
Abstract
The electron transport layer (ETL) is a critical component in perovskite quantum dot (PQD) solar cells, significantly impacting their photovoltaic performance and stability. Low-temperature ETL deposition methods are especially desirable for fabricating flexible solar cells on polymer substrates. Herein, we propose a room-temperature-processed tin oxide (SnO2) ETL preparation method for flexible PQD solar cells. The process involves synthesizing highly crystalline SnO2 nanocrystals stabilized with organic ligands, spin-coating their dispersion, followed by UV irradiation. The energy level of SnO2 is controlled by doping gallium ions to reduce the energy level mismatch with the PQD. The proposed ETL-based CsPbI3-PQD solar cell achieves a power conversion efficiency (PCE) of 12.70%, the highest PCE among reported flexible quantum dot solar cells, maintaining 94% of the initial PCE after 500 bending tests. Consequently, we demonstrate that a systemically designed ETL enhances the photovoltaic performance and mechanical stability of flexible optoelectronic devices.
Similar content being viewed by others

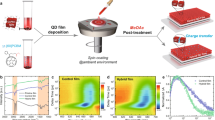

Introduction
Perovskite quantum dots (PQDs) have shown remarkable potential for application in optoelectronic devices, such as photodetectors1,2, light-emitting diodes3,4, and solar cells5,6,7, due to their extraordinary photoelectrical properties. Among the PQDs, CsPbI3-PQD has emerged as a promising material for next-generation solar cells, because of its appropriate band-gap energy (~1.73 eV), making it suitable for single- and multi-junction solar cells8,9,10. Accordingly, intensive and extensive studies have been conducted to harness CsPbI3-PQD for photovoltaic applications, with results indicating a power conversion efficiency (PCE) exceeding 16%11,12. These achievements have been realized through optimization strategies such as ligand engineering and additive engineering13,14. A distinct advantage of CsPbI3-PQD is its ability to be easily deposited on any substrate at desired nanoscale thicknesses using a layer-by-layer (LBL) method at room temperature15,16,17. Such a facile and low-temperature process facilitates the fabrication of devices even on lightweight and flexible polymer-based substrates. It has recently been demonstrated that a flexible solar cell employing CsPbI3-PQD as a light absorber can achieve the highest PCE among flexible colloidal quantum dot solar cells18. Moreover, CsPbI3-PQD films are expected to provide better mechanical stability than bulk perovskite films owing to their nanoscale grain boundaries and soft surface ligands, thus underscoring their potential as flexible device components.
The fabrication of CsPbI3-PQD solar cells necessitates the sequential deposition of layers to construct the device architecture comprising transparent conducting oxide (TCO), an n-type electron transport layer (ETL), CsPbI3-PQD, a p-type hole transport layer (HTL), and a metal electrode6. In order to realize flexible photovoltaic devices on plastic substrates, all procedures must employ a low-temperature process, below 150 °C, compatible with polymer substrates19,20,21. Among these, n-type metal oxides traditionally used for ETLs necessitate high-temperature thermal annealing to enhance crystallinity and interparticle connectivity22,23. Specifically, to prepare high-performance TiO2 and SnO2 ETL, the fabrication process typically necessitates temperatures of 450–550 °C and 180–200 °C, respectively24. The primary challenge in fabricating high-performance, flexible CsPbI3-PQD solar cells lies in the preparation of an ETL capable of forming high-quality thin films even at low temperatures (<150 °C). A potential solution involves the use of low-temperature-processable colloidal tin oxide (SnO2) nanocrystals (SnO2 NP) ETL, which are commonly used in flexible perovskite solar cells25,26. However, SnO2 NP may not be the ideal ETL because low-temperature processing is known to induce surface defects and low crystallinity19,20,27. Additionally, the compatibility of CsPbI3-PQDs with ETLs, initially designed for bulk perovskite solar cells, may result in energy level mismatches. Thus, it becomes imperative to develop a customized ETL with suitable energy levels that can be prepared at room temperature to attain high-performance flexible CsPbI3-PQD solar cells. The design of an ETL specifically for flexible CsPbI3-PQD solar cells can offer several benefits, including: (1) improved photovoltaic performance through energy level control, (2) enhanced device stability by providing an appropriate interface with the adjacent ETL, and (3) minimized damage to the polymer substrate through the consistent use of a low-temperature process during the entire fabrication process.
In this study, we present a SnO2-based ETL using a UV sintering method designed to fabricate high-performance flexible CsPbI3-PQD solar cells23. Initially, we synthesized colloidal SnO2 curved nanorods (SnO2 CNRs), which were capped with oleic acid (OA) and oleylamine (OAm) to ensure quality, dispersibility, and stability28. For the application of SnO2 CNRs as an ETL, they were dispersed in hexane, after which the organic ligands were removed at a low temperature (<50 °C) via UV irradiation following a spin-coating process. We further managed to control the energy level of SnO2 CNRs by doping them with gallium ions (Ga3+), thereby reducing the energy level mismatch with the CsPbI3-PQD. This led to a remarkable enhancement in the PCE of CsPbI3-PQD solar cells, from 10.39 to 15.06%, with the introduction of Ga3+ doped SnO2 CNRs (Ga:SnO2 CNRs) when compared that obtained using commercial SnO2 NPs. Eventually, we implemented the proposed ETL on a polymer-based flexible substrate, which exhibited the highest PCE, 12.70%, amongst all reported flexible quantum dot solar cells. Moreover, we affirmed that flexible CsPbI3-PQD solar cells, utilizing the proposed ETL, maintained excellent mechanical performance by retaining 94% of the initial PCE even after 500 bending cycles with a curvature radius of 7.5 mm.
Results
Synthesis and characterization of Ga3+ doped SnO2 nanocrystals
A significant energy level mismatch exists between CsPbI3-PQD absorbers and SnO2 ETLs, with a conduction band edge (CBE) difference of 0.7 V29. This large potential loss at the interface can lead to open circuit voltage (VOC) loss when the generated photoelectrons are transferred to the SnO2 ETL30. Therefore, to enhance the performance of CsPbI3-PQD solar cells, it is necessary to regulate the energy level of the ETL. Previous studies suggest that the energy level shifts upward toward the vacuum direction when SnO2 is doped with trivalent metal atoms31,32,33,34,35. This shift is attributed to the role of trivalent metal atoms, acting as shallow acceptors upon substituting Sn atoms within SnO2. Experimental results and density functional theory calculations have validated this finding. Among the trivalent metal ions, Ga3+ is believed to substitute Sn4+ easily with minimal lattice distortion, given that the radius of Ga3+ (0.62 Å) closely matches that of Sn4+ (0.71 Å). To empirically demonstrate the impact of Ga3+ doping on SnO2, we introduced a synthetic method that allows for the uniform control of the doping ions within a simple solution system28. The long-chain organic ligands (OA and OAm), which function as surface stabilizers for colloidal dispersion, were eliminated by UV irradiation in the thin film state, as they can act as an electrical barrier for charge transport. The removal of organic ligands via UV irradiation was verified by attenuated total reflectance–Fourier transform infrared (ATR–FTIR) analysis (Supplementary Fig. 1). To evaluate the effectiveness of UV irradiation in removal of organic ligands, we performed ATR–FTIR measurements while varying both UV power and irradiation time. These results revealed that the peaks associated with hydrocarbon vibrations in the SnO2 CNR film disappeared after ~20 min at 500 W power and around 30 min at 250 W power.
We carried out Atomic Force Microscopy (AFM) analysis to compare the uniformity of the SnO2 CNR film both before and after exposure to UV irradiation (Supplementary Fig. 2a). Following UV irradiation, we observed an improved film uniformity without the presence of nanopores or film shrinkage. Additionally, the X-ray diffraction (XRD) results after UV irradiation indicated a decrease in the intensity of the (101) plane relative to the (110) plane (Supplementary Fig. 2b). According to our previous research, UV-generated hydroxyl radicals decompose organic ligands capping the nanocrystals into gaseous components (H2O and CO2)23. Simultaneously, the reduction in film roughness primarily arises from the detachment of organic ligands from the (101) plane, resulting in their spontaneous coalescence onto this plane.
To evaluate the defect densities (Nt) of SnO2 CNRs before and after UV irradiation, we conducted space-charge-limited current (SCLC) measurements using the structure of ITO/SnO2/CsPbI3-PQD/Phenyl-C61-butyric acid methyl ester (PCBM)/Au (Supplementary Fig. 3a). No significant differences were observed in Nt values, indicating that both organic ligands and the UV irradiation process have a negligible effect on the defect density. Additionally, the change of defect state of SnO2 CNRs after UV treatment was investigated by performing photoluminescence (PL) measurements using a high excitation energy wavelength (250 nm, 5.0 eV) (Supplementary Fig. 3b). These results further confirmed the noticeable change of defect density did not occur, as indicated by the ~375 nm (3.3 eV) and 430 nm (2.9 eV) defect PL signals36.
To characterize the synthesized SnO2 CNRs, we compared them with commercially available SnO2 NPs, which are typically used as an ETL for lead halide perovskite solar cells. We obtained the morphology of SnO2 nanocrystals using high-resolution transmission electron microscopy (HR-TEM). The SnO2 CNRs and Ga:SnO2 CNRs displayed a curved nanorod shape, irrespective of Ga3+ ions doping (Supplementary Fig. 4). These images unveiled that the diameter of SnO2 CNRs is ~1.5 nm, significantly smaller than that of SnO2 NP (>5 nm). As previously reported, the quantum size effect resulting from the small diameter (<2.7 nm; Bohr radius) of the nanocrystals can further increase the transmittance of SnO2 CNRs23. The UV–Vis transmittance of each SnO2 film on Glass/indium tin oxide (ITO) substrates is displayed in Supplementary Fig. 5. The SnO2 CNR and Ga:SnO2 CNR films exhibited marginally higher transmittance than the SnO2 NP film at 350–500 nm, suggesting that more near-UV light can reach the light absorbing layer. Supplementary Fig. 4 shows that the lattice distance of all SnO2 nanocrystals is ~3.3 Å, arising from the (110) plane of the rutile structure. As illustrated in Supplementary Fig. 6, XRD patterns of SnO2 films indicated that all reflection peaks can be indexed to the rutile SnO2 crystal structure (PDF # 41-1445). These results confirm that all SnO2 nanocrystals possess the same crystal structure, and no unexpected peaks, such as Ga2O3, were observed. This indicates that Ga ions are incorporated into the SnO2 lattice37.
To further characterize the crystal structures of SnO2 films, we employed grazing-incidence wide-angle X-ray scattering (GIWAXS). As depicted in Fig. 1a–c, all the SnO2 films exhibit nearly isotropic Bragg rings at q ≈ 1.85 Å–1, corresponding to the (110) plane of rutile SnO2, and no unexpected peaks, such as Ga2O3, were observed. However, the high response of Bragg diffraction dots at q ≈ 0.81 (i), 1.57 (ii), 1.82 (iii), 2.04 (iv), 2.18 (v), and 2.27 Å–1 (vi) in the SnO2 NP film was identified. These diffraction dots can be assigned to the (010), (020), (011), (101), (021), and (030) reflection of Sn2O3, which is an oxygen (O)-deficient structure of SnO2 crystals. Additionally, we conducted line profiling by azimuthal angles across the diffraction dots (Supplementary Fig. 7). These results revealed that the unwanted crystal structures of Sn2O3 are randomly present in the SnO2 NP film, irrespective of the out-of-plane (90°) and in-plane (0°) orientation.

a−c GIWAXS patterns for SnO2 NP, SnO2 CNR, and Ga:SnO2 CNR films, respectively. d XPS core level spectra of Ga 2p and Sn 3d for each SnO2 film. e Deconvoluted XPS O 1 s spectra of each SnO2 film. f HR-TEM image and schematic representation of Ga:SnO2 CNR morphology and chemical structure.
We employed X-ray photoelectron spectroscopy (XPS) to investigate the chemical composition differences among each SnO2. Figure 1d presents two peaks at 1145.2 eV and 1118.2 eV, corresponding to Ga 2p1/2 and Ga 2p3/2 transitions, respectively. We confirmed the effectiveness of our designed doping strategy by verifying the increasing Ga 2p peak in the XPS spectra of the products with the rise in Ga3+ concentration in the reactants (Supplementary Fig. 8). Furthermore, Fig. 1d indicates the tin core levels of Sn 3d3/2 and Sn 3d5/2, which are observed with a peak-to-peak separation of 8.4 eV, corresponding to the tin oxidation state38. The binding energy of Sn 3d3/2 for SnO2 CNRs (495.6 eV) and Ga:SnO2 CNR (495.3 eV) becomes noticeably higher when compared to that of SnO2 NPs (494.8 eV), implying enhanced chemical stability of the SnO228. Interestingly, Ga:SnO2 CNR exhibits a relatively lower binding energy of Sn 3d than SnO2 CNR. The cause of the XPS binding energy shift in heterogeneous MA − O − MB bonds has been extensively investigated, where Mi represents the metal species35,39,40. When metal species B is incorporated into the A − O lattice, the formation of B − O bonds with a higher degree of ionicity enhances the covalency of the A − O bond compared to its state when only A − O − A bonds are present. This enhanced covalency results from the redistribution of the electron cloud toward the B direction through the O bridge. Consequently, the lower ionicity of the A − O bond leads to a decrease in the binding energy of A in A − O − B type compared to the A − O − A type. In our case, the binding energy of Sn was lowered when Ga was doped into the SnO2 lattice. This is attributed to the higher ionicity of the Ga−O bond when compared to the Sn−O bond, which arises from the difference in electronegativity between Ga and Sn (electronegativity: Sn = 1.96, Ga = 1.81).
We conducted an XPS analysis of the O 1 s spectrum for each type of SnO2, as depicted in Fig. 1e. The O signal of SnO2 NP can be deconvoluted into two sub-spectra centered at 530.15 and 531.70 eV by using the Lorentzian–Gaussian function. By calculating the area under these sub-spectra, we can estimate the contribution of lattice O (OL, 69.4%) and O vacancies (OV, 30.6%)41,42,43. We also deconvoluted the O 1 s spectra of SnO2 CNRs and noted a definite decrease in OV compared to SnO2 NP. Moreover, we conducted Raman spectroscopy on each SnO2 sample to identify the OV within the sample (Supplementary Fig. 9). An OV peak appeared at ~570 cm–1 for SnO2 NP, while it was challenging to identify a distinct peak for the SnO2 CNRs24,44. The presence of fewer O vacancies in SnO2 CNRs when compared to SnO2 NP suggests that the stoichiometry of SnO2 CNRs is closer to that of stoichiometric SnO224,27. Figure 1f presents the HR-TEM image and schematic of the Ga:SnO2 CNR atomic structure.
Charge transport property from CsPbI3-PQD to customized SnO2
We prepared SnO2 NP (with 150 °C annealing) and SnO2 CNRs (with UV-sintering) films to evaluate their effectiveness as ETL. We calculated the conductivity (σ) of SnO2 films using the Eq. 1.
Here, I, d, and A denote the current, film thickness, and film area, respectively. The conductivity of both SnO2 CNR (1.82 × 10–3 mS cm–1) and Ga:SnO2 CNR (1.78 × 10–3 mS cm–1) exhibited higher values compared to that of SnO2 NP (0.79 × 10–3 mS cm–1). These measurements represent average values derived from evaluating the current density versus voltage (J − V) characteristic of ten ITO/SnO2/gold (Au) devices, respectively (Fig. 2a). Furthermore, we investigated the electrical characteristics of the film surface using conductive atomic force microscopy (C-AFM). The average conductivities for SnO2 NP, SnO2 CNR, and Ga:SnO2 CNR films were 19 pA, 95 pA, and 87 pA, respectively (Supplementary Fig. 10). Moreover, it is observed that SnO2 CNR films formed multiple conduction pathways for the transport of charge carriers, compared to SnO2 NP. The superior conductivity of SnO2 CNR and Ga:SnO2 CNR ETL could significantly enhance the fill factor (FF) of solar cells by minimizing the series resistance45.

a J − V characteristics and calculated conductivity for each SnO2-based device. b Energy band diagrams for the configurations of the CsPbI3-PQD solar cell. c Mott–Schottky plots of CsPbI3-PQD solar cell with each SnO2 ETL. d Steady-state PL spectra of the CsPbI3-PQD films deposited on each SnO2 film. e TRPL spectra with excitation wavelength of 463 nm for CsPbI3-PQD films deposited on each SnO2 film. f Dark J − V curves of the electron-only devices (ITO/SnO2/CsPbI3-PQD/PCBM/Au) with VTFL kink points.
Energy levels for each SnO2 film were determined via Kelvin probe force microscopy (KPFM) and ultraviolet photoelectron spectroscopy (UPS) measurements. The work functions for SnO2 NP, SnO2 CNR, and Ga:SnO2 films were established at −4.52, −4.44, and −4.33 eV, respectively (Supplementary Fig. 11). CsPbI3-PQDs, which are uniformly sized, were synthesized employing hot-injection synthetic techniques, corroborated by HR-TEM images (Supplementary Fig. 12). The lattice parameter for CsPbI3-PQDs was determined to be 6.3 Å, matching the (100) plane of the cubic-phase CsPbI3 perovskite structure46. We dispersed these CsPbI3-PQDs in octane, subsequently prepared films using the LBL method with the NaOAc ligand solution. Supplementary Fig. 13 showcases the energy level diagram for each material. The disparity between the CBE of CsPbI3-PQD and the Fermi level of ETL decreases in the sequence of SnO2 NP, SnO2 CNR, and Ga:SnO2 CNR. This aligns with previous reports indicating SnO2 with fewer O vacancies has a Fermi level closer to the CBE27,47. The Fermi level difference between SnO2 NP and SnO2 CNR would be attributed to the variation in the number of O vacancies, which act as traps below the CBE48,49. Additionally, we experimentally confirmed that Ga3+ doping triggers an upward shift in the energy level of SnO2. Consequently, these results allowed us to illustrate the energy band diagrams for the configurations of the CsPbI3-PQD solar cell, as displayed in Fig. 2b. The difference between electron and hole quasi-Fermi levels would determine the theoretical maximum VOC of the solar cell device45,50,51. Hence, the Fermi level of Ga:SnO2 is closer to the CBE of CsPbI3-PQD compared to others, which potentially can boost VOC. Moreover, the built-in-potential (Vbi), which is deeply related to VOC in solar cells, was confirmed for each SnO2-based device (ITO/SnO2/CsPbI3-PQD/Spiro-OMeTAD/Au) by capacitance–voltage (C − V) measurement. Vbi is identified as the intercept of the linear regime to the x-axis in Mott–Schottky plots (C–2 vs. V). A substantial Vbi implies an extension of the depletion region, which assists in charge separation and mitigates the back reaction of electrons from SnO2 to the CsPbI3-PQD. The increase in Vbi is ascribed to the upward shift of the Fermi level of ETL, ultimately enhancing the VOC of the solar cells51,52. In Fig. 2c, we verified that the Vbi values align with the trends of the energy band structures of SnO2. This outcome is also expected to contribute to a higher voltage output in CsPbI3-PQD solar cells with Ga:SnO2 CNR ETL.
We investigated the charge transfer ability at the interface of each SnO2 and CsPbI3-PQD using steady-state photoluminescence (PL) spectra and time-resolved PL (TRPL) decay measurements. Figure 2d illustrates that devices using SnO2 CNR/PQD and Ga:SnO2 CNR/PQD demonstrate more effective PL quenching than those utilizing SnO2 NP/PQDs. This outcome suggests that charge extraction from CsPbI3-PQD to UV-sintered SnO2 films is more efficient than that of SnO2 NP. Moreover, the PL emission peaks of CsPbI3-PQD on UV-sintered SnO2 CNRs were located at 680.0 nm, representing a blue shift compared to the 682.5 nm emission of the SnO2 NP/PQD sample. The radiative recombination originated from trap states results in a red shift in the PL peak, compared to that of the band edge transition50,52,53. Therefore, the CsPbI3-PQD film with UV-sintered ETLs exhibits a less red-shifted PL peak, reflecting a lower trap density within SnO2. Figure 2e displays the PL decay transients of CsPbI3-PQD coated on each SnO2 ETL. The corresponding PL lifetimes and amplitudes are determined by fitting with a bi-exponential decay function and are detailed in Supplementary Table 1. In general, the fast decay component (τ1) originates from the quenching of charge carriers at the interface, while the slow decay component (τ2) results from the radiative recombination of free charge carriers due to traps in the absorption layer54. Compared to the only PQD film, the samples employing ETL exhibited a shorter τ1 with an increasing amplitude of fast decay (A1), indicating the dominance of charge transfer from the CsPbI3-PQD film to the ETL. The average lifetime for the SnO2 NP/PQD sample was 8.84 ns, which was reduced to 5.31 and 4.67 ns for SnO2 CNR and Ga:SnO2, respectively. Compared to the SnO2 NP/PQD sample, τ1 also decreased from 2.77 to 2.32 and 2.26 ns in UV-sintered SnO2-based samples with an increasing A1. To further investigate the quality of the interface, we performed electrochemical impedance spectroscopy (EIS) measurements, and interfacial electrochemical parameters were obtained by fitting the impedance data using Z-view software (Supplementary Fig. 14). The series resistance (Rs) and recombination resistance (Rrec) of the device can be evaluated from the Nyquist plot18. The SnO2 CNRs-based device presents smaller Rs and larger Rrec than the SnO2 NP-based device, indicating that the recombination process could be effectively suppressed.
Additionally, we fabricated electron-only devices with the structure of ITO/SnO2/CsPbI3-PQD/PCBM/Au to evaluate the trap-state density (Nt) of each ETL-based device (Fig. 2f). It is evident that the SnO2 CNR-based device (Nt = 2.02 × 1015 cm−3) and Ga:SnO2 CNR-based device (Nt = 1.63 × 1015 cm−3) possess much lower Nt than the SnO2 NP-based device (Nt = 5.83 × 1015 cm−3). The reduced Nt of the Ga:SnO2-based device, attributable to relatively low O vacancies and Ga3+ doping, indicates fewer charge recombination sites at the interface and within the ETL24,34. Hence, these results suggest that the SnO2 CNR-based ETLs can amplify the photovoltaic performance of CsPbI3-PQD solar cells by enhancing the charge transfer properties at the interface.
Enhanced performance and stability of CsPbI3-PQD solar cells via customized ETL
CsPbI3-PQD solar cells based on SnO2 CNR are fabricated using the UV-sintering method for the ETL and LBL method for PQD deposition, as demonstrated in Fig. 3a. To examine the thickness and uniformity of each SnO2 film, we conducted scanning electron microscopy (SEM) and AFM measurements (Supplementary Figs. 15 and 16). We confirmed that each SnO2 layer used in the device has an almost constant thickness (~30 nm) and smooth surface. We also found that UV-sintered SnO2 CNR films (a root mean square (RMS) of ~0.85 nm) displayed slightly more uniformity than the SnO2 NP film (RMS of 1.05 nm). Additionally, we compared the wettability of each SnO2 ETL using contact angle measurements (Supplementary Fig. 17). No significant differences in water contact angle were observed among the individual SnO2 films.

a Schematic illustration representing the UV sintering process for formation of SnO2 CNR thin film and fabrication method for CsPbI3-PQD solar cells. b J − V characteristics of best-performing CsPbI3-PQD solar cells using various SnO2 as ETL (inset: device configuration measured by SEM). c IPCE spectra of CsPbI3-PQD solar cells with SnO2 ETLs and integrated JSC calculated from IPCE. d Normalized PCE obtained by stability tests of each SnO2-based device under ambient condition (25 °C, 20% RH) for 10 days (inset: XRD data of the CsPbI3-PQD film exfoliated from each SnO2 after 1 day and 7 days, respectively).
We initially explored the impact of Ga3+ doping on SnO2 by assessing the photovoltaic properties of CsPbI3-PQD solar cells (Supplementary Fig. 18). We observed a steady increase in VOC as Ga3+ were doped with SnO2 from 0–10 mol%. The optimal concentration was identified as 10 mol% because highly doped SnO2 resulted in reduced performance. This decline in PCE can be attributed to the decreasing difference in the conduction band edge between 15% Ga:SnO2 CNR and CsPbI3-PQD (Supplementary Fig. 19). This indicates that excessive doping reduces the driving force for electron injection from CsPbI3-PQD to Ga:SnO2 CNR, consequently lowering the photovoltaic performance55. Figure 3b presents the best J − V curves for each SnO2 ETL-based solar cell, with their photovoltaic performances summarized in Table 1. The optimized Ga:SnO2 CNR-based device outperformed the other SnO2-based devices, achieving a PCE of 15.06% (VOC = 1.305 V, JSC = 15.73 mA cm–2, FF = 73.32%). The SnO2 CNR-based device had a PCE of 11.18% (VOC = 1.160 V, JSC = 15.08 mA cm–2, FF = 63.87%), while the SnO2 NP-based device exhibited the lowest performance, with a PCE of 10.39% (VOC = 1.107 V, JSC = 14.82 mA cm–2, FF = 62.33%). Notably, the VOC increased significantly in the Ga:SnO2 CNR-based device when compared to the SnO2 NP-based device. This confirms our hypothesis that the voltage output of solar cells can be enhanced by upshifting the Fermi level. We measured incident-photon-to-current conversion efficiency (IPCE) spectra to verify the charge collection ability of each SnO2 ETL on the photocurrent of CsPbI3-PQD solar cells. Figure 3c presents the IPCE spectra of PQD solar cells based on different ETLs, matching well with the values obtained from the J − V curves in Fig. 3b. The SnO2 CNR- and Ga:SnO2 CNR-based solar cells exhibit higher IPCE values with a strong spectral response from 350 to 500 nm. Photons with shorter wavelengths would have a shallower penetration depth, and tend to be absorbed around the front region of CsPbI3-PQD layer. Therefore, the observed increase in IPCE values for short wavelengths implies a more efficient electron extraction at the interface between SnO2 and CsPbI3-PQD, alongside the improved optical transparency of UV-sintered SnO2 ETLs56. The hysteresis of solar cells also decreased in the order of SnO2 NP, SnO2 CNR, and Ga:SnO2 CNR (Supplementary Fig. 20). This suggests that the Ga:SnO2 CNR-based solar cell reduced charge accumulation at the CsPbI3-PQD/ETL interface due to its efficient electron extraction capability. Additionally, we confirmed the reproducibility of performance by averaging the data from 30 individual devices for each ETL (Supplementary Fig. 21).
To evaluate the stability of the devices, we tracked the maximum power point (MPP) to monitor the steady-state efficiency of each SnO2 ETL-based solar cell. The PCEs of the SnO2 NP-, SnO2 CNR-, and Ga:SnO2 CNR-based devices stabilized at 8.87, 12.24, and 14.61%, respectively (Supplementary Fig. 22). The SnO2 CNR- and Ga:SnO2 CNR-based devices retained over 95% of the maximum PCE, highlighting the excellent photovoltaic stability of UV-sintered SnO2 CNRs. Further, we evaluated the long-term stability of each SnO2-based device under consistent ambient conditions (25 °C, 20% RH) without any additional encapsulation (Fig. 3d). Compared to the SnO2 NP-based device, the UV-sintered SnO2-based devices demonstrated improved long-term stability. We additionally confirm the stability of each device under higher humidity conditions (25 °C, 40% RH and 25 °C, 60% RH) and elevated temperatures (40 °C and 60 °C in N2). These results reveal that SnO2 CNR- and Ga:SnO2 CNR-based devices exhibit improved stability against degradation induced by humidity rather than by temperature (Supplementary Fig. 23).
To investigate the crystalline phase of the CsPbI3-PQD film adjacent to each SnO2, we conducted SEM and XRD analysis on the CsPbI3-PQD film exfoliated from SnO2 using adhesive epoxy (Supplementary Fig. 24). Initially, we inspected the morphology of the interface for damage during the exfoliation process through planar-view SEM images. All samples showed no unintended damage due to improper exfoliation, such as unidirectional regular cracks57. In contrast to the UV-sintered SnO2-based samples, which did not show any pinholes, we observed randomly present pinholes at the SnO2 NP/CsPbI3-PQD interface. The inset of Fig. 3d shows that unfavorable phase peaks in XRD patterns only appeared at the SnO2 NP/CsPbI3-PQD interface after 7 days. This result suggests that the CsPbI3-PQDs on the SnO2 NP film are more prone to a phase transformation from the cubic phase (α) to the orthorhombic phase (δ) after a certain period. The hydrated CsPbI3-PQDs, which are susceptible to transformation with an inappropriate bandgap, can progressively generate at the ETL/CsPbI3-PQD interface due to surface defects in ETL27,29. Based on these results, we concluded that SnO2 CNR ETLs improve device stability due to fewer interfacial defect sites27,58.
Expanded application of SnO2 ETL-based CsPbI3-PQD solar cells to flexible devices
To verify the advantages of the annealing-free UV-sintering process, we employed each SnO2 as an ETL for flexible PQD solar cells using an ITO/ Polyethylene naphthalate (PEN) substrate. The best J − V curves for each SnO2-based CsPbI3-PQD solar cell, as well as a photograph of the flexible device, are presented in Fig. 4a, with the photovoltaic performance summarized in Table 2. Remarkably, the Ga:SnO2 CNR-based flexible device reached the highest PCE amongst the flexible QD solar cells reported to date, as summarized in Supplementary Table 2. The PCEs of the Ga:SnO2 CNR- and SnO2 CNR-based devices were 12.70% (VOC = 1.305 V, JSC = 14.17 mA cm–2, FF = 68.84%) and 10.85% (VOC = 1.157 V, JSC = 14.02 mA cm–2, FF = 66.90%), respectively. The SnO2 NP-based device demonstrated the lowest performance, with a PCE of 9.20% (VOC = 1.076 V, JSC = 13.93 mA cm–2, FF = 61.36%). The hysteresis behavior of the flexible solar cells based on each SnO2 ETL was similar to that of rigid devices (Supplementary Fig. 25). VOC and FF of SnO2-based flexible devices also exhibited similar trends to those of the rigid devices, although there was a minor difference in JSC. It was confirmed that the absorption of near-UV light by the PEN/ITO substrate resulted in a reduction in current density (Supplementary Fig. 26).

a J − V characteristics of best-performing flexible CsPbI3-PQD solar cells using various SnO2 as ETL (inset: photograph of the fabricated flexible solar cell). b In-situ relative resistance change (ΔR/R0) of each SnO2-based device (ITO/SnO2/Au) during bending test with 7.5 mm of curvature of radius (RC) (inset: after recovery from bending at various values of RC). c Normalized PCE of the flexible CsPbI3-PQD solar cell after repeated bending at RC = 7.5 mm for 500 bending cycles (inset: top-view SEM image of SnO2 film on a flexible substrate before and after bending test).
We performed a bending test using a custom-made bending machine to assess the mechanical stability of SnO2-based devices59,60. Initially, we subjected the Ga:SnO2 CNR-based solar cell to 100 consecutive bending cycles, gradually reducing the curvature of the bending radius (RC) values from 10 to 7.5, 5.0, and finally 4.5 mm. The J − V characterization performed after these cycles showed a decline in PCE from an initial 10.27% (flat, RC = ∞) to 10.21, 9.64, 8.78, and 6.07% for RC values of 10, 7.5, 5.0, and 4.5 mm, respectively. This result reveals a significant reduction in PCE starting from RC = 5.0 mm (Supplementary Fig. 27). Additionally, we evaluated the relative resistance changes (ΔR/R0) using the PEN/ITO/SnO2/Au device after the same 100 bending cycles at each RC. The ΔR/R0 started to rise rapidly from RC = 5.0 mm. As previously reported, plastic deformation and ITO broken can occur from RC = 5.0 mm in our devices23,60. Consequently, we set the RC at 7.5 mm for the bending test to ascertain mechanical stability, excluding any influence of flexible substrate damage. Additionally, we calculated Young’s modulus of all SnO2 films and CsPbI3-PQD film. The Young’s modulus was determined by measuring the force-distance curves at five individual points (Supplementary Fig. 28)61. These results indicate no significant difference in modulus among the SnO2 films, implying that the modulus of SnO2 itself does not significantly affect the mechanical stability of our devices18.
In Fig. 4b, the SnO2 NP-based device demonstrated a steady increase in in-situ ΔR/R0 during repeated cycles of 1.5 s release and 1.5 s bending (Supplementary Fig. 29 and Supplementary Video 1). Conversely, UV-sintered SnO2 CNR-based devices showed negligible changes in ΔR/R0 even after 500 cycles. To investigate whether these results were influenced by the annealing process of the flexible substrate, we performed bending tests under identical conditions on the pristine substrate, annealed substrate, and UV-sintered substrate. As shown in Supplementary Fig. 30, the annealed PEN/ITO substrate, treated at 150 °C for 30 min, revealed a noticeable increase in in-situ ΔR/R0 over 500 bending cycles in comparison to the pristine PEN/ITO and UV-sintered PEN/ITO62,63. The rise in resistance across bending cycles can be attributed to the difference in the thermal expansion coefficients of PEN and ITO. These findings underscore the importance of avoiding an annealing process to preserve the mechanical stability of flexible devices. To further investigate the morphological changes of the SnO2 films after the bending test by SEM, as depicted in Fig. 4c. Previous reports noted partial swelling on the surface of SnO2 NP film and detachment from the substrate20. However, we did not observe any significant morphological alterations in the UV-sintered SnO2 films after the bending test (Supplementary Fig. 31). Figure 4c also displays the normalized PCE change of each SnO2-based CsPbI3-PQD flexible solar cell throughout the 500-cycle bending test, with their photovoltaic parameters presented in Supplementary Fig. 32. The SnO2 CNR and Ga:SnO2 CNR-based devices showed a minor decline in PCE by only 5 and 6%, respectively, in comparison to the initial PCE after 500 bending cycles. As anticipated, the SnO2 NP-based device exhibited a substantial decrease of 25% from the initial PCE after the same number of bending cycles, suggesting the mechanical stability advantage offered by the UV-sintering process.
Moreover, we fabricated a thin-film perovskite solar cell utilizing SnO2 CNR as an ETL and performed a bending test. Following the 500 bending tests, the thin-film perovskite solar cell exhibited an 84% reduction in its initial PCE, indicating comparatively lower mechanical stability in contrast to CsPbI3-PQD solar cells (Supplementary Fig. 33a). Additionally, we confirmed the morphology of the perovskite thin-film deposited on the SnO2 CNR film using SEM (Supplementary Fig. 33b). As reported previously, the perovskite thin-films gradually formed random cracks as the number of bends increased, highlighting the advantageous mechanical flexibility of QD films18. In conclusion, we verified that our proposed ETL is particularly beneficial for flexible device applications. The UV-sintering process offers the advantage of strengthening the adhesion between the SnO2 layer and bottom substrate without inflicting damage to flexible substrates. Hence, the proposed SnO2 thin film can be effectively implemented in multi-layer devices requiring a low-temperature process.
Discussion
In this study, we presented the fabrication of Ga:SnO2 CNR thin films using a thermal annealing-free UV-sintering method, as a promising ETL for high-performance flexible CsPbI3-PQD solar cells. Our elaborately designed SnO2 aims to minimize O vacancies, positively impacting the device’s performance. Furthermore, we addressed the energy level mismatch between CsPbI3-PQD light absorbers and the ETL by doping SnO2 with Ga3+. This strategy led to an up-shifted energy level of the SnO2, increasing the built-in potential at the junction between the Ga:SnO2 CNR layer and CsPbI3 PQD layer. Hence, we observed enhanced charge carrier extraction to the Ga:SnO2 CNR layers, thereby improving device performance. The customized ETL-based flexible CsPbI3-PQD solar cells achieved the highest PCE of 12.70% among colloidal QD solar cells reported to date, demonstrating the superiority of Ga:SnO2 CNR as an ETL for flexible photovoltaic devices. Additionally, we determined the flexible substrate’s susceptibility to the conventional high-temperature annealing process in comparison to the UV-sintering method by observing the in-situ relative resistance change (ΔR/R0) during a bending test. Hence, the flexible CsPbI3-PQD solar cells, fabricated using a low-temperature process, demonstrated enhanced durability against the bending test. These results emphasize the importance of low-temperature processing for flexible devices, considering the vulnerability of polymer substrates to heat. Thus, our study indicates that the proposed fabrication method for SnO2 thin films can be effectively applied to flexible optoelectronic devices requiring low-temperature processes.
Methods
Materials
Tin(IV) oxide (SnO2 NPs, 15% in H2O Colloidal dispersion) and gallium(III) ethoxide (Ga(OC2H5)3), lead (II) iodide (PbI2, 99.999%), and 1-octadecene (ODE, 90%) were purchased from Alfa Aesar. Tin(IV) tert-butoxide (Sn(OtBu)4, 99.99%), cesium carbonate (Cs2CO3, 99.99%), n-hexane (anhydrous, 95%), n-octane (extra pure, ≥99%), Oleic acid (OA, 90%), Oleylamine (OAm, 70%), sodium acetate (NaOAc, 99.995%), chlorobenzene (CB, anhydrous, 99.8%), N,N-dimethylformamide (DMF, anhydrous, 99.8%), dimethyl sulfoxide (DMSO, >99.5%), acetonitrile (ACN, anhydrous, 99.8%), and Li-bis(trifluoromethanesulfonyl) imide (Li-TFSI) were purchased from Sigma Aldrich. Phenethylammonium iodide (PEAI) was purchased from GreatcellSolar. Ethyl alcohol (EtOH, Pure), methyl acetate (MeOAc, ≥99.5%), and ethyl acetate (EtOAc, ≥99.5%) were purchased from Duksan. 2,20,7,70-Tetrakis(N,N-di-p-methoxyphenylamine)-9,90-spirobifluorene (Spiro-OMeTAD) and tris(2-(1Hpyrazol-1-yl)-4-tert-butylpyridine)-cobalt(III) tris(bis(trifluoromethylsulfonyl))imide (FK209) were purchased from Lumtec. 2-amylpyridine (≥98.0%) was purchased from TCI. Phenyl-C61-butyric acid methyl ester (PCBM) and poly(triarylamine) (PTAA) were purchased from Nanostructured Carbon (nano-c).
Synthesis of colloidal SnO2 curved nanorods (SnO2 CNRs)
SnO2 CNRs were synthesized by modifying the previously reported one-step synthetic method. We began the synthesis process by mixing 7 mmol of Sn(OtBu)4, 6 mL of OAm, 8.75 mL of OA, and 1 mL of deionized water in a 3-neck round-bottom flask, all under an argon (Ar) gas flow. This mixture was then heated to 180 °C for 4 h with consistent vigorous stirring. Upon completion of the reaction, we cooled the mixture to room temperature and added 5 mL of n-hexane to dissolve the resulting product. To obtain Ga-doped SnO2 CNRs, we introduced Ga(OC2H5)3 powder under the same experimental conditions, varying the amount added to obtain 5, 10, and 15 mol% Ga concentration relative to Sn. The as-grown SnO2 CNRs were purified by washing with EtOH and n-hexane to remove any residual side products. This was followed by centrifugation of the NCs suspension at 8000 rpm for 15 min to ensure all residual side products were removed. The final product, dissolved in n-hexane (30 mg ml−1), was stored under an N2 gas flow within a glove box.
Preparation of UV-sintered SnO2 CNR and Ga:SnO2 CNR ETL
A highly dispersed SnO2 CNRs solution in n-hexane (30 mg ml−1) was dropped onto TCO substrates. Immediately after this, the substrates were spin-coated at a speed of 5000 rpm for 30 s. The SnO2 CNRs-coated substrates were then subjected to UV irradiation from a UV cure machine (JHCI-051B, JECO) equipped with an Hg lamp with a peak wavelength of 365 nm. In this experiment, UV irradiation lasted for 30 min at a power of 500 W. Throughout the UV exposure process, we maintained the temperature of the samples at ~45 °C.
Synthesis of colloidal CsPbI3-PQDs
The Cs-oleate solution was prepared by adding Cs2CO3 (0.407 g), ODE (20 mL), and OA (1.25 mL) to a 100 mL 3-neck round-bottom flask, which was then degassed for 30 min at 120 °C under vacuum. This process of filling the flask with Ar gas and subsequent degassing was repeated thrice before storing the Cs-oleate solution under Ar. To synthesize CsPbI3-PQDs, PbI2 (0.5 g) and ODE (25 mL) were added to a 100 mL 3-neck round-bottom flask, followed by a 30 min degassing period at 115 °C. Degassed OA (2.5 mL) and OAm (2.5 mL) were then added to the flask, and the system was further degassed for 30 min at 115 °C. After the flask was filled with Ar, the flask was heated to 185 °C, and Cs-oleate (2 mL) was rapidly injected. Ten seconds post-injection, the flask was rapidly cooled to room temperature in an ice bath. To isolate the desired CsPbI3-PQDs, MeOAc (35 mL) was added to the synthesized CsPbI3-PQD solution (15 mL) as a polar anti-solvent, followed by centrifugation at 5000 rpm for 3 min. The supernatant was discarded, and the precipitated CsPbI3-PQDs were redispersed in n-hexane (5 mL). MeOAc (7 mL) was subsequently added, and the solution was centrifuged again at 5000 rpm for 3 min, followed by the removal of the supernatant. The CsPbI3-PQDs were then re-dispersed in n-hexane (15 mL), and the solution was centrifuged at 5000 rpm for 3 min to remove any aggregated CsPbI3-PQDs. The supernatant was collected and stored in the dark at 4 °C for 48 h, after which the precipitated products were removed. The remaining n-hexane was dried under vacuum, and the dried CsPbI3-PQD pellets were redispersed in n-octane at a concentration of ~75 mg ml−1.
CsPbI3-PQD solar cell device fabrication
The patterned indium tin oxide (ITO, 8 Ω sq−1) substrates were sequentially cleaned by sonicating in detergent water, deionized water, acetone, and isopropyl alcohol for 10 min each. For the preparation of SnO2 ETLs, the SnO2 NPs were diluted with deionized water at a weight ratio of 1:6. This solution was then deposited onto UV-ozone treated ITO substrates via spin-coating at a speed of 3000 rpm for 30 s, followed by annealing at 150°C for 30 min. The synthesized SnO2 and Ga:SnO2 CNRs were subsequently deposited onto ITO substrates using the above-mentioned UV curing method. For flexible device fabrication, ETLs were deposited onto PEN/ITO (15 Ω sq−1, Nano Clean Tech) substrates, employing the same deposition procedure used for glass substrates. For the fabrication of CsPbI3-PQD solids based on a solid-state ligand exchange process, a solution of CsPbI3-PQD in n-octane (75 mg ml−1) was spin-coated onto the SnO2/ITO substrates at 1000 rpm for 20 s, followed by 2000 rpm for 5 s. The CsPbI3-PQD coated film was then immersed in a 1 mg ml−1 NaOAc ligand solution in MeOAc and spin-dried. The ligand-treated film was further soaked and washed with MeOAc before being spin-dried. This process was repeated 5 times to realize the optimal thickness of approximately 300 nm under a controlled humidity condition of approximately 25–30% to enable effective ligand exchange. Following this, the thick film of CsPbI3-PQDs was soaked in a 1 mg ml−1 PEAI solution in EtOAc for 10 s for post-treatment and spin-dried. This PEAI-treated CsPbI3-PQD film was then washed and soaked in MeOAc before being spin-dried again. The Spiro-OMeTAD layer, used as a HTL in solar cells, was spin-coated at 4000 rpm for 30 s. The Spiro-OMeTAD solution was prepared by doping 90 mg ml−1 Spiro-OMeTAD in CB with 39 µL 2-amylpyridine, 23 µL Li-TFSI (520 mg ml−1 in ACN), and 5 µL FK209 (180 mg ml−1 in ACN). Finally, an Au electrode was deposited using a thermal evaporator to a thickness of 80 nm. For electron-only devices, PCBM was applied on top of the CsPbI3-PQD layer with a device configuration of ITO/SnO2/CsPbI3-PQD/PCBM/Au. PCBM dissolved in CB (20 mg ml−1) was spin-coated at 3000 rpm for 30 s under a N2-filled glove box. For the thermal stability test, a thermally stable PTAA layer with a concentration of 10 mg ml−1 in 1 mL toluene mixed with 2 µL 2-amylpyridine and 1.5 µL Li-TFSI solution was spin-coated on the CsPbI3-PQD film at 3000 rpm for 20 s.
Perovskite thin-film fabrication
Perovskite thin-films were prepared according to reported methods with slight modifications64. The perovskite precursor solution was prepared by dissolving FAPbI3 powder (1.62 M) and CsPbI3 powder (0.18 M) in a mixture solvent of DMF and DMSO (4:1 v/v). The precursor solution was spin-coated on the PEN/ITO substrate at 5000 rpm for 40 s, where 200 μL CB was quickly dropped onto the center of the spinning substrate at 15 s prior to the end. The film was immediately annealed at 100 °C for 30 min.
Characterization
The morphology of SnO2 nanocrystals (NCs) and CsPbI3-PQDs was analyzed via transmission electron microscope (TEM, JEM2100F, JEOL), and their films were analyzed via a scanning electron microscope (FE-SEM, Verios G4 UC, FEI) and atomic force microscopy (AFM, XE-100, Park Systems). Grazing-incidence wide-angle X-ray scattering (GIWAXS) measurements were performed at the 3 C beamline of the Pohang Accelerator Laboratory, Republic of Korea. The C-AFM measurements of the SnO2 film were characterized by AFM (XE-100, Park Systems) in C-AFM mode with CONTSCPt cantilever to evaluate the current distribution of film surface by applying a bias of 2 V. The surface modulus was obtained with probes from Park Systems (PPP-FMR, resonant frequency = ~75 kHz, k (spring constant) = ~ 2.8 N m−1, tip radius ~10 nm). The work function of SnO2 NCs was analyzed by Kelvin prove force microscopy using a NSC14/Cr-Au cantilever (XE-100, Park Systems). High resolution X-ray diffractometer (XRD, D8 ADVANCE, BRUKER), using Cu-Kα radiation of 1.541 Å, was used to characterize the crystal structure of SnO2 NCs. The DC conductivity of ITO/SnO2/Au and space-charge-limited-current (SCLC) of electron-only devices were measured by J − V measurements using Keithley 2400 source meter. Trap-state density (Nt) was calculated from trap-filled limit voltage (VTFL), obtained from the SCLC curve, using the following equation: VTFL = qNtL2/2εε0. In the equation, q denotes the elementary charge, L denotes the thickness of CsPbI3-PQD layer, ε denotes the relative permittivity of CsPbI3 (ε = 6.32ε0), and ε0 denotes the vacuum permittivity, respectively. The XPS and UPS measurements were performed with a scanning XPS microprobe (PHI 5000 Versa Probe, Ulvac-PHI) using an Al-Kα monochromator (1486.6 eV) for XPS and a HeI (21.2 eV) for UPS. Raman scattering measurements were performed via micro-Raman spectrometer (DXR-3xi, Thermo Fishier Scientific) with a 532 nm laser excitation source. The optical properties of SnO2 thin films were examined using a UV–Vis spectrometer (Shimadzu, UV-2600i). The absorption coefficient (α) of direct bandgap semiconductor near the band edge can be obtained by αhν = A(hν–Eg)2, where A denotes the proportionality constant, hν denotes the photon energy, and Eg denotes the optical bandgap. Specifically, Eg was estimated by extrapolating the line tangent to the plotted curve. ATR–FTIR spectroscopy (Nicolet iS50, Thermo Fisher Scientific) was employed to measure the ligand removal after UV treatment. Mott–Schottky plots were executed at fixed frequency (1 kHz) under dark conditions (SP-300, BioLogic). EIS analysis was performed on CsPbI3-PQD solar cells employing different SnO2 ETLs at 1.0 V under dark conditions (SP-300, BioLogic). The steady-state PL and TRPL spectra were performed on a Fluoromax-4 (iHR320, HORIBA Scientific) spectrometer. The excitation wavelength was 463 nm, and TRPL measurements were conducted to determine the PL maximum (~680 nm) of the CsPbI3-PQD film. Contact angle measurements were conducted using a Phoenix 150 (Surface Electro Optics) contact angle measurement equipment. Under dry conditions (20 ± 5 °C, below 15% RH), the current density–voltage (J − V) curve and MPP tracking characteristics of devices were measured using a Keithley 2400 source under a Xenon-lamp-based solar simulator (Newport 91160 s, AAA class). A Si solar cell calibrated by the National Renewable Energy Laboratory (NREL) was employed to adjust the light intensity to the AM 1.5 G 1sun condition (100 mW cm−2). The voltage sweep rate was 20 mV with a delay of 20 ms for the reverse (forward) sweeping direction from 1.4 V (−0.1 V) to −0.1 V (1.4 V). The devices were measured with SUS304 aperture with an area of 0.09 cm2. IPCE measurement system (PV Measurement, Inc.) was used to characterize incident photon-to-current conversion efficiency (IPCE). Long-term stability of CsPbI3-PQD solar cells was conducted by storing devices according to the experimental conditions using a constant temperature and humidity chamber (20 − 95 RH%, −20−150 °C range, Jeiotech Co. Ltd., TH-ME-025). The mechanical stability of flexible devices under various conditions was assessed using a 2-axis motion controller (STM-2-USB, ST1) to ensure a well-controlled bending state. This was linked to a bending machine system (ST-BJS-0810-SSU, ST1) (Supplementary Fig. 29 and Supplementary Video 1). To measure the in-situ relative resistance change during bending tests, the Au electrodes of the devices and copper wires were soldered using indium.
Data availability
The data that support the findings of this study are available from https://doi.org/10.6084/m9.figshare.25284907.v165.
References
Ahmadi, M., Wu, T. & Hu, B. A review on organic–inorganic halide perovskite photodetectors: device engineering and fundamental physics. Adv. Mater. 29, 1605242 (2017).
Wang, H. & Kim, D. H. Perovskite-based photodetectors: materials and devices. Chem. Soc. Rev. 46, 5204–5236 (2017).
Veldhuis, S. A. et al. Perovskite materials for light-emitting diodes and lasers. Adv. Mater. 28, 6804–6834 (2016).
Liu, X. K. et al. Metal halide perovskites for light-emitting diodes. Nat. Mater. 20, 10–21 (2021).
Chen, J. X. et al. Emerging perovskite quantum dot solar cells: feasible approaches to boost performance. Energy Environ. Sci. 14, 224–261 (2021).
Yuan, J. Y. et al. Metal halide perovskites in quantum dot solar cells: progress and prospects. Joule 4, 1160–1185 (2020).
Zhang, J. R., Hodes, G., Jin, Z. W. & Liu, S. Z. All-inorganic CsPbX3 perovskite solar cells: progress and prospects. Angew. Chem. Int. Ed. 58, 15596–15618 (2019).
Zhao, Q. et al. Colloidal quantum dot solar cells: progressive deposition techniques and future prospects on large-area fabrication. Adv. Mater. 34, 2107888 (2022).
Hazarika, A. et al. Perovskite quantum dot photovoltaic materials beyond the reach of thin films: full-range tuning of A-site cation composition. ACS Nano 12, 10327–10337 (2018).
Yang, S., Duan, Y., Liu, Z. & Liu, S. Recent advances in CsPbX3 perovskite solar cells: focus on crystallization characteristics and controlling strategies. Adv. Energy Mater. 13, 2201733 (2022).
Jia, D. L. et al. Tailoring solvent-mediated ligand exchange for CsPbI3 perovskite quantum dot solar cells with efficiency exceeding 16.5%. Joule 6, 1632–1653 (2022).
Jia, D. L. et al. Surface matrix curing of inorganic CsPbI3 perovskite quantum dots for solar cells with efficiency over 16%. Energy Environ. Sci. 14, 4599–4609 (2021).
Liu, L. et al. Perovskite quantum dots in solar cells. Adv. Sci. 9, 2104577 (2022).
Yuan, J. et al. Perspective on the perovskite quantum dots for flexible photovoltaics. J. Energy Chem. 62, 505–507 (2021).
Swarnkar, A. et al. Quantum dot-induced phase stabilization of alpha-CsPbI3 perovskite for high-efficiency photovoltaics. Science 354, 92–95 (2016).
Kim, J. et al. Alkali acetate-assisted enhanced electronic coupling in CsPbI3 perovskite quantum dot solids for improved photovoltaics. Nano Energy 66, 104130 (2019).
Kim, J. et al. Single-step-fabricated perovskite quantum dot photovoltaic absorbers enabled by surface ligand manipulation. Chem. Eng. J. 448, 137672 (2022).
Hu, L. et al. Flexible and efficient perovskite quantum dot solar cells via hybrid interfacial architecture. Nat. Commun. 12, 466 (2021).
Chung, J. et al. Record-efficiency flexible perovskite solar cell and module enabled by a porous-planar structure as an electron transport layer. Energy Environ. Sci. 13, 4854–4861 (2020).
Paik, M. J. et al. SnO2-TiO2 hybrid electron transport layer for efficient and flexible perovskite solar cells. ACS Energy Lett. 7, 1864–1870 (2022).
Jung, H. S., Han, G. S., Park, N. G. & Ko, M. J. Flexible perovskite solar cells. Joule 3, 1850–1880 (2019).
Ye, T. et al. Temperature effect of the compact TiO2 layer in planar perovskite solar cells: an interfacial electrical, optical and carrier mobility study. Sol. Energy Mater. Sol. Cells 163, 242–249 (2017).
Jeong, I. et al. A tailored TiO2 electron selective layer for high-performance flexible perovskite solar cells via low temperature UV process. Nano Energy 28, 380–389 (2016).
Yoo, J. J. et al. Efficient perovskite solar cells via improved carrier management. Nature 590, 587–593 (2021).
Yang, L. et al. Record-efficiency flexible perovskite solar cells enabled by multifunctional organic ions interface passivation. Adv. Mater. 34, 2201681 (2022).
Dong, Q. et al. Flexible perovskite solar cells with simultaneously improved efficiency, operational stability, and mechanical reliability. Joule 5, 1587–1601 (2021).
Lee, J. H. et al. Interfacial α-FAPbI3 phase stabilization by reducing oxygen vacancies in SnO2−x. Joule 7, 380–397 (2023).
Wang, G. et al. V-shaped tin oxide nanostructures featuring a broad photocurrent signal: an effective visible-light-driven photocatalyst. Small 2, 1436–1439 (2006).
Lim, S. et al. Suppressed degradation and enhanced performance of CsPbI3 perovskite quantum dot solar cells via engineering of electron transport layers. ACS Appl. Mater. Interfaces 13, 6119–6129 (2021).
Hu, X. et al. 22% efficiency inverted perovskite photovoltaic cell using cation-doped brookite TiO2 top buffer. Adv. Sci. 7, 2001285 (2020).
Duan, Y. D. et al. Enhancing the performance of dye-sensitized solar cells: doping SnO2 photoanodes with Al to simultaneously improve conduction band and electron lifetime. J. Mater. Chem. A 3, 3066–3073 (2015).
Ma, Z. et al. Negligible hysteresis planar perovskite solar cells using Ga-doped SnO2 nanocrystal as electron transport layers. Org. Electron. 71, 98–105 (2019).
Song, J. et al. Colloidal synthesis of Y-doped SnO2 nanocrystals for efficient and slight hysteresis planar perovskite solar cells. Sol. Energy 185, 508–515 (2019).
Singh, A. K., Janotti, A., Scheffler, M. & Van de Walle, C. G. Sources of electrical conductivity in SnO2. Phys. Rev. Lett. 101, 055502 (2008).
Duan, Y. D. et al. Effects of Ga doping and hollow str`ucture on the band-structures and photovoltaic properties of SnO2 photoanode dye-sensitized solar cells. RSC Adv. 5, 93765–93772 (2015).
Xu, X., Zhuang, J. & Wang, X. SnO2 quantum dots and quantum wires: controllable synthesis, self-assembled 2D architectures, and gas-sensing properties. J. Am. Chem. Soc. 130, 12527–12535 (2008).
Roose, B. et al. A Ga-doped SnO2 mesoporous contact for UV stable highly efficient perovskite solar cells. J. Mater. Chem. A 6, 1850–1857 (2018).
Choi, W. K., Jung, H. J. & Koh, S. K. Chemical shifts and optical properties of tin oxide films grown by a reactive ion assisted deposition. J. Vac. Sci. Technol. A 14, 359–366 (1996).
Barr, T. L. Recent advances in x‐ray photoelectron spectroscopy studies of oxides. J. Vac. Sci. Technol. A 9, 1793–1805 (1991).
Li, J. & Zeng, H. C. Hollowing Sn-doped TiO2 nanospheres via ostwald ripening. J. Am. Chem. Soc. 129, 15839–15847 (2007).
Gu, F. B., Wang, H. T., Han, D. M. & Wang, Z. H. Enhancing the sensing performance of SnO2 inverse opal thin films by In and Au doping. Sens. Actuators B: Chem. 245, 1023–1031 (2017).
Babu, B. et al. Bandgap tuning and XPS study of SnO2 quantum dots. Mater. Lett. 221, 211–215 (2018).
Hilal, M. & Yang, W. A dual-functional flexible sensor based on defects-free Co-doped ZnO nanorods decorated with CoO clusters towards pH and glucose monitoring of fruit juices and human fluids. Nano Converg. 9, 14 (2022).
Lju, L. Z. et al. Determination of surface oxygen vacancy position in SnO2 nanocrystals by raman spectroscopy. Solid State Commun. 151, 811–814 (2011).
Xie, J. S. et al. Enhanced electronic properties of SnO2 via electron transfer from fraphene quantum dots for efficient perovskite solar cells. ACS Nano 11, 9176–9182 (2017).
Protesescu, L. et al. Nanocrystals of cesium lead halide perovskites (CsPbX3, X = Cl, Br, and I): novel optoelectronic materials showing bright emission with wide color gamut. Nano Lett. 15, 3692–3696 (2015).
Lee, J. H. et al. Band alignment engineering between planar SnO2 and halide perovskites via two-step annealing. J. Phys. Chem. Lett. 10, 6545–6550 (2019).
Ayguler, M. F. et al. Influence of fermi level alignment with tin oxide on the hysteresis of perovskite solar cells. ACS Appl. Mater. Interfaces 10, 11414–11419 (2018).
Park, S. Y. & Zhu, K. Advances in SnO2 for efficient and stable n–i–p perovskite solar cells. Adv. Mater. 34, 2110438 (2022).
Xiang, W., Liu, S. & Tress, W. Interfaces and interfacial layers in inorganic perovskite solar cells. Angew. Chem. Int. Ed. 60, 26440–26453 (2021).
Khan, J. et al. Low-temperature-processed SnO2-Cl for efficient PbS quantum-dot solar cells via defect passivation. J. Mater. Chem. A 5, 17240–17247 (2017).
Li, Z. et al. Annealing free tin oxide electron transport layers for flexible perovskite solar cells. Nano Energy 94, 106919 (2022).
Shao, Y. et al. Origin and elimination of photocurrent hysteresis by fullerene passivation in CH3NH3PbI3 planar heterojunction solar cells. Nat. Commun. 5, 5784 (2014).
Yang, D. et al. High efficiency planar-type perovskite solar cells with negligible hysteresis using EDTA-complexed SnO2. Nat. Commun. 9, 3239 (2018).
Song, S. et al. Systematically optimized bilayered electron transport layer for highly efficient planar perovskite solar cells (η = 21.1%). ACS Energy Lett. 2, 2667–2673 (2017).
Jeong, I. et al. Solution-processed ultrathin TiO2 compact layer hybridized with mesoporous TiO2 for high-performance perovskite Solar cells. ACS Appl. Mater. Interfaces 9, 36865–36874 (2017).
Chen, S. et al. Crystallization in one-step solution deposition of perovskite films: upward or downward? Sci. Adv. 7, eabb2412 (2021).
Yang, X. et al. Buried interfaces in halide perovskite photovoltaics. Adv. Mater. 33, 2006435 (2021).
Park, D. H. et al. Highly stretchable, high-mobility, free-standing all-organic transistors modulated by solid-state elastomer electrolytes. Adv. Funct. Mater. 29, 1808909 (2019).
Kim, B. J. et al. Highly efficient and bending durable perovskite solar cells: toward a wearable power source. Energy Environ. Sci. 8, 916–921 (2015).
Mela, I. et al. Revealing nanomechanical domains and their transient behavior in mixed-halide perovskite films. Adv. Funct. Mater. 31, 2100293 (2021).
Hamasha, M. M. et al. Stability of ITO thin film on flexible substrate under thermal aging and thermal cycling conditions. J. Disp. Technol. 8, 383–388 (2012).
Machinaga, H. et al. Effects of annealing temperature on mechanical durability of indium-tin oxide film on polyethylene terephthalate substrate. Thin Solid Films 559, 36–39 (2014).
Kim, G. et al. Impact of strain relaxation on performance of α-formamidinium lead iodide perovskite solar cells. Science 370, 108–112 (2020).
Kim, W. & Ko, M. J. Completely annealing-free flexible perovskite quantum dot solar cells employing UV-sintered Ga-doped SnO2 electron transport layers data sets. Figshare https://doi.org/10.6084/m9.figshare.25284907.v1 (2024).
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant (NRF- 2022M3J1A1064315, NRF-2021R1A2C2094794) and ERC Center funded by the Korea government (MSIT) (NRF-2022R1A5A1033719).
Author information
Authors and Affiliations
Contributions
M.J.K. and Y.K. designed and supervised the project. M.J.K., W.K. and J.K. constructed the concept, analyzed the data, and prepared the manuscript. M.J.K., W.K., D.K., and B.K. designed and characterized the Ga:SnO2 CNR. W.K., J.K., and S.Y. synthesized the CsPbI3-PQD and optimized the solar cell. M.J.K., Y.K., W.K., J.K., B.K., and Y.L. reviewed and edited the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, W., Kim, J., Kim, D. et al. Completely annealing-free flexible Perovskite quantum dot solar cells employing UV-sintered Ga-doped SnO2 electron transport layers. npj Flex Electron 8, 20 (2024). https://doi.org/10.1038/s41528-024-00305-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41528-024-00305-3
