
Abstract
The French concept developed to dispose high-level radioactive waste in geological repository relies on glassy waste forms, isolated from the claystone host rock by steel containers. Understanding interactions between glass and surrounding materials is key for assessing the performance of a such system. Here, isotopically tagged SON68 glass, steel and claystone were studied through an integrated mockup conducted at 50 °C for 2.5 years. Post-mortem analyses were performed from nanometric to millimetric scales using TEM, STXM, ToF-SIMS and SEM techniques. The glass alteration layer consisted of a crystallized Fe-rich smectite mineral, close to nontronite, supporting a dissolution/reprecipitation controlling mechanism for glass alteration. The mean glass dissolution rate ranged between 1.6 × 10−2 g m−2 d−1 to 3.0 × 10−2 g m−2 d−1, a value only 3–5 times lower than the initial dissolution rate. Thermodynamic calculations highlighted a competition between nontronite and protective gel, explaining why in the present conditions the formation of a protective layer is prevented.
Similar content being viewed by others

Introduction
In France, like in the other countries using nuclear power plants, plans for disposing high-level radioactive waste (HLW) in a deep geological repository are being developed. The selected rock is the Callovo–Oxfordian claystone (Cox) located in the North-East of the Parisian basin and the concept under consideration is based on the vitrified HLW poured in a stainless-steel canister and placed in a carbon steel overpack. The resulting waste package should be placed in horizontal tunnels dug into the low permeability claystone. However it is expected that groundwater will corrode the metallic envelopes and eventually reach the glass. This system essentially aims at delaying water arrival during the first period of the disposal1. From the safety standpoint the performance of each component must be evaluated, as well as that of the global system. Owing to the amount of radioactive elements that would be disposed of (roughly 20000 metric tons of glass with ∼18 wt% waste loading made of fission products and minor actinides), such a disposal must be safe for a period of ∼1 My. Performance assessment primarily relies on the calculation of the waste form durability and the migration of radionuclides in the geosphere. The release of radionuclides from the waste form depends on physicochemical parameters (chemical interactions, diffusion transport) and interactions between materials (glass/steel/clay) in contact with water2,3. The expected scenario would then be resaturation of clay, corrosion of the canisters, glass leaching and the release of radioelements4. Their dispersion/migration in the geosphere depends on many parameters such as clay sorption capacity, formation of secondary phases, microbial activity…5
Nuclear glass alteration has been studied for decades. SON68 glass, the inactive surrogate of R7T7 glass produced at La Hague is certainly one of the most studied borosilicate glass to date6. Several dissolution mechanisms were observed when glass is placed in contact with water7. The first step concerns coupled mechanisms, i.e., glass hydration, where water molecules penetrate the borosilicate network8, and interdiffusion which exchanges protons contained in solution with alkali elements (i.e., Na, Li, Cs) in the glass. In parallel, hydrolysis of covalent bonds forming the glass network linkages (mostly Si-O-X (X = Si, Al, Zr, B) induces depolymerization of the silicate network. This reaction takes place at the hydrated layer/solution interface and releases of orthosilicic acid (H4SiO4) into solution. Sparingly soluble elements can either condense or precipitate at the glass surface, leading to the formation of a glass alteration layer (GAL), generally composed of Si, Al, Zr, Ca, and highly depleted in Na and B. Hydrolysis is characterized by the highest alteration rate, named initial dissolution rate (r0), which can decrease by several orders of magnitude when an amorphous protective gel is formed at the glass/solution interface. The gel is formed either by in situ condensation of hydrolyzed Si within the gel9, or by precipitation of aqueous species, depending on the glass composition and the alteration conditions10,11,12,13,14,15. The gel self-reorganizes and densifies16. Glass alteration is generally considered as isovolumetric, i.e., the gel replaces the glass maintaining the same volume of solids17,18. When the gel porosity is partly clogged at the gel/solution interface19,20, it acts as a diffusion barrier that limits exchanges and slows down the global glass dissolution rate. Gels formed in circum-neutral pH conditions are stable in solution containing 50–100 ppm of dissolved Si21,22,23,24,25,26,27, meaning that Si from the gel does not exchange with that from the solution in such conditions. In addition to the gel, secondary phases can precipitate (phyllosilicate, calcium-silicate-hydrates, zeolites) and impact both the gel properties and the solution chemistry20,28,29. In this case, the dissolved constituents of the glass, especially the Si, undergo both in situ condensation and precipitation30.
Whatever the mechanism at play, i.e., hydrolysis/condensation or dissolution/reprecipitation, the fate of Si during glass alteration is crucial as it controls the glass behavior31. A few studies have focused on glass alteration mechanisms following Si migration in isotopic 29Si leaching experiments and secondary ion mass spectrometry (SIMS) 29Si/28Si ratio detection30,32,33. They observed that the gel had a 29Si/28Si ratio intermediate between glass and solution, and an outer phyllosilicate-rich layer with a 29Si/28Si ratio close to the one detected in solution, clear evidence that the hydrolysis/condensation or dissolution/reprecipitation can take place simultaneously. A recent study demonstrated that the two mechanisms can control sequentially glass corrosion10.
The above-mentioned studies were conducted in the absence of iron. Iron ions as a result of steel corrosion can precipitate with Si to form Fe-Si-O phases. These phases are assimilated to phyllosilicates and could impact the glass alteration mechanisms and kinetics26,34,35,36,37,38,39. As an example, Neill et al.26 altered ISG glass at 90 °C and pH 7 in a 29Si saturated solution and after 70 days—a duration allowing the formation of a dense and protective gel layer—magnetite (Fe3O4) was added leading to a drop in the 29Si concentration, followed by a subsequent resumption of alteration. Although global trends were observed, current models such as GM2001 or GRAAL2,40,41, cannot predict quantitatively the glass behavior in such complex environments. For that reason, a better experimental understanding of the interactions between glass, steel and clay is necessary.
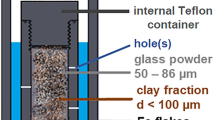
Here, we studied the influence of iron and claystone on glass alteration mechanisms and kinetics. We focused on a SON68 nuclear glass/steel/claystone system, as presented in Fig. 1. A piece of SON68 glass doped with 57Fe and 29Si, with one face in near-contact with a piece of iron was altered for 2.5 years at 50 °C in a core of water saturated Cox claystone (Fig. 1). Post-mortem characterization techniques were used to identify the alteration products and assess the fate of Si. We demonstrate that Fe from the iron piece reacted with Si from the glass to form nontronite, leading to a high glass dissolution rate and a low dispersion of Si released by the glass.

Arrangement of the two SON68 glass coupons pressed against a piece of iron in the Cox claystone cylinder (a), and cross section obtained on the system (b and c).
Results
Glass/steel interface
A large area view of the glass/iron/claystone system using Scanning Electron Microscopy (SEM), focusing on the reaction products at the doped glass/metallic iron interface, with the metallic iron on the top and the doped pristine glass (PG) on the bottom is shown in Fig. 2a. On the right-hand side (where the iron corrosion products (ICP) are thickest), a layer is present in the embedding resin (in black) inserted between the two materials, which is part of the GAL separated from the PG. The GAL of the left-hand side is close to the PG. Note that the right-sided GAL separated during sample preparation (freeze-drying, cutting and/or polishing): the similarity between the GAL surface appearance of the right side with PG clearly supports this hypothesis (see the SEM micrograph with the appropriate scale, Supplementary Fig. S1a).

a SEM micrograph of the doped nuclear glass/iron interface. The rectangles indicate the areas studied in detail (i.e., GAL and ICP). b SEM micrograph and EDS elemental mapping of the doped glass surface. White squares indicate areas of EDS analyses (Table 1).
Glass alteration layer characterization
Along the pristine glass specimen, SEM coupled with Energy Dispersive Spectroscopy (SEM-EDS) analyses show an inner and outer glass alteration layer, hereafter referred to as i-GAL and o-GAL, respectively (Fig. 2b). Both the i-GAL and o-GAL thicknesses are regular and measure 1 µm (σ = 0.2 µm, ten measurements) and 5 µm (σ = 1 µm, ten measurements), respectively (Fig. 2b). The main elements detected in the GALs and PG by SEM-EDS are shown in Table 1 and mapped in Fig. 2b. The GALs contain mainly Fe, Si, O, Al, and Zr. No Na was detected in the GALs (Na < detection limit of the EDS), and heavy elements such as Mo, Cs, Ba and lanthanides (La, Ce, Pr, Nd) are not detected. Conversely, PG contains 4.5 wt.% of Na and 10.5 wt% of the heavy elements. The Si/Zr ratio decreases by a factor of 2 in the GALs (4–5) compared to the PG (∼10), mainly attributed to the Si content of the PG being twice that of the GALs, as the Zr content is similar since it is not soluble in water2,9,42. In contrast, GALs are enriched in Fe, as the Fe/Si ratios are equal to 0.1 and 2 in PG and GALs, respectively. Note that the incorporation of Fe in GALs has been already reported in previous studies35,36,38,39,43. Furthermore, the o-GAL sometimes corresponds to a stack of micrometric layers (Supplementary Fig. S1b), with a total thickness of 10 µm. Each layer has the same chemical composition as the one described hereafter.
Transmission Electron Microscopy (TEM) with Energy Dispersive Spectroscopy (TEM-EDS) analyses (Fig. 3a, b and c) show that the i-GAL consists of a stack of thin alteration layers of 0.2 µm (total 1–1.5 µm thickness), which are composed of foliated aggregates (Fig. 3c) compatible with the poorly-ordered smectite morphology44. Also pores are evidenced in the alteration layer, of the order of 10–50 nm in diameter, much larger than that observed in dense passivating GALs10,45. Note that the PG/i-GAL interface is precisely located by TEM-EDS due to the change in the Si/Zr ratio and Fe content (Fig. 3d). TEM analyses performed on the o-GAL shows the presence of crystallized sheets (Fig. 4), mainly composed of Si, Fe, O, Al (Table 2). High-resolution TEM (HRTEM) revealed characteristic morphology of well-ordered clay sheets, with reticular distances of 10.4 Å (σ = 0.6, 15 measurements) between the sheets (Fig. 4c), corresponding to the d001 distance. This value is compatible with partially dehydrated smectites46,47,48, whose reticular distance could be modified through the freeze-drying preparation process.

a TEM micrographs of the i-GAL evidencing several strata and location of the profile composition (red line) in the first stratum (b). In c, HRTEM in the first stratum of the i-GAL showing foliated aggregates compatible with the smectite morphology. In d, TEM-EDS profile composition (wt.%) obtained in the first stratum of the i-GAL highlighting the PG/i-GAL interface.

High resolution TEM micrographs (a + b + c) highlighting the presence of crystalline phases with inter-reticular distance of 10.4 Å (σ = 0.6) compatible with smectite group. TEM-EDS spectrum (d) corresponding to the crystalline phase (composition in Table 2).
Carrière et al.37 demonstrated the possibility to identify nanometric silicates (crystallized or not) contained in glass matrix with Scanning Transmission X-ray Microscopy (STXM) at Si K and Fe L-edges. The same comparative approach is led for both edges using the same database of silicate reference spectra. STXM spectra at the Si K-edge from the PG, i-GAL and o-GAL are shown in Fig. 5a. For the PG, the spectrum is characteristic of amorphous SiO2 (SiO2(am))37. Spectra obtained from the i-GAL and o-GAL differ from that of the PG, also having peaks B and C at around 1850 and 1855 eV, respectively. A qualitative comparison with the Si K-edge database supports the presence minerals from the smectite group (peaks in A, C and D positions), and excludes the presence of minerals from chlorites, mica, and serpentines-kaolinite groups for the GALs. Linear combination fitting of the GALs spectra using the Si K-edge database suggests that the i-GAL is around 75% nontronite and 25% SiO2(am) (expressed in equivalent thickness) and that the o-GAL is about 90% nontronite and 10% SiO2(am) (Fig. 5b, Table 3). The higher content of SiO2(am) for the i-GAL than for the o-GAL is consistent with a less-ordered nontronite.

a Si K-edge spectra obtained in pristine glass, i-GAL and o-GAL and superposition of i-GAL and o-GAL spectra with corresponding fitted spectra (b). Fe L-edge spectra obtained in pristine glass, i-GAL and o-GAL (c).
Fe L-edge spectra are used to assess the Fe valence state in the PG and GALs, and are presented in Fig. 5c. Linear combination fitting of the PG and GALs Fe spectra, using reference spectra from pure Fe(II) (siderite) and Fe(III) (maghemite) compounds49,50,51,52, are shown in Table 4. The Fe valence in PG is 100% Fe(III). On the contrary, mixed valence is observed in the GALs, 50% Fe(II) and 50% Fe(III) for i-GAL and 10% Fe(II) and 90 % Fe(III) for o-GAL. This Fe valence ratio corresponds to a well-ordered nontronite for the o-GAL37, but for i-GAL the Fe valence ratio highlights a nontronite in formation, poorly ordered, as evoked by the calculated amorphous part (25%).
The SON68 glass was initially doped with 29Si and 57Fe - two isotopes at low concentration at the natural abundance (4.68% and 2.2%, respectively)—to monitor the distribution of these elements in the GAL, and thus better understand how this material forms. Note the i-GAL was divided into two parts (internal, adjacent to PG, and external) to extract the Time-of-Flight Secondary Ion Mass Spectrometry (ToF-SIMS) data. Table 5 presents the ToF-SIMS Si and Fe isotopic content after extraction of the mass spectra from the PG, i-GAL (internal and external), and o-GAL regions. ToF-SIMS analyses performed in the GAL of the undoped glass (right glass in Fig. 1) does not evidence any 29Si enrichment (results not shown here), i.e., Si isotopic contents correspond to the natural abundance of the primal glass matrix. This strongly suggests that the two compartments of the experimental cell can be considered as isolated without interactions. The undoped compartment is not considered further in the rest of the paper. Note also that the 30Si+ and 54Fe+ contents correspond to the natural abundance and are not discussed hereafter53.
An overlay mapping of 56Fe+ and B+ ions is represented to localize at the submicrometric scale the PG/i-GAL interface (Fig. 6b). Indeed, boron is only present in PG, as it is one of the first element to solubilize while glass alters9,40. On the contrary, 56Fe+ is quasi-exclusively present in GAL (Fig. 6a, Table 5) and comes from another source (corrosion of iron, see ICP characterization part). PG contains 52% and 45% of 29Si+ and 28Si+, respectively, similar to that of the internal i-GAL. There is a slight enrichment in the 28Si+ content in the external i-GAL (54%) compared to the internal i-GAL (46%). The Cox water initially contains silicon in the following proportions: 28Si (92.2%), and 29Si (4.7%) in accordance with the natural abundance53. This suggests that some of the Si in the external i-GAL originates from the Cox water. The 29Si+ and 28Si+ isotope content measured in the o-GAL is 20% and 78%, respectively, considerably different from that of the PG and i-GALs. Thus, a good portion of the Si content in the o-GAL originates from the Cox water. The origin of the Si in the external i-GAL and in the o-GAL could be estimated precisely assuming the following equations Eqs. (1) and (2):
where \({\,}^{29}{\mathrm{Si}}_{{\mathrm{PG}}}\) and \({\,}^{29}{\mathrm{Si}}_{{\mathrm{sol}}}\) correspond to the isotope content of 29Si originating from the PG and Cox solution respectively, and the percentage of \({\,}^{29}{\mathrm{Si}}_{{\mathrm{PG}}}\) and \({\,}^{29}{\mathrm{Si}}_{{\mathrm{sol}}}\) are noted \(x_{PG}\) and \(y_{sol}\), respectively. \({\,}^{29}{\mathrm{Si}}_{{\mathrm{GAL}}}\), \({\,}^{28}{\mathrm{Si}}_{{\mathrm{GAL}}},{\,}^{29}{\mathrm{Si}}_{{\mathrm{PG}}}\) and \({\,}^{28}{\mathrm{Si}}_{{\mathrm{PG}}}\) correspond to the isotopic contents estimated by ToF-SIMS and \({\,}^{29}{\mathrm{Si}}_{{\mathrm{sol}}}\) and \({\,}^{28}{\mathrm{Si}}_{{\mathrm{sol}}}\) are related to the natural abundance. Solving this system yields that about 85% of Si contained in the external i-GAL comes from the glass, and 15% from the solution. Regarding o-GAL, about one third of Si (32%) comes from the glass, and two thirds from solution (68%).

ToF-SIMS image of 56Fe+ (a) and overlay of 56Fe+ and B+ maps (b).
Most of the Fe in the PG corresponds to the 57Fe (96%). On the contrary, the internal i-GAL contains 49% of 56Fe, coming from corrosion of the iron metal (discussed thereafter). The external i-GAL is further enriched with 56Fe+ (76%) and the o-GAL is almost entirely 56Fe (91%), close to the Fe natural abundance53. Thus, Fe migrated from the iron metal toward the glass surface.
Iron corrosion product characterization
The ICP on the left-hand side of the metallic part (Fig. 2a) were investigated with SEM-EDS and Raman spectroscopy (Fig. 7a, b, and c). On this side, the ICP layer is about 5 µm thick (σ = 1 µm, 20 measurements), with a maximum value of 10 µm. Only O, Fe, and Ca are detected in the ICP (Fig. 7b and Table 1). µRaman spectra (Fig. 7c) obtained at this location show two typical vibration bands at 281 and 1082 cm−1, attributed to siderite (FeCO3)54,55. Previous research of iron corrosion in carbonated and anoxic environments showed that ICP are formed by precipitation of aqueous carbonates with Fe(II), and are associated with generalized corrosion35,37,39,43,52,56,57,58,59,60. ICP on the right-hand side of the metallic zone are thicker, with an average thickness of about 100 µm (σ = 10 µm, 15 measurements), reaching a thickness of 140 µm in some areas. Two types of corrosion products are detected at this location. The first one seems crystalized with a needle shape (Fig. 7d and e) and contains mainly O and Fe (63%) (Table 1), with a small amount of Si (4%), while the second ICP contains more Si (18%) and less Fe (47%) and few quantities of Na and Ca (1%) (Table 1). Raman Spectroscopy identifies an iron carbonate, chukanovite Fe2(OH)2CO3 (Supplementary Fig. S2)61, corresponding to the needles. However it was not possible to identify the Si-containing ICP with this technique due to high fluorescence.

a SEM micrograph, b SEM-EDS elemental mapping and c µRaman analyses of ICP on the left side, without Si. d SEM micrograph and e TEM micrograph of ICP on the right side, i.e., containing Si or needle-shaped ICP. f STXM spectra at Si K-edge on ICP with silicon (blue) and fitted spectrum (brown). g quantitative Fe redox nanomapping obtained with STXM at Fe L-edge from micrograph in e.
ToF-SIMS was performed on the second ICP containing high silica to determine the origin of the Si (Table 5). The Si isotope contents are very similar to that observed for o-GAL, i.e., 19% and 78% for 29Si+, and 28Si+ respectively, which suggests that one third of the Si contained in ICP comes from the glass, and two thirds from the solution, based on GAL analysis, Eqs. 1 and 2. These ICP contain amounts of 57Fe+ and 56Fe+ ions similar to the iron metal, (meaning natural abundance of Fe)53 and not from the 57Fe doped PG. Therefore, negligible Fe in the ICP came from the glass alteration.
STXM analysis at the Si K-edge was carried out on the ICP to identify the Si-containing phases. Only one Si species was apparent in the ICP with high Si (between the needles—in blue, Fig. 7f). Based on the spectrum shape, it is characteristic of smectites37. Linear combination fitting of the Si K-edge spectrum (Fig. 7f) using reference spectra from our library indicates that the Si species is nontronite (97%), with a small amount of SiO2 (3%) (Fig. 7, Table 3). Note that the presence of SiO2 could be caused by the amorphization of each faces of the FIB foil during sample preparation62,63. Contrary to the nontronite identified in the o-GAL, this one is poorly crystallized (diffused rings on the selected area electron diffraction pattern in Supplementary Fig. S3). STXM at Fe L-edge is sensitive to the iron valence49,50,64. Fig. 7g displays an Fe(II)/∑Fe nanomapping obtained with STXM at Fe L-edge, according to the procedure described by Bourdelle et al.49. The needles displayed in red have an Fe(II)/∑Fe close to 80%, while Si containing phases, identified as nontronite, features an Fe(II)/∑Fe close to 20%. These values correspond to those expected for chukanovite and nontronite respectively, with an obvious bias of Fe valence as these two phases may overlap.
Discussion
Our study shows that glass alteration at 50 °C, in close proximity to iron metal, in an anoxic clayey environment forms Na-free, Si-depleted and Fe-rich alteration layer. This GAL consists of a stack of many sublayers parallel to the glass surface, with a total thickness ranging from 6 to 10 µm. Authors usually interpret the depletion of Na by ion-exchange between protons dissociated from water molecules with alkalies acting as glass modifiers (Na, Li…)8,34,65. Moreover, a decrease in the Si/Zr ratio by a factor of two from the PG attests to a significant release of Si from the glass, suggesting that hydrolysis of the silicate network drives the alteration of the glass2,66. In general, ion-exchange, hydrolysis and condensation reactions lead to the formation of an amorphous hydrated layer on the glass surface, called gel, whose transport-limiting capability depends on the density of the material16,67,68,69. Highly protective gels are characterized by a high retention factor of Si and the presence of micropores poorly connected together45,70. However, in the present study, ordered precipitates of an Fe(III)-rich phyllosilicate, nontronite, belonging to the smectite group of clay minerals, forms at the glass surface, with pore sizes ranging from 10 to 50 nm in the layer closest to the glass (i.e., i-GAL) (Fig. 3). The nontronite constituting the porous i-GAL takes up a majority of the silicon released by the glass (H4SiO4), while the long-ordered nontronite (o-GAL) uses mainly the silicon from the solution (i.e., Cox water). Even though the solution composition was not analyzed at the end of the experiment (i.e., 2.5 yr), the 29Si/28Si ratio in the ordered nontronite is presumably close to the solution in equilibrium with claystone containing also hydrolyzed silicon. Hence the observations suggest that: i) the GAL is not or poorly protective, and ii) a dissolution/reprecipitation mechanism controls glass alteration under the study conditions.
In the present study, the alteration conditions (1 bar, 50 °C, neutral pH in an anoxic and carbonated medium) correspond to the geochemical environment favorable for nontronite precipitation46,71,72,73,74,75,76,77,78. However temperature has a strong influence on iron silicate formation. Indeed, iron/claystone or glass/iron/claystone experiments performed at 90 °C showed that serpentine formation occurs, whereas a temperature close to 50 °C tends to destabilize serpentine35,48,56,57,79,80,81,82,83,84. This could explain the absence of serpentine in our study. Moreover, saponite (belonging to the smectite group of clay minerals) is not observed here since it contains mainly Fe(II) and preferentially precipitates at higher pH than nontronite76,85,86,87.
Despite initial reducing conditions, nontronite contains mostly Fe(III), as already shown in this iron phyllosilicate88,89. The literature suggests that iron oxidation from Fe(II) to Fe(III) in reducing conditions could be explained by water reduction71,90,91, but could also result from a charge transfer or charge compensation within the silicate structure77,92. Another explanation would be to consider the glass as an oxidizer. Fe2+ coming from iron corrosion, and adsorbed at the glass surface, could be oxidized in Fe3+ by Fe2O3 (i.e., an Fe(III) oxide) of the glass, while the Fe(III) of glass would be reduced. A redox front could form between pristine glass, nontronite and solution, with two Fe(III)-rich ends, i.e., pristine glass and nontronite in contact with the solution, and a Fe2+/Fe3+ gradient between glass and nontronite. This hypothesis requires further investigations.
As a reminder, the GAL consists of a succession of several nontronite strata (Fig. 3 and Supplementary Fig. S1). These observations were already modeled93, suggesting a mechanism by cyclic locally pH modification. Indeed, glass dissolution rises the pH by releasing sodium or boron into solution94,95, while smectite precipitation tends to decrease the pH, by consuming hydroxyl groups contained in solution75, and releasing protons96,97,98. Moreover, redox variations cannot be excluded to justify these strata.
To estimate glass alteration rates, quantification of boron in leaching solution is commonly used9. Unfortunately, the solution could not be periodically sampled as the reactor remained a closed system for the whole duration of the experiment. Previous studies have shown that the GAL thickness could be used to estimate glass alteration rates40. This method assumes the glass alteration to be isovolumetric. In aggressive conditions such as used in this study (i.e., glass in contact with iron) some researchers consider that the glass alteration is isovolumetric35,99, while others describe it as non-isovolumetric due to the incorporation of Fe in the GAL and to glass density modification18,100. Consequently, the alteration rate calculated from the GAL thickness could be over or underestimated. Nevertheless, it was established that the difference between these two calculation methods was a factor of two, when silicon retention in a gel, named f(Si), is low (<15%)9. Another method, based on the f(Si) in the GAL, is proposed in this paper for a complementary estimation of the mean alteration rate.
In this study, the average rate of glass dissolution is estimated at 1.8 × 10−2 g m−2 d−1 to 3.0 × 10−2 g m−2 d−1, for a GAL thickness of 6–10 µm, respectively, assuming a constant dissolution rate and an isovolumetric process. This is five to three times lower than the initial dissolution rate of SON68 glass measured in previous experiments in synthetic Cox poral fluid at 50 °C, referred to as \(r_{0\,Cox}^{50\,^\circ C}\) = 9.3 × 10−2 g m–2 d−1. However, it is several orders of magnitude higher than the residual rate (rr) estimated at around 6 × 10−5 g m–2 d−1 at 50 °C101,102.
A second estimation of the alteration rate can be made from the retention factor of Si, f(Si), in the GAL. As Zr is supposed to remain undissolved according to previous studies2,9,42, silicon retention obtained with Eq. 3 enables the estimation of the alteration rate103.
Two empirical curves (Fig. 8) showing glass alteration rate in pure water at 50 °C vs f(Si) (Fig. 8a) or vs [H4SiO4] in solution (Fig. 8b) were drawn from literature data (Supplementary Table S1)104,105. To our knowledge, these values do not exist in literature for glass alteration in Cox water. Therefore this method is used to approximate the alteration rate and complements the first method. Looking in detail at the curve, from f(Si) = 0–0.6, the glass dissolution rate is divided by 10. From 0.6 to maximal Si retention at 0.8, the rate reaches r0/1000, due to the formation of a protective gel at the glass surface. The Si retention factor calculated for the GAL, i.e., 0.5 in the present study, suggests that hydrolyzed Si is partially retained in the altered layer. The glass alteration rate associated with these value is 1.6 × 10−2 g m−2 d−1, and is in fair agreement with the rate obtained from GAL thickness (i.e., \(r_{0\,Cox}^{50\,^\circ C}/5\)).

According to the f(Si) = 0.5 in GAL and the [H4SiO4] = 0.062 mol l−1 in solution, the glass alteration rates associated with these values are respectively 1.6 × 10−2 g m−2 j−1 (a) and 2.3 × 10−2 g m−2 j−1 (b).
Alteration rate calculated from this second method yields a mean concentration of H4SiO4 in the leachate of 6.2 × 10-5 mol l−1 (Fig. 8b). This concentration is far from saturation with a protective gel (about 10−3 mol l−1)21,22,23,24,25,26. The glass alteration rate can be determined using Eq. (4), where \(k^ + = 1.2.10^8{\mathrm{g}}.{\mathrm{m}}^{ - 2}.{\mathrm{d}}^{ - 1}\) is the kinetic constant in pure water, n = −0.4 the coefficient of the pH-dependence of the reaction rate, Ea = 77 kJ mol−1 the activation energy of the glass dissolution reaction, aH4SiO4 the H4SiO4 activity in solution, and K the solubility constant of the protective gel (corresponding to amorphous silica)2,66. Hence a concentration of 6.2 × 10−5 mol l−1of H4SiO4 in solution lead to a dissolution rate estimated at 2.3 × 10- 2 g m−2 d−1, i.e., \(r_{0\,Cox}^{50\,^\circ C}/4\), with the Eq. (4).
The two methods used to estimate the glass alteration rate give consistent results. Glass alters at a high rate, releasing large amount of Si, which then co-precipitates with Fe. It can then be proposed that the presence of Fe in the vicinity of glass favors a mechanism of dissolution/reprecipitation instead of the formation of a passivating layer by in-situ reorganization of the silicate network.
The affinity between Si and Fe was evidenced with concentration diagram calculated with the physicochemical alteration conditions, i.e., temperature of 50 °C, atmospheric pressure, redox conditions of −0.14 V vs.SHE, Cox water composition (Supplementary Table S2). Phases observed experimentally in this study (mainly nontronite, and small amounts of amorphous silica SiO2(am)) were included in the model (see “Methods”). Figure 9 illustrates the predominance domains of precipitate phases (in yellow) of nontronite(s) and SiO2(am). The activities of the H4SiO4 and the ratio Fe2+/Fe3+, are the two variables that control the formation and stability of the main phases observed in the system. The Fe2+/Fe3+ ratio was calculated using Nernst equation. Al is required to form nontronite. Although its concentration in solution is not known, it was fixed at 4.6 × 10−6 mol l−1 in the simulation. This concentration corresponds to the amount of Al in solution assuming that Al and Si are dissolved congruently from the glass. It has been verified that the activity diagrams are not modified for an order of magnitude smaller and higher than our Al concentration (not shown).

Eh = −0.14 V vs. SHE and pH = 6.86 at atmospheric pressure. In blue dissolved species, and in yellow stability domains of nontronite and amorphous silica (SiO2(am)). Blue lines present initial concentration of Fe and H4SiO4 in solution. Red line shows the concentration of H4SiO4 estimated in solution from Fig. 8b.
At pH = 6.9, the blue and red lines in Fig. 9 indicate the initial H4SiO4 concentration in Cox solution in equilibrium with claystone, and the estimated concentration of 6.2 × 10−5 mol l−1 deduced from Fig. 8, respectively. It suggests that these concentrations are high enough to form nontronite, but do not allow SiO2(am) to form. Because the protective gel has a solubility product close to that of SiO2(am), this result strongly suggests that, in the studied conditions, the presence of iron in solution delays or prevents the formation of a protective gel, reinforcing the idea that glass alters following a dissolution/reprecipitation process. However, it must be kept in mind that a small fraction of SiO2(am) was also evidenced in the GAL, in addition to nontronite. This amorphous part was attributed to nontronite in formation, but it could also result from a local iron depletion at the vicinity of glass. Indeed, local iron concentration can decrease as nontronite precipitates, leading to an increase of [H4SiO4] due to glass alteration. Presumably saturation condition can be reached locally to form SiO2(am), i.e., a gel, until iron arrival to the glass and resumption of nontronite precipitation.
Owing to the high affinity between Si and Fe, and the low solubility limit and high precipitation rate of nontronite in our conditions, such porous phase forms at the expense of a protective gel. However it cannot be excluded that gradients of chemical species could appear near the surface of the glass, allowing some gel to form.
In the present study, the glass was altered by the solution in contact with steel and claystone. Owing to the high Si/Fe affinity, both Si released by the glass and that present in the pore water, precipitate with the aqueous Fe(II) species coming from steel corrosion to form Fe(III)-rich smectite layer (assimilated to nontronite), as illustrated in Fig. 10. Near the glass surface, a thin poorly-ordered and non-protective nontronite layer starts to precipitate mainly from species coming from glass dissolution. This material was associated to amorphous silica in the STXM spectrum deconvolution, but it does not relate to the protective gel described in other studies conducted in the absence of Fe. In the present work, we demonstrate that Fe prevents or delay the formation of a protective gel. As a consequence of nontronite formation, glass alters following a dissolution/reprecipitation mechanism, likely limited by glass dissolution31.

It remains to be seen whether such a precipitated nontronite layer could serve as a barrier limiting exchanges at the glass/solution interface. A recent study evidences that both glass alteration mechanisms, i.e., dissolution/precipitation and hydrolysis/condensation can lead to a pore closure, reducing water diffusion through the GAL10. Indeed, a precipitated layer can be protective, slowing down water transport. Hence, in their experimental conditions, authors conclude that the two mechanisms are not systematically antagonistic. It seems to depend on the geochemical conditions, and thus of the Fe-silicate group formation and stability in such conditions106,107,108. Furthermore, local and cyclic concentrations evidenced in the present study, and pH/potential modifications are likely to initiate a gel formation, even though Fe in solution behaves as a silicon pump under our conditions.
Methods
Glass/Iron/claystone system
The glass/iron/claystone system reactor consists of a cylindrical core (Φ ∼ 4 cm, L ∼ 4 cm) of Cox claystone, sampled from the Underground Research Laboratory of Bure (URL), cut into two half-cylinders and machined in order to place in the middle two coupons of SON68 glass (composition in Supplementary Table S3), pressed against a 1 mm thick piece of iron (Fig. 1). 400 µm separates the coupon and the piece of iron. One of the glass coupons is initially doped with 29Si (55 wt%) and 57Fe (100 wt%) to follow the migration of these two elements, while the other is not doped. Dimensions of doped glass and nondoped glass are respectively 10 mm × 10 mm × 2 mm and 20 mm × 10 mm × 5 mm (height × width × thickness). The iron (low alloy steel mainly ferritic) piece is prepared from a 450 yr old nail recovered from corrosion products (the Glinet archeological site, Normandy), such that pristine iron is on one face and on the opposite face is Glinet iron corrosion products. The pristine iron face is placed adjacent to the doped and undoped PG, butt-ended to each other. The undoped glass is used for reproducibility, and same analyses are performed on both glass coupons. The aim of Glinet corrosion products in contact with claystone is to limit the reactivity and flux of iron at the iron/claystone interface since old corrosion products are stable and less reactive than a “fresh” surface of iron39,58. Once assembled the reactor is placed in a membrane and resaturated with the deoxygenated synthetic Cox water (Supplementary Table S2) under 26 bar of pressure to rehydrate the medium and eliminate the gaseous oxygen present in the reactor38. The reactor is placed vertically and water circulation is from bottom to top. The system is closed once it is re-saturated, then the water circulation is stopped. The system is aged in an oven at 50 °C for 2.5 yr, after which the system is frozen and lyophilized to eliminate water, embedded in a bi-component epoxy resin (Epothin Epoxy BuehlerTM) and then hot mounted in a tri-component Flucka resin at 40 °C under vacuum. This fluidized second resin enabled impregnation to the core of the system. The embedded system is then cut and polished down to 1 µm roughness under a N2 atmosphere in a glove box to prevent oxidation. Cross section obtained at the doped glass/iron interface is presented in Fig. 2a.
Analytical techniques
Cross section, and especially the doped SON68 glass/metal iron interface is observed and characterized at different scales (macro-micro-nano) using different analytical techniques.
Scanning electron microscopy coupled with dispersive energy spectroscopy (SEM-EDS) was performed on a Jeol JSM-7001 F Field Effect at 15 kV and a beam time current of 12 nA. A carbon-coating layer of 15 nm was applied on the sample surface before analyses.
MicroRaman spectroscopy was carried out on an Invia Renishaw microspectrometer equipped with a doubled Nd-YAG laser at an excitation wavelength of 532 nm, and equipped with a CCD detector. The spatial resolution was about 1 µm3 with the 50× lens. The laser power was filtered down to 0.5 mW and spectra were recorded for 30 sec with a resolution of 2 cm−1. Spectra obtained in ICP are acquired and processed with the Wire 3.4 software.
Time-of-flight secondary ion mass spectrometry (ToF-SIMS), led on a TOF.SIMS 5 spectrometer (IONTOF, Munster, Germany) provides information on the migration of Si and Fe during glass and iron alteration (reminder: one SON68 coupon was initially doped with 29Si and 57Fe). Pulsed Bi+ are used as primary ions (25 kV) in burst mode, i.e., ion beam is emitted with a series of short pulses to avoid detector saturation109,110, enabling good mass resolution (up to 7000) and a good lateral resolution (up to 200 nm). This technique as also a high sensitivity with a detection threshold in the range of ppb. Positive ions ToF-SIMS images (Supplementary Fig. S4) are recorded in the regions of interest. Then isotopic contents are calculated from the mass spectra reconstructed from the selected areas.
Cross sections of 100 nm and 1 µm thick are cut in the regions of interest in the glass/iron/claystone system (i.e., mainly GAL and ICP) using a FEI Helios Nanolab 660 scanning electron microscope/focused ion beam (SEM-FIB). The acceleration voltage is from 0.5 to 30 kV and ion current from 1 pA to 65 nA. A “cleaning” at 5, 2, and 1 kV of the two faces of the FIB foils punctuates the preparation to eliminate the layer of damage and contamination.
To identify iron-silicate formed on GAL and ICP at nanometer scale, X-ray microscopy (STXM) imaging and spectroscopy is carried out on the spectromicroscopy (SM) beamline 10ID-1 at the Canadian Light Source (Saskatoon, Canada). Image sequences (stacks) of Near-Edge X-ray Absorption Fine Structure are collected on 100 nm and 1 µm thick cross sections at the Fe L- and Si K-edges, respectively. The measured transmitted signals (I) are converted into absorbance values using the incident flux (I0) measured in the absence of the sample. STXM at Fe L-edge is sensitive to the iron valence49,50,64, and Si K-edge probes atomic environment and nanostructure around the absorber atom at a distance greater than 5 or 6 Å111,112,113,114. The spectra derived from the stacks are decomposed using a least squares method with standard reference spectra, from our Si K- and Fe L-edges database37,115, normalized to an absolute linear absorbance corresponding to a phase thickness of 1 nm (expressed in optical density). Fe(II)/∑Fe nanomapping at Fe L-edge is conducted according to the procedure described by Bourdelle et al.49. Analyses and data processing are achieved with the aXis2000 software.
Transmission electron microscopy on FIB cross sections is performed for iron-silicate identification on the GAL using a Jeol 2010 F electron gun at 200 kV, equipped with a diode EDAX X-ray microanalysis for chemical analyses or electron diffraction. The d001-spacing of silicate sheets are measured from the HRTEM images using ImageJ software. The error is around 0.05 Å due to pixel resolution.
Geochemical modeling
Thermodynamic modeling is performed with the Geochemist’s Workbench 12.0 software and the thermodynamic database “Thermochimie v10a electron” of Andra. The Cox water composition is used as input to plot the activity diagrams, with the Act2 mode. This mode establishes the diagram of the most thermodynamically stable phases, considering different approaches (equilibrium dissolution, speciation, precipitation reaction)116. The phases observed experimentally are included in the model (i.e., nontronite, SiO2(am)), and a small amount of aluminum is also added ([Al] = 4.6 × 10−6 mol.l-1). Precipitated phases and species in solution are indicated in yellow and blue, respectively. The thermodynamic constants from the database and reactions used for the geochemical modeling are summarized in Supplementary Tables S4 and S5. Initial concentration of iron in solution at 50 °C was deducted from Gailhanou et al.117.
Data availability statement
The datasets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Andra. Safety Options Report—Operating Part (DOS-Expl). 521 (2016). https://international.andra.fr/sites/international/files/2019-03/Safety%20Options%20Report%20-%20Operations_2.pdf
Frugier, P. et al. SON68 nuclear glass dissolution kinetics: current state of knowledge and basis of the new GRAAL model. J. Nucl. Mater. 380, 8–21 (2008).
Grambow, B. Nuclear waste glasses—how durable? Elements 2, 357–364 (2006).
Vernaz, É. Y. Estimating the lifetime of R7T7 glass in various media. Comptes. Rendus. Phys. 3, 813–825 (2002).
Gin, S., Jollivet, P., Tribet, M., Peuget, S. & Schuller, S. Radionuclides containment in nuclear glasses: an overview. Radiochim. Acta 105, 927–959 (2017).
Jollivet, P., Gin, S. & Schumacher, S. Forward dissolution rate of silicate glasses of nuclear interest in clay-equilibrated groundwater. Chem. Geol. 330–331, 207–217 (2012).
Advocat, T., Jollivet, P., Crovisier, J. L. & Del Nero, M. Long-term alteration mechanisms in water for SON68 radioactive borosilicate glass. J. Nucl. Mater. 298, 55–62 (2001).
Bunker, B. C., Arnold, G. W., Day, D. E. & Bray, P. J. The effect of molecular structure on borosilicate glass leaching. J. Non Cryst. Solids 87, 226–253 (1986).
Gin, S., Ribet, I. & Couillard, M. Role and properties of the gel formed during nuclear glass alteration: importance of gel formation conditions. J. Nucl. Mater. 298, 1–10 (2001).
Gin, S. et al. A general mechanism for gel layer formation on borosilicate glass under aqueous corrosion. J. Phys. Chem. C. 124, 5123–5144 (2020).
Gin, S. et al. The controversial role of inter-diffusion in glass alteration. Chem. Geol. 440, 115–123 (2016).
Geisler, T., Dohmen, L., Lenting, C. & Fritzsche, M. B. K. Real-time in situ observations of reaction and transport phenomena during silicate glass corrosion by fluid-cell Raman spectroscopy. Nat. Mater. 18, 342–348 (2019).
Stroncik, N. A. & Schmincke, H. Evolution of palagonite: crystallization, chemical changes, and element budget. Geochem. Geophys. Geosyst. 2, 45–88 (2001).
Hellmann, R. et al. Nanometre-scale evidence for interfacial dissolution–reprecipitation control of silicate glass corrosion. Nat. Mater. 14, 307 (2015).
Ducasse, T. et al. Alteration of synthetic basaltic glass in silica saturated conditions: analogy with nuclear glass. Appl. Geochem. 97, 19–31 (2018).
Cailleteau, C. et al. Insight into silicate-glass corrosion mechanisms. Nat. Mater. 7, 978 (2008).
Donzel, N., Gin, S., Augereau, F. & Ramonda, M. Study of gel development during SON68 glass alteration using atomic force microscopy. comparison with two simplified glasses. J. Nucl. Mater. 317, 83–92 (2003).
Reiser, J. T. et al. The use of positrons to survey alteration layers on synthetic nuclear waste glasses. J. Nucl. Mater. 490, 75–84 (2017).
Jollivet, P. et al. Investigation of gel porosity clogging during glass leaching. J. Non Cryst. Solids 354, 4952–4958 (2008).
Gin, S. et al. The fate of silicon during glass corrosion under alkaline conditions: a mechanistic and kinetic study with the international simple glass. Geochim. Cosmochim. Acta 151, 68–85 (2015).
Ferrand, K., Abdelouas, A. & Grambow, B. Water diffusion in the simulated French nuclear waste glass SON 68 contacting silica rich solutions: experimental and modeling. J. Nucl. Mater. 355, 54–67 (2006).
Gin, S. Protective effect of the alteration gel: a key mechanism in the long-term behavior of nuclear waste glass. MRS Online Proc. Libr. Arch. 663, 207–216 (2000).
Gin, S. et al. Atom-probe tomography, TEM and ToF-SIMS study of borosilicate glass alteration rim: a multiscale approach to investigating rate-limiting mechanisms. Geochim. Cosmochim. Acta 202, 57–76 (2017).
Icenhower, J. P. & Steefel, C. I. Experimentally determined dissolution kinetics of SON68 glass at 90 °C over a silica saturation interval: evidence against a linear rate law. J. Nucl. Mater. 439, 137–147 (2013).
Neeway, J., Abdelouas, A., Grambow, B. & Schumacher, S. Dissolution mechanism of the SON68 reference nuclear waste glass: new data in dynamic system in silica saturation conditions. J. Nucl. Mater. 415, 31–37 (2011).
Neill, L. et al. Various effects of magnetite on international simple glass (ISG) dissolution: implications for the long-term durability of nuclear glasses. npj Mater. Degrad. 1, 1 (2017).
Fournier, M. et al. Effect of pH on the stability of passivating gel layers formed on international simple glass. J. Nucl. Mater. 524, 21–38 (2019).
Fournier, M. et al. Glass dissolution rate measurement and calculation revisited. J. Nucl. Mater. 476, 140–154 (2016).
Crovisier, J.-L., Advocat, T. & Dussossoy, J.-L. Nature and role of natural alteration gels formed on the surface of ancient volcanic glasses (Natural analogs of waste containment glasses). J. Nucl. Mater. 321, 91–109 (2003).
Valle, N. et al. Elemental and isotopic (29Si and 18O) tracing of glass alteration mechanisms. Geochim. Cosmochim. Acta 74, 3412–3431 (2010).
Grambow, B. Nuclear waste glass dissolution: mechanism, model and application. (1987).
Bouakkaz, R., Abdelouas, A., El Mendili, Y., Grambow, B. & Gin, S. SON68 glass alteration under Si-rich solutions at low temperature (35–90 °C): kinetics, secondary phases and isotopic exchange studies. RSC Adv. 6, 72616–72633 (2016).
Verney-Carron, A. et al. Understanding the mechanisms of Si–K–Ca glass alteration using silicon isotopes. Geochim. Cosmochim. Acta 203, 404–421 (2017).
Gin, S. et al. Origin and consequences of silicate glass passivation by surface layers. Nat. Commun. 6, 6360 (2015).
De Combarieu, G. et al. Glass–iron–clay interactions in a radioactive waste geological disposal: an integrated laboratory-scale experiment. Appl. Geochem. 26, 65–79 (2011).
Rébiscoul, D. et al. Glass-iron-clay interactions in a radioactive waste geological disposal: a multiscale approach. MRS Proc. 1518, 185–190 (2013).
Carrière, C. et al. Use of nanoprobes to identify iron-silicates in a glass/iron/argillite system in deep geological disposal. Corros. Sci. 158, 108104 (2019).
Burger, E. et al. Impact of iron on nuclear glass alteration in geological repository conditions: a multiscale approach. Appl. Geochem. 31, 159–170 (2013).
Dillmann, P., Gin, S., Neff, D., Gentaz, L. & Rebiscoul, D. Effect of natural and synthetic iron corrosion products on silicate glass alteration processes. Geochim. Cosmochim. Acta 172, 287–305 (2016).
Grambow, B. & Müller, R. First-order dissolution rate law and the role of surface layers in glass performance assessment. J. Nucl. Mater. 298, 112–124 (2001).
Frugier, P., Minet, Y., Rajmohan, N., Godon, N. & Gin, S. Modeling glass corrosion with GRAAL. npj Mater. Degrad. 2, 1–13 (2018).
Brendebach, B., Altmaier, M., Rothe, J., Neck, V. & Denecke, M. A. EXAFS study of aqueous ZrIV and ThIV complexes in alkaline CaCl2 solutions: Ca3 [Zr (OH) 6] 4+ and Ca4 [Th (OH) 8] 4+. Inorg. Chem. 46, 6804–6810 (2007).
Michelin, A. et al. Silicate glass alteration enhanced by iron: origin and long-term implications. Environ. Sci. Technol. 47, 750–756 (2012).
Grauby, O., Petit, S., Decarreau, A. & Baronnet, A. The nontronite-saponite series: an experimental approach. Eur. J Mineral. 6, 99–112 (1994).
Ngo, D. et al. Spectroscopic ellipsometry study of thickness and porosity of the alteration layer formed on international simple glass surface in aqueous corrosion conditions. npj Mater. Degrad. 2, 1–9 (2018).
Changela, H. & Bridges, J. Alteration assemblages in the nakhlites: variation with depth on Mars. Meteorit. Planet. Sci. 45, 1847–1867 (2010).
Moore, D. M. & Hower, J. Ordered interstratification of dehydrated and hydrated Na-smectite. Clays Clay Miner. 34, 379–384 (1986).
Mosser-Ruck, R. et al. Effects of temperature, pH, and iron/clay and liquid/clay ratios on experimental conversion of dioctahedral smectite to berthierine, chlorite, vermiculite, or saponite. Clays Clay Miner. 58, 280–291 (2010).
Bourdelle, F. et al. Quantification of the ferric/ferrous iron ratio in silicates by scanning transmission X-ray microscopy at the Fe L2,3 edges. Contrib. Mineral. Petrol. 166, 423–434 (2013).
Dynes, J. J. et al. Speciation and quantitative mapping of metal species in microbial biofilms using scanning transmission X-ray microscopy. Environ. Sci. Technol. 40, 1556–1565 (2006).
van Aken, P. A., Liebscher, B. & Styrsa, V. J. Quantitative determination of iron oxidation states in minerals using Fe L2,3-edge electron energy-loss near-edge structure spectroscopy. Phys. Chem. Miner. 25, 323–327 (1998).
Michelin, A. et al. Effect of iron metal and siderite on the durability of simulated archeological glassy material. Corros. Sci. 76, 403–414 (2013).
Lide, D. R. CRC handbook of chemistry and physics, internet version 2005. (2005).
Bernard, M. C. et al. Analysis of corrosion products beneath an epoxy-amine varnish film. Prog. Org. Coat. 45, 399–404 (2002).
Saheb, M. et al. Multisecular corrosion behaviour of low carbon steel in anoxic soils: characterisation of corrosion system on archaeological artefacts. Mater. Corros. 60, 99–105 (2009).
Schlegel, M. L. et al. Metal corrosion and argillite transformation at the water-saturated, high-temperature iron–clay interface: a microscopic-scale study. Appl. Geochem. 23, 2619–2633 (2008).
Schlegel, M. L. et al. Corrosion of metal iron in contact with anoxic clay at 90 C: characterization of the corrosion products after two years of interaction. Appl. Geochem. 51, 1–14 (2014).
Leon, Y. et al. Interfacial layers at a nanometre scale on iron corroded in carbonated anoxic environments. RSC Adv. 7, 20101–20115 (2017).
Saheb, M., Berger, P., Raimbault, L., Neff, D. & Dillmann, P. Investigation of iron long-term corrosion mechanisms in anoxic media using deuterium tracing. J. Nucl. Mater. 423, 61–66 (2012).
Azoulay, I., Rémazeilles, C. & Refait, P. Determination of standard Gibbs free energy of formation of chukanovite and Pourbaix diagrams of iron in carbonated media. Corros. Sci. 58, 229–236 (2012).
Saheb, M., Neff, D., Bellot-Gurlet, L. & Dillmann, P. Raman study of a deuterated iron hydroxycarbonate to assess long-term corrosion mechanisms in anoxic soils. J. Raman Spectrosc. 42, 1100–1108 (2011).
Rajsiri, S., Kempshall, B., Schwarz, S. & Giannuzzi, L. FIB damage in silicon: amorphization or redeposition. Microsc. Microanal. 8, 50–51 (2002).
Giannuzzi, L. A., Geurts, R. & Ringnalda, J. 2 keV Ga+ FIB milling for reducing amorphous damage in silicon. Microsc. Microanal. 11, 828 (2005).
Michelin, A. et al. Investigation at the nanometre scale on the corrosion mechanisms of archaeological ferrous artefacts by STXM. J. Anal. Spectrom. 28, 59–66 (2013).
Vernaz, E. Y. & Dussossoy, J. L. Current state of knowledge of nuclear waste glass corrosion mechanisms: the case of R7T7. glass Appl. Geochem. 7, 13–22 (1992).
Gin, S. & Mestre, J. SON 68 nuclear glass alteration kinetics between pH 7 and pH 11.5. J. Nucl. Mater. 295, 83–96 (2001).
Rebiscoul, D. et al. Morphological evolution of alteration layers formed during nuclear glass alteration: new evidence of a gel as a diffusive barrier. J. Nucl. Mater. 326, 9–18 (2004).
Gin, S. et al. An international initiative on long-term behavior of high-level nuclear waste glass. Mater. Today 16, 243–248 (2013).
Vienna, J. D., Ryan, J. V., Gin, S. & Inagaki, Y. Current understanding and remaining challenges in modeling long‐term degradation of borosilicate nuclear waste glasses. Int. J. Appl. Glas. Sci. 4, 283–294 (2013).
Gin, S. et al. Dynamics of self-reorganization explains passivation of silicate glasses. Nat. Commun. 9, 2169 (2018).
Chevrier, V., Poulet, F. & Bibring, J.-P. Early geochemical environment of Mars as determined from thermodynamics of phyllosilicates. Nature 448, 60 (2007).
Müller, G. & Förstner, U. Recent iron ore formation in Lake Malawi, Africa. Miner. Depos. 8, 278–290 (1973).
Cole, T. G. The nature and origin of smectite in the Atlantis II Deep, Red Sea. Can. Mineral. 26, 755–763 (1988).
Bischoff, J. L. A ferroan nontronite from the Red Sea geothermal system. Clays Clay Miner. 20, 217–223 (1972).
Harder, H. Nontronite synthesis at low temperatures. Chem. Geol. 18, 169–180 (1976).
Farmer, V. C., Krishnamurti, G. S. R. & HtJANG, P. M. Synthetic allophane and layer-silicate formation in SiO2-Al2O3-FeO-Fe2O3-MgO-H2O systems at 23 °C and 89 °C in a calcareous environment. Clays Clay Miner. 39, 561–570 (1991).
Merola, R. B., Fournier, E. D. & McGuire, M. M. Spectroscopic investigations of Fe2+ complexation on nontronite clay. Langmuir 23, 1223–1226 (2007).
Petit, S., Baron, F. & Decarreau, A. Synthesis of nontronite and other Fe-rich smectites: a critical review. Clay Miner. 52, 469–483 (2017).
Lantenois, S. et al. Experimental study of smectite interaction with metal Fe at low temperature: 1. Smectite destabilization. Clays Clay Miner. 53, 597–612 (2005).
Perronnet, M. et al. Evidence of a critical content in Fe (0) on FoCa7 bentonite reactivity at 80 °C. Appl. Clay Sci. 38, 187–202 (2008).
Pignatelli, I. et al. A multi-technique characterization of cronstedtite synthesized by iron–clay interaction in a step-by-step cooling procedure. Clays Clay Miner. 61, 277–289 (2013).
Pignatelli, I. et al. Iron–clay interactions: Detailed study of the mineralogical transformation of claystone with emphasis on the formation of iron-rich T–O phyllosilicates in a step-by-step cooling experiment from 90 °C to 40 °C. Chem. Geol. 387, 1–11 (2014).
Wilson, J. C., Benbow, S., Sasamoto, H., Savage, D. & Watson, C. Thermodynamic and fully-coupled reactive transport models of a steel–bentonite interface. Appl. Geochem. 61, 10–28 (2015).
Carrière, C. et al. New insights of Auger spectroscopy for the identification of Fe-Si compounds in iron/glass corrosion systems at nanoscale. J. Electron Spectros. Relat. Phenom. 235, 51–59 (2019).
Aréna, H. et al. Impact of iron and magnesium on glass alteration: characterization of the secondary phases and determination of their solubility constants. Appl. Geochem. 82, 119–133 (2017).
Thien, B. M. J., Godon, N., Ballestero, A., Gin, S. & Ayral, A. The dual effect of Mg on the long-term alteration rate of AVM nuclear waste glasses. J. Nucl. Mater. 427, 297–310 (2012).
Christidis, G. E. M. J. Wilson (2013) rock-forming minerals, Vol. 3c, sheet silicates–clay minerals, 2nd edition. The Geological Society, London, 724 pp. £120. Clay Miner. 49, 765–766 (2014).
Manceau, A., Chateigner, D. & Gates, W. P. Polarized EXAFS, distance-valence least-squares modeling (DVLS), and quantitative texture analysis approaches to the structural refinement of Garfield nontronite. Phys. Chem. Miner. 25, 347–365 (1998).
Manceau, A. et al. Oxidation-reduction mechanism of iron in dioctahedral smectites: I. Crystal chemistry of oxidized reference nontronites. Am. Mineral. 85, 133–152 (2000).
Schikorr, G. Über Eisen (II)-hydroxyd und ein ferromagnetisches Eisen (III)-hydroxyd. Z. f.ür. Anorg. Allg. Chem. 212, 33–39 (1933).
Odziemkowski, M. S., Schuhmacher, T. T., Gillham, R. W. & Reardon, E. J. Mechanism of oxide film formation on iron in simulating groundwater solutions: Raman spectroscopic studies. Corros. Sci. 40, 371–389 (1998).
Eggleton, R. A. Nontronite topotaxial after hedenbergite. Am. Mineral. 60, 1063–1068 (1975).
Wang, Y., Jove-Colon, C. F. & Kuhlman, K. L. Nonlinear dynamics and instability of aqueous dissolution of silicate glasses and minerals. Sci. Rep. 6, 30256 (2016).
Ribet, S. & Gin, S. Role of neoformed phases on the mechanisms controlling the resumption of SON68 glass alteration in alkaline media. J. Nucl. Mater. 324, 152–164 (2004).
Aréna, H. et al. Impact of Zn, Mg, Ni and Co elements on glass alteration: additive effects. J. Nucl. Mater. 470, 55–67 (2016).
Viviano, C. E., Moersch, J. E. & McSween, H. Y. Implications for early hydrothermal environments on Mars through the spectral evidence for carbonation and chloritization reactions in the Nili Fossae region. J. Geophys. Res. Planets 118, 1858–1872 (2013).
Fernández-Caliani, J. C., Crespo, E., Rodas, M., Barrenechea, J. F. & Luque, F. J. Formation of nontronite from oxidative dissolution of pyrite disseminated in Precambrian felsic metavolcanics of the Southern Iberian Massif (Spain). Clays Clay Miner. 52, 106–114 (2004).
Worden & Morad. Clay mineral cements in sandstones Richard H. Worden and Sadoon Morad. Special Publication Number 34 of the International Association of Sedimentologists, 2003; 489 pages. Clays Clay Miner. 52, 139–140 (2004).
De Echave, T., Tribet, M., Gin, S. & Jégou, C. Influence of iron on the alteration of the SON68 nuclear glass in the Callovo-Oxfordian groundwater. Appl. Geochem. 100, 268–278 (2019).
Rebiscoul, D., Van Der Lee, A., Frugier, P., Ayral, A. & Gin, S. X-ray reflectometry characterization of SON 68 glass alteration films. J. Non Cryst. Solids 325, 113–123 (2003).
Ribet & Godon. Altération par l’eau des verres borosilicatés Exemple des verres nucléaires. Tech. l’ingénieur Matériaux résistance à la Corros. au Vieil. (2014).
Advocat, T., Dussossoy, J.-L. & Petitjean, V. Vitrification des déchets radioactifs. Tech. l’ingénieur Cycle du Combust. nucléaire Combust. usés déchets Radioact. (2008).
Jollivet, P., Minet, Y., Nicolas, M. & Vernaz, É. Simulated alteration tests on non-radioactive SON 68 nuclear glass in the presence of corrosion products and environmental materials. J. Nucl. Mater. 281, 231–243 (2000).
Delage, F. Etude de la fonction cinétique de dissolution d’un verre nucléaire, Université de Montpellier 2 (PhD thesis) (1992).
Fillet, S. Mécanismes de corrosion et comportement des actinides dans le verre nucléaire” R7T7”, Université de Montpellier 2 (PhD thesis) (1987).
Francisco, P. C. M. et al. Interaction of FeII and Si under anoxic and reducing conditions: Structural characteristics of ferrous silicate co-precipitates. Geochim. Cosmochim. Acta 270, 1–20 (2020).
Saldi, G. D. et al. Mineralogical evolution of Fe–Si-rich layers at the olivine-water interface during carbonation reactions. Am. Mineral. 100, 2655–2669 (2015).
Daval, D. et al. Mechanism of wollastonite carbonation deduced from micro-to nanometer length scale observations. Am. Mineral. 94, 1707–1726 (2009).
Holzlechner, G., Kubicek, M., Hutter, H. & Fleig, J. A novel ToF-SIMS operation mode for improved accuracy and lateral resolution of oxygen isotope measurements on oxides. J. Anal. Spectrom. 28, 1080–1089 (2013).
Vanbellingen, Q. P. et al. Time-of-flight secondary ion mass spectrometry imaging of biological samples with delayed extraction for high mass and high spatial resolutions. Rapid Commun. Mass Spectrom. 29, 1187–1195 (2015).
Cabaret, D., Sainctavit, P., Ildefonse, P. & Flank, A.-M. Full multiple scattering calculations of the X-ray absorption near edge structure at the magnesium K-edge in pyroxene. Am. Mineral. 83, 300–304 (1998).
Cabaret, D. et al. Medium range structure of borosilicate glasses from Si K-edge XANES: a combined approach based on multiple scattering and molecular dynamics calculations. J. Non Cryst. Solids 289, 1–8 (2001).
Fontaine, A. Spectroscopie d’absorption X (EXAFS et XANES) Application du rayonnement synchrotron. Tech. l’ingénieur (1989).
Levelut, C., Cabaret, D., Benoit, M., Jund, P. & Flank, A.-M. Multiple scattering calculations of the XANES Si K-edge in amorphous silica. J. Non Cryst. Solids 293, 100–104 (2001).
Michelin, A. et al. Investigation at the nanometer scale on the corrosion mechanisms of the archaeological ferrous artifacts by STXM. J. Anal. Spectrom. 28, 59–66 (2013).
Carrillo-Chavez, A. et al. Geochemistry and mineralogy of mine-waste material from a “skarn-type” deposit in central Mexico: modeling geochemical controls of metals in the surface environment. J. Geochem. Explor. 144, 28–36 (2014).
Gailhanou, H. et al. Effects of a thermal perturbation on mineralogy and pore water composition in a clay-rock: an experimental and modeling study. Geochim. Cosmochim. Acta 197, 193–214 (2017).
Calas, G., McMillan, P. F. & Bernier-Latmani, R. Environmental mineralogy: new challenges, new materials. Elements 11, 247–252 (2015).
Bailey, L., Lekkerkerker, H. N. W. & Maitland, G. C. Smectite clay—inorganic nanoparticle mixed suspensions: phase behaviour and rheology. Soft Matter 11, 222–236 (2015).
Acknowledgements
We thank the French National Centre for Scientific Research (CNRS), the French Alternative Energies and Atomic Energy Commission (CEA Saclay, CEA Marcoule) and the French National Radioactive Waste Management Agency (Andra) for the financial support of this work. The CIMAP laboratory is thankful to the French state managed by the National Research Agency through the “Investissements d’Avenir” program which reference is “ANR-11-EQPX-0020”, by the Normandie Region and the “Fonds Européen de Développement Régional” FEDER. Part of the research described in this paper was performed at the Canadian Light Source, a national research facility of the University of Saskatchewan, which is supported by the Canada Foundation for Innovation (CFI), the Natural Sciences and Engineering Research Council (NSERC), the National Research Council (NRC), the Canadian Institutes of Health Research (CIHR), the Government of Saskatchewan, and the University of Saskatchewan.
Author information
Authors and Affiliations
Contributions
C.C. wrote the paper, contributed to all experimental solid characterizations. P.D., D.N., and S.G. conceived the project and designed the experiment. L.G. and F.B. set up the experiment. E.G. and I.M. respectively machined the FIB foils and performed the TEM analyses in the GAL and in ICP. STXM analyses were conducted by E.F., J.D., C.C., P.D., and D.N. N.N. performed the ToF-SIMS analyses. M.S. and A.D. conducted the geochemical calculations. C.C., P.D., S.G., D.N., N.M., and C.M. interpreted the results. All the authors revised and approved the final paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Carriere, C., Dillmann, P., Gin, S. et al. The fate of Si and Fe while nuclear glass alters with steel and clay. npj Mater Degrad 5, 16 (2021). https://doi.org/10.1038/s41529-021-00160-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-021-00160-x
This article is cited by
-
Glass/steel/clay interactions in a simulated radioactive waste geological disposal system
Scientific Reports (2023)
-
Long-term interactive corrosion between International Simple Glass and stainless steel
npj Materials Degradation (2022)
