
Abstract
Next-generation sequencing (NGS) has been proven to be one of the most powerful diagnostic tools for rare Mendelian disorders. Several studies on the clinical application of NGS in unselected cohorts of Middle Eastern patients have reported a high diagnostic yield of up to 48%, correlated with a high level of consanguinity in these populations. We evaluated the diagnostic utility of NGS-based testing across different clinical indications in 1436 patients from Iran, representing the first study of its kind in this highly consanguineous population. A total of 1075 exome sequencing and 361 targeted gene panel sequencing were performed over 8 years at a single clinical genetics laboratory, with the majority of cases tested as proband-only (91.6%). The overall diagnostic rate was 46.7%, ranging from 24% in patients with an abnormality of prenatal development to over 67% in patients with an abnormality of the skin. We identified 660 pathogenic or likely pathogenic variants, including 241 novel variants, associated with over 342 known genetic conditions. The highly consanguineous nature of this cohort led to the diagnosis of autosomal recessive disorders in the majority of patients (79.1%) and allowed us to determine the shared carrier status of couples for suspected recessive phenotypes in their deceased child(ren) when direct testing was not possible. We also highlight the observations of recessive inheritance of genes previously associated only with dominant disorders and provide an expanded genotype–phenotype spectrum for multiple less-characterized genes. We present the largest mutational spectrum of known Mendelian disease, including possible founder variants, throughout the Iranian population, which can serve as a unique resource for clinical genomic studies locally and beyond.
Similar content being viewed by others

Introduction
Single-gene Mendelian disorders affect millions of individuals worldwide and account for an important public health burden as predominantly life-threatening or chronically debilitating conditions. Genetic diagnosis can benefit patients and their families by leading to appropriate therapy and providing insights into the prognosis and recurrence risk of the disorder within the family. However, establishing a definitive genetic diagnosis in many cases is a complicated and extended process, and a significant number of patients with presumed genetic disorders remain undiagnosed after receiving conventional clinical evaluation and genetic testing1,2.
In recent years, next-generation sequencing (NGS) has been proven to be one of the most powerful diagnostic tools for rare Mendelian disorders, especially genetically heterogeneous conditions. There are currently two general approaches in the clinical application of NGS assays. When a specific phenotype associated with a number of genes is suspected, targeted gene panel sequencing is applied, whereas exome sequencing (ES) is commonly implemented in the diagnostic evaluation of patients with a wide range of differential diagnoses or uncharacterized genetic diseases.
While the current diagnostic rate of NGS-based testing in unselected cohorts of patients generally ranges from 24% to 34%3,4,5,6,7,8,9, several studies in Middle Eastern populations have reported a higher yield of up to 48%10,11,12,13. The relatively higher yield in Middle Eastern patients is correlated with a high level of parental consanguinity and the predominance of autosomal recessive (AR) diagnoses in these populations. This signifies the implementation of NGS testing as a primary test of choice in countries within the so-called “consanguinity belt,” including Iran, with a 40% rate of consanguineous marriages14,15.
Despite the anticipated high frequency of AR disorders in the Iranian population, the focus has been mainly on common monogenic disorders such as thalassemia, cystic fibrosis, phenylketonuria, spinal muscular atrophy, duchene muscular dystrophy, and fragile X syndrome, which are detectable through conventional genetic testing. However, comprehensive data on the prevalence and genetic diversity of rare Mendelian disorders in this region is currently lacking, primarily due to limited access to advanced genome sequencing methods16. Consistent with increased affordability and access to NGS technologies in developing countries in recent years, NGS testing is becoming widely ordered by clinicians across Iran. This represented a unique opportunity to evaluate the diagnostic utility of this approach across different clinical indications in Iranian patients and allowed us to create the most comprehensive view of the mutational spectrum of known Mendelian disease throughout the country. We report a retrospective analysis of data from 1436 consecutive cases referred to our clinical diagnostic laboratory for ES or targeted gene panel testing over an 8-year period.
Results
Patient demographics
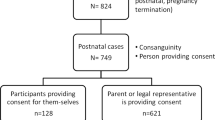
A total of 1436 Iranian index cases were referred for diagnostic NGS testing, with 1075 (74.9%) submitted for exome sequencing and 361 (25.1%) for targeted gene panel sequencing. NGS was performed on 1315 probands only (solo, 91.6%), 42 probands plus parents (trio, 2.9%), and 79 one or both healthy parents of the deceased affected child(ren) (parents, 5.5%). The majority of patient referrals (62.3%), encompassing 76.4% of ES cases (821/1075), were between 2018 and 2020. The breakdown of clinical indications in ES cases over time is presented in Supplementary Fig. 1.
The patient population comprised 777 males (54.1%) and 614 females (42.8%), in addition to 45 (3.1%) fetal cases from terminated pregnancies. The age of probands at testing, categorized into three age groups, ranged between prenatal to 72 years. Notably, parental consanguinity was present in the majority of cases (72.4%), while most patients (65%) had a negative family history (Table 1). Overall, 56% (805/1436) of patients presented with neurological conditions (Supplementary Fig. 2).
The distribution of patients’ major clinical indications based on HPO is presented in Table 1. The two most common testing indications, collectively accounting for more than half of all referrals, were abnormality of the nervous system (29.5%) and abnormality of the musculature (26.9%). Developmental delay and/or intellectual disability and seizures were the most frequent indications in patients grouped as an abnormality of the nervous system. Patients within the abnormality of the musculature group mainly presented with muscular dystrophy, myopathy, and myasthenic syndrome. The next most frequent indications for referral were abnormalities of the ear (8.1%), metabolism/homeostasis (6.4%), central motor function (6.2%), peripheral neuropathy (6.1%), skin (4.9%), and eye (3.8%). A complete list of the patient’s phenotypic and genotypic information is provided in Supplementary Table 2.
Spectrum of identified variants
A total of 1115 unique variants in phenotype-related genes were reported across 1005 cases with non-negative results, including 286 (25.7%) pathogenic variants, 374 (33.5%) likely pathogenic variants, and 455 (40.8%) variants of uncertain significance, classified according to the ACMG/AMP guidelines (Supplementary Table 2). Remarkably, about 55.6% (620/1115) of all the reported variants, including 241 (36.5%, 241/660) of the pathogenic and likely pathogenic variants, were novel as defined by not being previously reported in patients described in the literature. A broad spectrum of variant types was observed among all the reported variants, including 612 missense, 186 nonsense, 178 frameshift, 102 splice-site, 24 in-frame insertion or deletions, 8 initiation codon, 2 stop-loss, 2 promoter region, and 1 synonymous. Of 660P/LP variants, 406 (61.5%) were null variants (nonsense, frameshift, ±1 or 2 splice sites, initiation codon), 241 (36.5%) were missense variants, and 13 (2.0%) were other variant types (in-frame insertion or deletions, stop-loss, promoter region, synonymous) (Fig. 1a).

a Distribution of the identified variants classified according to the ACMG guidelines and variant types. VUS variants of uncertain significance, P/LP pathogenic and likely pathogenic variants, Null nonsense, frameshift, ±1 or 2 splice sites, and initiation codon variants; Other, in-frame insertions or deletions, stop-loss, synonymous and promoter region variants. b Diagnostic yield based on major clinical indications (top-level HPO term).
Diagnostic yield
Overall, across the 1436 cases, a positive molecular diagnosis was reported in 46.7% (n = 670), a VUS result was reported in 22.1% (n = 318), an unclear result was reported in 1.2% (n = 17), and 30% (n = 431) received a negative result. There was a higher diagnostic yield from patients referred for targeted testing (54.3%, 196/361) compared to ES patients (44.1%, 474/1075). The frequency of positive results was the highest among cases performed as parents (49.4%, 39/79), followed by solo (47%, 618/1315) and trio cases (31%; 13/42) (Table 1). In the analysis of the diagnostic outcome in phenotype groups, the yield was highest for abnormalities of the skin (67.6%), blood and blood-forming tissues (64.7%), the musculature (54.5%), the skeletal system (51.4%), the ear (50%), central motor function (46.1%), and metabolism/homeostasis (45.6%). In contrast, abnormalities of the cardiovascular system (25%) and prenatal development/birth (24.2%) had the lowest diagnostic yield (Fig. 1b).
The distribution of the presumed mode of inheritance in positive cases is presented in Table 2. Autosomal recessive inheritance accounted for the majority of positive cases (79.1%, 530/670), followed by autosomal dominant inheritance (14.6%, 98/670) and X-linked inheritance (6.3%; 42/670). Of the 530 autosomal recessive diagnosed conditions, 441 (83.2%) were linked to homozygous variants, 50 (9.4%) demonstrated compound heterozygosity of two distinct variants, and 39 (7.4%) were cases of heterozygous variants identified in the parent(s) of a deceased patient. Among the 98 autosomal dominant diagnosed conditions, 46 (46.9%) were linked to a de novo variant, 9 (9.2%) were inherited from a symptomatic parent, 4 (4.1%) resulted from parental gonadal mosaicism, and 39 (39.8%) remained undetermined due to unavailability of parental samples. Of the 42 X-linked diagnosed disorders, 36 (85.7%) occurred in males and 6 (14.3%) in females; 15 X-linked alleles were inherited from healthy mothers, 7 resulted from de novo variants, 3 were inherited from a symptomatic parent, and 17 remained with unknown parental origin.
Dual diagnoses
Among the 474 cases with a positive ES result, 11 patients (2.3%) received a dual molecular genetic diagnosis (targeted cases were excluded considering the limitations of panel sequencing to detect dual diagnosis comprehensively). The clinical and genetic information of these patients is provided in detail in Table 3 and Supplementary Table 3. The majority of patients in this group were considered to have overlapping phenotypes based on the observation of one or more clinical features associated with both molecular diagnoses, while patients for whom no phenotypic features were shared between molecular diagnoses were categorized as having two distinct phenotypes. Consanguinity was reported for the majority of these cases (81.8%, 9/11), and autosomal recessive phenotypes were expectably the most frequent observation in this group.
Important medical implications of diagnostic NGS
Medically actionable secondary findings in the ACMG-recommended list of 59 genes were analysed in 270 cases referred for ES testing. Reportable findings were identified in nine cases (3.3%), which include P/LP variants in MYBPC3 (2), DSG2 (2), MUTYH, OTC, TNNI3, BRCA1 and BRCA2 (Supplementary Table 4). The positive diagnostic results had the potential to impact the treatment and clinical management of several patients. Selected examples included children diagnosed with Wilson disease (MIM:277900), DOPA-responsive Dystonia (MIM: 128230), Pyruvate dehydrogenase E1-alpha deficiency (MIM:312170), Pyridoxine-dependent Epilepsy (MIM: 266100), and Cerebral creatine deficiency syndrome 2 (MIM:612736). Furthermore, predictive testing for subsequent pregnancies (prenatal or preimplantation genetic diagnosis) was performed in 16% (n = 108) of families with positive diagnoses.
Recurrent molecular findings
The majority of the positive cases (68.2%, 457/670) had P/LP variants in a gene at least twice observed in this series (117 genes). These genes were predominantly associated with neuromuscular disorders, sensorineural hearing loss, skin disorders and various neurodevelopmental phenotypes, providing insight into the prevalence of these genetic conditions among Iranian patients. The recurrently mutated genes in general and in each phenotypic group are listed in Supplementary Table 5 and Fig. 3. The most commonly diagnosed disorders in our cohort, each accounting for 1–5% of positive results, were muscular dystrophies related to CAPN3 (n = 34), DYSF (n = 15), LAMA2 (n = 12), SGCA (n = 11), and DMD (n = 22), NF1-related neurofibromatosis (n = 12), ATM-related ataxia (n = 11), MYO7A-related hearing loss (n = 10), CHRNE-related myasthenic syndrome (n = 9), RYR1-related congenital myopathy (n = 8), SPG11-related spastic paraplegia (n = 8), and TYR-related Albinism (n = 7).
Furthermore, of the reported recessive P/LP variants, 59 were identified in two or more unrelated positive cases, and 35 were present in the heterozygous state in at least one healthy Iranian individual in the Iranome database17 (Supplementary Table 6). These variants, collectively accounting for 14.2% (94/660) of all the identified P/LP variants, suggest presumptive founder effects in the Iranian population that require additional haplotype and population-specific analysis for confirmation. Notably, among these presumed founder variants, 28 were absent from other population databases, including gnomAD and the Greater Middle East (GME) variome18. This subset encompasses 19 novel or rare recessive pathogenic variants exclusively reported in Iranian patients up to the present, which could be denoted as “Iranian-only” mutations. It is noteworthy that several of these variants were novel missense variants that could be classified as likely pathogenic only through observation in two unrelated cases with the same phenotypic presentation. These variants include MME c.499T>A (p.Trp167Arg), SGCA c.113A>G (p.His38Arg), FKRP c.1034G>C (p.Gly345Ala), ASAH1 c.109C>A (p.Pro37Thr)19, and OTOA c.1727T>C (p.Ile576Thr).
Unexpected inheritance patterns and molecular events
The predominance of consanguineous families in this cohort led to the identification of bi-allelic variants in 11 genes with only an autosomal dominant pattern of inheritance in the OMIM database at the time of reporting. These genes were either not previously associated with any AR disorders or were reported with recessive inheritance only in extremely rare cases in the literature (Table 4). The clinical presentations in the majority of cases in this series were either similar to or more severe than the established dominant disorder. For example, in three separate cases with variants in TBX4, GLI3 (both published elsewhere20,21), and BICC1 genes, a severe embryonic condition was observed in the homozygous fetuses, while the heterozygous parents were either mildly affected or unaffected. Other remarkable examples include recessive forms of DCTN1-related neurodegenerative disorder, KCNC3-related spinocerebellar ataxia, HARS1-related Charcot-Marie-Tooth disease22, and MITF-related hearing loss23. Unlike previous observations of biallelic PKD1 variants in severe pediatric Polycystic Kidney Disease24,25, a homozygous missense variant in this gene was identified in a 33-year-old patient presenting with bilateral cystic kidneys, hypertension, urinary issues, and an apparently negative family history. This finding is consistent with the published literature on biallelic hypomorphic PKD1 alleles leading to a mild adult-onset phenotype26. In addition, a distinct and only recently reported recessive phenotype for the UFSP2 gene was observed in a patient presenting with global developmental delay, intractable epilepsy, and brain atrophy27. Variants in this gene were previously associated with autosomal dominant skeletal system disorders without neurological dysfunction.
Furthermore, despite the initial assumption of a recessive genetic condition in 3 families with multiple similarly affected children and healthy parents, we identified heterozygous pathogenic variants in AD disease genes (SETBP1, CRYAA, SMCHD1) in the affected children but neither of the parents, indicating possible parental gonadal mosaicism. (patients 184, 794, 1003 in Supplementary Table 2). Consanguineous marriage suggestive of a typical AR pedigree was interestingly reported in two of these families. Identification of de novo variants in AR disease genes was another unusual molecular event observed in 3 patients who were each compound heterozygote for two P/LP variants in SGCA, ASPM, and ABCA4 genes. In these individuals, one of the variants was parentally inherited while the other had arisen de novo (patients 36, 253, 398 in Supplementary Table 2).
Expanding the mutational and phenotypic spectrum of less characterized genes
Variants in genes with limited mutational/phenotypic evidence in the literature or with OMIM phenotypes based on a single study were identified in 11 patients (MRPS34, FIBP, LRP4, ZBTB11, CRIPT, BCKDK, PLAA, CC2D1A, TUBB2A, SOD1). A detailed description of molecular findings and observed phenotypes in these patients are provided in Supplementary Table 7. These include the third so far described patients with FIBP-related Thauvin–Robinet–Faivre syndrome and LRP4-related congenital myasthenic syndrome, the third variant so far associated with SOD1-related spastic tetraplegia and axial hypotonia, and the first truncating variant associated with TUBB2A-related cortical dysplasia with other brain malformations. Notably, a possible expanded phenotype was observed in a patient with a homozygous MRPS34 variant presenting with chronic motor neuropathy, which deviates significantly from the delayed psychomotor development reported in a single study for this gene28. Similarly, a milder clinical presentation for PLAA-related disorder was observed in two siblings with developmental delay, seizures, and hypotonia.
Discussion
Large-scale clinical genomic studies have significantly contributed to our understanding of the clinical relevance of human genetic variation. While studies in outbred populations predominantly contribute to the better diagnosis of autosomal dominant genetic diseases, Middle Eastern populations with high rates of consanguinity have been proven as a valuable resource for the genetic analysis of recessive disorders. Despite the abundance of published research studies using NGS in Iranian patients, especially for novel disease gene discovery29,30, the clinical utilization of this technology on an unselected group of patients and the overall distribution of known Mendelian disorders in this ethnically diverse population is lacking.
We present the results of diagnostic NGS-based testing in 1436 consecutive cases from a clinically heterogenous cohort of patients in Iran. Establishing the molecular diagnosis of over 342 known disorders in 670 cases, we achieved an overall diagnosis rate of 46.7%. Furthermore, in 22.1% of cases, we identified clinically relevant VUS with strong evidence supporting pathogenicity but lacking sufficient data to be classified as P/LP based on ACMG/AMP criteria. Segregation analysis in the family and additional clinical and functional information could provide the evidence required to re-classify these variants either as P/LP or likely benign. Our overall diagnostic yield is similar to previously reported large heterogenous clinical series in consanguineous Middle Eastern patients10,11,12,13, while it is higher than that of studies in outbred populations3,4,5,6,7,8,9, further supporting the positive impact of consanguinity on diagnostic yield. Other possible contributing factors include the early use of NGS testing in the diagnostic process in the majority of cases, different categories of indications, differences in sample size, the population’s genetic background, or other properties of the cohort.
As expected, targeted sequencing of small gene panels, most commonly ordered for highly specific conditions such as epidermolysis bullosa, osteogenesis imperfecta, albinism, non-syndromic ichthyosis, etc., had a higher diagnosis rate than ES testing, which was typically ordered for more complex phenotypes and highly heterogenous groups of disorders. Furthermore, among patients with neurological conditions, those presenting with more specific neurological symptoms (e.g., ataxia, spastic paraplegia, sensory and motor and neuropathies) had the highest molecular diagnosis rate (46.9%, 139/296), likely due to facilitated genotype–phenotype correlation analysis4 (Supplementary Fig. 2). In line with previous reports6,12, we observed a statistically higher diagnostic yield in patients with positive family history (53.6% vs. 42.9%, p = 0.0002) and consanguinity (48.6% vs. 41.7%, p = 0.024). However, a lower diagnosis rate was achieved in our trio exome analysis compared to proband-only cases, which could be explained by the bias toward ordering trio testing for the nonconsanguineous families, and usually as a second-tier test.
Although the numbers are still modest for some clinical indications, a diagnostic yield of at least 24% is observed in all phenotypic groups. While disorders of the skin, blood, musculature, skeletal system, ear, metabolism, central motor function, peripheral nervous system, and eye have a high diagnostic yield (41–68%), other indications such as abnormality of prenatal development and the cardiovascular system show low diagnostic yield (<30%). The lack of sufficient phenotyping, as well as the higher likelihood of the presence of disease-causing structural genetic variations that remain undetectable by the NGS technique, might explain the lower yield obtained in patients in the latter two groups. The same trend and a low detection rate for isolated/less characterized phenotypes have been reported by previous studies6,8. Moreover, due to the shifts in referral patterns and the changing composition of high and low-yield phenotypes, we observed a decrease in the general diagnostic yield of ES over time. Specifically, referrals related to Musculature decreased from 79% to 20%, while cases associated with prenatal development increased from 0% to 4% of our patient cohort in the first to the last two years of the study period (Supplementary Fig. 1). This observation underscores that while the growing number of established disease genes over time can increase the diagnosis rate of ES in specific patient groups, a significant portion of diagnostic success within certain phenotypes relies on already-known genes.
The highly consanguineous nature of this cohort led to several significant outcomes. First, it resulted in the diagnosis of autosomal recessive disorders in the majority of patients (79.1%), with the enrichment for homozygous P/LP variants (83.2%). Interestingly, we identified rare homozygous P/LP variants in 37 ostensibly non-consanguineous cases, which could be attributed to either a founder effect or hidden consanguinity in populations such as Iran with a historical prevalence of intrafamily and interclan marriages, where familial relationships may be too distant to appear in the pedigree. The majority of seemingly unrelated parents within this group originated from the same rural region or town, suggesting regional clustering of private recessive pathogenic variants30,31. Moreover, we observed the recurrence of the identical recessive P/LP variants in multiple unrelated patients, indicating presumed founder variants in the Iranian population, which could potentially serve as a unique resource for population-based screening programs. Second, it facilitated determining the shared carrier status of the majority of couples (49.4%, 39/79) referred for the diagnosis of a suspected recessive phenotype observed in their deceased child(ren), when there was no access to their sample for direct testing. The practical and high-yield advantage of duo parental analysis in investigating the cause of fetal demise or premature death (referred to as molecular autopsy by proxy) has been previously reported in consanguineous populations25,32. Consistent with our results, the diagnostic rate in this group of cases has been reported to be higher compared to the general diagnostic yield in the same population, reaching 63% through reanalysis and novel gene discovery methodology25. Therefore, this approach holds promise in providing accurate genetic counseling and informed reproductive choices among comparable inbred families. Nevertheless, the lack of direct confirmation of the candidate variant in the deceased index remains a notable limitation of this method. Third, it led to the observation of several instances of homozygous variants in genes previously associated only with dominant disorders. These cases have the potential to advance our understanding of the mutational mechanisms (loss of function, gain of function, dominant negative) of associated phenotypes, which can further improve variant interpretation and genotype–phenotype correlation analysis33. Finally, we identified novel variants in ultra-rare recessive disorders, expanding the genetic and phenotypic spectrum of less-characterized genes and phenotypes. The value of ES in characterizing patients with multiple diagnoses and those with clinically actionable secondary findings has been previously emphasized34,35. In this study, we observed dual molecular diagnoses in 2.3% of positive ES cases and identified secondary findings in 3.3% of the analyzed cases, a frequency comparable to previously reported studies10,36.
While AR disorders accounted for the majority of diagnoses in this cohort, we observed autosomal dominant conditions in 14.6% of patients. An increase in this rate is expected upon parental testing and confirmation of the de novo occurrence of additional heterozygous variants reported as VUS in several patients. It is noteworthy that we unexpectedly identified dominant de novo causative variants in 17 cases with positive parental consanguinity, two of which had occurred in typical AR pedigrees due to possible germline mosaicism in either parent. In addition, among consanguineous cases with a dual molecular diagnosis, two patients were diagnosed with both dominant and recessive phenotypes, and in one patient two de novo causative variants in different dominant disease genes were identified. These observations highlight the importance of the application of all inheritance patterns when testing patients with a known family history of consanguinity.
Proband-only analysis performed in 91.6% of our cases demonstrated the high diagnostic utility of this strategy in a consanguineous population. While trio testing is the most commonly recommended strategy that allows the identification of de novo variants and phasing of recessive variants, its high cost remains a major financial constraint in countries like Iran, where there is no medical insurance coverage for genetic testing. The superiority of parent–child trios over solo analysis is mainly demonstrated in outbred populations where the majority of genetic diagnoses are attributed to de novo dominant or recessive compound heterozygous disease alleles. While in inbred populations with enrichment for homozygous recessive disorders, variant phasing at the time of interpretation is not considered an important determinant factor for better diagnosis. However, it is noteworthy that solo testing misses the potential preventive utility of parent–child trios in informing consanguineous parents about the 3% residual risk for additional autosomal recessive disorders in their offsprings37.
The comparable diagnostic yields of first-tier and reflex exome sequencing in our study (365/835 (43.7%) vs. 109/240 (45.4%), p = 0.67) highlight the cost-saving benefit of this testing strategy when applied as a first-line diagnostic approach. Moreover, while previous studies have suggested the application of broadly designed panels or sub-exome approach (medical exome) as cost-effective solutions in countries with limited resources7,13, performing ES and limiting the analysis to established disease genes offers an advantage by providing an opportunity for more resolved cases upon reanalysis of existing sequencing data through a combined diagnostic/research approach.
As a reference laboratory, we lacked access to post-test clinical follow-up data necessary to evaluate clinical management changes in patients with positive diagnoses. Additionally, the diagnostic yield in our center would have been affected by the low quality and quantity of available clinical information in test orders and the lack of communication between clinicians and interpreters. This information can facilitate clinicopathologic correlation analysis and variant interpretation by enabling comparisons with previously reported cases. The partnership of the clinician with the molecular laboratory has been previously shown to increase diagnostic yield38. Therefore, post-test diagnostic assessments, including biochemical and radiological tests, complementary molecular tests (e.g., deletion and duplication analysis by multiplex ligation-dependent probe amplification (MLPA)), and segregation analysis within the extended family, can help diagnose numerous additional cases. For instance, in two families with single P/LP variants identified by exome sequencing in AR genes, large multi-exon deletions on the second allele were detected by further MLPA analysis (patients 288 and 1335 in Supplementary Table S2). In addition, in two patients with heterozygous likely pathogenic variants in the SMCHD1 gene, identified by ES, additional haplotype and DNA methylation analysis confirmed the digenic inheritance and thereby the diagnosis of Facioscapulohumeral muscular dystrophy 2 (FSHD2) (patients 184 and 744 in Supplementary Table 2).
Technical limitations may also account for a small but considerable fraction of our nondiagnostic cases. The presence of pathogenic copy number variation (CNV) may have been missed due to the non-optimized pipeline for CNV analysis. Considering the recent developments in algorithms for exon-level CNV detection by NGS, we anticipate an increase in the number of diagnosed patients upon reanalysis of the current cohort. This increase is especially expected for patients who received unclear diagnoses and who were carriers of P/LP variants in clinically relevant recessive genes, while a second variant was not detected. Moreover, considering the increasing number of newly discovered disease genes, improvements in bioinformatics algorithms, and new clinical manifestations in patients, we expect the annual reanalysis of ES data will continue to provide additional diagnoses. Nevertheless, other genetic changes such as translocations or inversions, repeat expansions, and alterations in intergenic, intronic, or regulatory regions remain undetected by exome or targeted sequencing. Our pipeline was also not designed or validated for detecting somatic mutations and mtDNA variants.
In conclusion, our data demonstrated the high diagnostic utility of NGS-based testing for a wide range of unselected phenotypes in Iranian patients. We identified recurrently diagnosed rare Mendelian disorders, shedding light on the prevalence of these genetic conditions in Iran. We show that proband-only genomic testing is an efficient and cost-effective diagnostic strategy in resource-limited countries with a high rate of consanguineous marriage and homozygous recessive genetic diseases. In addition to providing an expanded genotype–phenotype spectrum for less-characterized genes, we report observations of recessive inheritance of genes previously associated only with dominant disorders. Moreover, we highlight the potential of consanguineous populations for the diagnosis of suspected recessive conditions in deceased children by duo analysis of their parents. Our cohort’s genotypic and phenotypic data can serve as a unique resource for clinical genomic studies locally and beyond.
Methods
Patient population
This study includes 1546 sequenced samples comprising 1436 cases with suspected Mendelian disorders referred for clinical NGS testing at the Kariminejad-Najmabadi Pathology & Genetics Center, Tehran, Iran, from July 2012 to July 2020. Patients were referred by their clinicians either for ES or targeted gene panel sequencing. The study included 89 (6%) previously published cases.
For the majority of patients, proband-only testing was requested, whereas, for non-consanguineous families with negative family histories, clinicians were encouraged to consider trio-based sequencing. For couples (most commonly consanguineous) with suspected recessive phenotypes in their deceased children, NGS was performed on either or both individuals to determine their shared carrier status. In the earlier stages of the study, to minimize the financial burden on families, we exclusively performed targeted and exome sequencing on the mother, while the father’s analysis involved investigating the maternal candidate variant(s) through Sanger sequencing. This approach was later replaced by duo parental exome sequencing.
Samples were collected from index cases and available family members in the form of peripheral blood, tissue for fetal tissue samples obtained at autopsy, or extracted DNA samples. Pre- and post-test genetic counseling was provided by certified genetic counselors and/or a clinical geneticist. The clinical information provided by the referring clinicians, along with a family pedigree and test histories, e.g., MRI or genetic/metabolic, were carefully reviewed and collected.
We categorized patients’ clinical indications using top-level branching of Human Phenotype Ontology (HPO) nomenclature. In cases with multiple clinical features, the most medically impactful feature was considered for phenotypic categorization. Patients with developmental delay/intellectual disability often co-presented with other congenital anomalies, and multiple/specific neurological diagnoses such as seizures and autism spectrum disorder were categorized as “Abnormality of the nervous system.” Suspected motor neuron disease and movement disorders such as Spinal Muscular Atrophy (SMA), Amyotrophic lateral sclerosis (ALS), Hereditary Spastic Paraplegia (HSP), Ataxia, and Parkinson’s disease were grouped as “Abnormal central motor function.” For fetuses with multiple malformations, “Abnormality of prenatal development or birth” was selected. The referred patients either had previously undergone non-diagnostic genetic testing (karyotype, whole genome array CGH, molecular testing for specific genes) or were considered for NGS testing as a first-tier diagnostic approach.
Ethics approval and consent to participate
This study was approved in Iran by the ethics committee of the Kariminejad-Najmabadi Pathology and Genetics Center. Written informed consent was obtained from all patients or their guardians. The study was carried out in accordance with the declaration of Helsinki.
Exome/targeted sequencing and data analysis
Genomic DNA was extracted, and sequencing was performed by applying exome or targeted NGS protocols. For 1075 ES cases, the exome target regions were captured using Agilent SureSelect Human All Exome V4, V5, V6, and V7 Kits (Agilent Technologies, Inc., Santa Clara, CA, USA) or Twist Human Core Exome (Twist Bioscience, San Francisco, CA, USA). For 312 targeted cases, a custom-designed SureSelect hybrid capture panel targeting the coding regions of 685 confirmed disease-causing genes was used. For 49 targeted hearing loss cases, the OtoSCOPE® v6, v7, and v8 genetic testing platforms were used39. Paired-end sequencing was performed on Illumina sequencers (HiSeq 2000/2500/4000 and NovaSeq 6000) (Illumina, San Diego, CA, USA) according to the manufacturer’s protocol.
FASTQ files were mapped to the GRCh37/hg19 human reference sequence using Burroughs Wheeler Aligner (BWA)40. BAM processing, quality control, and coverage assessment were performed using Picard tools and the Genome Analysis ToolKit (GATK), adhering to the GATK best practices recommendations41. Variant calling was performed using GATK Haplotype Caller. An average coverage depth of 126X and 522X was obtained for exome and targeted sequencing samples, respectively, with 97.5% of the targeted regions (protein-coding exons based on CCDS and ±10 bp of flanking intronic sequence) sequenced at 10X and higher.
Variant analysis, interpretation, and reporting
Variants were annotated and filtered using Annovar42 and an in-house bioinformatics tool. For visual verification of alignments, Integrative Genomics Viewer43 was used. Common variants (≥1% in the general population) and recurrent artifact variant calls were filtered out based on the latest available versions of 1000G (http://www.1000genomes.org), the Exome Variant Server (http://evs.gs.washington.edu), the Exome Aggregation Consortium database (EXAC) (http://exac.broadinstitute.org), the Genome Aggregation Database (gnomAD) (https://gnomad.broadinstitute.org), Iranome (http://www.iranome.ir) and internal databases.
All intronic variants located outside the boundaries of 10 bp from the exons and synonymous variants, except those located at exon/intron boundaries, were filtered out. Variants were then prioritized considering inheritance patterns, phenotype compatibility, population frequencies, variant types, and in silico prediction scores based on the information obtained from several resources, including the Online Mendelian Inheritance in Man (OMIM), ClinVar, HGMD, PubMed and using a number of in silico prediction tools (SIFT, Polyphen-2, MutationTaster, CADD, dbscSNV among others).
For targeted testing, only the variants within the requested gene panel, out of the 30 offered clinically themed gene panels, were analyzed (Supplementary Table 1). For ES analysis, variants in the internally developed phenotype-associated gene lists were considered initially, and if no candidate variant was identified, continued analysis for all known disease-causing genes in the OMIM database was performed. For determination of the shared carrier status of a couple for a suspected recessive phenotype in their deceased child (ren) within the duo parental sequencing group, only the variants identified within mutual genes between both parents were considered for analysis, with additional consideration given to X-linked recessive (XLR) genes in the female parent. In single-parent sequencing cases, our analysis was restricted to variants within clinically relevant AR and XLR genes identified in the female parent, and the candidate variant(s) were further investigated through Sanger Sequencing in the male parent.
The analysis of secondary findings began in September 2019, coinciding with a delay in the accreditation of clinical exome sequencing in Iranian laboratories. ES results were analyzed for known or expected pathogenic variants in 59 medically actionable genes in accordance with the latest recommendations of the ACMG (ACMG SF v2.0)35. Prior to the specified variants in these genes were exclusively analyzed when relevant to the patient’s phenotype.
The selected list of candidate variants in each patient was re-evaluated by a local clinician to determine those most relevant to the patient’s phenotype, and the final candidate variants were classified according to the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) guidelines and the Clinical Genome (ClinGen) recommendations for using ACMG/AMP criteria44,45. The results were classified into the following four categories.
-
1.
Positive: Pathogenic or likely pathogenic (P/LP) variant(s) in an established disease-causing gene associated with the reported clinical indication and present in the expected zygosity. Results including assumed compound heterozygous variants with at least one variant classified as P/LP, and those with pathogenic or likely pathogenic variant(s) in more than one gene (Dual diagnoses), are also included in this category.
-
2.
VUS: Variant(s) of uncertain significance (VUS) in an established disease-causing gene associated/possibly associated with the reported clinical indication and present in the expected zygosity. This category includes VUS strongly suspected to be pathogenic but currently lacking sufficient evidence to be classified as P/LP by ACMG/AMP criteria.
-
3.
Unclear: Single heterozygous P/LP variant in an established disease-causing gene with autosomal recessive phenotype compatible with the patient’s clinical indication.
-
4.
Negative: No VUS/P/LP variants were identified in genes associated with the reported clinical indication.
Variant confirmation and segregation analysis
All the candidate variants detected by NGS were confirmed by conventional PCR amplification and Sanger sequencing. Parents and available healthy/affected family members were also tested by Sanger sequencing for segregation analysis and determination of the origin and phase of variants. Short tandem repeat testing was additionally performed to confirm parentage for apparent de novo variants.
Statistical analysis
The significance of the differences in diagnostic rate was analyzed with the Pearson chi-square test using SPSS. A p-value of 0.05 was used as a significance threshold.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request. All P/LP variants identified in this study have been submitted to the ClinVar database (Submission IDs: SUB10350875, SUB13967151), and accession numbers are provided in Supplementary Table 8.
References
Liu, Z., Zhu, L., Roberts, R. & Tong, W. Toward clinical implementation of next-generation sequencing-based genetic testing in rare diseases: where are we? Trends Genet. 35, 852–867 (2019).
Boycott, K. M., Vanstone, M. R., Bulman, D. E. & MacKenzie, A. E. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat. Rev. Genet. 14, 681–691 (2013).
Lee, H. et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 312, 1880–1887 (2014).
Yang, Y. et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 312, 1870–1879 (2014).
Farwell, K. D. et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet. Med. 17, 578–586 (2015).
Trujillano, D. et al. Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur. J. Hum. Genet. 25, 176–182 (2017).
Hu, X. et al. Proband-only medical exome sequencing as a cost-effective first-tier genetic diagnostic test for patients without prior molecular tests and clinical diagnosis in a developing country: the China experience. Genet. Med. 20, 1045–1053 (2018).
Hartman, P. et al. Next generation sequencing for clinical diagnostics: five year experience of an academic laboratory. Mol. Genet. Metab. Rep. 19, 100464 (2019).
Retterer, K. et al. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 18, 696–704 (2016).
Monies, D. et al. The landscape of genetic diseases in Saudi Arabia based on the first 1000 diagnostic panels and exomes. Hum. Genet. 136, 921–939 (2017).
Monies, D. et al. Lessons learned from large-scale, first-tier clinical exome sequencing in a highly consanguineous population. Am. J. Hum. Genet. 105, 879 (2019).
Al-Dewik, N. et al. Clinical exome sequencing in 509 Middle Eastern families with suspected Mendelian diseases: the Qatari experience. Am. J. Med. Genet. A 179, 927–935 (2019).
Saudi Mendeliome Group. Comprehensive gene panels provide advantages over clinical exome sequencing for Mendelian diseases. Genome Biol. 16, 134 (2015).
Saadat, M., Ansari-Lari, M. & Farhud, D. D. Consanguineous marriage in Iran. Ann. Hum. Biol. 31, 263–269 (2004).
Jalal Abbasi-Shavazi, M., McDonald, P. & Hosseini-Chavoshi, M. Modernization or cultural maintenance: the practice of consanguineous marriage in Iran. J. Biosoc. Sci. 40, 911–933 (2008).
Behnam, B. & Zakeri, M. Genetics and genomic medicine in Iran. Mol. Genet. Genom. Med. 7, e00606 (2019).
Fattahi, Z. et al. Iranome: a catalog of genomic variations in the Iranian population. Hum. Mutat. 40, 1968–1984 (2019).
Scott, E. M. et al. Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat. Genet. 48, 1071–1076 (2016).
Karimzadeh, P. et al. Five patients with spinal muscular atrophy-progressive myoclonic epilepsy (SMA-PME): a novel pathogenic variant, treatment and review of the literature. Neuromuscul. Disord. 32, 806–810 (2022).
Kariminejad, A. et al. Homozygous null TBX4 mutations lead to posterior Amelia with pelvic and pulmonary hypoplasia. Am. J. Hum. Genet. 105, 1294–1301 (2019).
Kariminejad, A. et al. A GLI3 variant leading to polydactyly in heterozygotes and Pallister-Hall-like syndrome in a homozygote. Clin. Genet. 97, 915–919 (2020).
Galatolo, D. et al. Bi-allelic mutations in HARS1 severely impair histidyl-tRNA synthetase expression and enzymatic activity causing a novel multisystem ataxic syndrome. Hum. Mutat. 41, 1232–1237 (2020).
Thongpradit, S. et al. MITF variants cause nonsyndromic sensorineural hearing loss with autosomal recessive inheritance. Sci. Rep. 10, 12712 (2020).
Al-Hamed, M. H. et al. Bialleleic PKD1 mutations underlie early-onset autosomal dominant polycystic kidney disease in Saudi Arabian families. Pediatr. Nephrol. 34, 1615–1623 (2019).
Shamseldin, H. E. et al. Lethal variants in humans: lessons learned from a large molecular autopsy cohort. Genome Med. 13, 161 (2021).
Pandita, S., Khullar, D., Saxena, R. & Verma, I. C. Autosomal dominant polycystic kidney disease: presence of hypomorphic alleles in PKD1 gene. Indian J. Nephrol. 28, 482–484 (2018).
Ni, M. et al. A pathogenic UFSP2 variant in an autosomal recessive form of pediatric neurodevelopmental anomalies and epilepsy. Genet. Med. 23, 900–908 (2021).
Lake, N. J. et al. Biallelic mutations in MRPS34 lead to instability of the small mitoribosomal subunit and Leigh syndrome. Am. J. Hum. Genet. 101, 239–254 (2017).
Najmabadi, H. et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 478, 57–63 (2011).
Hu, H. et al. Genetics of intellectual disability in consanguineous families. Mol. Psychiatry 24, 1027–1039 (2019).
Kahrizi, K. et al. Effect of inbreeding on intellectual disability revisited by trio sequencing. Clin. Genet. 95, 151–159 (2019).
Shamseldin, H. E. et al. Molecular autopsy in maternal-fetal medicine. Genet. Med. 20, 420–427 (2018).
Monies, D. et al. Autozygosity reveals recessive mutations and novel mechanisms in dominant genes: implications in variant interpretation. Genet. Med. 19, 1144–1150 (2017).
Posey, J. E. et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N. Engl. J. Med. 376, 21–31 (2017).
Kalia, S. S. et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet. Med. 19, 249–255 (2017).
Balci, T. B. et al. Debunking Occam’s razor: diagnosing multiple genetic diseases in families by whole-exome sequencing. Clin. Genet. 92, 281–289 (2017).
AlAbdi, L. et al. Residual risk for additional recessive diseases in consanguineous couples. Genet. Med. 23, 2448–2454 (2021).
Baldridge, D. et al. The Exome Clinic and the role of medical genetics expertise in the interpretation of exome sequencing results. Genet. Med. 19, 1040–1048 (2017).
Shearer, A. E. et al. Comprehensive genetic testing for hereditary hearing loss using massively parallel sequencing. Proc. Natl Acad. Sci. USA 107, 21104–21109 (2010).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Auwera, G. A. et al. From FastQ data to high‐confidence variant calls: the genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 43, 11.10.1–11.10.33 (2013).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Harrison, S. M., Biesecker, L. G. & Rehm, H. L. Overview of specifications to the ACMG/AMP variant interpretation guidelines. Curr. Protoc. Hum. Genet. 103, e93 (2019).
Acknowledgements
We are grateful to the patients and their families for participating in this study, and to the physicians who referred their patients to our center. This study received no funding.
Author information
Authors and Affiliations
Contributions
H.N. initiated and directed this project with contributions from A.K. A.Y.A. participated in the design of the study, performed data analysis, reviewed the literature and drafted the manuscript. Z.F. established and supervised the next-generation sequencing bioinformatics pipeline, performed sequencing data analysis and provided intellectual input to the manuscript. M.A.B. coordinated data collection, performed clinical interpretation of the molecular evidence, and provided support in statistical analysis. A.K. clinically evaluated results and supervised the genetic counselling group. R.V. provided support for drafting the manuscript, performed sequencing data analysis and participated in data collection and reevaluation. M.F., F.A., S.D., M.F.Z., and F.P. performed sequencing data analysis and participated in data collection and reevaluation. M.O.B., E.P. and Z.K. participated in sequencing data and segregation analysis. M.M.N. was a major contributor to data collection. K.N., F.F. and S.Z.N. provided support in data collection. A.K., H.Y., B.B., A.Z.A., S.S.M., P.N., F.F., P.J., M.R.A., P.K., H.H., K.K. and S.N. recruited the patients and provided expertise regarding the clinical details of the patients. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abolhassani, A., Fattahi, Z., Beheshtian, M. et al. Clinical application of next generation sequencing for Mendelian disease diagnosis in the Iranian population. npj Genom. Med. 9, 12 (2024). https://doi.org/10.1038/s41525-024-00393-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-024-00393-0
