (Fig. S3e). The enhancement of gene expression of Trap and Ctsk was correspondingly decreased with fecal transplantation of NO mice (Fig. 3p). Furthermore, the serum content of RANKL, but not TRAP, in NO+FMT mice was significantly lower than in HIO+FMT mice (Fig. S3f). Thus, HIO mice transplanted with NO gut microbiota acquired SCFA-producing microbiome that efficiently ameliorated bone loss from HFD-induced obesity.
NO gut microbiome affects CD4+CD25+Foxp3+ Treg proliferation
SCFAs can regulate the number and function of colonic Tregs in mice. Recent studies have shown a close relationship between immune cells and bone cells, which share regulatory molecules such as cytokines and their receptors, signaling molecules, and transcription factors41,61,62. The above results indicated that the lack of obesity in NO mice was modulated via SCFAs from the gut microbiota, resulting in significant modulation of OC differentiation. The next set of measurements were made to determine the involvement of Treg-related factors. Our results showed a dramatically higher percentage of both CD4+CD25+Foxp3+ Tregs and CD4+CD25+ T cells in the NO mice compared with those in the HIO mice (Fig. 4a–d). Expectedly, Young’s modulus, BV/TV of cancellous bone, and SMI of trabecular bone showed a significant positive correlation with CD4+CD25+ Foxp3+ Tregs and CD4+CD25+ T cells (Fig. 4e and S4a–f). However, Tb.sp of trabecula was negatively correlated with CD4+CD25+ Foxp3+ Tregs and CD4+CD25+ T cells (Fig. S4g, h). No correlation was found between mean/density of cortical bone or Tb.N of trabecula and the above two parameters. (Fig. 4e). Tregs that are characterized as expression of forkhead box p3 (Foxp3) secretes cytokines interleukin 10 (Il10) and transforming growth factor (Tgf-β1) can regulate OC differentiation41. We further demonstrated that gavaged SCFAs enhanced Foxp3 and Il10 but not Tgf-β1 mRNA expression in the colon and small intestine (Fig. S4i–k), which supports our hypothesis that SCFAs were involved in Tregs regulation by NO.

a CD4+CD25+ Foxp3 Treg population from mesenteric lymph nodes. b Percentage of CD4+CD25+ Foxp3 Tregs. c Percentage of CD4+CD25+ T cell. d CD4+CD25+ T cells population. e Correlation between CD4+CD25+ Foxp3 Tregs and CD4+CD25+ T cells against the bone parameters of Young’s modules, Mean/Density of cortical bone, BV/TV of cancellous, Tb.N of trabecula, Tb.sp of trabecula and SMI of trabecula. n = 4 mice per group. Correlations were performed using Pearson test between two different locations of recipients and kinds of genus by “R” packet. Data are presented as means ± SEM. One-way ANOVA was used to analyze significant differences. *p < 0.05, **p < 0.01.
To confirm the effects of Tregs on bone cellular activity, we performed an ex vivo experiment, using T cells from spleen cocultured with the murine mononuclear macrophages RAW 264.7 cells (Fig. 5a). We found that T cells obtained from NO mice inhibited the differentiation of RAW 264.7 macrophages, as indicated by a reduction in purple reaction and reduced numbers of multinucleated cells after 10 days of direct coculture (Fig. 5b). T cells extracted from NO mice significantly downregulated the relative RNA expression of Rankl and Trap in RAW 264.7 macrophages. Furthermore, substantial down-regulation of the ratio of Rankl/Opg was observed in the NO group (Fig. 5c). Additionally, supernatants obtained from cultured cells showed significant higher levels of IL-10 in the NO cultures during 9 days coculture compared with those of HIO mice, however, the TGF-β1 level was not significance (Fig. 5d, e).

a Study design. T cells obtained from CD, HIO, and NO were cocultured with RAW264.7 macrophages for 9 days. b TRAP staining. c Total RNA was extracted from the RAW264.7 macrophages, and the relative expression of Rankl, Opg, Rankl/Opg and Trap was determined. d–e Assessment of cytokines of IL-10 and TGF-β levels in cell supernatants. n = 3 per group. Data are presented as means ± SEM. Two-way ANOVA was used to analyze the significance of differences. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To confirm whether the effect on CD4+CD25+ Foxp3+ Tregs was caused by the transplanted microbiota, we measured Treg-relative factors in recipient mice. The recurrent microbiota transplantation of NO microbiota significantly promoted the proliferation of Foxp3+ Tregs in the colon tissue of mice and increased the expression of IL-10 (brown-yellow spots in Fig. 6a), suggesting increased intestinal immunoregulation that could inhibit OC differentiation. We also examined the relative gene expression of Foxp3, IL-10, and TGF-β1 in the systemic immune system. Foxp3 expression in the NO+FMT groups was markedly increased in the lymph nodes and colon (Fig. 6b), and the levels of Il10, a marker for bone OC differentiation, were increased in the lymph nodes (Fig. 6c). Of note, TGF-β1 showed no significant change (Fig. S4l). Overall, the Tregs immune response via IL-10 related to NO gut microbiota transplantation not only affected the function of the colon and small intestine but also influenced the more distant organ of lymphatic tissues of the immune system.

a Transplanted fecal material enhanced the expression of the colon Treg-related immune factors Foxp3 and IL-10 and TGF-β1, as measured by immunohistochemistry (400×), length of the scale bar is 200 μm. b, c Relative mRNA expression levels of Foxp3 and Il10 measured in the colon, lymph nodes, or small intestine. d, e Relative mRNA expression levels of Hdacs, including Hdac2 and Hdac7 measured in the colon and spleen. f Expression of histone acetyltransferases, including P300, Crebbp and Pcaf, in the colon. g Claudin1 and Occludin mRNA expression in the colon. h Figure abstract of the regulatory mechanism of acetylated Foxp3. Two-way ANOVA was used to analyze the significance of differences in the relative mRNA expression. Data are presented as means ± SEM (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
The expression of Hdac-2 and Hdac-7 in the colon and spleen of NO+FMT mice was significantly downregulated (Fig. 6d, e). In the colon, NO mice fecal microbiota transfer upregulated the expression of the acetylation-related genes P300, Crebbp, and Pcaf (Fig. 6f). P300 is an important acetylation-related gene that functions in the colon and small intestine. The interaction between P300 and Foxp3 can promote the acetylation of Foxp3 (Fig. 6h). Acetylated Foxp3 enhances the stability of Foxp3 and promotes the normal functioning of Tregs in the colon63,64,65. The relative expression of the Foxp3 gene in the colon and small intestine observed in our present study is also consistent with this. Furthermore, NO FMT recovered HIO-associated intestinal barrier damage by elevating Claudin1 mRNA expression (Fig. 6g).
Discussion
Excessive fat intake causes obesity and microinflammation, resulting in bone fragility and bone loss66,67. Although the relationship between obesity and bone loss is controversial, some studies have found that under HFD stimulation, with the occurrence of obesity, bone metabolism occurs in the femur and induces abnormalities in bone structure and bone strength20,24. However, whether HFD alone preferentially drives bone loss with or without obesity and by what mechanism, other than a simple causal relationship, has not been studied. Our data show that physical and biomechanical properties of bone are largely affected by the obesity itself, but not by the HFD, and that HFD-fed NO mice had normal trabecular and cancellous bone structure and strength. To our knowledge, these datasets clarify the distinction between HFD-induced obese mice and HFD-fed NO mice regarding bone properties for the first time. Interestingly, NO mice did not have the negative effects on bone that HFD-induced obese (HIO) mice had. Many people who have high-fat and high-caloric food are still slim and healthy along with consequently healthy bones68,69,70. There could be several explanations regarding the origin of these individual differences, including gut microbiota, and it has been proposed that genetics and metabolic factors as well as ethnic factors, play significant roles71,72,73,74. Without excluding any of the above-mentioned causes, our data analysis supports the notion that compensatory gut microbiota prevents obesity and gut dysbiosis75,76 suggesting that HFD-independent effects of bone loss due to HFD-induced obesity itself can be ameliorated through altering gut microbiota based on the NO type assemblage. In addition, our work shows that HFD-induced bone loss could be alleviated through FMT of NO microbiota, suggesting a major protective effect for the overall microbiota.
Studies have shown that SCFAs are regulators of OC metabolism and bone mass in vivo36,77. Of note, further studies indicate that SCFA-producing bacteria and the SCFA metabolic products, specifically, acetate propionate and butyrate found in the NO group, are involved in gut microbiota modulation. Therefore, we applied a SCFA mix and confirmed that it exerts bone protective effects in HIO mice. Consistent with this finding, our results found that transplanted fecal microbiota from NO mice enhanced bone strength and microstructure, as well as decreased relevant osteoclast genes, by altering the microbiome, which was positively correlated with Ffar2. We also identified purines including 5-hydroxyisourate, xanthine and hypoxanthine, as obesity-related metabolites that may reflect bone health. Excessive purines can lead to hyperuricemia, which is a known factor for gouty arthritis78,79,80. The gut microbiota is involved in purine oxidative metabolism by secreting active enzymes81,82. However, increases in SCFA levels can inhibit xanthine oxidase activity and finally alleviate hyperuricemia83,84. Hence, our results suggested that the NO microbiome might exert its bone-protective effects through SCFAs produced by their reshaped microbiota.
SCFAs have the ability to influence Tregs development and promote bone metabolism by inhibiting the occurrence, differentiation, and function of OCs, thus blocking bone destruction85,86. Ffar2, as one of the major SCFA receptors, has been shown to combine with SCFAs to promote Treg development and impede the progression of colitis87,88. SCFAs alter gene expression by regulating the activity of Hdacs as well as the histone acetyltransferase P300 through epigenetic mechanisms87,88. The interaction between Foxp3 and P300 induces Foxp3 acetylation to enhance Foxp3 stability, thus boosting Treg immune function89,90. Notably, our work shows that after NO+FMT, the expression of Hdac2 and Hdac7 was decreased, while the expression of Ffar2, P300, Crebbp and Pcaf was increased, which contributed to SCFA-induced Treg development. Foxp3 expression, as well as Il10 expression, increased not only in the colon, but also in the lymph nodes, suggesting that systemic immunoregulation was involved in this regulation. Systemic Treg regulation facilitated Foxp3+ Treg proliferation and the production of the Treg factor IL-10, which inhibited OC differentiation.
In summary, this study indicates that HFD-indued obesity and not HFD itself leads to bone fragility and bone loss through homeostasis disequilibrium of the gut microbiota. SCFAs produced by gut bacteria, as significant metabolites, can affect HDACs directly or through activating Ffar2 to regulate IL-10 Tregs function, leading to balanced maintenance of osteoclastogenesis via the gut-bone axis in NO mice despite the HFD. Enhanced gut SCFAs may serve to explain the bone quality differences between obese and NO individuals after HFD and is a promising strategy to address bone loss with obesity. These findings and insights suggest that whether or not an HFD is good or bad diet for an individual is not decided by the dietary ingredients but by the individual’s gut microbiome, which may prove valuable for precisely tailoring nutrition strategies in the future.
Methods
Animal model
Animal experimental procedures followed the National Institutes of Health guidelines for the care and use of laboratory animals and were approved by the ethics committee of the 2nd Affiliated Hospital of Harbin Medical University (Approval No. SYXK-(HEI) 2019-001). Five-week-old, specific pathogen-free (SPF), male BALB/c mice were purchased from the Animal Experimental Centre of the 2nd Affiliated Hospital of Harbin Medical University (Harbin, Heilongjiang, China) and housed in a controlled-environment animal facility at 22 ± 2 °C with a 12-h light/dark cycle. The animals were allowed to acclimate for 1 week prior to experiments and then provided a HFD (45% of calories from fat; Research Diet D12451) and water ad libitum for 10 weeks to induce obesity. The normal control group received a standard chow diet (CD, 10% of calories from fat; Research Diet D12450B). The HFD and CD were isocaloric. The mice were divided into two groups of HFD-induced obesity mice (HIO) or non-obesity mice (NO) after 10 weeks HFD feeding according to body weight of 20% higher or less than 10% difference from that of the CD group, respectively. After that, the HIO and NO mice were continued to be fed with HFD for 10 weeks. The HIO or SCFAs group was fed an HFD diet for extended 10 weeks and simultaneously administered saline or SCFA mix, composed with 67.5 mM acetate, 40 mM butyrate, and 25.9 mM propionate, by gavage at 300 mmol/L every day, respectively. Control mice received normal saline intragastrically. The body weight was recorded every 3 days during the study. Blood was collected from the retro orbital socket prior to sacrifice. Adipose and visceral tissues, as well as the femur, were obtained, weighed, snap frozen and stored at −80 °C until use. The fresh feces were stored at −80 °C for high-throughput sequencing for bacteria and metabolomics analysis.
Fecal transplantation
Feces were freshly collected from donor mice of CD, HIO, and NO group daily for 10 weeks. 10 weeks CD fed mice which received feces from donor CD group was grouped as CD+FMT. The recipient obese mice of HIO+FMT or NO+FMT were received feces from HIO or NO group, respectively, along with continued HFD diet. The donor mice in each group were allowed to freely defecate between 8 and 10 in the morning to get the mixed feces daily, and 100 mg of feces was resuspended in 1 ml of sterile saline. The solution was vigorously mixed for 10 s with a benchtop vortex (XH-5, Hangzhou Yuexin Company, China) before centrifugation at 800 g for 3 min. The supernatant was collected and used as transplant material. Fresh transplant material was prepared on the day of transplantation within 10 min before oral gavage to prevent changes in the bacterial composition. Male recipient mice were fed a CD or HFD were inoculated daily with fresh transplant material using oral gavage, for 10 weeks.
Biochemical analyses
Serum was isolated by centrifugation (2000g, 15 min) and frozen at −20 °C. Analyses were performed according to the manufacturer’s protocols for serum triglyceride (TG), total cholesterol (TC), and serum alkaline phosphatase (ALP) (Nanjing Jiancheng Bioengineering Institute, China). Determination of serum tartrate-resistant acid phosphatase (TRAP) levels was carried out with commercially available assay kits (Beyotime, China). As an important indicator of bone mass, serum levels of receptor activator for nuclear factor-κ B ligand (RANKL) were assessed with an ELISA kit (Cloud-Clone Corp., Houston, USA).
Histology and immunohistochemistry
Serial sagittal plane 5-μm sections were obtained from the whole medial compartment of a joint subjected to standard histological preparation, including decalcification, dehydration, and paraffin embedding. Bone samples were stained with HE or Masson trichrome, and subsequently evaluated using a modified Mankin scoring system (SI Appendix, Table 2). For bone immunohistochemistry, after dehydration, bone tissue slices were placed in EDTA antigen repair buffer (pH 8.0) (G1206, Servicebio, Wuhan, China) and goat serum and incubated for 30 min, followed by overnight incubation 4 °C with anti-asteoprotegerin rabbit pAb primary antibody (GB11151, Servicebio, Wuhan, China) and anti-rankl rabbit pAb (GB11235, Servicebio, Wuhan, China). Slices were then washed and incubated with corresponding Cy3 conjugated goat anti-rabbit second antibody (GB21303, Servicebio, Wuhan, China) at room temperature for 50 min. The slices were then counterstained with diamidino phenyl indole (DAPI) (G1012, Servicebio, Wuhan, China), and incubated with spontaneous fluorescence quenching reagent (G1221, Servicebio, Wuhan, China) for 5 min. After washed with running tap water for 10 min, the slices were then mounted using anti-fade mounting medium (G1401, Servicebio, Wuhan, China). Images were collected by fluorescent microscopy (DS-U3, Nikon, Tokyo, Japan). For bone immunohistochemistry, after paraffin embedding, 5-μm-thick colon tissues were repaired with an antigen repair fluid (Shunobai Biotechnology Co., Ltd., Shanghai, China) and sequentially incubated in H2O2 followed by 10% goat serum (SL038, Solarbio life sciences, Beijing, China) for 30 min for each solution. The tissue sections were next incubated with antibodies of anti-Foxp3 (wl00721, Wanleibio, Shenyang, China), anti-IL-10 (wl01124, Wanleibio, Shenyang, China), and anti-TGF beta 1 (wl02998, Wanleibio, Shenyang, China), diluted in PBS overnight at 4 °C, and then with a horseradish peroxidase-labeled secondary antibody (31460, ThermoFisher Scientific, Rockford, USA) for 30 min. After coloration with diaminobenzidine, the sections were dehydrated, cleared, sealed, and observed under a microscope (BX53, Olympus, Tokyo, Japan) and camera (DP73, Olympus, Tokyo, Japan). Results of histology staining and immunohistochemical labeling were assessed in a blinded manner by three technicians who were unaware of the findings on bone loss and inflammation.
Evaluation of physical and biomechanical properties of the femur
The femoral breaking force was measured according to a three-point bending method using a universal material testing machine (Instron 5569, USA) at a constant load speed of 1 mm/min and span length of 6 mm. The maximum flexural stress (MPa), maximum flexural load (N), and elastic modulus (MPa) values were obtained from the curve.
Micro-computed tomography (μ-CT)
Right femur bone samples were fixed for 48 h in 4% polyformaldehyde, and rinsed three times with PBS. Femur images were reconstructed with a SCANCO μ-CT 100 in vitro system (Hangzhou Yue Bo Biological Technology Co., China). The femur was imaged with an X-ray tube voltage of 70 kV and current of 200 μA. The femur images were analyzed with SCANCO μ-CT 100 analysis software to measure density (mg HA/ccm), total volume (TV), bone volume (BV), relative bone volume (BV/TV), trabecular number (Tb. N, n), mean trabecular thickness (Tb. Th, μm), trabecular separation/spacing (Tb. Sp), bone surface (BS) and the BS/BV ratio.
RNA isolation and real-time PCR
Total RNA was isolated with TRNZOL-A + total RNA extraction reagent (Tian Gen Biochemical Technology Co., Ltd., Beijing, China). Equal amounts of total RNA were used to synthesize cDNA with a cDNA synthesis kit (Transgen Biotech, Beijing, China). Amplifications were performed on an ABI Prism 7300 Real-Time PCR System (Applied Biosystems, America), with an initial denaturation step at 94 °C for 30 s, followed by 40 cycles of 94 °C for 5 s and 55 °C for 30 s. Relative quantification was performed using the 2-△△CT method. Data are reported as fold changes relative to the expression levels of the housekeeping gene. All the primers used in our present study are shown in Supplementary Table 2.
High-throughput sequencing of the 16S rDNA gene V3-V4 regions of fecal bacteria
Fecal DNA was extracted using a Mag-Bind® Soil DNA Kit (OMEGA), For the first round of amplification, 16S rRNA gene V3-V4 regions were amplified using the following specific primers with barcodes: 338F (ACTCCTACGGGAGGCAGCA) and 806 R (GGACTACHVGGGTWTCTAAT). The samples were incubated with Illumina bridge PCR compatible primers for the second round of amplification. Samples were then distinguished by barcode and analyzed based on operational taxonomic unit (OTU) clustering and species classification, after controlling and filtering for the quality of the sample sequence.
Fecal metabolomics
Sample preparation
The collected and frozen feces from individual animal were thawed on ice and grounded to a fine powder with liquid nitrogen. Proteins were precipitated with 50% precooled methanol under vigorous shaking for 1 min, and incubated at room temperature for 10 min, followed by overnight at -20 °C. After centrifugation at 4,000 g for 20 min, the supernatants were transferred into a 96-well plate. The samples were stored at 80 °C prior to the LC-MS analysis.
UPLC-MS
All samples acquired by the LC-MS system followed machine orders. All chromatographic separations were performed using an UPLC system (SCIEX, Warrington, UK). An ACQUITY UPLC T3 column (100 mm×2.1 mm, 1.8 μm, Waters, UK) was used for the reversed-phase separation with a mobile phase consisting of solvent A (water, 0.1% formic acid) and solvent B (Acetonitrile, 0.1% formic acid). The extract was gradient eluted as follows: 0-5-0.5 min, 5% B; 0.5-7 min, 5-100% B; 7-8 min, 100% B; 8-8.1 min, 100-5% B and 8.1-10 min, 5% B. A high-resolution tandem mass spectrometer (Triple TOF 5600 plus, SCIEX, Warrington, UK) was used to detect metabolites eluted from the column. The Q-TOF was operated in both positive and negative ion modes with ionspray voltage floating at 5000 V and −4500 V, respectively. Mass spectrometry data were acquired in IDA mode, in the rage from 60-1200 Da with 150 ms.
Data analysis
LC-MS raw data files were converted into mzXML format and then processed by the XCMS, CAMERA and metaX toolbox implemented with the R software. The online KEGG, HMDB database was used to annotate the metabolites by matching the exact molecular mass data (m/z) of samples with those from the database. The intensity of peak data was further preprocessed by metaX. The P value was adjusted for multiple tests using an FDR (Benjamini–Hochberg). Supervised PLS‐DA was conducted through metaX to discriminate the different variables between groups. The VIP value was calculated. A VIP cut‐off value of 1.0 was used to select important features.
Targeted SCFAs measurement
Fecal SCFA contents were analyzed with a Thermo Scientific Q Exactive Focus (Wilmington, DE, USA). 1 mg of samples prepared as described above was added to 50% acetonitrile aqueous solution in the ratio of 1∶200 and centrifuged at 12000 × g for 10 min. 40 μL supernatant of the sample was mixed with 20 μL 200 mM 3-NPH (dissolved in 50% acetonitrile water) and 20 μL 120 mM EDC-6% pyridine solution (dissolved in 50% acetonitrile water). The samples were then placed in aqueous solution at 40°C for 30 min and then taken out and placed on ice for 1 min after derivatization and diluted it to 2 mL with 10% acetonitrile water. After mixing with isotope internal standard at 1∶1, sample loading was carried out.
Flow cytometry
A single-cell suspension of mesenteric lymph nodes was obtained by grinding fresh tissue through a 70 µm cell strainer, and then gently washing the tissue with PBS. The obtained cells were washed twice and then resuspended in RPMI medium. For Treg analysis, lymphocytes (2 × 106) were labeled with CD4-FITC and CD25-PE for 30 min at 4 °C and then washed with fixation-permeabilization buffers. Next, precooled PBS was added to the surface-stained cells, followed by the addition of intracellular staining permeabilization wash buffer. After centrifugation (1500 g), the cells were labeled intracellularly with Pacific Blue-conjugated antibodies specific for Foxp3. For assessment of CD3, 4, and 8 expression, cells were labeled with FITC anti-mouse CD3, PE anti-mouse CD4, and APC anti-mouse CD8, and incubated for 30 min at 4 °C. The cells were then resuspended in PBS. Cells were analyzed using an ACEA NovocyteTM (Agilent, USA) Flow cytometry and NovoExpress software. All antibodies and buffers were purchased from Biolegend (San Diego, CA) (SI Appendix, Table 3).
TRAP staining of RAW264.7 microphages
Mice were sacrificed and sterilized with 75% ethanol. The spleens were removed and ground to obtain the lymphocytes. The obtained lymphocytes were then added into lymphocyte separation medium and centrifuged at 1500 rpm/ min for 20 min. The supernatants were then aspirated, and monocytes were washed with PBS. The cells were resuspended in RPMI 1640 medium supplemented with 20% BSA, and then added into a nylon wool fiber column for 1-h incubation. T cells were then obtained by rinsing with HANKS and BSA. RAW264.7 macrophages, used as a preosteoclast model, were incubated in 89% DMEM medium until they reached 80% confluence, and were then cocultured for 10 days with T cells extracted from the treated mice. The stimulated RAW264. 7 macrophages were stained using a TRAP kit (Beijing Leagene biotech, Beijing, China). Treatment with 100 ng/mL RANK added into the cell culture medium, was used as positive control. In contrast, DMEM medium without RANKL was used to culture negative-control cells.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are openly available in the MetaboLights database for the metabolomics mass spectrometry data with the URL http://www.ebi.ac.uk/metabolights/MTBLS5432, reference number MTBLS5432, 16S rDNA gene sequencing data were deposited in the SRA repository and can be accessible with the link: https://www.ncbi.nlm.nih.gov/sra/PRJNA882941, reference number PRJNA882941.
References
Arandjelovic, S. et al. Elmo1 signaling is a promoter of osteoclast function and bone loss. Nat. Commun. 12, 4974 (2021).
Tysoe, O. Melatonin prevents diabetes mellitus-induced bone loss. Nat. Rev. Endocrinol. 17, 707–707 (2021).
Yu, W. et al. Bone marrow adipogenic lineage precursors promote osteoclastogenesis in bone remodeling and pathologic bone loss. J. Clin. Investig. 131, e140214 (2021).
Bashiruddin, N. K. et al. Development of cyclic peptides with potent in vivo osteogenic activity through rapid-based affinity maturation. Proc. Natl Acad. Sci. USA 117, 31070–31077 (2020).
Chevalier, C. et al. Warmth prevents bone loss through the gut microbiota. Cell Metab. 32, 575–590 (2020).
Li, X. Q. et al. Targeting actin-bundling protein L-plastin as an anabolic therapy for bone loss. Sci. Adv. 6, eabb7135 (2020).
Pagnotti, G. M. et al. Combating osteoporosis and obesity with exercise: leveraging cell mechanosensitivity. Nat. Rev. Endocrinol. 15, 339–355 (2019).
Estell, E. G. & Rosen, C. J. Emerging insights into the comparative effectiveness of anabolic therapies for osteoporosis. Nat. Rev. Endocrinol. 17, 31–46 (2021).
Guo, D. J. et al. Dietary interventions for better management of osteoporosis: an overview. Crit. Rev. Food Sci. Nutr. 63, 125–144 (2021).
Tanski, W., Kosiorowska, J. & Szymanska-Chabowska, A. Osteoporosis—risk factors, pharmaceutical and non-pharmaceutical treatment. Eur. Rev. Med Pharm. Sci. 25, 3557–3566 (2021).
Wang, Y. et al. Alpha-ketoglutarate ameliorates age-related osteoporosis via regulating histone methylations. Nat. Commun. 11, 5596 (2020).
Yang, Y. S. et al. Bone-targeting aav-mediated silencing of schnurri-3 prevents bone loss in osteoporosis. Nat. Commun. 10, 2958 (2019).
Feng, W. L., Wang, X. P., Huang, D. Q., Lu, A. Q. Role of diet in osteoporosis incidence: umbrella review of meta-analyses of prospective observational studies. Crit. Rev. Food Sci. Nutr. 1–10 (2021).
Rizzoli, R., Biver, E. & Brennan-Speranza, T. C. Nutritional intake and bone health. Lancet Diabetes Endocrinol. 9, 606–621 (2021).
Song, S. S., Guo, Y. Y., Yang, Y. H. & Fu, D. H. Advances in pathogenesis and therapeutic strategies for osteoporosis. Pharmacol. Ther. 237, 108168 (2022).
Buckley, J. Availability of high-fat foods might drive the obesity epidemic. Nat. Rev. Endocrinol. 14, 574–575 (2018).
Kumar, A. et al. High-fat diet-induced upregulation of exosomal phosphatidylcholine contributes to insulin resistance. Nat. Commun. 12, 213 (2021).
Pan, X. F., Wang, L. M. & Pan, A. Epidemiology and determinants of obesity in China. Lancet Diabetes Endocrinol. 9, 373–392 (2021).
Lian, W. S. et al. Microrna-29a in osteoblasts represses high-fat diet-mediated osteoporosis and body adiposis through targeting leptin. Int. J. Mol. Sci. 22, 9135 (2021).
Qiao, J., Wu, Y. W. & Ren, Y. Z. The impact of a high fat diet on bones: potential mechanisms. Food Funct. 12, 963–975 (2021).
Silva, M. J. et al. Effects of high-fat diet and body mass on bone morphology and mechanical properties in 1100 advanced intercross mice. J. Bone Miner. Res. 34, 711–725 (2019).
Varley, I., James, L. J., Willis, S. A., King, J. A. & Clayton, D. J. One week of high-fat overfeeding alters bone metabolism in healthy males: a pilot study. Nutrition 96, 111589 (2022).
Harasymowicz, N. S. et al. Intergenerational transmission of diet-induced obesity, metabolic imbalance, and osteoarthritis in mice. Arthritis Rheumatol. 72, 632–644 (2020).
Hou, J. et al. Obesity and bone health: a complex link. Front. Cell Dev. Biol. 8, 600181 (2020).
Lopez-Gomez, J. J. et al. Influence of obesity on bone turnover markers and fracture risk in postmenopausal women. Nutrients 14, 1617 (2022).
Cao, W. X. et al. The role of gut microbiota in the resistance to obesity in mice fed a high fat diet. Int J. Food Sci. Nutr. 71, 453–463 (2020).
Ding, C., Guo, J. & Su, Z. The status of research into resistance to diet-induced obesity. Horm. Metab. Res. 47, 404–410 (2015).
Keipert, S. & Ost, M. Stress-induced Fgf21 and Gdf15 in obesity and obesity resistance. Trends Endocrinol. Metab. 32, 904–915 (2021).
Durack, J. & Lynch, S. V. The Gut Microbiome: relationships with disease and opportunities for therapy. J. Exp. Med. 216, 20–40 (2019).
Fan, Y. & Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 19, 55–71 (2021).
Seely, K. D., Kotelko, C. A., Douglas, H., Bealer, B. & Brooks, A. E. The human gut microbiota: a key mediator of osteoporosis and osteogenesis. Int. J. Mol. Sci. 22, 9452 (2021).
Ding, K., Hua, F. & Ding, W. E. Gut microbiome and osteoporosis. Aging Dis. 11, 438–447 (2020).
Peng, J. et al. The influence of gut microbiome on bone health and related dietary strategies against bone dysfunctions. Food Res. Int. 144, 110331 (2021).
Yan, J., Takakura, A., Zandi-Nejad, K. & Charles, J. F. Mechanisms of gut microbiota-mediated bone remodeling. Gut Microbes 9, 84–92 (2018).
Li, J. Y. et al. Parathyroid hormone-dependent bone formation requires butyrate production by intestinal microbiota. J. Clin. Investig. 130, 1767–1781 (2020).
Lucas, S. et al. Short-chain fatty acids regulate systemic bone mass and protect from pathological bone loss. Nat. Commun. 9, 55 (2018).
McHugh, J. Wnt signalling in the gut microbiota-bone axis. Nat. Rev. Rheumatol. 15, 4–4 (2019).
Rosser, E. C. et al. Microbiota-derived metabolites suppress arthritis by amplifying aryl-hydrocarbon receptor activation in regulatory B cells. Cell Metab. 31, 837–851 (2020).
Hou, H. Q. et al. Gut microbiota-derived short-chain fatty acids and colorectal cancer: ready for clinical translation? Cancer Lett. 526, 225–235 (2022).
Koh, A., De Vadder, F., Kovatcheva-Datchary, P. & Backhed, F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell 165, 1332–1345 (2016).
Smith, P. M. et al. The microbial metabolites, short-chain fatty acids, regulate colonic T-reg cell homeostasis. Science 341, 569–573 (2013).
Jia, L. M. et al. Probiotics ameliorate alveolar bone loss by regulating gut microbiota. Cell Prolif. 54, e13075 (2021). 2021.
Santinon, F. et al. Involvement of tumor necrosis factor receptor type II in Foxp3 stability and as a marker of treg cells specifically expanded by anti-tumor necrosis factor treatments in rheumatoid arthritis. Arthritis Rheumatol. 72, 576–587 (2020).
Xie, M. X. et al. Nf-kappa B-driven Mir-34a impairs Treg/Th17 balance via targeting Foxp3. J. Autoimmun. 102, 96–113 (2019).
Kondegowda, N. G. et al. Osteoprotegerin and denosumab stimulate human beta cell proliferation through inhibition of the receptor activator of NF-κB ligand pathway. Cell Metab. 22, 77–85 (2015).
Turnbaugh, P. J. et al. A core gut microbiome in obese and lean twins. Nature 457, 480–484 (2009).
Barczynska, R., Kapusniak, J., Litwin, M., Slizewska, K. & Szalecki, M. Dextrins from maize starch as substances activating the growth of Bacteroidetes and Actinobacteria simultaneously inhibiting the growth of Firmicutes, responsible for the occurrence of obesity. Plant Foods Hum. Nutr. 71, 190–196 (2016).
Massier, L. et al. Adipose tissue derived bacteria are associated with inflammation in obesity and type 2 diabetes. Gut 69, 1796–1806 (2020).
Chavez-Carbajal, A. et al. Gut microbiota and predicted metabolic pathways in a sample of mexican women affected by obesity and obesity plus metabolic syndrome. Int. J. Mol. Sci. 20, 438 (2019).
Galaris, A. et al. Obesity reshapes the microbial population structure along the gut-liver-lung axis in mice. Biomedicines 10, 494 (2022).
Liu, B. N., Liu, X. T., Liang, Z. H. & Wang, J. H. Gut microbiota in obesity. World J. Gastroenterol. 27, 3837–3850 (2021).
Xu, H. et al. The ameliorative effect of the Pyracantha Fortuneana (Maxim.) H. L. Li extract on intestinal barrier dysfunction through modulating glycolipid digestion and gut microbiota in high fat diet-fed rats. Food Funct. 10, 6517–6532 (2019).
Duan, J. J. et al. Characterization of gut microbiome in mice model of depression with divergent response to escitalopram treatment. Transl. Psychiatry 11, 303 (2021).
Russell, J. T. et al. Genetic risk for autoimmunity is associated with distinct changes in the human gut microbiome. Nat. Commun. 10, 3621 (2019).
Zheng, P. et al. The gut microbiome from patients with schizophrenia modulates the glutamate-glutamine-gaba cycle and schizophrenia-relevant behaviors in mice. Sci. Adv. 5, eaay2759 (2019).
Akhtar, M. et al. Gut microbiota-derived short chain fatty acids are potential mediators in gut inflammation. Anim. Nutr. 8, 350–360 (2022).
Nogal, A., Valdes, A. M. & Menni, C. The role of short-chain fatty acids in the interplay between gut microbiota and diet in cardio-metabolic health. Gut Microbes 13, 1–24 (2021).
Xu, Y. Q., Zhu, Y., Li, X. T. & Sun, B. G. Dynamic balancing of intestinal short-chain fatty acids: the crucial role of bacterial metabolism. Trends Food Sci. Technol. 100, 118–130 (2020).
Hijova, E. Synbiotic supplements in the prevention of obesity and obesity-related diseases. Metabolites 12, 313 (2022).
Tavella, T. et al. Elevated gut microbiome abundance of christensenellaceae, porphyromonadaceae and rikenellaceae is associated with reduced visceral adipose tissue and healthier metabolic profile in Italian elderly. Gut Microbes 13, 1–19 (2021).
Tyagi, A. M. et al. The microbial metabolite butyrate stimulates bone formation via T regulatory cell-mediated regulation of Wnt10b expression. Immunity 49, 1116–1131 (2018).
El-Jawhari, J. J., Jones, E. & Giannoudis, P. V. The roles of immune cells in bone healing; what we know, do not know and future perspectives. INJURY 47, 2399–2406 (2016).
Beier, U. H., Akimova, T., Lu, Y. J., Wang, L. Q. & Hancock, W. W. Histone/protein deacetylases control Foxp3 expression and the heat shock response of T-regulatory cells. Curr. Opin. Immunol. 23, 670–678 (2011).
Beier, U. H. et al. Histone deacetylases 6 and 9 and sirtuin-1 control Foxp3(+) regulatory T cell function through shared and isoform-specific mechanisms. Sci. Signal. 5, ra45 (2012).
Dahiya, S. et al. Hdac10 deletion promotes Foxp3(+) T-regulatory cell function. Sci. Rep. 10, 424 (2020).
Duan, Y. H. et al. Inflammatory links between high fat diets and diseases. Front. Immunol. 9, 2649 (2018).
Oliveira, M. C., Vullings, J. & van de Loo, F. A. J. Osteoporosis and osteoarthritis are two sides of the same coin paid for obesity. Nutrition 70, 110486 (2020).
Clamp, L., Hehir, A. P. J., Lambert, E. V., Beglinger, C. & Goedecke, J. H. Lean and obese dietary phenotypes: differences in energy and substrate metabolism and appetite. Br. J. Nutr. 114, 1724–1733 (2015).
Germain, N. et al. Specific appetite, energetic and metabolomics responses to fat overfeeding in resistant-to-bodyweight-gain constitutional thinness. Nutr. Diabetes 4, e126 (2014).
Ling, Y. et al. Resistance to lean mass gain in constitutional thinness in free-living conditions is not overpassed by overfeeding. J. Cachexia Sarcopenia Muscle 11, 1187–1199 (2020).
Liu, H. C. et al. Different effects of high-fat diets rich in different oils on lipids metabolism, oxidative stress and gut microbiota. Food Res. Int. 141, 110078 (2021).
Lotta, L. A. et al. Human gain-of-function Mc4r variants show signaling bias and protect against obesity. Cell 177, 597–607 (2019).
Orthofer, M. et al. Identification of alk in thinness. Cell 181, 1246–1262 (2020).
Stryjecki, C., Alyass, A. & Meyre, D. Ethnic and population differences in the genetic predisposition to human obesity. Obes. Rev. 19, 62–80 (2018).
Song, W. et al. Lactobacillus M5 prevents osteoarthritis induced by a high-fat diet in mice. J. Funct. Foods 72, 104039 (2020).
Song, W. et al. (2021). Lactobacillus alleviated obesity induced by high-fat diet in mice. J. Food Sci. 86, 5439–5451 (2021).
Montalvany-Antonucci, C. C. et al. Short-chain fatty acids and Ffar2 as suppressors of bone resorption. Bone 125, 112–121 (2019).
C’Abau, G., Crisan, T. O., Kluck, V., Popp, R. A. & Joosten, L. A. B. Urate-induced immune programming: consequences for gouty arthritis and hyperuricemia. Immunol. Rev. 294, 92–105 (2020).
Dehlin, M., Jacobsson, L. & Roddy, E. Global epidemiology of gout: prevalence, incidence, treatment patterns and risk factors. Nat. Rev. Rheumatol. 16, 380–390 (2020).
Zhao, H. Y., Lu, Z. X. & Lu, Y. J. The potential of probiotics in the amelioration of hyperuricemia. Food Funct. 13, 2394–2414 (2022).
Wang, J. et al. The gut microbiota as a target to control hyperuricemia pathogenesis: potential mechanisms and therapeutic strategies. Crit. Rev. Food Sci. Nutr. 62, 3979–3989 (2022).
Yun, Y. et al. Intestinal tract is an important organ for lowering serum uric acid in rats. PLoS ONE 12, e0190194 (2017).
Ni, C. X. et al. Lactic acid bacteria strains relieve hyperuricaemia by suppressing xanthine oxidase activity via a short-chain fatty acid-dependent mechanism. Food Funct. 12, 7054–7067 (2021).
Guo, Y. J. et al. Inulin supplementation ameliorates hyperuricemia and modulates gut microbiota in Uox-knockout mice. Eur. J. Nutr. 02160, 2217–2230 (2021).
Bao, M. Y. et al. Therapeutic potentials and modulatory mechanisms of fatty acids in bone. Cell Prolif. 53, e12735 (2020).
Wallimann, A. et al. Butyrate inhibits osteoclast activity in vitro and regulates systemic inflammation and bone healing in a murine osteotomy model compared to antibiotic-treated mice. Mediators Inflamm. 2021, 8817421 (2021).
Furusawa, Y. et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450 (2013).
Yang, W. J. et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell Il-22 production and gut immunity. Nat. Commun. 11, 4457 (2020).
Castillo, J. et al. Cbp/P300 drives the differentiation of regulatory T cells through transcriptional and non-transcriptional mechanisms. Cancer Res. 79, 3916–3927 (2019).
Liu, Y. J. et al. Inhibition of P300 impairs Foxp3(+) T regulatory cell function and promotes antitumor immunity. Nat. Med. 19, 1173–1177 (2013).
Acknowledgements
This study was supported by the projects of the National Natural Science Foundation of China (31901701) and the National Key Research and Development Program of China (2019YFC1606704).
Author information
Authors and Affiliations
Contributions
W. Song and Q.L.S. contributed equally. W. Song conceived this study, contributed to experimental designs, interpreted the results, generated figures and wrote the manuscript; Q.L. Sheng interpreted the results, and wrote the manuscript; Y.Y.B. performed the research, contributed new analytic tools, analyzed the data and wrote the paper; L.L. performed the research and analyzed the data; X.N. performed the research, analyzed the data and wrote the paper; Y.G.L. and C. Song conducted the experiments and contributed new analytic tools; T.Y.W. and X.H.D. provided technical support; Y.E.L. proofread the manuscript; J.H.H., L.N.Z. performed the research; X.L.C. and B.C. analyzed the data and arranged the figures. L.L.L. conducted the experiment. C.L.C., and H.B.C. analyzed the data; T.L.Y. conceived and supervised the project. All authors discussed the results and approves the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Song, W., Sheng, Q., Bai, Y. et al. Obesity, but not high-fat diet, is associated with bone loss that is reversed via CD4+CD25+Foxp3+ Tregs-mediated gut microbiome of non-obese mice. npj Sci Food 7, 14 (2023). https://doi.org/10.1038/s41538-023-00190-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41538-023-00190-6
