
Abstract
Considering the complex coupling of steel corrosion in partially saturated concrete filled with water, the quantitative description of control mechanisms is still under debate. This work provides new experimental evidence supporting that diffusion control (relative diffusion coefficient) is the dominant mechanism in controlling corrosion rate by limiting the ferrous ion migration in unsaturated concrete. Furthermore, a new mechanism-based kinetic model is developed to predict the corrosion rate in different cementitious materials and corrosion conditions. In addition, the proposed kinetic model can quantify the variation of critical [Cl−]/[OH−] with degree of saturation, classify corrosive conditions, and predict the electrical resistivity and corrosion rate relationships.
Similar content being viewed by others


Introduction
Corrosion of steel and alloys causes economical loss equivalent to roughly 3–4% of a nation’s gross domestic product (GDP)1, and the extra CO2 emissions associated with replacing corroded steel are estimated to be 4.1–9.1% of the total2. A substantial proportion of the corrosion is related to the corrosion of steel in concrete infrastructure, such as bridges, buildings, dams, ports, and roads. The corrosion issue is particularly serious when concrete infrastructure is served in a variety of complex and harsh environments, such as salt-laden atmosphere with high temperature and relative humidity3,4, wet-dry cycle5,6, seaside environment7,8, and underground infrastructure9. Although electrochemical mechanisms of corrosion of metal in various electrolyte solutions, e.g., chloride attack on the passive film, product precipitation at high alkalinity, and product oxidation by dissolved oxygen, have been revealed10,11,12,13,14,15,16,17,18, it needs to be identified and confirmed the role of these mechanisms in steel corrosion in partially saturated concrete exposed in above complex and varying service environments. Because the corresponding corrosion evaluation of steel in unsaturated concrete becomes more complicated, involving the reduction of the contact area of steel with electrolyte in concrete pores (active area)19,20, drastic change of gas and ion diffusion coefficients of concrete21,22,23,24, and electrochemical reaction and precipitation of corrosion products15,25,26. Nevertheless, few investigations establish the complete description and underlying mechanism of corrosion kinetics from the electrolyte solution to partially saturated porous media filled with water in cementitious materials due to the lack of systematic experimental investigations.
Although the corrosion behaviors of steel in concrete have been extensively investigated, the electrochemical mechanisms in unsaturated concrete are still under debate. Specifically, there is no consensus on the exact control condition governing corrosion kinetics in different environments, i.e., ohmic resistivity control, cathodic control (e.g., oxygen control), and anodic control (e.g., ion diffusion control). Ohmic resistivity control is commonly considered to provide a partial and secondary contribution to corrosion rate restriction in some particular occasions, e.g., superhydrophobic concrete with high bulk electrical resistivity27 and the initial phase of pit growth28. Earlier literature reported that the corrosion rate of specimens immersed in seawater or exposed to high relative humidity over 90% is restrained by the limited availability of oxygen in concrete1,29. However, the recent findings28,30,31 claimed that anodic control, including the limitation of active area (contact area of steel with pore solution) and dissolution rate of iron, is the main mechanism restraining the corrosion propagation in most cases including saturated concrete. The inconsistent findings necessitate more extensive comparative studies and in-depth investigations of steel corrosion mechanisms in different environments. Moreover, the fundamental compositional and microstructural parameters of cementitious media that govern corrosion rate of steels remain to be determined and quantified, especially in a partially saturated state.
Many corrosion phenomena of steel in unsaturated concrete and the underlying mechanisms still lack a rational explanation. For instance, the critical ratio of chloride ion concentration to hydroxide ion concentration in pore solution (critical [Cl−]/[OH−]) is an essential parameter to determine the start point of corrosion initiation (refer to the critical corrosion rate of 0.1 μA/cm2). However, the critical [Cl−]/[OH−] is not a threshold with a specific value, but is sensitively influenced by material-related parameters, e.g., chloride binding capacity and steel-concrete interface defects, as well as environment-related parameters, e.g., moisture content32,33,34. Moreover, electrical resistivity is commonly used to classify corrosion conditions and assess corrosion risk based on the empirical linear relationship of concrete electrical resistivity and steel corrosion rate (R-C relationship) for a given binder type and corrosion condition35,36. However, considerable deviations of R-C relationships37,38 are observed among reinforced concrete with different binders and serving environments in extensive published experimental studies29,30,35,37,38,39,40,41,42,43, which obstructs the accurate classification of corrosion conditions. Unfortunately, those phenomena have not been explained and quantified because of unclear mechanisms of steel corrosion.
This work explores the underlying electrochemical mechanisms and quantifies the key parameters governing corrosion rate of steels based on systematic experiments from corrosion in simulated pore solution to corrosion in partially saturated cementitious porous media with water, as shown in Fig. 1. Moreover, a mechanistic-based kinetics model for corrosion prediction was proposed based on the data of chloride-induced steel corrosion in small-size mortar specimens and further verified by the published data involving chloride-induced or/and carbonation-induced steel corrosion in full-size concrete elements. The proposed model could be used to explain and quantify relevant corrosion phenomena, including the dependence of critical [Cl−]/[OH−] on degree of saturation and the drift of R-C (corrosion rate versus concrete resistivity) relationship lines in various environments.

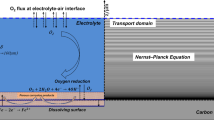
The specimens are exposed to three typical corrosion conditions a corrosion behaviors in simulated pore solution, b corrosion behaviors in saturated mortar at RH 97%, and c corrosion behaviors in partially saturated mortar at RH 84%, 69%, and 43%.
Results and Discussion
Kinetic model of steel corrosion
The proposed mechanistic-based kinetic model of steel corrosion is illustrated in Fig. 2, established by bridging the steel corrosion in an electrolytic solution to its corrosion in partially saturated cementitious porous media. More specifically, the corrosion rate of steel in unsaturated concrete, \({i}_{{corr}}\), is deduced from the corrosion rate of steel in pore solution, \({i}_{{corr},{pore}}\), by two multiplied coefficients, including reduction coefficient of pore structure, \({\eta }_{{pore}}\), and reduction coefficient of degree of saturation, \({\eta }_{{DoS}}\). Correspondingly, the role of key influential factors in steel corrosion, i.e., pore solution chemical composition, porosity and pore structure, and relative diffusion coefficient (related to degree of saturation), are systematically analyzed in the proposed corrosion model. It should be noted that the developed kinetic model primarily focuses on average corrosion rate of the entire steel rebar in the propagation period of corrosion in reinforced concrete, that is, from the steel rebars starting to corrosion to the concrete cracking. In this period, corrosion of steel bars is relatively uniform and severe pitting corrosion resulting from concrete cracking rarely occurs. Therefore, these prerequisites and application scope need to pay attention to when using this model.

The corrosion models with/without consideration of relative diffusion coefficient are compared with different pore tortuosity.
To develop the kinetic model, the potential mechanisms governing the corrosion rate in partially saturated concrete, i.e., ohmic control, cathodic control (e.g., oxygen control), and anodic control (e.g., reduction of active area and ion diffusion control) are comprehensively discussed and analyzed in Supplementary Note 1. Ohmic control is unlikely to dominate corrosion rate evidenced by the order of magnitude difference between the anticipated corrosion rate and experimental results27,28,30,31. In most service conditions, e.g., atmospheric environment, wet-dry cycle, and seaside environment, oxygen control rarely constrains corrosion rate because oxygen around steel rebars is readily available. It is supported by the negative correlation of corrosion rate and oxygen transport rate and the opposite open circuit potential change with corrosion behavior in simulated pore solution (see Supplementary Fig. 2 and Supplementary Table 1)42,43,44,45. Even in the saturated conditions, there is no significant reduction in corrosion rate observed due to potential oxygen shortage although specimens are exposed at a high RH of 97% for a long-term of 5 months in this study (see Supplementary Figure. 3). Besides, the chloride ion and hydroxide ion are unlikely to be responsible for the reduction of corrosion rate in partially saturated concrete because their concentration change with degree of saturation is not significant.
As comprehensively discussed in Supplementary Note 1 based on extensive experimental data, corrosion rate in partially saturated concrete is governed by anodic control in two aspects, i.e., reduction of active area and ion diffusion control. On the one hand, at a low degree of saturation, the reduction of contact area of steel with electrolytic pore solution, i.e., active area, limits the corrosion rate because the potential anodic area in micro-battery corrosion cell decreases (see Fig. 2), which is quantified using the reduction coefficient of pore structure, \({\eta }_{{pore}}\). On the other hand, the dissolution rate of iron is constrained by the rising ferrous ion concentration because of the low migration rate (larger ion diffusion coefficient) of ferrous ions at low degree of saturation (see Fig. 2). It is evidenced by multiple observed corrosion behaviors, i.e., the negative correlation of open circuit potential with corrosion rate (Supplementary Fig. 2), the reduction of corrosion rate with the dropping degree of saturation (Supplementary Table 1), and the quantitative linear and quadratic relation of corrosion rate and diffusion coefficient (elaborated in the following section). Thus, the reduction coefficient of degree of saturation, \({\eta }_{{DoS}}\), is introduced to clarify and quantify this dual impact of degree of saturation. The detailed mechanism and quantitative subexpression involving key factors will be elaborated on later.
Electrochemical mechanism of steel corrosion in pore solution
The corrosion rate of a given steel rebar immersed in a pore solution, \({i}_{{corr},{pore}}\) [μA/cm2], is primarily controlled by the chemical composition of pore solution, which lays the foundation for establishing the corrosion kinetic model in cementitious materials. Except for the chemical compositions of steel alloy, corrosion behaviors are also affected by the microstructures, metallography, and heat treatments of chemically similar rebar steels46,47,48. As such, to determine corrosion kinetics in pore solution as precisely as possible, the same batch of steel rebars and the electrolytic solutions with similar composition as the pore solution of cementitious mortars are adopted. As a common finding49, the corrosion rate of steel increases with the rising [Cl−]/[OH−], as shown in Fig. 3a. The mathematical expression of corrosion kinetics for the specific steel rebars is determined as follows:

a Corrosion rate versus [Cl−]/[OH−] in simulated pore solution. b Corrosion rate in saturated mortar versus corrosion rate in simulated pore solution. c Relationship between the reduction coefficient of saturation degree \({\eta }_{{DoS}}\) and degree of saturation \(S\). d Relationship between the reduction coefficient of saturation degree, \({\eta }_{{DoS}}\), and the relative diffusion coefficient, \({D/D}_{0}\). The regressive mathematical curves are also shown. (The data of NH-A5-Cl0.6 were excluded in regression because it was in a state of passive at each RH due to low chloride content).
Corrosion rate in saturated cementitious materials
The ratio of corrosion rate of steel in simulated pore solution and saturated mortar is defined as the reduction coefficient of pore structure \({\eta }_{{pore}}\). The reduction of corrosion rate is attributed to the direct limitation of the contact area between the steel rebars and pore solution liquid by the pore structure. Moreover, the existence of pore sturcutre significantly restrains the migration of corrosion products thereby reducing the dissolution rate of iron. Technically, this parameter is not only related to the pore structure of cementitious porous media30 but also inevitably affected by the local microstructure of the steel–mortar interface because of its remarkable inhomogeneity in porosity and pore connectivity around steel50,51,52,53,54, as Supplementary Note 2 shows. To determine the parameter as accurately as possible in our work, the reduction coefficient of pore structure is directly and experimentally obtained by the slope of fitting lines between corrosion rates of steel rebars in saturated mortars and simulated pore solution, as shown in Fig. 3b. It is observed that the experimentally determined \({\eta }_{{pore}}\) is close among mixtures and approximately equal to the water-permeable porosities of mortars because the porosities among different mixtures concentrate on a narrow range of 0.20 to 0.25 (listed in Table 1). Thus, the average of reduction coefficient of pore structure \({\eta }_{{pore}}\) with the value of 0.193 are used in this case to simplify the calculation.
Decisive influence of relative diffusion coefficient on corrosion rate in partially saturated mortar
The ratio of corrosion rate in partially saturated cementitious materials, \({i}_{{corr}}\), and corrosion rate in saturated cementitious materials, \({i}_{{corr},0}\), is defined as the reduction coefficient of saturation degree, \({\eta }_{{DoS}}\). This parameter is impacted by the reduction of active area and diffusion coefficient in cementitious materials at different degrees of saturation, as discussed in Supplementary Note 3. Technically, the proportion of active area of steels in partially saturated cementitious materials and saturated cementitious materials is equal to degree of saturation in value even without considering the steel-concrete interface. However, the reduction coefficient of saturation degree, \({\eta }_{{DoS}}\), is significantly smaller than degree of saturation, as shown in Fig. 3c. It implies that considering the reduction of active area alone is insufficient and incomplete to correct corrosion rate of steel in unsaturated concrete.
Therefore, ion diffusion control, as another potential mechanism of limitation to corrosion rate, is introduced particularly in partially saturated cementitious materials. As Fig. 2 shows, the low ion diffusion coefficient constraints ferrous ion migration in porous channels of cementitious materials and leads to the ferrous ion accumulation around steel rebars, which hinders the further dissolution of ferrous ions. In a dry environment, the remarkably decreasing diffusion coefficient with the drop of degree of saturation accelerates the accumulation of ferrous and eventually dominates the reduction of corrosion rate. This mechanism is supported by the consistency in electrochemistry with the reducing corrosion rate with dropping RH accompanied by the increase of open circuit potential50,55,56.
To further quantify the ion diffusion control mechanism, relative diffusion coefficient, \(D/{D}_{0}\), defined by the ratio of diffusion coefficient of partially saturated cementitious materials, \(D\), and saturated cementitious materials, \({D}_{0}\), is used to develop the relation between corrosion rate and degree of saturation in a partially saturated system. Quantitatively, there is a linear relation between the reduction coefficient of saturation degree, \({\eta }_{{DoS}}\), and relative diffusion coefficient for OPC with a slope of 1.0 and \({R}^{2}\) of 0.9499, as shown in Fig. 3d. Differently, the regressive curve for NH (except for NH-A5-Cl0.6) is a power function with an index of 2.1 and \({R}^{2}\) of 0.9871. The different regressive functions imply that reduction coefficient of saturation degree of AAS is more sensitive to the relative diffusion coefficient, which is related to its dense pore structure. Thus, the corrosion rate in partially saturated cementitious materials could be calculated as follows:
where a is a parameter related to the average pore diameter of cementitious materials, as explained later.
In practice, the relative diffusion coefficient of unsaturated cementitious materials is commonly calculated by readily measured electrical resistivity according to Supplementary Note 3. Otherwise, the relative diffusion coefficient can be predicted by the Archie equation57 if the material model has been established referring to Supplementary Note 4.
where \({\rho }_{0}\) is the electrical resistivity of saturated cementitious materials; \(\rho\) is the electrical resistivity of partially saturated cementitious materials (degree of saturation \(S\)); \(n\) is a material parameter related to pore structure [unitless]. The mathematical expression explains why the relationship between corrosion rate and degree of saturation in Fig. 3d is a power function with a high exponent.
Model verification by chloride-induced corrosion in small-size AAS and OPC mortar specimens
The data of AAS mortar specimens with different chloride content, activator type, and alkali dosage, as well as the reference OPC mortar specimens, were used to verify the developed kinetic model. The detailed cementitious material parameters, e.g., pore solution chemical composition, porosity and pore structure parameters, and electrochemical parameters, are listed in Table 2 Utilizing the equations of the developed corrosion model in Fig. 2, the predicted corrosion rate can be calculated based on the measured properties of cementitious materials.
As shown in Fig. 4a, there is a good linear relation between the predicted and measured corrosion rate with a slope of 0.989 and \({R}^{2}\) of 0.9598, which proves that the proposed corrosion model works well for cementitious materials regardless of binder type. Nevertheless, it should be noted that our previous works mainly involve chloride-induced corrosion in small-size mortar specimens in various quasi-stable environments independent of time. The proposed corrosion model needs further verification for more practical full-size reinforced concrete components, as well as complex environmental conditions changing with time. In this paper, the term “small-scale” refers to deliberately designed small specimens utilized in laboratory settings to ensure precise control over variables and facilitate in-depth exploration of underlying mechanisms. On the other hand, “full-scale” refers to the dimensions of components used in experimental studies that closely resemble or are comparable to actual engineering structures.

a Chloride-induced corrosion in hardened AAS and OPC materials and b chloride-induced and carbonation-induced corrosion in hardened OPC-based materials.
Model verification by chloride-induced and carbonation-induced corrosion in full-size OPC concrete elements
To further verify the validity and applicability of the proposed corrosion kinetic model for concrete elements under more complex service environments, published corrosion data and corresponding concrete property results are collected, including the full-size concrete elements exposed to varying relative humidity and carbonated mortar specimens20,30,39. Afterward, the predicted corrosion rate is calculated as per the equations in Fig. 2 based on pore solution chemical composition, porosity and pore structure parameters, and electrical resistivity (for evaluating relative diffusion coefficient).
These relevant concrete properties and electrochemical parameters are summarized in Table 3 Although most concrete properties are included in these papers, some of parameters need to be assumed according to the common value or experimental value in similar situations. For the full-size concrete elements39, some data of pore solution chemical composition is assumed. Specifically, the chloride ion concentration of pore solution without calcium chloride addition is adopted as 0.005 mol/L according to reference39. For the elements with carbonation depth over the thickness of cover concrete, the hydroxide ion concentration of pore solution is adopted as 0.0005 mol/L (pH value of around 10.7) based on the experimental value of OPC specimens after 5% CO2 accelerated carbonation58. In the situation of partially carbonated concrete, that is the carbonation depth is close to the thickness of cover concrete, the hydroxide ion concentration of pore solution is assumed as 0.005 mol/L (pH value of around 11.7). Reduction coefficient of pore structure \({\eta }_{{pore}}\) is adopted as the value of mortar porosities according to the previous analysis. Differently, in the publications20,30, the reduction coefficient of pore structure, \({\eta }_{{pore}}\), and corrosion rate in bulk pore solution, \({i}_{{corr},{pore}}\), have been directly and experimentally determined. Besides, saturation coefficient of 3.5 for OPC concrete without admixtures and 5.0 for OPC-based concrete with clinker replacement, e.g., slag, limestone, and burnt oil shale, is used according to the commonly reported value21,59.
The predicted corrosion rate and measured corrosion rate from other reported publications are compared in Fig. 4b. Although the slope of fitting line is 0.831 smaller than 1 meaning the overestimation of corrosion rate by the proposed corrosion model to some extent likely owing to the assumed parameters, the predicted corrosion rate is consistent with the measured corrosion rate in general with \({R}^{2}\) of 0.8508. The comparing results support that the proposed model is capable of predicting the corrosion rate in the full-size reinforced concrete elements exposed to complex environments with changing ambient relative humidity, as well as chloride-contaminated and carbonated corrosive conditions.
Key factors determining corrosion rate and classifying corrosion conditions
As described in Fig. 2, the corrosion rate of steel in unsaturated cementitious pore media is determined by pore solution chemical composition, porosity and pore structure parameters, and relative diffusion coefficient. Among these parameters, reduction coefficient of pore structure \({\eta }_{{pore}}\), saturation coefficient \(n\), and index of \(a\) are related to the properties of cementitious materials, noted as material-related parameters. On the other hand, the ratio of chloride to hydroxide ion concentration [Cl−]/[OH−] and relative diffusion coefficient \({D/D}_{0}\) is highly dependent on the various environmental conditions, e.g., chloride invasion, carbonation, and wet-dry cycle, noted as environment-related parameters.
For given cementitious materials, the corrosion conditions can be accurately classified based on two key environment-related factors, that is relative diffusion coefficient \({D/D}_{0}\) and the ratio of chloride to hydroxide ion concentration [Cl−]/[OH−], which is quantitatively presented in Fig. 5. In this case, the different pore structure parameters, i.e., \({\eta }_{{pore}}=0.193,{a}=2.1\) for AAS and \({\eta }_{{pore}}=0.193,{a}=1.0\) for OPC, contribute to their distinct corrosion behaviors. Specifically, the corrosion rate of AAS is more sensitive to relative diffusion coefficient at high [Cl−]/[OH−] than OPC, meaning its corrosion rate decreases faster with the drop of degree of saturation, which is attributed to its denser pore structure. Moreover, corrosion rate is more sensitive to [Cl−]/[OH−] at a high relative diffusion coefficient but remains relatively stable at a low relative diffusion coefficient for both AAS and OPC. This is because the corrosion rate at low degree of saturation is mainly restrained by ion diffusion control.

a AAS mortar (\({\eta }_{{pore}}=0.193,{a}=2.1\)) and b OPC mortar (\({\eta }_{{pore}}=0.193,{a}=1.0\)).
Additionally, it should be noted that the critical [Cl−]/[OH−] corresponding to the critical corrosion rate of icorr = 0.1 μA/cm2 decreases with the increase of relative diffusion coefficient, implying that steels need higher [Cl−]/[OH−] to achieve the active state in a dry environment (low degree of saturation). It explains why the critical [Cl−]/[OH−] is not a fixed threshold but varies with the environmental RH and pore structure of cementitious materials32,33. This phenomenon has been reported by several researchers34; however, the quantitative description is determined for the first time in this work.
Prediction of R-C relationship lines
Based on the equations in Fig. 2, the formula between electrical resistivity of concrete, \(\log \left(\rho \right)\), and corrosion rate of steel, \(\log \left({i}_{{corr}}\right)\), in a logarithmic coordinate system, that is R-C relationship lines, can be deduced as follows:
where the slope is \(-a(1+\frac{1}{n})\), which is related to the pore structure parameters; the intercept is \(\log ({i}_{{corr},{pore}})+a\left(1+\frac{1}{n}\right)\log \left({\rho }_{0}\right)+\log \left(\phi \right)\), which is related to the pore solution chemical composition, electrical resistivity of saturated cementitious matrices, and porosity.
To verify the deduced equation, the concrete properties summarized in
Table 2 were incorporated into the corrosion model to predict R-C relationship lines and then compared with the experimental data60, as shown in Fig. 6. There is good an agreement between the predicted R-C relationship line and the experimental data points with the close parameters in Table 4. It indicates that R-C relationship lines, as a material- and environment- related empirical formula, can be predicted by the corrosion kinetic model established in this work.

Additionally, the proposed corrosion model can explain the difference in the slope and intercept of R-C relationship lines. On the one hand, the slope of R-C relationship lines is mainly impacted by the parameters of \(a\) and related to pore structure, which is supported by its positive relation with the average pore diameter detected by MIP61, as shown in Fig. 6b. On the other hand, the change in chloride ion concentration has a limited impact on the drift of R-C relationship lines, however, the drop in hydroxide ion concentration is mainly responsible for the increase of intercept. Specifically, the increase of chloride ion concentration leads to the simultaneous rising corrosion rate and dropping electrical resistivity in electrochemistry, as listed in Table 2. The two impacts offset the most change in intercept as per Eq. (5). Therefore, there is no drift of R-C relationship lines (the same intercept) observed in Fig. 6 among R-C relationship lines of NH with various incorporated chloride contents. Differently, the drop in hydroxide ion concentration leads to the increase of corrosion rate and the reduction of electrical resistivity, as listed in Table 2. These two impacts jointly work and lead to the drift of R-C relationship lines towards left and up (increasing intercept), supported by comparing R-C relationship lines of NH and NC/NSi. These mechanisms underlying R-C relationship lines were proposed in our previous work60 and now could be quantitatively predicted based on Eq. (5) developed from the corrosion model proposed in this study.
In summary, this work revealed the role of ion diffusion coefficient as the key microstructural parameter in steel corrosion in partially saturated cementitious porous material based on systematic experimental data. At low degree of saturation, the limited migration and accelerated accumulation of ferrous ions around anodic areas owing to the low diffusion coefficient, as well as the reduction of active area, jointly govern corrosion rate. Besides, the connection in electrochemistry between steel corrosion in electrolyte solution and in cementitious porous media is linked. On this basis, a quantitative corrosion kinetic model was proposed to predict the corrosion rate of steel in partially saturated cementitious materials, in which the relative diffusion coefficient \({D/D}_{0}\) and pore solution chemistry [Cl−]/[OH−] are the fundamental microstructural and compositional parameters that govern corrosion rate. A corrosion model was developed based on the experimental data of steel corrosion in AAS and OPC mortars with small size exposed in stable environments. Its reliability and accuracy were further verified by the good agreement between predicted and measured corrosion rate from published data including chloride-induced and carbonation-induced corrosion of steels in full-size concrete elements exposed in varying environments. Furthermore, the proposed corrosion model could quantitatively determine the relation between the critical [Cl−]/[OH−] and degree of saturation, classify corrosive conditions, and predict the empirical R-C relationship lines for different cementitious materials and environmental conditions.
Methods
Systematical electrochemical tests in different corrosion conditions were designed to establish the link of corrosion behaviors in various environmental conditions. As shown in Fig. 1, three typical corrosion conditions, (a) corrosion in simulated pore solution, (b) corrosion in saturated mortar, and (c) corrosion in partially saturated mortar, were applied using the same batch of samples and cement materials to ensure the comparability of results.
Corrosion in the simulated pore solution
First, the corrosion behaviors in simulated pore solution were characterized to link the relation of corrosion rate and electrolyte chemical composition as the baseline for the corrosion kinetics in cementitious materials. To ensure the consistency and comparability of experimental data, the simulated pore solutions were prepared according to the measured chemical compositions of pore liquid in matured mortars. It covered the typical range of chloride ion and hydroxide ion concentrations for non-carbonated AAS and OPC materials60, as shown in Table 5. To ensure the reproducibility of results, three duplicates of steel rebars samples were polished, cleaned, and processed with epoxy resin to leave a contact area of 2.513 cm2 exposed in the electrolytic solution. For each simulated pore solution, a three-electrode cell filled with 150 ml simulated solution was prepared for electrochemical measurements, installed with an Ag/AgCl electrode as a reference electrode, a platinum wire as a counter electrode, and three steel rebar samples as working electrodes. After being soaked for 24, 48, and 72 h to reach stable conditions (determined when the open circuit potential change was less than 1 mV/min in 10 min before tests), the corrosion rate of steel was measured through the polarization resistance method at ±10 mV around the open circuit potential with a scanning rate of 0.167 mV/s.
Corrosion in saturated mortars
In the second part, the same batch of polished and cleaned steel rebars was embedded in mortar specimens and then exposed to saturated conditions to characterize their corrosion behaviors. The plate mortar specimens were designed with a small thickness to accurately and quickly control the moisture condition inside specimens, as introduced in our previous work60. For each specimen, an Ag/AgCl sintered electrode as the reference electrode, a stainless steel mesh as the counter electrode, and six steel rebar samples as working electrodes were arranged as shown in Fig. 1, which allows six independent corrosion rate tests and five independent electrical resistivity tests in electrochemical measurements.
A total of eleven different mixtures were designed to collect their electrochemical corrosion data, including different cement systems, i.e., ordinary Portland cement (OPC) or alkali-activated slag (AAS), as well as various chloride contents, alkali dosages, and activator types. For convenience, the mixture proportioning parameters of the studied mortars were summarized in Table 6 and the detailed information was presented in refs. 60,61. Those specimens were cast and cured in the sealed plastic film at 25 ± 1 °C for 28 days and then exposed to an environmental container of 97% RH controlled by saturated K2SO4 solution. After being exposed for 28 days and 56 days until the moisture inside specimens reaches equilibrium with the external relative humidity, electrochemical tests were performed to measure the open circuit potential, electrical resistivity of mortar, and corrosion rate of steel using the electrochemical workstation of Gamry Reference 600+. The open circuit potentials were measured with respect to the Ag/AgCl reference electrode and the results were converted to the SCE scale based on the calibration results. The corrosion rate, i.e., corrosion current density, was measured using the linear polarization resistance method by applying a scan voltage to a carbon steel rebar in the three-electrode configuration, ranging from -10 mV to +10 mV around the open circuit potential with a scanning rate of 0.1667 mV/s. The electrical resistance of mortars was measured using the electrochemical impedance spectroscopy method by applying a ± 10 mV voltage to two adjacent carbon steel rebars in a two-electrode circuit with a scanning frequency from 105 Hz to 1 Hz. The minimum frequency of 1 Hz has been proven to be adequate in accurately determining the resistivity of the concrete while simultaneously mitigating the polarization effect on the surface of steel rebars from applied low-frequency voltage. The electrical resistivity was calculated by multiplying the tested electrical resistance with a geometry factor of 66.667 mm based on the tested section with a cross-sectional area of 100 mm × 10 mm and a length of 15 mm.
Corrosion in partially saturated mortar
For corrosion in partially saturated cementitious material, other three duplicates of plate specimens (see Fig. 1) were prepared using the same batch of fresh mortar and cured in the same condition for 28 days, and then were exposed at different RH levels of 84%, 69%, and 43% respectively. The different RH levels were maintained using sealed containers with different saturated salt solutions of KCl, KI, and NaBr, respectively, at 25 ± 0.2 °C as per ASTM E104. Meanwhile, three duplicates of mini-bar specimens were prepared for monitoring the change in degree of saturation, which were exposed to the same environment and experienced the same temperature and RH history as the corresponding plate specimens. Due to the small size of specimens, the hardened AAS mortar plate specimens could quickly reach a rapid hygroscopic equilibrium with the ambient relative humidity RH, identified as the daily mass change of less than 0.1% by the ASTM C1498 standard. The electrochemical tests were performed using the same measuring parameters as those in the saturated specimens. Moreover, the Tafel curves of carbon steel rebars were measured for both saturated and partially saturated specimens after being exposed to different RHs for 5 months. Specifically, a voltage of scan range from -250 mV to 250 mV versus open circuit corrosion potential was applied at a scan rate of 0.1667 mV/s to obtain the reacted current. The large applied voltage in Tafel curve tests inevitably leads to the disturbance to the electrochemical cell, therefore, these measurements were only performed after other electrochemical tests had been completed.
Microstructural characterization of steel-mortar interface
To uncover the mechanism of steel corrosion in cementitious materials, microstructural and compositional characterizations of the corroded steel-mortar interface were detected. After being exposed at 97% RH for 5 months, the mortar specimens embedded with corroded steel rebars were first segmented perpendicular to the longitudinal direction of steel rebars for further steel-mortar interface characterization. The segmented specimens were first immersed in isopropyl alcohol for at least one week followed by vacuum drying at 40 °C for at least 3 days and then impregnated with low-viscosity epoxy under vacuum to solidify the microstructure. After 24 h of epoxy resin curing, the samples were polished using diamond suspensions down to 1 μm and carbon-coated prior to imaging. The microstructure and corrosion rust distribution around the steel-mortar interface was characterized by the Hitachi S-3400N scanning electron microscopy equipped with energy-dispersive X-ray spectroscopy (SEM-EDS).
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Bertolini, L., Elsener, B., Pedeferri, P., Redaelli, E. & Polder, R. B. Corrosion of steel in concrete: prevention, diagnosis, repair. (John Wiley & Sons, 2013).
Iannuzzi, M. & Frankel, G. S. The carbon footprint of steel corrosion. npj Mater. Degrad. 6, 101 (2022).
Andrade, C., Alonso, C. & Sarrı́a, J. Corrosion rate evolution in concrete structures exposed to the atmosphere. Cem. Concr. Compos. 24, 55–64 (2002).
Dong, B. et al. Corrosion failure analysis of low alloy steel and carbon steel rebar in tropical marine atmospheric environment: Outdoor exposure and indoor test. Eng. Fail. Anal. 129, 105720 (2021).
Hong, K. & Hooton, R. D. Effects of cyclic chloride exposure on penetration of concrete cover. Cem. Concr. Res. 29, 1379–1386 (1999).
Castro, P., De Rincon, O. T. & Pazini, E. J. Interpretation of chloride profiles from concrete exposed to tropical marine environments. Cem. Concr. Res. 31, 529–537 (2001).
Monticelli, C. et al. Corrosion behavior of steel in alkali-activated fly ash mortars in the light of their microstructural, mechanical and chemical characterization. Cem. Concr. Res. 80, 60–68 (2016).
Tian, Y., Wen, C., Wang, G., Hu, J. & Mai, Z. Marine field test for steel reinforcement embedded in mortar: Coupled influence of the environmental conditions on corrosion. Mar. Struct. 73, 102788 (2020).
Melchers, R. E. Predicting long-term corrosion of metal alloys in physical infrastructure. npj Mater. Degrad. 3, 1–7 (2019).
Page, C. L. & Treadaway, K. W. J. Aspects of the electrochemistry of steel in concrete. Nature 297, 109–115 (1982).
Zhang, B. et al. Unmasking chloride attack on the passive film of metals. Nat. Commun. 9, 2559 (2018).
Xue, F., Wei, X., Dong, J., Wang, C. & Ke, W. Effect of chloride ion on corrosion behavior of low carbon steel in 0.1 M NaHCO3 solution with different dissolved oxygen concentrations. J. Mater. Sci. Technol. 35, 596–603 (2019).
Li, Z. et al. Adaptive bidirectional extracellular electron transfer during accelerated microbiologically influenced corrosion of stainless steel. Commun. Mater. 2, 67 (2021).
Xiao, K. et al. Effect of concentrations of Fe2+ and Fe3+ on the corrosion behavior of carbon steel in Cl− and SO42− aqueous environments. Met. Mater. Int. 27, 2623–2633 (2021).
Stefanoni, M., Zhang, Z., Angst, U. & Elsener, B. The kinetic competition between transport and oxidation of ferrous ions governs precipitation of corrosion products in carbonated concrete. RILEM Tech. Lett. 3, 8–16 (2018).
Liu, P., Zhang, Q.-H., Watanabe, Y., Shoji, T. & Cao, F.-H. A critical review of the recent advances in inclusion-triggered localized corrosion in steel. npj Mater. Degrad. 6, 1–17 (2022).
Hayden, S. C. et al. Localized corrosion of low-carbon steel at the nanoscale. npj Mater. Degrad. 3, 1–9 (2019).
Bland, L. G. & Locke, J. S. (Warner) Chemical and electrochemical conditions within stress corrosion and corrosion fatigue cracks. npj Mater. Degrad. 1, 1–8 (2017).
Huet, B., L’hostis, V., Santarini, G., Feron, D. & Idrissi, H. Steel corrosion in concrete: determinist modeling of cathodic reaction as a function of water saturation degree. Corros. Sci. 49, 1918–1932 (2007).
Stefanoni, M., Angst, U. M. & Elsener, B. Electrochemistry and capillary condensation theory reveal the mechanism of corrosion in dense porous media. Sci. Rep. 8, 1–10 (2018).
Weiss, J., Snyder, K., Bullard, J. & Bentz, D. Using a saturation function to interpret the electrical properties of partially saturated concrete. J. Mater. Civ. Eng. 25, 1097–1106 (2013).
Spragg, R. et al. Factors that influence electrical resistivity measurements in cementitious systems. Transportation Res. Rec. 2342, 90–98 (2013).
Rajabipour, F. & Weiss, J. Electrical conductivity of drying cement paste. Mater. Struct. 40, 1143–1160 (2007).
Spragg, R., Villani, C. & Weiss, J. Electrical properties of cementitious systems: formation factor determination and the influence of conditioning procedures. Adv. Civ. Eng. Mater. 5, 124–148 (2016).
Zhao, Y., Wu, Y. & Jin, W. Distribution of millscale on corroded steel bars and penetration of steel corrosion products in concrete. Corros. Sci. 66, 160–168 (2013).
Angst, U., Elsener, B., Jamali, A. & Adey, B. Concrete cover cracking owing to reinforcement corrosion - theoretical considerations and practical experience: Concrete cover cracking owing to reinforcement corrosion. Mater. Corros. 63, 1069–1077 (2012).
Shevtsov, D. S., Zartsyn, I. D. & Komarova, E. S. Relation between resistivity of concrete and corrosion rate of reinforcing bars caused by galvanic cells in the presence of chloride. Cem. Concr. Compos. 119, 104026 (2021).
Angst, U., Elsener, B., Larsen, C. K. & Vennesland, Ø. Chloride induced reinforcement corrosion: rate limiting step of early pitting corrosion. Electrochim. Acta 56, 5877–5889 (2011).
López, W. & González, J. A. Influence of the degree of pore saturation on the resistivity of concrete and the corrosion rate of steel reinforcement. Cem. Concr. Res. 23, 368–376 (1993).
Stefanoni, M., Angst, U. M. & Elsener, B. Kinetics of electrochemical dissolution of metals in porous media. Nat. Mater. 18, 942–947 (2019).
Stefanoni, M., Angst, U. & Elsener, B. The mechanism controlling corrosion of steel in carbonated cementitious materials in wetting and drying exposure. Cem. Concr. Compos. 113, 103717 (2020).
Angst, U., Elsener, B., Larsen, C. K. & Vennesland, Ø. Critical chloride content in reinforced concrete — A review. Cem. Concr. Res. 39, 1122–1138 (2009).
Cao, Y. et al. Critical chloride content in reinforced concrete — An updated review considering Chinese experience. Cem. Concr. Res. 117, 58–68 (2019).
Chloride induced corrosion of reinforcing steel evaluated by concrete resistivity measurements. Electrochim. Acta 49, 4447–4453 (2004).
Hornbostel, K., Larsen, C. K. & Geiker, M. R. Relationship between concrete resistivity and corrosion rate – A literature review. Cem. Concr. Compos. 39, 60–72 (2013).
Sengul, O. Use of electrical resistivity as an indicator for durability. Constr. Build. Mater. 73, 434–441 (2014).
Morris, W., Vico, A., Vazquez, M. & de Sanchez, S. R. Corrosion of reinforcing steel evaluated by means of concrete resistivity measurements. Corros. Sci. 44, 81–99 (2002).
Chávez-Ulloa, E., Camacho-Chab, R., Sosa-Baz, M., Castro-Borges, P. & Pérez-López, T. Corrosion process of reinforced concrete by carbonation in a natural environment and an accelerated test chamber. Int. J. Electrochem. Sci. 8, 9015–9029 (2013).
Bertolini, L. & Polder, R. B. Concrete resistivity and reinforcement corrosion rate as a function of temperature and humidity of the environment. TNO Report 97-BT-R0574, TNO Building and Construction Research, Delft (1997).
González, J. A., Miranda, J. M. & Feliu, S. Considerations on reproducibility of potential and corrosion rate measurements in reinforced concrete. Corros. Sci. 46, 2467–2485 (2004).
Feliu, S., González, J. A., Feliu, S. & Andrade, C. Relationship between conductivity of concrete and corrosion of reinforcing bars. Br. Corros. J. 24, 195–198 (1989).
Glass, G. K., Page, C. L. & Short, N. R. Factors affecting the corrosion rate of steel in carbonated mortars. Corros. Sci. 32, 1283–1294 (1991).
Stefanoni, M., Angst, U. & Elsener, B. Corrosion rate of carbon steel in carbonated concrete – a critical review. Cem. Concr. Res. 103, 35–48 (2018).
Gonzalez, J. A., Algaba, J. S. & Andrade, C. Corrosion of reinforcing bars in carbonated concrete. Br. Corros. J. 15, 135–139 (1980).
Alonso, C., Andrade, C. & González, J. A. Relation between resistivity and corrosion rate of reinforcements in carbonated mortar made with several cement types. Cem. Concr. Res. 18, 687–698 (1988).
Clover, D., Kinsella, B., Pejcic, B. & De Marco, R. The influence of microstructure on the corrosion rate of various carbon steels. J. Appl. Electrochem. 35, 139–149 (2005).
BETONA, S. P. V. Corrosion properties of different forms of carbon steel in simulated concrete pore water. Mater. Tehnol. 48, 51–57 (2014).
Katiyar, P. K., Behera, P. K., Misra, S. & Mondal, K. Comparative corrosion behavior of five different microstructures of rebar steels in simulated concrete pore solution with and without chloride addition. J. Mater. Eng. Perform. 28, 6275–6286 (2019).
Goñi, S. & Andrade, C. Synthetic concrete pore solution chemistry and rebar corrosion rate in the presence of chlorides. Cem. Concr. Res. 20, 525–539 (1990).
Yonezawa, T., Ashworth, V. & Procter, R. P. M. Pore Solution Composition and Chloride Effects on the Corrosion of Steel in Concrete. CORROSION 44, 489–499 (1988).
Soylev, T. A. & François, R. Quality of steel–concrete interface and corrosion of reinforcing steel. Cem. Concr. Res. 33, 1407–1415 (2003).
Ann, K. Y. & Buenfeld, N. R. The effect of calcium nitrite on the chloride-induced corrosion of steel in concrete. Mag. Concr. Res. 59, 689–697 (2007).
Rodrigues, R., Gaboreau, S., Gance, J., Ignatiadis, I. & Betelu, S. Reinforced concrete structures: a review of corrosion mechanisms and advances in electrical methods for corrosion monitoring. Constr. Build. Mater. 269, 121240 (2021).
Kenny, A. & Katz, A. Steel-concrete interface influence on chloride threshold for corrosion – Empirical reinforcement to theory. Constr. Build. Mater. 244, 118376 (2020).
Environmental factors affecting the corrosion behavior of reinforcing steel II. Role of some anions in the initiation and inhibition of pitting corrosion of steel in Ca(OH)2 solutions. Corrosion Sci. 11 (2010).
Liu, G., Zhang, Y., Ni, Z. & Huang, R. Corrosion behavior of steel submitted to chloride and sulphate ions in simulated concrete pore solution. Constr. Build. Mater. 115, 1–5 (2016).
Archie, G. E. The electrical resistivity log as an aid in determining some reservoir characteristics. Trans. AIME 146, 54–62 (1942).
Anstice, D. J., Page, C. L. & Page, M. M. The pore solution phase of carbonated cement pastes. Cem. Concr. Res. 35, 377–383 (2005).
Tian, Z. & Ye, H. Electrical resistivity of partially-saturated alkali-activated slag containing sodium nitrite admixture. Cem. Concr. Compos. 120, 104053 (2021).
Tian, Z. & Ye, H. Mechanisms underlying the relationship between electrical resistivity and corrosion rate of steel in mortars. Cement Concrete Res. 159, 06867 (2022).
Tian, Z., Chen, Z. & Ye, H. Relationship of electrical resistivity and corrosion rate in alkali-activated mortars with varying alkalinity and nitrite inhibitor. Constr. Build. Mater. 409, 134211 (2023).
Acknowledgements
The work was funded by the Hong Kong Research Grants Council (Project number: 27204818). Any opinions, findings and conclusions, or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the sponsors. The publication was made possible in part by support from the HKU Libraries Open Access Author Fund sponsored by the HKU Libraries.
Author information
Authors and Affiliations
Contributions
Zushi Tian and Hailong Ye conceived and planned the experiments. Zushi Tian carried out the experiments. Zushi Tian and Hailong Ye contributed to the interpretation of the results. Zushi Tian, Chuanqing Fu, and Hailong Ye took the lead in writing the manuscript. All authors provided critical feedback and helped shape the research, analysis, and manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tian, Z., Fu, C. & Ye, H. Mechanisms and kinetic model for steel corrosion in unsaturated cementitious materials. npj Mater Degrad 8, 20 (2024). https://doi.org/10.1038/s41529-024-00441-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-024-00441-1
