
Abstract
The emergent secondary phases and surface altered layer (SAL) during the aqueous corrosion of borosilicate glass have a great impact on its chemical durability. However, the formation and evolution of these structures are still unclear. Here, by studying the borosilicate glass altered at 90 °C in pore water, the water in pore space between glass powders, the formation of secondary phases could follow two ways: 1. the consumption of aqueous ions forms analcime, zeolite, calcium silicate and barite at the surface of glass; 2. the reorganization of silica aggregates leads to smectite within the SAL. Small-angle X-ray scattering and cross-sectional scanning electron microscopy results show that the release of soluble elements and the formation of smectite within the SAL significantly increase the porosity of SAL. Furthermore, the layer containing smectite reorganizes inwardly and the crystallinity of smectite is gradually increased over time. The observations of transmission electron microscopy reveal that the dissolution of glass potentially goes through an interface-coupled dissolution-reprecipitation process.
Similar content being viewed by others

Introduction
The chemical durability of silicate/borosilicate glasses in contact with aqueous fluids over geological scale raises challenging scientific issues, especially for predicting the long-term radionuclide retention potential in geologic repositories1,2,3. Furthermore, the weathering of basaltic glass, as the main composition of oceanic crust, by seawater is vital for carbonate precipitation and sedimentation in the oceans4. Archeological glass corrosion is also important for sample dating and preservation5.
Two models prevailed in the past for the formation of surface altered layer (SAL) during glass corrosion: (i) the congruent dissolution of glass followed by the formation of a passivation layer by interface-coupled dissolution-precipitation mechanism (ICDP)6,7. (ii) the incongruent dissolution, mainly B, Na, and Ca, followed by the reorganization of the relict silicate network processes8,9,10,11. The SAL is predominantly composed of amorphous hydrated silicate with different degrees of polymerization, which forms from the glass surface at the expense of the pristine glass and undergoes structural evolution over time12,13,14. In addition, the secondary phases originated from the reorganization of SAL and within the SAL is part of the SAL. Hydrolysis of glass results in the existence of a rapid initial dissolution rate and then a dramatic decrease of the so-called steady-state corrosion rate, as the formation of SAL2,3,15. The SAL could passivate the glass dissolution by limiting water and ion diffusion10,11,12,16,17. The densification-induced pore closure of the SAL could sharply decrease the corrosion rate. The chemical durability of the passivation layer is improved by the strengthening effect of Al on the hydrolysis of Si-O bonds12,18. Furthermore, the dissolution kinetics of glass are correlated to glass composition and experimental conditions such as pH, temperature, glass-surface-area-to-solution-volume (S/V) ratio, and solution chemistry19,20,21. Ojovan et al. reported that the formation of a gap in SAL, resulting in a freshly formed glass surface, led to an effective increase of leaching rate22. As aqueous silica became saturated in solution, secondary phases such as zeolite might form at the surface of SAL23. The precipitation of secondary phases could lead to a resumption of glass corrosion since the formation of secondary phases consumes aqueous silica in solution, causing the destabilization of SAL19,24,25,26,27.
A complete understanding of the formation and evolution of SAL and secondary phases is essential for predicting long-term behaviors of glass10,20,27,28. However, the formation and evolution of SAL and secondary phases and their impacts on glass corrosion have not been fully unraveled. Therefore, we used scanning electron microscopy (SEM), (scanning) transmission electron microscopy ((S)TEM) with a high-angle annular dark-field (HAADF) detector, laboratory and synchrotron X-ray diffraction, including both small-angle X-ray scattering (SAXS) and wide-angle X-ray scattering (WAXS), and Geochemist’s Workbench code to study the formation and evolution of secondary phases and SAL during borosilicate glass (10.00 g glass powders and 1 glass coupon) alteration in Milli-Q pore water (3.11 mL) with a surface area-to-solution volume ratio (S/V) of 225000 m−1 at 90 °C in a static mode at free pH. The main components of borosilicate glass are 44.9% SiO2, 12.3% B2O3, and 11.6% Na2O, and the other components can be found in METHODS, which is a typical composition of glass used in China geological disposal. This volume of Milli-Q pore water is representative of the saturation of high-level nuclear waste glasses in the geological disposal environment29,30. Subsequent aqueous and solid characterizations were done after glass corrosion for 3, 7, 20, 90, 180, 380, 540, and 760 d. This work explicitly demonstrates the progress of long-term glass corrosion, especially for high-level nuclear waste glasses.
Results and discussion
Leaching kinetics
The dissolution rate of glass was evaluated by aqueous analysis. The calculated pH, the measured aqueous concentrations of Si and B, the equivalent thickness (Eth) calculated from B concentration, and the dissolution rate (r) of glass during corrosion are shown in Fig. 1. The corrosion of glass in pore water would impact the pH of pore suspension, but it could not be measured directly. The Geochemist’s Workbench® code31 was adopted to calculate the pH of suspension during glass corrosion in pore water and the calculated result is shown in Fig. 1a. As we can see, the calculated pH increased initially and then decreased with the reaction process. The initial rise of the calculated pH could be explained by the release of cations, e.g., the calculated Na+, Mg2+, and Ca2+ concentrations in Supplementary Fig. 1, during glass dissolution coupling with a decrease of H+ in solution to maintain charge balance. The decrease of calculated pH at the following reaction process was probably correlated to the formation of smectite to produce H+ 32,33. Figure 1b shows that the measured concentrations of Si increased rapidly within 180 d, and then achieved a slightly increasing concentration for the rest of the test in addition to the data at 540 d, which can be considered as the saturation of silica in solution with respect to the passivating layer on the surface of glass20,34. A plausible explanation for the higher concentration of Si at 540 d was probably related to the breakdown of the passivating layer to some extent, which could lead to the release of hydrous amorphous silica that partially filled in the gap parallel to SAL (in Formation and evolution of surface altered layers part), but it should be noted that drying of altered glass could form and broaden the gap. Gin et al. reported that despite the saturation of amorphous silica, the glass still dissolved and released Si into the solution due to the lack of an efficient passivation layer until oversaturation, thus allowing precipitation to occur35. Furthermore, the released concentration of B is usually selected to monitor the dissolution rate of glass as it is not retained in SAL and precipitated36. The aqueous concentration of B in Fig. 1b readily increased within 90, and then achieved a moderate increase from 90 to 760 d. The equivalent thickness of altered glass (Eth) (Fig. 1c) calculated from aqueous B concentration reached 2.81 μm at 90 d and increased to 8.35 μm at 760 d. The glass dissolution rate, determined by the release time of B, shows that the rate decreased ~11 times from 3 d (~ 58.5 nm d−1) to 760 d (~5.5 nm d−1) (Fig. 1d), and the average corrosion rate was about 11.0 nm d−1, which is higher than that (6.3 nm d-1) of SON68 glass under Si-rich solutions at 90 °C37. The discrepancy could be due to the static conditions with a sufficiently high surface area-to-solution volume (S/V) ratio (225,000 m−1) and different glass compositions in our study12,20,38.

a The calculated pH with reaction process, b time evolution of the measured concentrations of Si and B, c the equivalent thickness of altered glass (Eth), calculated from B released into solution, and d the glass dissolution rate determined by aqueous B concentration. The concentrations of B and Si were given with an uncertainty of approximately ±25%, according to Fournier et al.64.
Formation and evolution of the secondary phases
The secondary phases formed during glass powder corrosion were identified by XRD. As shown in Fig. 2, no obvious crystalline phases were present at 20 d, which has been confirmed by SEM (presented later in this part). Here we should note that the detection limit of XRD was about 1%. However, a series of diffraction patterns appeared for the glass altered for 90 d. The reflections at 2θ around 12.5, 21.7, 28.1, and 33.4 indicated the formation of zeolite (Na6Al6 Si10O32·12H2O) (PDF 39-0219), and the reflections at around 15.9, 26.1, and 30.6 suggested the formation of analcime (NaSi2AlO6·H2O) (PDF 41-1478). Previous studies reported that different types of glass altered in alkaline solution, the pH higher than our calculated pH (Fig. 1a), could induce variations of zeolite phases23,39,40. In addition, the emergence of reflection at 2θ around 19.6 and 60.8 indicated the primary formation of smectite (possibly nontronite, Ca0.1Fe2(Si,Al)4O10(OH)2·4H2O, PDF 34-0842, and saponite, Ca0.2Mg3Al0.33Si3.67O10(OH)2, PDF 29-1491) at 180 d and the gradual precipitation of analcime was observed. The formation of secondary phases was a consequence of silica saturation. Smectite, as the main secondary phase, was also present in experiments of glass corroded by deionized water and 0.03–0.1 M NaOH solutions at 150 °C41. A lesser extent of calcium silicate (CaSiO3) (PDF 45-0156) was identified with reflections at 2θ around 17.6, 28.1, and 29.5 after 380 d. However, the absence of main lattice planes at 2θ around 12.5, 17.6, and 21.7 suggests that zeolite was probably dissolved or transformed to other phases, such as analcime at 760 d. CaSi2O5 phases (CSH) precipitated during CJ8 glass corrosion in deionized water after 12.8 y, and the glass has the same content of Ca as our glass42. Although the precipitation of CSH and zeolite by consuming Si, Al, and/or Ca and destabilizing the passivating layer dramatically drove the dissolution of glass24, the formation of the secondary phases did not trigger a sharp but relatively modest dissolution of glass evaluated by B concentration (Fig. 1b) in our system. This could be due to the limitation of glass corrosion to some extent as the persistence of saturation of silica in solution in accelerated corrosion experiment with static mode.

XRD patterns of secondary phases.
SEM images of glass powders at different corrosion times are shown in Fig. 3. No obvious particles except corrosion-induced imprinting at the edge of glass powder altered for 20 d were observed in Fig. 3a. With corrosion time increased to 90 d, the glass powders were covered by different morphologies (Fig. 3b) of precipitates and EDS results showed that analcime (point 1) with chemical composition NaAlSiO4 was identified and the particle (point 2) with an enrichment of Ca was probably CaSiO3. The significant precipitated phases with μm size (point 3) were comprised of Si, Na, Al, and Mg. The main elements of bright nanoparticles (point 4) were Ba, S, Si, Mg, and Na. The two particles (points 3 and 4) were probably zeolite and barite (BaSO4). The content of Mg in points 3 and 4 could originate from the SAL of glass. Barite was not identified by XRD as its low content (<1%, XRD detection limit) and the overlapping with the main diffraction peak at 2θ around 25.9 from analcime. At 90 d, the honeycomb morphology was observed in Fig. 3c, which was Fe and Mg-containing smectite within the SAL identified by WAXS and cross-sectional SEM in the formation and evolution of surface altered layer (SAL) part. After 180 d corrosion, both the size and precipitation of BaSO4 were increased (Fig. 3d). With reaction time increased to 380 d, the gap perpendicular to SAL occurred, and their size increased from nm to µm with corrosion progressing (Fig. 3e), which was formed in SAL when altered glass dried out in post-experiment. In general, although post-mortem analysis (e.g., by SEM) provides evidence for structural change of the altered glasses, cracking in the SAL due to sample preparation, i.e., drying and polishing, has to be taken into account. By increasing corrosion time to 540 and 760 d, the honeycomb morphology, as shown in Fig. 3f, h, was observed, and the main elements of the structure were Na, Mg, Al, Si, and Ca, which could be primarily the amorphous gel layer with smectite identified by XRD in Fig. 1. The typical structural formulas for saponite and nontronite are Ca0.2Mg3Al0.33Si3.67O10(OH)2 and Ca0.5(Si7Al0.8Fe0.2)(Fe3.5Al0.4Mg0.1)O20(OH)4, respectively43. A higher content of Si (20.80 atomic%) than Mg (4.76 atomic%) and Al (2.09 atomic%) in the honeycomb morphology based on EDS analysis probably suggests that it still inherited part of the relict silicon glass network. Generally, smectite was one of the main minerals formed during Na and Ba borosilicate glass corrosion44. Furthermore, the thickness of the layer with smectite was several hundred nanometers through the observation from a gap in Fig. 3f. Combining with the evolution of smectite size from 90 to 760 d and EDS analysis, it seemed that smectite formed within the SAL and it is part of the SAL, not by the precipitation from saturated ions in solution, which was different from the formation of other secondary phases in our cases. EDS analysis of point 6 indicated that the particle contains Ca and Si, with a tiny amount content of Na, confirmed as CaSiO3 by XRD. Moreover, we also found that smaller particles (Supplementary Fig. 2) came off from the glass powders after corrosion for 540 and 760 d, which was probably the SAL with smectite. The formation of smectite could induce the decrease of pH as the presence of hydroxyl groups in their composition32,45,46, and lead to the increase of corrosion rate by ions consumption from solution and diminishing passivating layer protective properties47,48.

a The morphology of the glass surface at 20 d, b, c the formation of different morphological precipitates and honeycomb morphology at 90 d, d the formation of plenty of barite nanoparticles at 180 d, e the morphology of gap formed on the surface of glass powder at 380 d, f the thickness of the layer observed though the gap at 540 d, g, h the morphology of calcium silicate and magnification of smectite at 760 d, and i the relative EDS analysis of particles from point 1 to 7.
The Geochemist’s Workbench® code31 was used to simulate the formation of secondary phases during glass corrosion at 90 °C and 1.01 bar, which emulated the experimental conditions. The simulated secondary phases shown in Fig. 4 included quartz (SiO2), wollastonite (CaSiO3), nontronite (Na0.3Fe2(Si,Al)4O10(OH)2·nH2O), analcime (NaSi2AlO6·H2O), sanbornite (BaSi2O5), mesolite (Na2Ca2(Al2Si3O10)3·8H2O) and saponite (Ca0.2Mg3Al0.33Si3.67O10(OH)2). Quartz is thermodynamically favored but has slow formation kinetics49. Thus, it was not identified by XRD and SEM. The simulated precipitation of wollastonite, nontronite, saponite (smectite group), analcime, and mesolite (zeolite group) are in agreement with experiment observations, except sanbornite. Barite, with a trace little amount, was also predicted in our calculation. In conclusion, it seems there are two paths for the formation of the secondary phases: one was through the consumption of aqueous Si, Al, Mg, Ca, Ba, Na, and S elements from solution to form analcime, zeolite, calcium silicate and barite on the surface of glass powders; another was though the reorganization of leached layer after depletion of soluble elements such as B and Na to form smectite.

The predicted secondary phases calculated by Geochemist’s Workbench® code during glass corrosion at 90 °C and 1.01 bar.
Formation and evolution of surface altered layer (SAL)
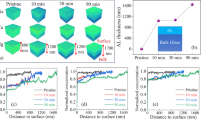
A fundamental understanding of the structure and stability of the SAL is essential for predicting the long-term performance of glass. Scanning X-ray microdiffraction was performed to identify the secondary phases and to monitor the evolution of SAL on dried altered glass powders. The overall morphologies of glass powders with different corrosion times are shown in Fig. 5a. Analcime, zeolite, calcium silicate, and barite were identified (Fig. 5b), which is consistent with the SEM and XRD results. Smectite led to a diffraction pattern in the WAXS range of q from 1.32 Å to 1.50 Å, which enabled us to determine the reorganization of smectite within SAL. The test zones, without interference from visible secondary phases showing the hot color, were marked by red circles in Fig. 5a. The WAXS results with q from 1.32 Å to 1.50 Å in Fig. 5d suggest that no phase was identified at 20 d and smectite was initially present after 90 d and the crystallinity of smectite was gradually increased with corrosion time. The formation of smectite could lead to two antagonistic effects: 1. the decrease of corrosion rate caused by the clogging of the passivating layer50; 2. the increase of corrosion rate due to the depletion of the passivating layer47,48. The SAXS results from the red circle zones were used to determine the porosity and roughness of these areas by using Porod’s law. The power law of SAXS curves of samples in Fig. 5e had different evolution. The variation of power of q for different samples indicates a structural reorganization of the SAL during corrosion. Decreasing of size of pores could increase the roughness on a 100 nm scale and lead to the power of q increasing from −4 to −2. Details of fitting Porod’s law have been discussed in METHODS. Figure 5e shows a significant increase of porosity for SAL altered for 90 d. This is probably caused by the release of soluble species for SAL to form a porous structure and then the reorganization of the relict silica in SAL by hydrolysis and recondensation. After 90 d, the power of q of SAXS showing an overall upward trend indicates that the sizes of pores on the surface of glass were decreasing with the restructuring of SAL during corrosion.

a The glass powders with different corrosion times were raster scanned with spatial resolution of 2.5 µm, the hot color in the scan represented high crystalline phases, b, c 1D XRD intensity profiles of secondary phases with different corrosion times were composition of different crystalline phases. The secondary phases had been numbered 1–7 in (a), d the intensity profiles of q ranging from 1.32 to 1.50 Å of red circle zones in (a) without visible secondary phases interference were enlarged for tracking smectite in SAL during corrosion; e the power of q of SAXS intensity profiles (ranging from 0.03 to 0.1 Å−1) from patterns within red circle zones in (a) was determined by Porod’s law to evaluate the roughness and porosity of SAL during corrosion. The power of q with reaction time was shown in the upper right part.
The cross-sectional SEM images of glass powders to study the formation and evolution of SAL and the results are shown in Fig. 6 and Supplementary Fig. 3. The thickness of SAL formed at 20 d shown in Fig. 6a and Supplementary Fig. 3a was about 126 nm. This leached layer was composited by packed silica aggregates with the size of several tens nanometers, which was also observed in previous studies51,52. Since interfacial water has different physical and chemical properties with bulk water53,54. Amorphous silica could nucleate and precipitate in the corrosion zone even in unsaturated concentrations (34.3 mg L−1). Besides, the aggregation of silica grew in alkaline solutions (the calculated pH > 8 in our system) by direct precipitation of silica onto silica particles by Ostwald ripening, which formed a silica gel51. Both paths for the formation of silica aggregates are followed by the interface-coupled dissolution-reprecipitation (ICDP) mechanism. Therefore, ICDP could elucidate the formation of silica aggregates in the altered glass powders at 20 d. At 90 d, the thickness of SAL increased to 2.36 μm, and two different morphologies, named as external layer and central layer, were present in the SAL (Fig. 6b). The external layer, including smectite, had a size of ~423 nm with regular shape, but the central layer had a larger size with the irregularly shaped dissolution front. The variations of morphologies between 20 and 90 d indicate the structural reorganization of SAL during corrosion. An EDS mapping performed on a cross-sectional SEM image of glass powder altered for 90 d corrosion in Supplementary Fig. 4 shows that Na was completely leached out in SAL, Ca and Al exhibited limit retention, and Si still inherited, in contrast, Fe and Mg were enriched. Fe could precipitate with silicate in the altered layer from Fe-silicate as their large affinity55,56. Mg and Ca could be incorporated as charge compensators for AlO4− in SAL47,57. Previous studies reported that the passivating layer enriched Al and Ca in SON68 nuclear glass, and Ca was enriched in the passivating layer of Complex Glass23,58. The morphologies of the external layer, silica aggregates, and the interface of glass powders altered for 540 d could be clearly seen in Fig. 6g. The visible porous structure between silica aggregates was a consequence of the condensation reactions between reactive monomeric silica/surface silanol groups51. The EDS results of glass powder after reaction for 540 d in Supplementary Fig. 5 (points 4 and 5) and Supplementary Table 1 shows that the external layer contained Fe/Mg–smectite and the content of Si in this layer was lower than that of silica aggregates, indicating that Si could release into bulk solution as the result of aqueous Si concentration with a slight increase in Fig. 1b during corrosion. More importantly, the ratio (12.7) of Si (23.37 atomic%) to Fe (1.84 atomic%) and the ratio (4.8) of Si to Mg (4.83 atomic %) in external layer (point 5) was much higher than that (1.9) in the typical structural formula for nontronite (Ca0.5(Si7Al0.8Fe0.2)(Fe3.5Al0.4Mg0.1)O20(OH)4) and that (1.2) in saponite (Ca0.2Mg3Al0.33Si3.67O10(OH)2), respectively, suggesting that the external layer contained not only smectite but also the relict silicon glass network. Combining with the SEM image in Fig. 3c that the honeycomb morphology on the surface of SAL originated from the corrosion of glass powders and with EDS analysis in Fig. 3f and Supplementary Fig. 5 that smectite was formed within the external layer, the external layer with regular shape should be formed by the reorganization of packed silica aggregates in SAL. The depth of SAL at different corrosion times was collected by the hypothesis testing statistics of 120 positions of backscattered electron (BSE) images, and the representative images are shown in Supplementary Fig. 6. Statistical significance was defined as p < 0.05. The depth of SAL presented as the mean ± standard deviation in Fig. 6i increased with corrosion time, which was compatible with the Eth(B) result. A similar trend for the external layer with Mg/Fe–smectite (Fig. 6h) suggested the reorganization of the layer coupling in time and space inwardly growth from the SAL surface. The gap parallel to SAL began to form in the interface of SAL/pristine glass at 90 d, and the size of the gap increased with corrosion time. This is consistent with the previous reports of a water-rich zone in situ glass samples and a gap in the dried sample at the corrosion layer/glass interface, and the width of the gap in the dried sample was comparable to the water-rich zone in situ7,59. The formation of a gap could be a result by that the silica molecules in the reaction interface were energetically favored to attach pre-existing silica particles compared with the deposition onto the dissolving glass surface, followed by ICDR mechanism7,51. Remarkably, drying of the altered glass and then the shrinkage of the resin during drying could also lead to the possible formation and broadening of gaps at the SAL/glass interface. Thus, the SAL included two layers with different morphologies, and the inward reorganization of the external layer with Mg/Fe-smectite from the SAL surface was probably from packed silica aggregates.

a–f Cross-sectional SEM images of SAL of glass powders with different corrosion times, g magnification of the external with smectite, aggregates, and porosity in SAL, h the evolution of external layer with smectite as a function of corrosion time, and i the comparison of the corrosion depth by Eth(B) and cross-sectional SEM results. Error bars representing the standard deviation (n = 120) and statistical significance defined as p < 0.05.
Altered glass coupon by (S)TEM analysis
To further identify the structure of SAL and decipher the nanometer-scale evidence of the interface, the glass coupon altered for 760 d was characterized by (scanning) transmission electron microscopy ((S)TEM) with a high-angle annular dark-field (HAADF) detector. The width of SAL of the altered glass coupon was 3.2 μm in Fig. 7a, which was lower than that (9.0 μm) of glass powders with a faster leaching rate at the same time. This might be due to the fact that compared to the glass coupon, the enlargement of the special surface area increased the pH at the surface of glass powders, which may cause the synergistic increase of the dissolution rate of glass powders60. Furthermore, the porosity in the central layer was observed, and the pore size closer to the external layer and pristine glass was larger than that in the middle area of the central layer. The larger pore near the external layer could result from the ripening of silica aggregates, and silica molecules energetically favored to attach pre-existing silica particles could increase the size of the pore in the reaction interface7,51. Selected area electron diffraction (SEAD) patterns of the external layer in Fig. 7a show (100), (111), and (118) reflections belonging to smectite (possibly nontronite and saponite). The gap perpendicular to SAL caused by dehydration was in the presence of the external layer, which is in agreement with the cross-sectional SEM results of altered glass powders. The SAL displayed four sublayers based on the elemental profiles in Fig. 7e: external layer, central layer, and two gradient layers. Si, Al, Fe, and Mg were enriched in the external layer as its composition with Mg/Fe–smectite and the large affinity between Fe and Si55. Na profile was generally attributed to ion exchange between H3O+ and Na+ from the pristine glass14, while Ca was retained in the central layer by its incorporation as a charge compensator for fourfold coordinated Al species47,57. The interface between the external layer and the central layer analyzed by energy-filtered TEM (EFTEM) in Fig. 7b was distinct and very sharp. The width of this interface was ~12 nm. As the 6.0 nm step size, the width (~ 60 nm) of the chemical gradient in the external/central layer (Fig. 7e) was notably larger than the width derived from the EFTEM image. The structural interface between the pristine glass and SAL shown in Fig. 7c was not a distinct linear band, but the chemical gradients based on Si and Na in this interface were sharp (Fig. 7e). The sharp structural interface was generally attributed to ICDP mechanism6. Moreover, nanoparticles smaller than 5 nm were observed on the top of the external layer in Fig. 7d. Here, we did not analyze their composition due to the fine size. Gin et al., also found that the rounded crystallized grains but with larger size (30–50 nm) were present in the gel layer, and these grains were rich in Ag, Te, and S in the borosilicate glass altered for 26 y at 90 °C61.

a HAADF image showing the SAL and porosity, and SEAD patterns of the external layer insert suggesting smectite (possibly nontronite and saponite), b, c TEM images of interfaces in external/central layers and central layer/pristine glass respectively, d TEM image of nanoparticles on the top of external layer, and e elemental profiles.
In summary, understanding the formation and evolution of SAL and secondary phases during glass corrosion is crucial for determining its chemical durability10. Here, borosilicate glass altered in pore water at 90 °C was investigated. We found that, firstly, the secondary phases such as analcime (NaSi2AlO6·H2O), zeolite (Na6Al6 Si10O32·12H2O), calcium silicate (CaSiO3), and barite (BaSO4) were formed by the precipitation of the saturation ions in solution. However, the formation of Mg/Fe–smectite was probably followed by the reorganization of silica aggregates, and the crystallinity of smectite was gradually increased with corrosion time. Analcime, zeolite and smectite, as the common secondary phases, were also formed during other glasses corrosion23,45,47. While calcium silicate and barite were rarely reported. Secondly, there was a significant increase in the size of pores at 90 d as a consequence of the release of soluble elements and the formation of smectite within SAL, while the pores were experiencing closure with the reorganization of SAL from 90 to 760 d. These pore closure of the SAL with reaction time was also observed in previous studies12,14,50. Thirdly, the external layer with Mg/Fe–smectite coupling in time and space reorganized inwardly from the SAL surface. Moreover, the gap between pristine glass and SAL was formed after 90 d, and the size of the gap increased with corrosion time. The formation of a gap parallel to SAL is commonly found in dried samples from ex-situ experiments6,55. It should be noted that sample drying and the shrinkage of the resin during drying could also lead to structural changes, such as the formation and broadening of gaps. Finally, the corrosion of glass powders and coupons was followed by an ICDP process. We comprehensively studied the formation and evolution of secondary phases and SAL during borosilicate glass corrosion. Our analysis could be extended to silicate and natural glasses as well as minerals, such as on basaltic glasses or feldspars, as their similarities in secondary phases and SAL12,47,51. Furthermore, the glass corrosion under actual disposal conditions (T ≥ 90 °C, saturated with respect to water) with the extra low kinetics of movement of underground water could provide a fundamental assessment of the potential uncertainty for long-term high-level nuclear waste glasses62.
Methods
Glass leaching experiment
The borosilicate glass (44.9% SiO2, 12.3% B2O3, 11.6% Na2O, 5.1% Al2O3, 4.4% MgO, 6.7% CaO, 3.5% BaO, 3.4% Fe2O3, 0.1% ZrO2, and other compositions) was produced at 1150 °C for 3 h and then cast into graphite crucibles with 6 cm × 30 cm. The samples were annealed for 1 h at 500 °C and cooled to room temperature. The glass was ground with a grinder and fractionated by sieving with the size of 75–149 μm and the specific surface area of the glass powders, determined by the Brunauer–Emmett–Teller (BET) N2 absorption method, was 0.07 ± 0.02 m2/g. The glass coupon of 10 × 10 × 2 mm3 was cut from the glass blocks by a diamond saw and polished to obtain a surface roughness of less than 1 µm. The AFM result of the glass coupon surface after polishing is shown in Supplementary Fig. 7. The glass (powders and coupon) corrosion experiments were performed in Milli-Q pore water (18.2 MΩ cm) at 90 °C. The schematic diagram of the experimental reactor is shown in Fig. 8. The measurement method of Milli-Q pore water, the water contained in pore space between glass powders, was described in the following text: 10.00 g of glass powders, and 1 coupon was added in a sand core filter funnel with 5–15 μm pore size, and the total mass was A. Next, we added a certain amount of Milli-Q water, just covering the surface of glass powders and coupon, in the sand core filter funnel. The free water was filtered out by its gravity, but the pore water among glass powders was retained. The total mass was B, and the difference (B – A) was the mass of pore water. The average data of pore water in 10.00 g glass powders and 1 glass coupon was 3.11 g (3.11 mL) by six groups of parallel experiments. In order to reduce the evaporation effect, the reactor was tightly closed after filling with 10.00 g glass powders, one glass coupon 3.11 mL Milli-Q water (the volume of pore water) in polytetrafluoroethylene (PTFE), and then was placed in the stainless steel sealed tank containing a small amount of Milli-Q pore water. The tank was sealed with welding and put in the oven at 90 °C for reaction. To check the loss of water at 90 °C, the blank experiments, adding 3.11 Milli-Q water but without glass in PTFE, were conducted in the same setup up and the result suggests that the loss of water was within 7% in 380 d.

The schematic diagram of the experimental reactor.
After reaction for 3, 7, 20, 90, 180, 380, 540, and 760 d, 3.11 mL Milli-Q water was added into the samples and let equilibrate for 1 d at room temperature to get enough free liquid for aqueous analysis. Then the suspension was filtered through a 0.22 μm nitrocellulose membrane. Considering the low equilibrium constant of secondary phases, such as analcime (log K25 = −15.0)63, it was reasonable to neglect the effect of post-addition of Milli-Q water on the dissolution of the secondary phases in 1 d at room temperature. The total concentrations of B and Si in the filtrate were analyzed by inductively coupled plasma optical emission spectrometry (ICP-OES) with a Varian 720-ES apparatus. The calculation of the equivalent thickness (Eth) and the dissolution rate (r) followed by Gin et al. paper14, Eth(B) of altered glass powders was calculated from the measured concentration of B at t alteration as follows:
Where C(B) is B concentration, Vt is the volume of the solution at t, and it is 3.11 mL in our system, ρ is the density of glass (2.5 g cm−3), S is the special surface area of glass powders (0.07 ± 0.02 m2 g−1), and we neglected that of monolith, xB is the mass fraction of B in glass (3.19%). The dissolution rate (r) is calculated by:
The dried altered glass powders were characterized using SEM, WAXS, SAXS, and X-ray diffraction (XRD) (Bruker axs, D8 advance) with Vortex-EX detector (Hitachi) under Cu Kα radiation. The Geochemist’s Workbench® code was used to model the formation of secondary phases during glass corrosion. A couple of glasses altered for 760 d were measured by (S)TEM-HAADF.
Post-dissolution characterization glass
Scanning X-ray microdiffraction was carried out at the ID13 beamline at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. X-ray was focused to a size of 2 μm × 2 μm with energy of 13 keV and flux of ~2.0 × 1012 photons/s. 2D area detector Eiger 4 M was used to collect diffraction patterns covering reciprocal space q ranging from 0.02 to 4 Å−1, which contains both SAXS/WAXS signals. The altered glass powders were spread on the Kapton tape. A region of ~200 μm × 200 μm was scanned by micro X-ray with step size 2 μm. Scanning X-ray microdiffraction patterns were azimuthally averaged to a 1D intensity profile by a custom developed python package. Small-angle X-ray scattering was determined by the electronic contrast of the sample. Here, the porosity caused by the corrosion of glass introduced significant electronic contrast, which could be extrapolated in q range of 0.03–0.1, Porod region. According, to Porod’s law, intensity profile:
where A is constant, x could vary from 2-4 based on the alteration of the porosity of the glass. If we assume a spherical surface of pores within the glass, the increase in size of pores within the glass will lead to an increase of x.
For the thickness measurement of SAL, the glass powders altered for certain times were embedded into epoxy resin, and then the cross-section of the samples was polished to 9, 6, 3, 1, and 0.25 μm step-by-step. After polishing, the cross-section part was washed with Milli-Q water and ethanol six times to remove diamond particles and other possible pollutants during polishing. The dried cross-sectional part was covered with a thin carbon layer (~20 nm) for conductivity during SEM analysis. For the surface morphology and secondary phases analysis, the altered glass powders were directly dispersed on the carbon tape and covered with ~1 nm gold layer, and then analyzed by SEM. The SEM (ZEISS Ultra 55) was equipped with energy selected X-ray microanalysis system (EDS) and SDD Detector (BRUKER AXS-30 mm²). The average thickness of SAL of altered glass powders was obtained by the statistics of 120 positions of backscattered electron (BSE) images measured by SEM (Vega3 Tescan) with EDS and SDD Detector. For the study of the glass coupon altered for 760 d, an ultrathin electron transparent cross-sectional sample prepared by focused ion beam (FIB) was analyzed by TEM (JEM-2100F) with energy dispersive X-ray spectroscopy (EDS). (S)TEM images were collected at 200 keV. Gatan Digital Micrograph software was used to analyze the images.
Data availability
All data, including aqueous and solid analysis needed to evaluate the conclusions, are shown in the article, and further data is provided in the Supplementary Material. Additional raw data related to the paper are available from the authors upon reasonable request.
References
Grambow, B. Nuclear waste glasses—how durable? Elements 2, 357–364 (2006).
Grambow, B. Mini review of research requirements for radioactive waste management including disposal. Front. Nucl. Eng. 1, 1052428 (2022).
Ojovan, M. I. & Steinmetz, H. J. Approaches to disposal of nuclear waste. Energies 15, 7804 (2022).
Dessert, C., Dupre, B., Gaillardet, J., Francois, L. M. & Allegre, C. J. Basalt weathering laws and the impact of basalt weathering on the global carbon cycle. Chem. Geol. 202, 257–273 (2003).
Silvestri, A., Molin, G. & Salviulo, G. Roman and medieval glass from the Italian area: bulk characterization and relationships with production technologies. Archaeometry 47, 797–816 (2005).
Hellmann, R. et al. Nanometre-scale evidence for interfacial dissolution-reprecipitation control of silicate glass corrosion. Nat. Mater. 14, 307–311 (2015).
Geisler, T., Dohmen, L., Lenting, C. & Fritzsche, M. B. K. Real-time in situ observations of reaction and transport phenomena during silicate glass corrosion by fluid-cell Raman spectroscopy. Nat. Mater. 18, 342–348 (2019).
Geisler, T. et al. Aqueous corrosion of borosilicate glass under acidic conditions: a new corrosion mechanism. J. Non-Cryst. Solids 356, 1458–1465 (2010).
Gin, S. et al. The controversial role of inter-diffusion in glass alteration. Chem. Geol. 440, 115–123 (2016).
Gin, S. et al. Insights into the mechanisms controlling the residual corrosion rate of borosilicate glasses. Npj Mater. Degrad. 4, 41 (2020).
Gin, S. et al. Dynamics of self-reorganization explains passivation of silicate glasses. Nat. Commun. 9, 2169 (2018).
Cailleteau, C. et al. Insight into silicate-glass corrosion mechanisms. Nat. Mater. 7, 978–983 (2008).
Bunker, B. C. Molecular mechanisms for corrosion of silica and silicate glasses. J. Non-Cryst. Solids 179, 300–308 (1994).
Gin, S. et al. Origin and consequences of silicate glass passivation by surface layers. Nat. Commun. 6, 6360 (2015).
Frankel, G. S. et al. Recent advances in corrosion science applicable to disposal of high-level nuclear waste. Chem. Rev. 121, 12327–12383 (2021).
Kerisit, S., Ryan, J. V. & Pierce, E. M. Monte Carlo simulations of the corrosion of aluminoborosilicate glasses. J. Non-Cryst. Solids 378, 273–281 (2013).
Grambow, B. & Muller, R. First-order dissolution rate law and the role of surface layers in glass performance assessment. J. Nucl. Mater. 298, 112–124 (2001).
Damodaran, K., Gin, S., Naranayasamy, S. & Delaye, J. M. On the effect of Al on alumino-borosilicate glass chemical durability. npj Mater. Degrad. 7, 46 (2023).
Vienna, J. D., Ryan, J. V., Gin, S. & Inagaki, Y. Current understanding and remaining challenges in modeling long-term degradation of borosilicate nuclear waste glasses. Int. J. Appl. Glass Sci. 4, 283–294 (2013).
Gin, S., Frugier, P., Jollivet, P., Bruguier, F. & Curti, E. New Insight into the residual rate of borosilicate glasses: effect of S/V and glass composition. Int. J. Appl. Glass Sci. 4, 371–382 (2013).
McGrail, B. P., Ebert, W. L., Bakel, A. J. & Peeler, D. K. Measurement of kinetic rate law parameters on a Na-Ca-Al borosilicate glass for low-activity waste. J. Nucl. Mater. 249, 175–189 (1997).
Ojovan, M. I. On alteration rate renewal stage of nuclear waste glass corrosion. Mrs Adv. 5, 111–120 (2020).
Mercado-Depierre, S., Fournier, M., Gin, S. & Angeli, F. Influence of zeolite precipitation on borosilicate glass alteration under hyperalkaline conditions. J. Nucl. Mater. 491, 67–82 (2017).
Gin, S. et al. The fate of silicon during glass corrosion under alkaline conditions: a mechanistic and kinetic study with the International Simple Glass. Geochim. Cosmochim. Acta 151, 68–85 (2015).
Mann, C. et al. Influence of young cement water on the corrosion of the International Simple Glass. npj Mater. Degrad. 3, 5 (2019).
Muller, I., Ribet, S., Pegg, I., Gin, S. & Frugier, P. Characterization of alteration phases on HLW glasses after 15 years of PCT leaching. Environmental Issues and Waste Management Technologies in the Ceramic and Nuclear Industries XI, 189–199 (2006).
Frugier, P. et al. SON68 nuclear glass dissolution kinetics: current state of knowledge and basis of the new GRAAL model. J. Nucl. Mater. 380, 8–21 (2008).
Gong, Y. X., Xu, J. & Buchanan, R. C. The aqueous corrosion of nuclear waste glasses revisited: probing the surface and interfacial phenomena. Corros. Sci. 143, 65–75 (2018).
ANDRA. Dossier 2005 Argile: Tome Phenomenological evolution of a geological repository. https://www.andra.fr/download/andra-international-en/document/editions/269va.pdf (2006)
Wang, J. High-level radioactive waste disposal in China: update 2010. J. Rock. Mech. Geotech. Eng. 2, 1–11 (2010).
Bethke, C. M. The geochemist’s workbench. Release 10.0. GWB essentials guide. Aqueous Solutions LLC. http://www.gwb.com/pdf/GWB10/GWBessentials.pdf (2014).
Verney-Carron, A., Gin, S., Frugier, P. & Libourel, G. Long-term modeling of alteration-transport coupling: application to a fractured Roman glass. Geochim. Cosmochim. Acta 74, 2291–2315 (2010).
Wang, Y. F., Jove-Colon, C. F. & Kuhlman, K. L. Nonlinear dynamics and instability of aqueous dissolution of silicate glasses and minerals. Sci. Rep. 6, 30256 (2016).
Gin, S., Ryan, J. V., Schreiber, D. K., Neeway, J. & Cabie, M. Contribution of atom-probe tomography to a better understanding of glass alteration mechanisms: application to a nuclear glass specimen altered 25 years in a granitic environment. Chem. Geol. 349, 99–109 (2013).
Gin, S. et al. A general mechanism for gel layer formation on borosilicate glass under aqueous corrosion. J. Phys. Chem. C 124, 5132–5144 (2020).
Gin, S., Delaye, J. M., Angeli, F. & Schuller, S. Aqueous alteration of silicate glass: state of knowledge and perspectives. npj Mater. Degrad. 5, 42 (2021).
Bouakkaz, R., Abdelouas, A., El Mendili, Y., Grambow, B. & Gin, S. SON68 glass alteration under Si-rich solutions at low temperature (35-90 degrees C): kinetics, secondary phases and isotopic exchange studies. Rsc Adv. 6, 72616–72633 (2016).
Reiser, J. T. et al. Effects of Al:Si and (Al plus Na):Si ratios on the static corrosion of sodium-boroaluminosilicate glasses. Int. J. Appl. Glass Sci. 13, 94–111 (2022).
Ribet, S. & Gin, S. Role of neoformed phases on the mechanisms controlling the resumption of SON68 glass alteration in alkaline media. J. Nucl. Mater. 324, 152–164 (2004).
Trivelpiece, C. L. et al. Corrosion of ISG fibers in alkaline solutions. J. Am. Ceram. Soc. 100, 4533–4547 (2017).
Inagaki, Y. et al. Aqueous alteration of Japanese simulated waste glass P0798: effects of alteration-phase formation on alteration rate and cesium retention. J. Nucl. Mater. 354, 171–184 (2006).
Gin, S., Beaudoux, X., Angeli, F., Jegou, C. & Godon, N. Effect of composition on the short-term and long-term dissolution rates of ten borosilicate glasses of increasing complexity from 3 to 30 oxides. J. Non-Cryst. Solids 358, 2559–2570 (2012).
Gates, W. P., Slade, P. G., Manceau, A. & Lanson, B. Site occupancies by iron in nontronites. Clay Clay Miner. 50, 223–239 (2002).
Kumar, A., Shrivastava, J. P., Meenakshi, Chopra, S. & Chakraborty, S. Impact glass applied as a standard for long-term performance assessment of Na-Ba borosilicate glass forms in geological environment. Appl. Geochem. 114, 104477 (2020).
Arena, H. et al. Impact of iron and magnesium on glass alteration: characterization of the secondary phases and determination of their solubility constants. Appl. Geochem. 82, 119–133 (2017).
Narayanasamy, S. et al. Influence of composition of nuclear waste glasses on vapor phase hydration. J. Nucl. Mater. 525, 53–71 (2019).
Arena, H. et al. Impact of Zn, Mg, Ni and Co elements on glass alteration: additive effects. J. Nucl. Mater. 470, 55–67 (2016).
Fleury, B., Godon, N., Ayral, A. & Gin, S. SON68 glass dissolution driven by magnesium silicate precipitation. J. Nucl. Mater. 442, 17–28 (2013).
Knauss, K. G. & Wolery, T. J. The dissolution kinetics of quartz as a function of pH and time at 70 °C. Geochim. Cosmochim. Acta 52, 43–53 (1988).
Jollivet, P. et al. Investigation of gel porosity clogging during glass leaching. J. Non-Cryst. Solids 354, 4952–4958 (2008).
Geisler, T. et al. The mechanism of borosilicate glass corrosion revisited. Geochim. Cosmochim. Acta 158, 112–129 (2015).
Dohmen, L. et al. Pattern formation in silicate glass corrosion zones. Int. J. Appl. Glass Sci. 4, 357–370 (2013).
Fenter, P. & Sturchio, N. C. Mineral-water interfacial structures revealed by synchrotron X-ray scattering. Prog. Surf. Sci. 77, 171–258 (2004).
Marry, V., Rotenberg, B. & Turq, P. Structure and dynamics of water at a clay surface from molecular dynamics simulation. Phys. Chem. Chem. Phys. 10, 4802–4813 (2008).
Michelin, A. et al. Silicate glass alteration enhanced by iron: origin and long-term implications. Environ. Sci. Technol. 47, 750–756 (2013).
Galai, L., Marchetti, L., Godon, N., Remazeilles, C. & Refait, P. Kinetic study of Fe silicates formation during iron corrosion in deaerated and alkaline Si-containing solutions at 50 degrees C. Corros. Sci. 211, 110846 (2023).
Thien, B. M. J., Godon, N., Ballestero, A., Gin, S. & Ayral, A. The dual effect of Mg on the long-term alteration rate of AVM nuclear waste glasses. J. Nucl. Mater. 427, 297–310 (2012).
Guittonneau, C. et al. A 25-year laboratory experiment on French SON68 nuclear glass leached in a granitic environment—first investigations. J. Nucl. Mater. 408, 73–89 (2011).
Lenting, C. & Geisler, T. Corrosion of ternary borosilicate glass in acidic solution studied in operando by fluid-cell Raman spectroscopy. npj Mater. Degrad. 5, 37 (2021).
Ebert, W. L. & Bates, J. K. A comparison of glass reaction at high and low glass surface/solution volume. Nucl. Technol. 104, 372–384 (1993).
Gin, S. et al. Nuclear glass durability: new insight into alteration layer properties. J. Phys. Chem. C 115, 18696–18706 (2011).
Stuckless, J. S. & Dudley, W. W. The geohydrologic setting of Yucca Mountain, Nevada. Appl. Geochem. 17, 659–682 (2002).
Ma, B. & Lothenbach, B. Synthesis, characterization, and thermodynamic study of selected K-based zeolites. Cem. Concr. Res. 148, 106537 (2021).
Fournier, M. et al. Glass dissolution rate measurement and calculation revisited. J. Nucl. Mater. 476, 140–154 (2016).
Acknowledgements
The authors would like to thank the State Administration of Science. Technology and Industry for National Defense (PRC) for project funding related to this work. The authors also thank Roland Hellmann for the discussions. The synchrotron facility ESRF is acknowledged for the allocation of the beamtime (proposal IH-MA-205).
Author information
Authors and Affiliations
Contributions
Kaifeng Wang: Investigation, Methodology, Formal analysis, Data curation, Writing-original draft. Yang Chen: Methodology, Software, Writing-review & editing. Nathaniel Findling: Formal analysis, Writing-review & editing. Frederic Charlot: Formal analysis. Laurent Charlet: Investigation, Writing-review & editing. Jiliang Liu: Methodology, Formal analysis, Writing-review & editing. Zhentao Zhang: Investigation, Resources, Supervision, Funding acquisition. All authors read and approved the final paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, K., Chen, Y., Findling, N. et al. Formation and evolution of secondary phases and surface altered layers during borosilicate glass corrosion in pore water. npj Mater Degrad 8, 26 (2024). https://doi.org/10.1038/s41529-024-00444-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-024-00444-y
