
Abstract
Niemann-Pick type C (NPC) disease is a lysosomal storage disease (LSD) characterized by the buildup of endo-lysosomal cholesterol and glycosphingolipids due to loss of function mutations in the NPC1 and NPC2 genes. NPC patients can present with a broad phenotypic spectrum, with differences at the age of onset, rate of progression, severity, organs involved, effects on the central nervous system, and even response to pharmacological treatments. This article reviews the phenotypic variation of NPC and discusses its possible causes, such as the remaining function of the defective protein, modifier genes, sex, environmental cues, and splicing factors, among others. We propose that these factors should be considered when designing or repurposing treatments for this disease. Despite its seeming complexity, this proposition is not far-fetched, considering the expanding interest in precision medicine and easier access to multi-omics technologies.
Similar content being viewed by others

Introduction
Lysosomal storage diseases (LSDs) comprise a family of over 70 monogenic disorders. Most LSDs have an autosomal recessive pattern of inheritance. They are caused by defective lysosomal proteins, including soluble acidic hydrolases, integral membrane proteins, lipid and ion transporters, enzyme modifiers, activators, or non-lysosomal proteins crucial for lysosomal function. This results in the progressive accumulation of various metabolites within lysosomes, interfering with their integrity and function1,2. These rare disorders are generally classified according to the age of onset of the clinical symptoms1,3. Furthermore, in many LSDs, secondary storage of substrates unrelated to the primary genomic defect can result from defects in non-enzymatic lysosomal proteins, but the mechanisms are poorly understood. Regardless of the mechanism, continuous substrate accumulation within lysosomes can initiate cascades of cytotoxic effects, leading to cellular damage, cell death, and eventually, organ dysfunction and degeneration, especially in the central nervous system (CNS)4.
In LSDs, the genotype cannot entirely predict the severity of the phenotype or the clinical course. In fact, LSDs are notable for being highly heterogeneous despite having an eventually progressive course with varying degrees of neurological involvement. As such, the clinical phenotypes of LSDs are regarded to fall somewhere on a continuum spectrum that ranges from mild to severe or rapidly progressive forms3.
The biological factors accounting for the broad phenotypic spectrum of LSDs, including the different ages of symptoms onset, rates of disease progressions, severity, degrees of organ involvement, and response to the pharmacological treatments, remain poorly studied5,6.
Niemann-Pick disease type C (NPC): causes, subtypes, and clinical manifestations
NPC disease is a progressive neurovisceral autosomal recessive LSD caused by loss of function variants in the NPC1 or NPC2 genes7,8. NPC1 is located on chromosome 18q11-q12, spanning 56 kilobases (kb), and contains 25 exons. NPC2 is located on chromosome 14q24.3, spans 13,5 kb, and has five exons8. Most NPC patients (up to 95%) have mutations in NPC19, with the remaining few (less than 5%) in NPC2.
For a long time, NPC was reported to have an incidence of about 1/100,000–1/120,000 live births10,11,12. However, in recent years, several teams have suggested that such an incidence is a significant underestimation of its actual incidence. For instance, using next-generation sequencing data from four independent projects, Wassif and colleagues have estimated an incidence of the juvenile or classical forms of NPC of ~1:90,000, and even a higher estimated incidence of 1:19,000–36,000 for the late-onset phenotype (i.e., adult) subtype13.
NPC1 is a highly glycosylated protein of 1278 amino acids located in late endosomes and lysosomes (LE/Lys). It has 13 transmembrane domains (TMD), five of them (TMD 3–7) form the sterol detection domain (SSD), which is highly conserved within a subclass of membrane proteins that participate in cholesterol metabolism14,15,16. NPC1 also contains three luminal domains that play a role in protein-protein interactions: the C-terminal domain, the middle luminal domain, and the N-terminal domain, which is highly conserved and has a leucine-zipper motif. The N-terminal tail binds to the 3β-hydroxyl end of the cholesterol molecule with nanomolar affinity17,18. NPC2 is a cholesterol-binding glycoprotein of 132 amino acids also located in LE/Lys19. NPC2 recognizes the iso-octyl side chain of the cholesterol molecule and exposes the 3β-hydroxyl end to NPC1 for its export outside these organelles18,20. Historically, Winkler et al.21 used the Saccharomyces cerevisiae NPC system (composed of NCR1, the NPC1 equivalent, and NPC2 proteins) as a model to greatly contribute to the understanding of how sterols can be transferred through the lysosomal membrane: first, sterols would be transferred between the hydrophobic pockets of vacuolar NPC2 and the membrane protein NCR1. Second, sterols would pass through a tunnel formed in the central core of NCR1, bypassing the glycocalyx that covers the luminal side of the vacuolar membrane and mediating the insertion of the sterol into the membrane21.
In addition to cholesterol, other lipids, particularly glycolipids, have been shown to accumulate in several tissues22,23. Cholesterol and sphingomyelin are the major lipids stored in peripheral tissues. GM2 and GM3 gangliosides are observed in NPC brains, although glucosylceramide, lactosylceramide, and gangliotriaosylceramide are also known to build up in this organ. The accumulation of unesterified cholesterol in peripheral tissues (mainly in the liver and the spleen) is associated with the infiltration of activated macrophages, which leads to the production of proinflammatory cytokines and plays a critical role in cell death24,25. The buildup of gangliosides in the CNS causes structural and functional damage to neurons (formation of mega neurites, extensive growth of ectopic dendrites, formation of neurofibril tangles, neuroinflammation, and dystrophy), particularly in the cerebellar Purkinje neurons, triggering the characteristic motor dysfunction observed in the disease26,27. The dysfunction of non-neuronal cells in the brain, such as microglia and astrocytes, also contributes to neurodegeneration9,28,29,30,31.
Clinical presentations of NPC disease are highly heterogeneous and vary from a rapidly progressing neonatal form to an adult-onset chronic neurodegenerative condition. The life expectancy of the patients ranges from a few days until over 60 years of age, but most patients survive to 10–25 years of age32, although probably this is an underestimation According to data collected from a large cohort of French NPC patients, the age of onset of neurological symptoms predicts disease severity and determines life expectancy but current data suggests that NPC should be considered a continuum spectrum (Fig. 1, adapted from ref. 33). NPC is usually classified into four general categories according to the age of onset of neurological symptoms as follows33,34,35:

The main differences are in the degrees of hepatosplenomegaly, splenomegaly, and in neurological and psychiatric symptoms. This diagram shows the great phenotypic variation in NPC disease and within each subtype of it. Adapted from Vanier 201531.
Visceral-neurodegenerative forms
In utero and neonatal (newborn <3 months)
In this form of disease, the internal organs are the most affected30. Patients can present in-utero splenomegaly, in-utero hepatomegaly, in-utero ascites, and intrauterine growth retardation36. Placentomegaly has also been reported36. After birth, symptoms include congenital thrombocytopenia, congenital anemia, and petechial rash. Some patients present with fetal hydrops or fetal ascites30. Others suffer from a prolonged neonatal cholestatic icterus, associated with progressive hepatosplenomegaly37,38. Pulmonary alveolar lipoproteinosis has also been described39,40.
Early-infantile (3 months to <2 years)
Hepatosplenomegaly and/or prolonged neonatal jaundice are usually observed; however, hepatosplenomegaly can be the only symptom37,38. Other less frequent infantile forms with neurological involvement appearing by 12–18 months have been described41,42. This latest form is characterized by hypotonia and delay in developmental motor milestones, usually the first neurological symptoms. In these children, visceral disease is also generally present. The subsequent clinical course involves a loss of acquired motor skills, cognitive regression, and pronounced spasticity. In addition, patients may present with tremors, vertical supranuclear gaze palsy (VSGP), and seizures. Many children never learn to walk, and most die before age five.
Neurodegenerative forms (Classical form)
These are the most common forms of the disease, which correspond to ~60–70% of all cases. They are classified into two subgroups: the late-infantile and juvenile onsets. Patients with a late-infantile onset and intractable epilepsy tend to die the earliest. Nonetheless, many live until their teens, but others can survive into their twenties or thirties30.
Neurological symptoms, including ataxia, dysmetria, dysarthria, and dysphagia, among others, are associated with neurodegeneration, especially in the cerebellum, due to extensive and progressive Purkinje and cortical neuronal death, followed by hippocampal and autonomic nuclei dysfunction30.
Late-infantile (2–6 years)
These patients can also present with isolated hepatosplenomegaly or splenomegaly. The first neurological symptoms include gelastic cataplexy (frequent) and hearing loss, which becomes evident in the adult forms of the disease35. Patients can show clumsiness due to ataxia, gait disturbance, frequent falls, VSGP (vertical supranuclear gaze palsy, usually present but often unrecognized), fine motor skill impairments, speech delay, and epilepsy. As patients age, motor problems worsen, and they develop dysphagia, dysarthria, and dementia. Most patients with this form survive between 7 to 12 years of age35.
Juvenile (6–15 years)
This is the second most frequent form of the disease. The first sign is splenomegaly and, less commonly, hepatosplenomegaly, which also may be detected earlier (including the neonatal period). These patients suffer a significant motor impairment and a variable cognitive decline. They may manifest poor school performance, language and learning difficulties, impaired attention, coordination problems such as clumsiness, frequent falls, progressive ataxia, dystonia, and VSGP (often as the initial sign). Later, they may present with dysarthria, dysphagia, seizures (in about half of these patients), dystonic postures, and psychotic signs. Life expectancy is variable, but usually, they survive till their teens to young adulthood30,35.
Psychiatric-neurodegenerative forms
Adolescent/Adult (>15 years)
Adolescent and adult-onset NPC patients may represent up to a third of all NPC patients. This group may encompass a broad array of time to symptom onset/rate of disease progression. Neurological manifestations frequently occur in these patients. The typical presentation is a history of severe progressive ataxia, dystonia, VSGP, variable cognitive decline, and psychotic symptoms. Other commonly reported symptoms are dysarthria and dysphagia, which occur later. Patients with this form are less likely to present with seizures, gelastic cataplexy, and usually show mild visceral disease, which may have been known for years30,35.
Lately, large numbers of older NPC adults (>30 years) are being identified. Many of them are experiencing a relatively favorable quality of life. They have been able to complete school or be in professions as they present very slow disease progression, which usually starts with physical difficulties and memory impairment. They generally present with progressive hearing loss35.
The psychiatric symptoms include paranoid delusions, auditory or visual hallucinations, and interpretative thoughts. Other mental manifestations include depression, behavioral problems with aggressiveness, and social isolation. Several cases have also been reported with bipolar disorder and obsessive-compulsive30,43,44. Patients are frequently being treated for bipolar, schizophrenia, or psychosis ahead of the NPC diagnosis.
Diagnosis
The combination of biochemical and genetic studies can confirm NPC diagnosis, and the analysis of biomarkers should be preferred as a first-line test to screen for NPC. Currently, three biochemical markers can be used in combination: oxysterols (cholesterol oxidation products) and lyso-sphingomyelin45,46. However, in all cases, the diagnosis must be confirmed by genetic analysis and, if necessary, by a Filipin test, a fluorescent probe that binds free cholesterol in skin fibroblasts46.
Genotype-phenotype correlations in NPC disease
More than 500 disease-causing variants have been described in different regions of the NPC1 and NPC2 genes. It has been challenging to establish genotype–phenotype correlations, mainly due to the private nature of most NPC1 variants and because even the most common variants (I1061T, P1007A, and G992W) are found in low frequency. However, some studies have shown a correlation between carrying nonsense or frameshift variants and a more severe neurological course. In addition, homozygous variants in SSD are usually deleterious. Variants in the cysteine-rich luminal loop, where the three most frequent variants are found, show a variable clinical impact. Still, they seem to have a minor effect on cellular trafficking. Concerning NPC2, on the other hand, a relatively frequent nonsense variant (E20X) and many different variants that lead to a truncated protein have been associated with very severe clinical phenotypes. In addition, it has been seen that the missense variants led to more varied phenotypes. These have been reported in juvenile and adult-onset forms of the disease30,47,48.
Dardis and colleagues49 performed a multicentric study to characterize the molecular basis of NPC in Italy. In this study, 105 patients belonging to 83 unrelated families were included. Seventy-seven families presented variants in the NPC1 gene, while six in NPC2. The data obtained were consistent with previous reports: the mutational spectrum was highly heterogeneous, even in patients with the same causal variant and within the same family. Most patients with the early infantile severe lethal and the severe infantile forms were associated with the presence of null variants in both alleles (partial deletions, frameshift/nonsense variants, or splicing variants showed to be severe by functional analysis), while all patients affected by the juvenile and adult forms harbored missense variants in at least one allele49.
Unfortunately, the correlation between NPC1 mutations and clinical phenotypes has not yet been thoroughly investigated at the molecular level. Recently, Shammas studied the pathogenicity of common NPC1 gene variants by evaluating their effect on protein and cellular levels. They expressed NPC1-Flag tagged chimeras with and without variants in COS-1 cells and identified the biosynthetic forms of wild-type NPC1 by immunoprecipitation and endoglycosidase H (endo H) treatment. They found that mutant proteins in different domains of NPC1 had different trafficking patterns, with mutants that were partially trafficked or blocked in the ER associated with infantile clinical onset, late infantile, or juvenile-onset. However, the limited sample of mutants and the use of heterologous cellular models represent the limitations of this study50. Despite its limitations, however, this study established the concept of phenotypic heterogeneity of NPC1 variants at the biochemical and cellular levels and correlated trafficking classes along the secretory pathway of NPC1 variants with clinical phenotypes and symptom severity. This offers a potential method to assess the pathogenicity of different NPC1 mutants50.
Using the described methodology, the biochemical phenotypes of cultured fibroblasts from two NPC patients carrying different compound heterozygous NPC1 variants were assessed51. The fibroblasts from patient one had a delay in trafficking along the secretory pathway, while fibroblasts from patient two had normal trafficking. The lipid levels were also measured in the fibroblasts, and patient one had higher cholesterol levels and lower levels of one type of glycolipid compared to patient two. Sphingosine levels were higher in both NPC fibroblasts, but phospholipid levels were not significantly different from normal cells. The distribution of a protein called flotilin-2, a classical lipid raft marker, was found to be variant-dependent. Treatment with Miglustat, a drug used to treat NPC, reduced cholesterol, and glycolipid levels in both patients. However, it did not change phospholipid and sphingosine levels or was able to restore normal lipid raft structure in NPC cells. These findings help explain why treatment effectiveness with this drug varies among patients51.
Family cases with different severities
Though some variants are frequently detected as associated with specific phenotypic presentations, many exceptions exist, even within the same family, suggesting that additional genetic and/or non-genetic factors could act as disease modifiers.
In 2009, a case of phenotypic heterogeneity in two non-identical siblings, a male, and a female, with the adult-onset form of NPC disease-carrying the same causing variant, was reported by a different group52. They presented differential schizophrenia-like psychosis and showed dimorphism in illness course, clinical, and biochemical parameters. The male sibling presented with psychosis at age 16 and cognitive and motor disturbance at age 25, whereas his older sister showed less severe biochemical, neuroimaging, and ocular motor parameters. She presented with a similar schizophrenia-like illness with an associated cognitive and motor disturbance at the age of 31 years. In this case, the disease onset, course, and response to treatment replicate the sexual dimorphism seen in schizophrenia and suggest an interaction between the neurobiology of the metabolic disorder of these siblings and the sex differences in neurodevelopment53. In this respect, it is interesting to note that the sexual dimorphism in schizophrenia, concerning the onset and course of the disease, has been recognized since Kraepelin first described this disorder54.
As a result, it has been observed that most men experience their first schizoid episode in their late teens and early twenties, while most women become affected in their mid- to late- 20s. Moreover, men generally have greater severity of disease and worse long-term outcomes. Finally, women typically respond to lower doses of antipsychotic medication, although they show a higher rate of affective troubles in psychotic episodes53. At the molecular level, estrogen has been implicated in many of these variables because it can exert a protective effect on the structures involved in schizophrenia and the consequences of oxidative stress and excitotoxicity through its regulation of growth factors and ion channel function55. In addition, increased myelination in temporal areas has been observed in females during adolescence, suggesting a pro-myelination effect of female sex steroids on white matter development56. As these siblings showed a significant sexual dimorphism in the schizophrenia-like-psychosis that developed as the main symptom of adult-onset NPC, their case raised the possibility that gender can indeed influence the effects of NPC on the CNS, which could have implications for illness course, and treatment response. However, there is still insufficient data on humans on the effect of sex on the biochemical and clinical parameters of NPC disease.
In 2014, Benussi and colleagues57 reported the first case of extensive phenotypic variability in monozygotic twins carrying the same causal homozygous variant (c.2662 C > T; p.P888S) in the NPC1 gene. They were diagnosed at the age of 24. One of the twins presented with a history of obsessive-compulsive disorder and slowly progressive inferior limb clumsiness, dysphagia, dysarthria, and moderate splenomegaly. Neurological examination revealed a broad-based ataxic gait, limb dysmetria, downward vertical gaze palsy, brisk lower limb reflexes, and ankle clonus. The neuropsychological assessment revealed global cognitive deficits in multiple domains. He also presented intrusive recurrent, and persistent thoughts that caused significant stress and anxiety. The second twin showed mild obsessive-compulsive disorder without impact on daily life activities. The neurological and neuropsychological evaluation revealed mild neurological impairment without the involvement of cerebellar functions. Fibroblasts from both twins displayed similar filipin staining with significant intracellular accumulation of unesterified cholesterol and increased plasma cholestane-3b,5a,6b-triol57.
Other cases of NPC within familial contexts displaying varying symptomatology have been documented. In a consanguineous Turkish family, three cousins presented with hepatosplenomegaly and progressive neurodegeneration. Regrettably, some family members succumbed to similar complaints. Notably, vertical supranuclear gaze palsy was not consistently observed across all cases. Through molecular diagnosis, a homozygous c.1553+5 G > A intronic mutation in NPC1 was identified, resulting in exon 9 skipping and the production of an aberrant NPC1 protein58.
Another case involved two siblings who harbored compound heterozygous variants in exon 13 of the NPC1 gene (c.1955C>G, p.Ser652Trp and c.2107 T > A, p.Phe703Ile). While both individuals presented with nonspecific neurological symptoms, the age of onset differed between them. One sibling exhibited dyspraxia and motor clumsiness at the age of 7, whereas the other displayed a systemic manifestation characterized by hepatosplenomegaly noted at 2 months of age, followed by neurological symptoms emerging at 4 years, including speech disturbance42. Other cases of NPC siblings with discordant phenotypes have been reported34,59,60.
In conclusion, the presence of substantial phenotypic variability among familial NPC cases suggests a contribution from various factors such as epigenetic variations, post-zygotic mutagenesis, modifier genes, or environmental influences. To comprehensively comprehend the underlying molecular mechanisms, further research is warranted.
NPC mouse models of different genetic backgrounds for modifier gene mapping of disease severity
The term “modifier gene” refers to a locus where DNA sequence variation changes the phenotypes typically linked to “target genes” that act independently. The defining feature of modifier genetics is the interactions between specific alleles of the modifier and target genes61. Variations in modifier genes and pathways may explain why some patients with the same NPC1 mutation exhibit heterogeneous phenotypes and disease progression.
Variants that inactivate NPC1 have been described in other species, including mice. Two of the most widely used and well-characterized murine models of Npc1 disease, BALB/c npcnih 62 and C57BL/6Jspm (sphingomyelinosis or spm)63, arose by spontaneous mutation and led to a severe phenotype. However, recent reports describe other spontaneous or targeted alleles able to affect mice and induce an NPC phenotype. These include three alleles leading to a severe phenotype Npc1pf, Npc1imagine, and Npc1pioneer 64,65; two leading less severe phenotypes, Npc1nmf164 and Npc1I1061T 66,67; and one that allows tissue-specific deletion of Npc1, the Npc1flox 68.
In 1986, ref. 69 investigated whether the expression of the spm might be influenced by the genetic background. C57BL/KsJspm/spm mice show striking hepatosplenomegaly with a prominent accumulation of sphingomyelin and cholesterol due to a deficiency of sphingomyelinase. In this study, they constructed two congenic strains, C57BL/6Jspm (selected as a related substrain) and DBA/2 Jspm (selected as a strain of different origin). The degree of hepatic sphingomyelinase deficiency in these three mutant backgrounds was similar. Specifically, in C57BL/6Jspm and DBA/2Jspm mice, hepatosplenomegaly was not pronounced despite increased levels of hepatic lipids. Nonetheless, the lifespan of C57BL/6Jspm/spm and DBA/2Jspm/spm mice was shorter than that of C57BL/KsJspm/spm mice whilst the increase in hepatic lipid levels was faster in C57BL/6Jspm/spm and DBA/2Jspm/spm mice than in C57BL/KsJspm/spm mice, which could be associated with the presence or absence of hepatosplenomegaly and shortened lifespan. In general, the occurrence of hepatosplenomegaly appeared to have a protective effect against the accumulation of lipids. Finally, histological studies showed the formation of massive foam cell clusters of variable size in the liver and spleen of C57BL/KsJspm/spm mice, whereas, in the case of the C57BL/6Jspm/spm and DBA/2Jspm/spm mice, foam cells were diffusely distributed in smaller clusters of relatively uniform size. Taken together, these findings suggest that the appearance of hepatosplenomegaly in mutant mice depends on the genetic background69.
More recently, a study aimed at identifying modifier genes affecting the age of onset of neurological signs was performed70 by linkage analysis by crossing the Npc1nih/+ mutant on the BALB/cJ background with DBA/2J mice. Then, the F1 progeny were intercrossed, and 540 F2 mice were generated, of which 88 were affected mice (Npc1nih/nih). The onset of neurological symptoms, defined as when a continuous extensor tremor of the forelimbs occurs for more than 6–7 s, was used as a trait to perform QTL analysis. The age range of the disease onset of the 88 affected F2 mice was from 39 to 62 days. Of these 88 affected mice, 22 were identified as more severely affected (13 female, 9 male; onset less than 47 days), and 23 mice were mildly affected (6 female, 17 male; onset past 54 days). When their genome was sequenced, researchers found a significant peak on chromosome 19 (LOD score = 2.24) around D19Mit88 that accounted for 52% of the variance observed for the neurological onset in the affected F2 progeny (unfortunately, they did not have enough power resolution to identify specific gene variants)70.
In parallel to these studies, Parra and colleagues71 have also investigated the role of genetic background on NPC progression. The authors transferred the Npc1nih mutation from BALB/c to C57BL/6J genetic background by successive backcrossing with the offspring. The survival curves showed a striking difference between genetic backgrounds. BALB/c-Npc1−/− mice died between 70 and 85 days, while C57BL/6J- Npc1−/− mice died between 28 and 35 days. Interestingly, BALB/ c-Npc1−/− mice gained weight until 6–7 weeks old and then started to lose weight whilst C57BL/6J-Npc1−/− mice began to lose weight at 2–3 weeks old and were significantly smaller than BALB/c-Npc1−/− mice at all ages. As NPC disease is a lipidosis that affects multiple organs, the authors measured the lipid content of the brain, liver, and spleen of 4-week-old mice. Accordingly, they observed that liver and spleen cholesterol content were increased in Npc1−/− mice from both genetic backgrounds compared to the wild type. However, the relative increase in C57BL6/J-Npc1−/− mice was more considerable. In addition, they evaluated the brain, liver, and spleen phospholipid content. They found that BALB/c-Npc1−/− and C57BL/6J- Npc1−/− mice showed no differences in brain or liver phospholipid content but observed a fivefold difference in the spleen of C57BL/6J-Npc1−/− mice versus the wild type, which suggests that spleen damage due to lipid accumulation could be responsible for the phenotypic differences between these strains. Notably, they observed that the 4 weeks old brains and livers from both genetic backgrounds showed similar features. However, the spleens from C57BL/6J- Npc1−/− mice showed severe histological and morphological abnormalities compared to their controls. Accordingly, Npc1−/− mice from the C57BL/6 J genetic background showed significant spleen damage at early ages, whereas Npc1−/− mice from the BALB/c genetic background showed minor spleen alterations71. Finally, no Purkinje cell degeneration was observed at this age in both genetic backgrounds. Although no specific genes were mapped, these results indicate that genetic background can strongly influence NPC disease severity.
Finally, in 2020 Rodriguez-Gil and colleagues72 assessed the impact of genetic background on NPC disease severity in mice. In their approach, they used CRISPR/Cas9 to generate a new NPC1 mouse mutant, Npc1em1Pav/em1Pav (Npc1em), which has an in-frame deletion allele in the cysteine-rich domain of NPC1. These animals were generated and maintained on a C57BL/6J background and had many phenotypic features of NPC1 disease, such as reduced levels of NPC1 protein, lipid storage abnormalities (including accumulation of GSLs within visceral and neuronal tissue), visceral pathology (with the presence of foam cells in both the liver and spleen), Purkinje neuron loss in the cerebellum, motor impairment, neurodegeneration, and a reduced lifespan72. They then used marker-assisted selection/speed congenic techniques to establish a congenic strain of the Npc1em allele on a BALB/cJ genetic background. This experimental design allowed the production of Npc1em/em mice that showed ≥92% BALB/cJ homozygosity for all tested genetic markers by the N4 generation. When they analyzed these mutants at the N4 and N6 generations, the authors found that the new mice presented a significantly increased lifespan and less severe visceral pathology compared to the original C57BL/6J background. Analysis of N2 mice generated from a backcross using C57BL/6J and BALB/cJ found that Npc1em/em mutants also had an increased lifespan with greater variance, which suggests that strain-specific modifiers influenced disease severity and survival. This study included QTL analysis for the lifespan of 202 N2 mutants on a mixed C57BL/6J and BALB/cJ background and found markers associated with significant LOD scores on chromosome 1 (LOD = 5.57) and chromosome 7 (LOD = 8.91)72. Most importantly, these regions will provide candidate genes for future study as modifiers that could contribute to the variable phenotypes observed in NPC1 patients.
In conclusion, several NPC mouse models have been described in different genetic backgrounds. With the current advances in genetics and genomics, it is expected that they will represent very valuable resources for dissecting the genomic architecture underlying disease severity and progression of NPC73. Once identified, it is hoped that these modifier genes will facilitate the development of severity biomarkers and customized therapeutic approaches for subsets of patients.
Severity biomarkers
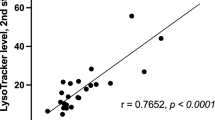
LSDs result in the expansion of the LE/Lys compartment in cells. Te Vruchte and colleagues74 showed that the relative lysosomal volume could serve as a potential biomarker for the age of onset and severity of these disorders. The assay uses the fluorescent probe Lysotracker, which accumulates selectively in the acidic compartments (LE/Lys). For their experiments, they stained B cells because they are a uniform population and do not change in response to infection. The assay was validated using resting splenic and circulating B cells from the NPC mice, and a progressive increase in relative acidic compartment volume was confirmed. Importantly, this expansion was seen at all ages (compared with age-matched controls) and was highly significant at each time point (3, 6, and 9 weeks) in both splenic and circulating B cells. Storage of GSLs in peripheral B cells occurred in parallel with the progressive storage of gangliosides and lactosylceramide in the forebrain of 2, 4, 7, and 9-week-old Npc1–/– mice compared with WT controls. This indicates that the lipid storage rate in peripheral B cells can be used as a proxy for the storage in the CNS74. Following these results, the B-cell lysotracker assay was validated and expanded to more than 100 NPC probands derived from multiple independent clinical cohorts, and this approach confirmed that this biomarker correlated with age-adjusted clinical severity scores and the levels of oxysterols, so these data support the use of Lysotracker for monitoring NPC disease75.
Previous reports have shown that the age of onset of neurological symptoms correlates with life expectancy and disease progression in NPC1 patients30,76. Therefore, knowing the age of onset of neurological symptoms in patients is crucial to predict the course of the disease and thus identify candidates who might benefit the most from earlier treatments. In this respect, Baxter and colleagues showed that the LysoTracker levels in skin fibroblasts of NPC patients could be used as a predictor for the age of onset and disease severity75. Their study found that LysoTracker levels were inversely correlated with the age of onset and the age of the first neurological symptom (the highest correlation). In addition, LysoTracker levels were directly associated with neurological severity scores. In the search for specific transcripts levels that may serve as biomarkers of severity, these researchers performed RNA-Seq in 41 NPC1 primary fibroblasts untreated and treated with 2-Hydroxypropyl-β-cyclodextrin (HPβCD) for 24 h. The gene expression data were correlated with the three clinical parameters: age of onset, age of first neurological symptoms, age-adjusted neurological severity score, and LysoTracker staining. The expression levels of 127 genes correlated with at least one clinical or cellular phenotype: ten genes were associated with the age of onset, 40 with the age of first neurological symptom, 43 with neurological severity score, 21 with LysoTracker levels in untreated cells, and 27 with changed in LysoTracker levels following HPβCD treatment. Most genes did not overlap among these correlations (only 14 genes correlated with two phenotypes), suggesting that phenotypes’ severity is affected by several genes/pathways rather than a small set of genes. Nonetheless, it is important to highlight that the pathways enriched in the total of 127 genes identified with this approach coincided with the role of NPC1 protein in cholesterol and GSL homeostasis.
Most importantly, Ingenuity Pathway Analysis identified 7 of the 127 genes as highly relevant biomarkers, which can be detected in human blood: GPNMB, HBEGF, HMGCR, PIK3CA, RSF1, STK11, and TLR4. Moreover, the drug–gene interaction database identified drug targets for several of the 127 genes, with ~53% of them that could be considered strong candidates for drug treatments. At present, treatments with these drugs are still in their infancy. All that can be said at present is that HPβCD treatment (ΔHPβCD values) did not change the gene expression of the mRNA biomarkers, suggesting that HPβCD did not affect many potentially clinically relevant genes in B cells. This study was the first to collect patient transcriptomic data, correlate it with clinical severity and cellular phenotypes, and suggest potential drug candidates for clusters of NPC patients. Variations in modifier genes and pathways could explain why some patients with the same NPC1 causal variant exhibit heterogeneous phenotypes and disease progression75.
Current therapeutic approaches and others under development
The primary molecular mechanism responsible for NPC symptomatology is the intra-lysosomal buildup of cholesterol and GSLs. Therefore, most current therapeutic interventions aim to lower their presence, either by inhibiting their synthesis or reducing their accumulation. Some drugs, such as Miglustat77, cyclodextrins (intravenous and intrathecally)78,79,80, and Vorinostat80, are the first drugs that have been tested in clinical trials for NPC. Still, several others have also been proposed, such as lovastatin, rapamycin, and arimoclomol, among others.
Miglustat
Miglustat (N-butyldeoxynojirimycin; NB-DNJ; Zavesca®), is a blood–brain barrier (BBB) permeable small iminosugar that acts as a competitive inhibitor of the glucosylceramide synthase enzyme, which catalyzes the first step in glycosphingolipid synthesis77.
Miglustat treatments in NPC cats reduced neuronal GSL accumulation, and the cellular pathology in the brain slowed the neurological progression of the disease and increased lifespan81. These animal models were followed by initial human clinical trials. Encouragingly, Miglustat-treated patients showed neurological improvements or stabilization82,83,84, although frequently reported adverse events included diarrhea, flatulence, weight loss, and tremors85.
The European Medicines Agency (EMA) and the Food and Drug Administration (FDA) had already approved Miglustat for the treatment of Gaucher disease86. In 2009, EMA extended Miglustat’s indication to treat the progressive neurological manifestations in NPC patients87. This extension was followed by other countries such as Japan and Canada. At the moment, this drug is the only treatment approved for NPC disease; however, the FDA has not approved its use for NPC disease88.
2-Hydroxypropyl-β-cyclodextrin (HPβCD)
HPβCD is a cyclic oligosaccharide with a hydrophobic center. HPβCD transports free cholesterol from the LE/Lys compartments to the cytosol, which reduces its accumulation24,89.
Either systemic or direct administration of HPβCD into the CNS of Npc1−/− mice normalized the biochemical abnormalities, reversed lysosomal cholesterol transport disturbances, increased their lifespans, significantly improved liver dysfunction, reduced lipid accumulation, and neurodegeneration24,90.
Clinical trials in NPC patients with NPC1 disease (NCT01747135, ClinicalTrials.gov) suggested that intrathecal treatment with HPβCD slowed disease progression91. Furthermore, a phase 1 trial involving intravenous administration of HPβCD demonstrated its biological activity in both the central nervous system and peripheral tissues of adult subjects79. The trial is registered under NCT02939547, and an open-label extension (NCT03893071), as well as a global pivotal trial (NCT04860960), are ongoing. The aforementioned phase 1 trial in patients was conducted subsequent to an extensive report on compassionate use of intravenous and intrathecal HPβCD, which concluded that the addition of intrathecal administration did not yield additional benefits, particularly considering the high doses used and the associated risks of neurotoxicities, including permanent and acute hearing loss91. In summary, these trials provide support for initiating longer-term clinical trials to assess the safety and efficacy of intravenous HPβCD in individuals with NPC92.
Vorinostat
Another approach explored as a potential treatment for NPC patients involves using histone deacetylase (HDAC) inhibitors. One of them, Vorinostat, is an FDA-approved drug for treating cutaneous T-cell lymphoma93.
In preliminary experiments, Munkacsi and colleagues administered Vorinostat intraperitoneally for 5 days at a 150 mg/kg body weight dose into the Npc1nih/nih mice93. Gene expression, liver function and pathology, serum and tissue lipid levels, body weight, and lifespan were used to follow disease progression. Studies showed a reversal of liver dysfunction. However, no positive effects on disease progression and animal survival were observed. Nonetheless, the metabolism of apolipoprotein B and the expression of the main components of lipid homeostasis in primary hepatocytes from mutant mice were altered by treatment with Vorinostat93. As a result, these results suggested that HDAC inhibitors could be applicable to the treatment of visceral NPC disease, but more work will be required to envision a future clinical trial with this compound.
In another similar study, Pipalia and colleagues treated NPC human fibroblasts with small HDAC inhibitors, which dramatically corrected the NPC phenotype in cells containing one or two copies of the NPC1 I1061T variant94. This effect was associated with increased NPC1 expression. Interestingly, the inhibition of HDACs was also identified as a candidate therapy for NPC in yeast92. The potential use of HDAC inhibitors for NPC treatment is high because they can cross the BBB and are being tested for mood behaviors95 in phase III clinical trials for several types of cancer96.
Arimoclomol
Arimoclomol is a hydroxamic acid that amplifies heat shock protein (HSP) gene expression to enhance the endogenous cytoprotective mechanisms in the presence of cellular stress97. This drug helps to preserve cellular function and to prevent cell death in cells experiencing lysosomal stress98. Arimoclomol can cross the BBB, and it can be detected in the cerebrospinal fluid of amyotrophic lateral sclerosis-treated patients98.
Regarding NPC disease, Arimoclomol-treated Npc1−/− mice re-activated the stress response in their brains, reducing neurodegeneration and improving locomotor coordination, but did not provide a rescue effect in the liver99. The treatment also enhanced myelination100. Furthermore, arimoclomol significantly reduced lysosomal storage and the buildup of unesterified cholesterol in NPC1 human fibroblasts101.
A Clinical trial showed that arimoclomol increased the likelihood of slowing disease progression102. Currently, the drug is available for its compassionate use (while collecting more safety data).
Activation of the Ca2+-permeable endo-lysosomal two-pore channel 2 (TPC2) by small molecules
Lysosomal Ca2+ release has significant physiological relevance because it regulates several cellular processes, such as autophagy, membrane trafficking, exocytosis, nutrient adaptation, membrane repair, and cell migration103. It has been reported that lysosomal Ca2+ content or Ca2+ release disturbances are associated with several diseases, mainly LSDs103,104. Mucolipidosis type IV (MLIV) represents the most direct link between defective lysosomal Ca2+ liberation and neurodegeneration. This disease is caused by dysfunction of the lysosomal cation channel TRPML1105. However, TRPML1 signaling or TRPML1-mediated Ca2+ release is also impaired in other LSDs such as NPC1, Niemann-Pick type A (NPA), and Fabry disease106. Accordingly, it has been observed that TRPML1 activation in cells can rescue lysosomal storage phenotypes106. However, the effects of activating the related two-pore channel 2 (TPC2) in LSDs have been less explored to this date. Recently, Rosato and colleagues107 hypothesized that TPC2 activation could modulate Ca2+ signaling and rescue LSD phenotypes, mainly in LSDs where TRPML1 is affected. They showed that small molecule activation of TPC2 ameliorates some cellular phenotypes associated with LSDs, such as cholesterol or lipofuscin accumulation and abnormal vacuoles. These rescue effects were evaluated in MLIV, NPC1, and Batten disease patient fibroblasts and neurons derived from isogenic human iPSC models of MLIV and Batten disease. For the in vivo proof of concept, they also tested TPC2 activation in the MLIV mouse model, rescuing neuroinflammation, accumulation of P62/SQSTM1 inclusions, and improving motor behaviors. The data suggested that TPC2 is a promising target for treating different types of LSDs107.
c-Abl inhibitors
The link between NPC neurodegeneration and the activation of the c-Abl/p73 apoptotic signaling has been studied in several disease models108,109,110,111. C-Abl is a kinase that phosphorylates and activates p73, a proapoptotic transcription factor. C-Abl and p73 are expressed in the cerebellum of NPC mice, and the levels of p73 target genes are increased. The expression of the active forms of p73 colocalizes with c-Abl and active caspase 3 in Purkinje cells. The inhibition of c-Abl with imatinib mesylate, an FDA-approved drug for treating chronic myelogenous leukemia, improves locomotor behaviors, reduces weight loss and neuronal apoptosis, and increases the survival of NPC mice [NO_PRINTED_FORM]. Thus, these inhibitors could be an excellent therapeutic alternative to attenuate the symptoms of NPC disease.
In parallel, it has been reported that histone deacetylation plays a relevant role in neuronal gene repression in neurodegenerative diseases94. In 2016, Contreras and colleagues112 demonstrated that c-Abl activity could also increase Histone deacetylase 2 (HDAC2) levels and activity, inducing neuronal gene repression in NPC models. As a result, c-Abl inhibition could prevent the increase of HDAC2 protein levels and activity in NPC neuronal models and the brain of mutant NPC mice112. Therefore, inhibition of c-Abl could be a pharmacological target for preventing the harmful effects of increased HDAC2 levels in NPC disease.
In addition to HDAC2, another report by Contreras and colleagues also explored the connection between c-Abl and the transcription factor EB (TFEB), a key element of lysosome signaling113. The study demonstrated that c-Abl phosphorylates TFEB on tyrosine and that its inhibition allowed TFEB translocation to the nucleus promoting the expression of its target genes. In addition, they found that c-Abl inhibitors promote a reduction in cholesterol accumulation in both in vitro and in vivo NPC models113. The TFEB-dependent clearance reveals the importance of the c-Abl/TFEB signaling as a therapeutic target for diseases with lysosomal dysfunction.
Alternative cholesterol exit from the lysosome
In recent years, several studies have focused on bypassing deficiencies in NPC1 and favor the release of cholesterol from the lysosome through alternative pathways. Interestingly, lysosomes, as well as other organelles and cellular compartments, such as the plasma membrane, lysosomes, endoplasmic reticulum (ER), mitochondria, Golgi apparatus, and lipid droplets, physically interact and communicate with each other, preserving their compartmentalization through Membrane Contact Sites (MCSs)114,115. The opposition between two organelles can form MCS through tethering proteins and lipids that allow the transfer of metabolites, such as calcium and lipids, thus modulating the function of one or both compartments114,115.
As a result, MCSs between the ER and lysosomes are vital for maintaining cholesterol homeostasis. Interestingly, NPC1 and the ER-localized Gramd1b sterol-transporter are part of the ER-lysosome MCS116. Thus, when lysosomes accumulate cholesterol, Gramd1b contact with NPC1 allows the transfer of cholesterol to the ER. In cells with NPC1 deficiency, this ER-lysosome MCS is disrupted, contributing to cholesterol buildup in lysosomes116. In line with the principle, a therapeutic approach for NPC could therefore be the functional recovery of ER-lysosome MCSs. With this idea, Hoglinger et al., 2019116 overexpressed a lysosomal membrane ORP1L mutant that cannot sense or transport sterol but can serve as an artificial tether expanding ER-lysosome contacts through constitutive interaction with VAP on the ER. Accordingly, this approach induced a decrease in lysosomal cholesterol accumulation in NPC1-deficient Hela cells 120114]. Along the same lines, Meneses-Salas and collaborators found a recovery in the percentage of the lysosome surface in contact with the ER in CHO cells with mutations in the Npc1 gene after silencing Annexin A6, which is involved in the regulation of cholesterol homeostasis by binding to membranes in a calcium-dependent manner.
These two studies suggest that it may be possible to postulate an increase of ER-lysosome MCSs as a potential strategy for new therapies for NPC disease117. Supporting this idea, the number of ER-lysosome MCSs was induced with agents that reduce cholesterol accumulation in NPC cells, such as hydroxypropyl- c-cyclodextrin (HPγCD) and hydroxypropyl-β-cyclodextrin (HPβCD) in NPC1 patient fibroblasts118.
N-acetyl-l-leucine
N-acetyl-l-leucine, a modified amino acid, is used in the treatment of vertigo. Its mechanism of action involves the activation of cerebral glucose metabolism in the cerebellum and other regions of the brain119,120. Notably, N-acetyl-l-leucine has also shown promise in delaying disease progression and extending the lifespan of Npc1−/− mice121.
Building upon these findings, an initial trial involving 12 NPC patients was conducted, wherein N-acetyl-l-leucine was administered for a duration of 1 month. The treatment resulted in improvements in ataxia, cognition, and overall quality of life for the patients122. Subsequently, a phase II clinical trial was carried out, demonstrating meaningful clinical improvements in symptoms, functioning, and quality of life after 6 weeks of treatment (NCT03759639)123.
Excitingly, a phase III randomized, double-blind, placebo-controlled, multi-center therapeutic study is currently underway (NCT05163288). This trial aims to evaluate the efficacy of N-acetyl-l-leucine in patients aged 4 and older, who have a confirmed diagnosis of NPC. Sixteen centers across Australia, the Czech Republic, Germany, the Netherlands, Slovakia, Switzerland, the UK, and the USA are participating in this study124. There is great anticipation surrounding the potential outcomes of this trial.
One-size does not fit all. Need for personalized therapies
As already discussed, NPC disease symptoms are highly heterogeneous; however, one-size-fits-all potential therapies are usually proposed. Emerging data show that biological differences among individuals should be considered for designing therapeutic approaches (Fig. 2).

Some potential modifying variables include the type of mutation, activation of lipid bypass exit pathways, epigenetics, environment, sex, modifier genes, RNA splicing defects, and the microbiome (the latter has not yet been studied for any of the LSDs).
Genomic variability in rapamycin response: a need for pharmacogenomics studies
Rapamycin activates autophagy and has immunosuppressive properties125. It is an FDA-approved drug and crosses the BBB126.
The effect of rapamycin in cellular models of NPC disease has shown contradictory results. On the one hand, autophagy induction with rapamycin increases cholesterol accumulation in human skin NPC cells, and its inhibition decreases, indicating that autophagy-derived cholesterol accumulates in NPC lysosomes127 Furthermore, NPC-induced mitochondrial fragmentation can be rescued by an autophagy inhibitor in human NPC neurons128. On the other hand, the induction of autophagy with rapamycin restored the autophagic flux in human NPC iPSC-derived hepatic and neuronal cells, increasing cell viability129. These data suggest that both inhibition and induction of autophagy could serve as therapeutic strategies for treating NPC disease.
Regarding this issue, Klein and Calderón tested the therapeutic outcomes of rapamycin on NPC disease progression in male Npc1−/− mice from C57BL6/J and FVB/NJ genetic backgrounds130. Mice were injected with mock or rapamycin 10 mg/kg daily from postnatal day 20. The average lifespan of PBS-treated FVB/NJ-NPC mice was ∼75 days, while C57BL6/J-NPC lived ∼30 days. Interestingly in the C57BL6/J-NPC mice, treatment with rapamycin increased survival by 100%, whereas in FVB-NPC mice, rapamycin was toxic and reduced their life expectancy to ∼65 days.
In addition to the contradictory published cell-based data, this paradoxical result indicates that other factors besides loss of NPC1 function influence NPC pathophysiology and therefore how patients should be treated. To better understand these apparent contradictions, how rapamycin exerts its differential effects in NPC models must be further interrogated in the future.
Other factors that could contribute to phenotypic variability in NPC
Sex
Reports observing sex differences in survival times have been published in NPC mice. For example, Bianconi’s group131 showed that female Npc1 mutant mice (BALB/c npcnih) live significantly longer (median survival of 78 days) than male Npc1 mice (median survival of 66 days). In 2020, Cougnoux’s group132 reported similar data on the C57BL/6J background. The mean survival was 65 ± 7 days and 55 ± 16 for C57BL/6J female and male mutant mice, respectively. Bianconi’s group also used a survival dataset to determine whether survival differences existed between male and female NPC1 patients. Using this dataset, it resulted that the median survival time for males and females was 12 and 14 years, respectively131.
This characteristic also seems to be evolutionarily conserved because sex differences have also been reported in animal models of NPC. In addition, male NPC mice show a more significant weight loss and impairments in locomotor activity, coordination, and balance, but not in cognition, compared to females133. In another study, Holzmann and colleagues134 compared the therapeutic effects of Miglustat, HPßCD, allopregnanolone, and combined therapy (Miglustat, HPßCD, and allopregnanolone) on the brain, body weight, and behavior in a large cohort of BALB/c npcnih mice, with emphasis on sex differences. All treatments in male and female mutant mice led to decreased loss of body weight and, partly, brain weight. Regarding motor coordination, assessed by the rotarod test, male mutant mice benefited from the combined treatment. In contrast, female mice benefited from the combined therapy and the unique Miglustat and HPßCD treatments. These results show that sex-dependent differences in response to therapies will become an important aspect to consider for treating patients in the best possible way.
Environmental factors
As previously shown, the genetic background significantly influences the disease progression and survival of NPC1 mouse models71,135. However, other elements, such as environmental factors, could also modify disease progression and survival in NPC mouse models.
The first indication of this effect came from Liu and colleagues135 reported that two colonies of NPC mice (Npc1nih/nih) of the BALB/c genetic background that had been isolated from one another for 9 years showed different mean survival: one colony lived 89 ± 1.4 days, while the other lived 80 ± 0.8 days. The average survival previously reported for these mice ranges between 70 and 90 days135. On top of this, Cougnoux and colleagues132 reported that in their facility, the Npc1nih/nih mice in the C57BL/6J genetic background lived 61 ± 12 days and had a predominantly neurological phenotype with loss of cerebellar Purkinje neurons in C57BL/6J Npc1nih/nih mice, like that observed in Npc1nih/nih BALB/c mice. However, as mentioned above, Parra and colleagues have reported that these mice lived 32 days71 and had a predominantly visceral phenotype.
Taken together, these results suggest that multiple environmental factors may explain some of these differences, such as pathogens in the facility, diet, accessibility to food, care or attention provided to animals, and genetic drift, among others. Considering some of the above observations and the fact that several reports have suggested that an acute immune response during pregnancy can affect brain development and have significant effects on the cerebellum136, Cougnoux’s group hypothesized that NPC disease pathology might be influenced by prenatal environmental factors, which could have long-term effects on the neuroinflammatory component of NPC pathology. This group evaluated the impact of maternal immune activation (MIA) on disease progression in an NPC1 mouse model132. To this end, they injected polyinosinic-polycytidylic acid (PolyI:C), an agonist of toll-like receptor 3 (TLR3) that mimics a viral, influenza-like infection, into 10- to 11-week-old Npc1+/nih BALB/c pregnant mice when developing embryos were at embryonic day 9.5. In this experiment, they observed that PolyI:C-induced MIA triggered cerebellar manifestations, including increased expression of inflammatory biomarkers and decreased Purkinje neuron density132. These are hallmarks of NPC pathology, and therefore these researchers hypothesized that PolyI:C-treatments could alter Npc1nih/nih disease progression. The results also supported that prenatal exposure to an acute immune response may worsen the motor function and the loss of Purkinje neurons (in lobules I/II and III) in female NPC mice without significantly affecting astrogliosis or microgliosis. In turn, these support previous observations showing that microglia in the Npc1nih mouse model could have ambivalent functions (protective and deleterious)25,27,137,138,139. The small but significant loss of Purkinje neurons caused by MIA in female Npc1 mutant mice was reflected in the impaired motor functioning on the 24 mm wide balance beam. Unexpectedly, in male NPC mice, MIA did not seem to affect the Purkinje cell number, possibly because the loss of Purkinje neurons was already significant by the time it was measured. Thus, MIA appears to eliminate the female advantages observed in Npc1 mutant mice132.
In conclusion, although intrinsically difficult to study in a highly controlled manner, all the data obtained from these studies suggest that prenatal environmental exposures may be a contributing factor to the NPC phenotypic variability and future work in this direction would certainly be justified.
mRNA processing defects
RNA processing involves several steps of maturation and quality control. Around 50% of the variants related to neurodegenerative diseases can affect pre-mRNA splicing140,141. For NPC, about half of the variants in the NPC1 gene are nonsense or frameshift and can generate a premature stop codon. Aberrant transcripts can be degraded through mRNA nonsense-mediated decay (NMD); therefore, prematurely truncated proteins cannot be produced142. The NPC1 mRNA transcript does not seem resistant to this rule because degradation via NMD of NPC1 transcripts harboring premature stop codons generated by nine nonsense or frameshift variants and one deletion has been reported. As a result, the mRNAs and the protein were undetectable143,144. In addition to frameshift variants, splicing defects exist in NPC and many other LSDs, such as Fabry, Pompe, and mucopolysaccharidosis type II145,146,147. Usually, variations that affect splicing occur in splicing regulatory sequences in the intronic and exonic regions145, and in this respect, it is important to note that even exonic variants that do not change the codon usage (i.e., same sense variants can also affect the splicing process148. Most of the reported NPC1 splicing variants are in the intronic regions, except for six variants located in exons 14, 17, 19, 22, and 24 (c.1553 G > A, c.2292 G > A c.2599c>T, c.2911 G > C, c.3422 T > G and c.3754 G > C)145. To this date, variants in the NPC2 gene capable of affecting pre-mRNA splicing are less frequent; nonetheless, three intronic splicing variants have been reported in one study (IVS1 + 2 T > C; IVS2 + 5 G > A; IVS3 + 6 T > G). whilst an additional variant has been reported in the second one (IVS4 + 1 G > A)142,149.
The study of these variants could be particularly important for the development of future therapeutic strategies with the aim of rescuing mRNA splicing abnormalities, as has been done for several LSDs150,151. The most common splicing-therapeutics include the use of antisense oligonucleotide (AONs) and of modified U1 small nuclear RNAs (U1snRNA) targeting splicing regulatory sequences in the RNAs of interest150. Successful examples with U1snRNA have been explored in Fabry disease, Sanfilippo Syndrome, and Mucopolysaccharidosis151,152,153.
Existence of complex alleles
Another factor that could partially explain phenotypic variability is the existence of complex alleles (CAs). CAs are defined as the presence of at least two genomic variants in the same parental allele. In most of the described cases, the functional effect of these variants is appreciated on the protein level. In addition, CAs can be challenging to detect due to the limitations of standard genetic screening methods. A few CAs that alter splicing have been reported and their effects could be predicted bioinformatically. For these cases, their functional analysis should be easier to determine than those that change the protein function. The reason is that variants in cis to native acceptor/donor splice sites could change their strength and modify the effect of other variants, which in turn could activate cryptic donor splice sites leading to pseudoexon inclusion or alter motifs of splicing enhancers/silencers and promote exon skipping154,155.
In this area, Bychkov and colleagues145 studied the impact of the frequent polymorphic variant c.2793 C > T, located in the donor splice site of NPC1 exon 18 on the rare synonymous variant c.2727 C > T, identified in two twins of 55 years old with adult-onset NPC. The variant leads to cryptic donor SS activation and frameshift deletion in the NPC1 exon 18. A minigene assay demonstrated that this exon shortening occurs only in the presence of the frequent polymorphic variant c.2793 C > T. Results of the transcript-specific qPCR showed that only the presence in the NPC1 exon 18 of both variants leads to a significant decrease in the WT transcript isoform149. However, since it is frequent among healthy controls and was not found in affected individuals, it is probably not pathogenetic13. Despite this conclusion, these results highlight the need to analyze raw sequencing data in cases where the identified variant could lead to cryptic SS activation. In fact, as observed in this study, any additional genetic variants nearby could affect the ratio of transcript isoforms and lead to a wrong genotype–phenotype correlation.
Concluding remarks
LSDs, including NPC, are genetically and clinically heterogeneous. In these disorders, a broad phenotypic spectrum has been described, with differences in the age of onset of symptoms, rate of progression, disease severity, organs affected, effects on the CNS, and response to pharmacological treatments. This article has reviewed the phenotypic variation described in NPC and discussed their possible causes. The results of the studies performed so far lead to the general conclusion that NPC phenotypic expression can be modified by many factors, which include, but are probably not limited to, such as the level of residual function of the defective protein, genetic background, sex, environment, and splicing factors. This conclusion highlights the need to examine and categorize carefully the differences between individuals, which should be taken into account when designing hopefully effective treatments. Several other factors could influence NPC disease severity and have not been explored yet. Among them are epigenetic factors, the 3D architecture of the genome, the microbiome, and others (Fig. 2).
Up to this moment, NPC one-size-fits-all potential therapies that do not consider the wide phenotypic variation between patients suffering from this disease have been proposed, but as discussed in this review, their success has been quite limited. All these emerging data point toward the conclusion that biological differences among individuals should be considered when designing therapeutic approaches. If, in previous years, this goal could have seemed unreachable, with the current multi-omics advances, we should soon strive to be ushered into the precision medicine era for NPC and other LSDs.
References
Platt, F. M., D’Azzo, A., Davidson, B. L., Neufeld, E. F. & Tifft, C. Lysosomal storage diseases. Nat. Rev. Dis. Prim. 4, 27 (2018).
Ballout, R. A. Niemann-Pick disease type C (NPC). Genetic Syndromes https://doi.org/10.1007/978-3-319-66816-1_1339-1 (2021).
Parenti, G., Andria, G. & Ballabio, A. Lysosomal storage diseases: from pathophysiology to therapy. Annu. Rev. Med. 66, 471–486 (2015).
Devlin, C. et al. Improvement in lipid and protein trafficking in Niemann-Pick C1 cells by correction of a secondary enzyme defect. Traffic 11, 601–615 (2010).
Gieselmann, V. What can cell biology tell us about heterogeneity in lysosomal storage diseases? Acta Paediatr. Suppl. 94, 80–86 (2005).
Beck, M. Variable clinical presentation in lysosomal storage disorders. J. Inherit. Metab. Dis. 24, 47–51 (2001).
Carstea, E. D. et al. Niemann-Pick C1 disease gene: Homology to mediators of cholesterol homeostasis. Science (1979) 277, 228–231 (1997).
Naureckiene, S. et al. Identification of HE1 as the second gene of Niemann-Pick C disease. Science 290, 2298–2301 (2000).
Wraith, J. E. et al. Recommendations on the diagnosis and management of Niemann-Pick disease type C. Mol. Genet. Metab. 98, 152–165 (2009).
Jiang, X. & Ory, D. S. Advancing diagnosis and treatment of Niemann-Pick C disease through biomarker discovery. Explor. Neuroprotective Ther. 1, 146–158 (2021).
Labrecque, M., Touma, L., Bhérer, C., Duquette, A. & Tétreault, M. Estimated prevalence of Niemann-Pick type C disease in Quebec. Sci. Rep. 11, 22621 (2021).
Burton, B. K. et al. Estimating the prevalence of Niemann-Pick disease type C (NPC) in the United States. Mol. Genet. Metab. 134, 182–187 (2021).
Wassif, C. A. et al. High incidence of unrecognized visceral/neurological late-onset Niemann-Pick disease, type C1, predicted by analysis of massively parallel sequencing data sets. Genet. Med. 18, 41–48 (2016).
Davies, J. P., Chen, F. W. & Ioannou, Y. A. Transmembrane molecular pump activity of Niemann-Pick C1 protein. Science 290, 2295–2298 (2000).
Kuwabara, P. E. & Labouesse, M. The sterol-sensing domain: multiple families, a unique role? Trends Genet. 18, 193–201 (2002).
Li, X. et al. 3.3 Å structure of Niemann-Pick C1 protein reveals insights into the function of the C-terminal luminal domain in cholesterol transport. Proc. Natl Acad. Sci. USA 114, 9116–9121 (2017).
Infante, R. E. et al. Purified NPC1 protein: II. Localization of sterol binding to a 240-amino acid soluble luminal loop. J. Biol. Chem. 283, 1064–1075 (2008).
Kwon, H. J. et al. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell 137, 1213–1224 (2009).
Xu, S., Benoff, B., Liou, H. L., Lobel, P. & Stock, A. M. Structural basis of sterol binding by NPC2, a lysosomal protein deficient in Niemann-Pick type C2 disease. J. Biol. Chem. 282, 23525–23531 (2007).
Vance, J. E. Lipid imbalance in the neurological disorder, Niemann-Pick C disease. FEBS Lett. 580, 5518–5524 (2006).
Winkler, M. B. L. et al. Structural insight into eukaryotic sterol transport through Niemann-Pick type C proteins. Cell 179, 485–497.e18 (2019).
Lloyd-Evans, E. et al. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 14, 1247–1255 (2008).
Malathi, K. et al. Mutagenesis of the putative sterol-sensing domain of yeast Niemann Pick C-related protein reveals a primordial role in subcellular sphingolipid distribution. J. Cell Biol. 164, 547–556 (2004).
Liu, B. et al. Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1-/- mouse. Proc. Natl Acad. Sci. USA 106, 2377–2382 (2009).
Lopez, M. E., Klein, A. D., Hong, J., Dimbil, U. J. & Scott, M. P. Neuronal and epithelial cell rescue resolves chronic systemic inflammation in the lipid storage disorder Niemann-Pick C. Hum. Mol. Genet. 21, 2946–2960 (2012).
Walkley, S. U. & Suzuki, K. Consequences of NPC1 and NPC2 loss of function in mammalian neurons. Biochim. Biophys. Acta 1685, 48–62 (2004).
Lopez, M. E., Klein, A. D., Dimbil, U. J. & Scott, M. P. Anatomically defined neuron-based rescue of neurodegenerative Niemann-Pick type C disorder. J. Neurosci. 31, 4367–4378 (2011).
Vanier, M. T. & Millat, G. Niemann-Pick disease type C. Clin. Genet. 64, 269–281 (2003).
Pallottini, V. & Pfrieger, F. W. Understanding and treating niemann–pick type c disease: Models matter. Int. J. Mol. Sci. 21, 1–37 (2020).
Vanier, M. Niemann-Pick disease type C. Orphanet J. Rare Dis. 5, 16 (2010).
Yu, T. & Lieberman, A. P. Npc1 acting in neurons and glia is essential for the formation and maintenance of CNS myelin. PLoS Genet. 9, e1003462 (2013).
Vanier, M. T. Complex lipid trafficking in Niemann-Pick disease type C. J. Inherit. Metab. Dis. 38, 187–199 (2015).
Evans, W. et al. International consensus on clinical severity scale use in evaluating Niemann-Pick disease Type C in paediatric and adult patients: results from a Delphi Study. Orphanet J. Rare Dis. 16, 482 (2021).
Vanier, M. T. et al. Type C Niemann-Pick disease: spectrum of phenotypic variation in disruption of intracellular LDL-derived cholesterol processing. Biochim. Biophys. Acta 1096, 328–337 (1991).
Bolton, S. C. et al. Clinical disease characteristics of patients with Niemann-Pick Disease Type C: findings from the International Niemann-Pick Disease Registry (INPDR). Orphanet J. Rare Dis. 17, 51 (2022).
Spiegel, R. et al. The clinical spectrum of fetal Niemann-Pick type C. Am. J. Med. Genet. A 149A, 446–450 (2009).
Kelly, D. A., Portmann, B., Mowat, A. P., Sherlock, S. & Lake, B. D. Niemann-Pick disease type C: diagnosis and outcome in children, with particular reference to liver disease. J. Pediatr. 123, 242–247 (1993).
Yerushalmi, B. et al. Niemann-Pick disease type C in neonatal cholestasis at a North American Center. J. Pediatr. Gastroenterol. Nutr. 35, 44–50 (2002).
Bjurulf, B. et al. Niemann-Pick disease type C2 presenting as fatal pulmonary alveolar lipoproteinosis: morphological findings in lung and nervous tissue. Med. Sci. Monit. 14, CS71–CS75 (2008).
Griese, M. et al. Respiratory disease in Niemann-Pick type C2 is caused by pulmonary alveolar proteinosis. Clin. Genet. 77, 119–130 (2010).
Yilmaz, B. S., Baruteau, J., Rahim, A. A. & Gissen, P. Clinical and molecular features of early infantile niemann pick type c disease. Int. J. Mol. Sci. 21, 1–34 (2020).
Soliani, L. et al. Neuropsychological and behavioral disorders as presentation symptoms in two brothers with early-infantile Niemann-Pick type C. Acta Biomed. 91, 1–8 (2020).
Lee, S. Y. et al. Two siblings with adolescent/adult onset Niemann-Pick disease type C in Korea. J. Korean Med. Sci. 31, 1168–1172 (2016).
Maubert, A., Hanon, C. & Metton, J. P. Adult onset Niemann-Pick type C disease and psychosis: literature review. Encephale 39, 315–319 (2013).
Porter, F. D. et al. Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann-Pick C1 disease. Sci. Transl. Med. 2, 56ra81 (2010).
Sitarska, D. & Ługowska, A. Laboratory diagnosis of the Niemann-Pick type C disease: an inherited neurodegenerative disorder of cholesterol metabolism. Metab. Brain Dis. 34, 1253–1260 (2019).
Kaminski, W. E. et al. Identification of novel mutations in the NPC1 gene in German patients with Niemann - Pick C disease. J. Inherit. Metab. Dis. 25, 385–389 (2002).
Fancello, T. et al. Molecular analysis of NPC1 and NPC2 gene in 34 Niemann-Pick C Italian Patients: Identification and structural modeling of novel mutations. Neurogenetics 10, 229–239 (2009).
Dardis, A. et al. Molecular genetics of niemann–pick type c disease in italy: An update on 105 patients and description of 18 NPC1 novel variants. J. Clin. Med. 9, 1–21 (2020).
Shammas, H., Kuech, E. M., Rizk, S., Das, A. M. & Naim, H. Y. Different Niemann-Pick C1 genotypes generate protein phenotypes that vary in their intracellular processing, trafficking and localization. Sci. Rep. 9, 1–12 (2019).
Brogden, G. et al. Different trafficking phenotypes of Niemann-Pick C1 gene mutations correlate with various alterations in lipid storage, membrane composition and miglustat amenability. Int. J. Mol. Sci. 21, 1–13 (2020).
Walterfang, M. et al. Gender dimorphism in siblings with schizophrenia-like psychosis due to Niemann-Pick disease type C. J. Inherit. Metab. Dis. 32, S221–S226 (2009).
Mendrek, A. & Mancini-Marïe, A. Sex/gender differences in the brain and cognition in schizophrenia. Neurosci. Biobehav. Rev. 67, 57–78 (2016).
Ebert, A. & Bär, K. J. Emil Kraepelin: a pioneer of scientific understanding of psychiatry and psychopharmacology. Indian J. Psychiatry 52, 191–192 (2010).
Hwang, W. J., Lee, T. Y., Kim, N. S. & Kwon, J. S. The role of estrogen receptors and their signaling across psychiatric disorders. Int. J. Mol. Sci. 22, 1–21 (2020).
Ho, T. C. et al. Sex differences in myelin content of white matter tracts in adolescents with depression. Neuropsychopharmacology 46, 2295–2303 (2021).
Benussi, A. et al. Phenotypic heterogeneity of Niemann–Pick disease type C in monozygotic twins. J. Neurol. 262, 642–647 (2014).
Klllç Ylldlrlm, G., Yarar, C., Aeker Yllmaz, B. & Ceylaner, S. Niemann-Pick type C disease with a novel intronic mutation: three Turkish cases from the same family. J. Pediatr. Endocrinol. Metab. 35, 535–541 (2021).
Alavi, A., Nafissi, S., Shamshiri, H., Nejad, M. M. & Elahi, E. Identification of mutation in NPC2 by exome sequencing results in diagnosis of Niemann-Pick disease type C. Mol. Genet. Metab. 110, 139–144 (2013).
Xiong, H. et al. Genotype/phenotype of 6 Chinese cases with Niemann-Pick disease type C. Gene 498, 332–335 (2012).
Riordan, J. D. & Nadeau, J. H. From peas to disease: modifier genes, network resilience, and the genetics of health. Am. J. Hum. Genet. 101, 177 (2017).
Loftus, S. K. et al. Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science 277, 232–235 (1997).
Miyawaki, S., Mitsuoka, S., Sakiyama, T. & Kitagawa, T. Sphingomyelinosis, a new mutation in the mouse: a model of Niemann-Pick disease in humans. J. Hered. 73, 257–263 (1982).
Xie, X. et al. Amino acid substitution in NPC1 that abolishes cholesterol binding reproduces phenotype of complete NPC1 deficiency in mice. Proc. Natl Acad. Sci. USA 108, 15330–15335 (2011).
Gómez-Grau, M. et al. New murine Niemann-Pick type C models bearing a pseudoexon-generating mutation recapitulate the main neurobehavioural and molecular features of the disease. Sci. Rep. 7, 41931 (2017).
Maue, R. A. et al. A novel mouse model of Niemann-Pick type C disease carrying a D1005G-Npc1 mutation comparable to commonly observed human mutations. Hum. Mol. Genet. 21, 730–750 (2012).
Praggastis, M. et al. A murine Niemann-Pick C1 I1061T knock-in model recapitulates the pathological features of the most prevalent human disease allele. J. Neurosci. 35, 8091–8106 (2015).
Elrick, M. J. et al. Conditional Niemann-Pick C mice demonstrate cell autonomous Purkinje cell neurodegeneration. Hum. Mol. Genet. 19, 837 (2010).
Miyawaki, S., Yoshida, H., Mitsuoka, S., Enomoto, H. & Ikehara, S. A mouse model for Niemann-Pick disease. Influence of genetic background on disease expression in spm/spm mice. J. Hered. 77, 379–384 (1986).
Zhang, J. & Erickson, R. P. A modifier of niemann Pick C 1 maps to mouse chromosome 19. Mamm. Genome 11, 69–71 (2000).
Parra, J. et al. Npc1 deficiency in the C57BL/6J genetic background enhances Niemann-Pick disease type C spleen pathology. Biochem. Biophys. Res. Commun. 413, 400–406 (2011).
Rodriguez-Gil, J. L. et al. Genetic background modifies phenotypic severity and longevity in a mouse model of Niemann-Pick disease type C1. Dis. Model. Mech. 13, dmm042614 (2020).
Olguín, V. et al. Genetic background matters: population-based studies in model organisms for translational research. Int. J. Mol. Sci. 23, 7570 (2022).
Te Vruchte, D. et al. Relative acidic compartment volume as a lysosomal storage disorder-associated biomarker. J. Clin. Investig. 124, 1320–1328 (2014).
Baxter, L. L. et al. Correlation of age of onset and clinical severity in Niemann-Pick disease type C1 with lysosomal abnormalities and gene expression. Sci. Rep. 12, 2162 (2022).
Imrie, J., Heptinstall, L., Knight, S. & Strong, K. Observational cohort study of the natural history of Niemann-Pick disease type C in the UK: a 5-year update from the UK clinical database. BMC Neurol. 15, 257 (2015).
Platt, F. M., Neises, G. R., Dwek, R. A. & Butters, T. D. N-butyldeoxynojirimycin is a novel inhibitor of glycolipid biosynthesis. J. Biol. Chem. 269, 8362–8365 (1994).
Ory, D. S. et al. Intrathecal 2-hydroxypropyl-β-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: a non-randomised, open-label, phase 1–2 trial. Lancet 390, 1758–1768 (2017).
Hastings, C., Liu, B., Hurst, B., Cox, G. F. & Hrynkow, S. Intravenous 2-hydroxypropyl-β-cyclodextrin (Trappsol® CycloTM) demonstrates biological activity and impacts cholesterol metabolism in the central nervous system and peripheral tissues in adult subjects with Niemann-Pick Disease Type C1: results of a phase 1 trial. Mol. Genet. Metab. 137, 309–319 (2022).
Pipalia, N. H. et al. Histone deacetylase inhibitor treatment dramatically reduces cholesterol accumulation in Niemann-Pick type C1 mutant human fibroblasts. Proc. Natl Acad. Sci. USA 108, 5620–5625 (2011).
Zervas, M., Somers, K. L., Thrall, M. A. & Walkley, S. U. Critical role for glycosphingolipids in Niemann-Pick disease type C. Curr. Biol. 11, 1283–1287 (2001).
Pineda, M. et al. Clinical experience with miglustat therapy in pediatric patients with Niemann-Pick disease type C: a case series. Mol. Genet. Metab. 99, 358–366 (2010).
Patterson, M. C. et al. Long-term survival outcomes of patients with Niemann-Pick disease type C receiving miglustat treatment: a large retrospective observational study. J. Inherit. Metab. Dis. 43, 1060–1069 (2020).
Wraith, J. E. et al. Miglustat in adult and juvenile patients with Niemann-Pick disease type C: long-term data from a clinical trial. Mol. Genet. Metab. 99, 351–357 (2010).
Champion, H. et al. Dietary modifications in patients receiving miglustat. J. Inherit. Metab. Dis. 33, 3 (2010).
Platt, F. M. & Jeyakumar, M. Substrate reduction therapy. Acta Paediatr. 97, 88–93 (2008).
Lyseng-Williamson, K. A. Miglustat: a review of its use in Niemann-Pick disease type C. Drugs 74, 61–74 (2014).
Wang, H., Shen, Y., Zhao, L. & Ye, Y. 1-Deoxynojirimycin and its derivatives: a mini review of the literature. Curr. Med. Chem. 28, 628–643 (2021).
Abi-mosleh, L., Infante, R. E., Radhakrishnan, A., Goldstein, J. L. & Brown, M. S. Cyclodextrin overcomes deficient lysosome-to-endoplasmic reticulum transport of cholesterol in Niemann-Pick type C cells. Proc. Natl Acad. Sci. USA, https://doi.org/10.1073/pnas.0910916106 (2009).
Davidson, C. D. et al. Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS ONE 4, e6951 (2009).
Hastings, C. et al. Expanded access with intravenous hydroxypropyl-β-cyclodextrin to treat children and young adults with Niemann-Pick disease type C1: a case report analysis. Orphanet J. Rare Dis. 14, 228 (2019).
Munkacsi, A. B. et al. An ‘exacerbate-reverse’ strategy in yeast identifies histone deacetylase inhibition as a correction for cholesterol and sphingolipid transport defects in human Niemann-Pick type C disease. J. Biol. Chem. 286, 23842–23851 (2011).
Munkacsi, A. B. et al. Normalization of hepatic homeostasis in the Npc1nmf164 mouse model of niemann-pick type C disease treated with the histone deacetylase inhibitor vorinostat. J. Biol. Chem. 292, 4395–4410 (2017).
Pipalia, N. H. et al. Histone deacetylase inhibitors correct the cholesterol storage defect in most Niemann-Pick C1 mutant cells. J. Lipid Res. 58, 695–708 (2017).
Pal, D., Sahu, P., Mishra, A., Hagelgans, A. & Sukocheva, O. Histone deacetylase inhibitors as cognitive enhancers and modifiers of mood and behavior. Curr. Drug Targets 24, (2022).
Bachy, E. et al. Romidepsin plus CHOP versus CHOP in patients with previously untreated peripheral T-cell lymphoma: results of the Ro-CHOP phase III study (Conducted by LYSA). J. Clin. Oncol. 40, 242–251 (2022).
Hargitai, J. et al. Bimoclomol, a heat shock protein co-inducer, acts by the prolonged activation of heat shock factor-1. Biochem. Biophys. Res. Commun. 307, 689–695 (2003).
Mengel, E. et al. Efficacy and safety of arimoclomol in Niemann-Pick disease type C: Results from a double-blind, randomised, of a novel treatment. J. Inherit. Metab. Dis. 44, 1463–1480 (2021).
Kirkegaard, T. et al. Heat shock protein-based therapy as a potential candidate for treating the sphingolipidoses. Sci. Transl. Med. 8, 355ra118 (2016).
Gray, J. et al. Heat shock protein amplification improves cerebellar myelination in the Npc1nih mouse model. EBioMedicine 86, 104374 (2022).
Pipalia, N. H. et al. HSP90 inhibitors reduce cholesterol storage in Niemann-Pick type C1 mutant fibroblasts. J. Lipid Res. 62, 100114 (2021).
Mengel, E. et al. Efficacy and safety of arimoclomol in Niemann-Pick disease type C: results from a double-blind, randomised, placebo-controlled, multinational phase 2/3 trial of a novel treatment. J. Inherit. Metab. Dis. 44, 1463–1480 (2021).
Medina, D. L. & Ballabio, A. Lysosomal calcium regulates autophagy. Autophagy 11, 970–971 (2015).
di Paola, S., Scotto-Rosato, A. & Medina, D. L. TRPML1: the Ca(2+)retaker of the lysosome. Cell Calcium 69, 112–121 (2018).
Waller-Evans, H. & Lloyd-Evans, E. Regulation of TRPML1 function. Biochem. Soc. Trans. 43, 442–446 (2015).
Zhong, X. Z. et al. BK channel agonist represents a potential therapeutic approach for lysosomal storage diseases. Sci. Rep. 6, 33684 (2016).
Scotto Rosato, A. et al. TPC2 rescues lysosomal storage in mucolipidosis type IV, Niemann–Pick type C1, and Batten disease. EMBO Mol. Med. 14, e15377 (2022).
Alvarez, A. R. et al. Imatinib therapy blocks cerebellar apoptosis and improves neurological symptoms in a mouse model of Niemann-Pick type C disease. FASEB J. 22, 3617–3627 (2008).
Klein, A. D., Alvarez, A. & Zanlungo, S. The unique case of the Niemann-Pick type C cholesterol storage disorder. Pediatr. Endocrinol. Rev. 12, 166–175 (2014).
Yañez, M. J. et al. Finding pathogenic commonalities between Niemann-Pick type C and other lysosomal storage disorders: Opportunities for shared therapeutic interventions. Biochim. Biophys. Acta Mol. Basis Dis. 1866, 165875 (2020).
Yañez, M. J. et al. c-Abl activates RIPK3 signaling in Gaucher disease. Biochim. Biophys. Acta Mol. Basis Dis. 1867, 166089 (2021).
Contreras, P. S. et al. Neuronal gene repression in Niemann-Pick type C models is mediated by the c-Abl/HDAC2 signaling pathway. Biochim. Biophys. Acta 1859, 269–279 (2016).
Contreras, P. S. et al. c-Abl inhibition activates TFEB and promotes cellular clearance in a lysosomal disorder. iScience 23, 101691 (2020).
Ballabio, A. & Bonifacino, J. S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 21, 101–118 (2020).
Prinz, W. A., Toulmay, A. & Balla, T. The functional universe of membrane contact sites. Nat. Rev. Mol. Cell Biol. 21, 7–24 (2020).
Höglinger, D. et al. NPC1 regulates ER contacts with endocytic organelles to mediate cholesterol egress. Nat. Commun. 10, 4276 (2019).
Enrich, C., Rentero, C., Grewal, T., Futter, C. E. & Eden, E. R. Cholesterol overload: contact sites to the rescue! Contact 2, 251525641989350 (2019).
Singhal, A., Szente, L., Hildreth, J. E. K. & Song, B. Hydroxypropyl-beta and -gamma cyclodextrins rescue cholesterol accumulation in Niemann-Pick C1 mutant cell via lysosome-associated membrane protein 1. Cell Death Dis 9, 1019 (2018).
Becker-Bense, S. et al. Acetyl-DL-leucine in cerebellar ataxia ([18F]-FDG-PET study): how does a cerebellar disorder influence cortical sensorimotor networks? J. Neurol. 270, 44–56 (2023).
Günther, L. et al. N-acetyl-l-leucine accelerates vestibular compensation after unilateral labyrinthectomy by action in the cerebellum and thalamus. PLoS ONE 10, e0120891 (2015).
Kaya, E. et al. Acetyl-leucine slows disease progression in lysosomal storage disorders. Brain Commun. 3, fcaa148 (2020).
Bremova, T. et al. Acetyl-dl-leucine in Niemann-Pick type C: a case series. Neurology 85, 1368–1375 (2015).
Bremova-Ertl, T. et al. Efficacy and safety of N-acetyl-L-leucine in Niemann-Pick disease type C. J. Neurol. 269, 1651–1662 (2022).
Fields, T. et al. N-acetyl-l-leucine for Niemann-Pick type C: a multinational double-blind randomized placebo-controlled crossover study. Trials 24, 361 (2023).
Garver, W. S. et al. The National Niemann-Pick type C1 disease database: correlation of lipid profiles, mutations, and biochemical phenotypes. J. Lipid Res. 51, 406–415 (2010).
Cloughesy, T. F. et al. Antitumor activity of rapamycin in a phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 5, 0139–0151 (2008).
Elrick, M. J., Yu, T., Chung, C. & Lieberman, A. P. Impaired proteolysis underlies autophagic dysfunction in Niemann-Pick type C disease. Hum. Mol. Genet. 21, 4876–4887 (2012).
Paulina Ordonez, M. et al. Disruption and therapeutic rescue of autophagy in a human neuronal model of Niemann Pick type C1. Hum. Mol. Genet. 21, 2651–2662 (2012).
Maetzel, D. et al. Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from Niemann-Pick Type C patient-specific iPS cells. Stem Cell Rep. 2, 866–880 (2014).
Calderón, J. F. & Klein, A. D. Controversies on the potential therapeutic use of rapamycin for treating a lysosomal cholesterol storage disease. Mol. Genet. Metab. Rep. 15, 135–136 (2018).
Bianconi, S. E. et al. Evaluation of age of death in Niemann-Pick disease, type C: utility of disease support group websites to understand natural history. Mol. Genet. Metab. 126, 466–469 (2019).
Cougnoux, A. et al. Maternal immune activation modifies the course of Niemann- Pick disease, type C1 in a gender specific manner. Mol. Genet. Metab. 129, 165–170 (2020).
Võikar, V., Rauvala, H. & Ikonen, E. Cognitive deficit and development of motor impairment in a mouse model of Niemann-Pick type C disease. Behav. Brain Res. 132, 1–10 (2002).
Holzmann, C., Witt, M., Rolfs, A., Antipova, V. & Wree, A. Gender‐specific effects of two treatment strategies in a mouse model of niemann‐pick disease type c1. Int. J. Mol. Sci. 22, 1–32 (2021).
Liu, B., Li, H., Repa, J. J., Turley, S. D. & Dietschy, J. M. Genetic variations and treatments that affect the lifespan of the NPC1 mouse. J. Lipid Res. 49, 663–669 (2008).
Aavani, T., Rana, S. A., Hawkes, R. & Pittman, Q. J. Maternal immune activation produces cerebellar hyperplasia and alterations in motor and social behaviors in male and female mice. Cerebellum 14, 491–505 (2015).
Cougnoux, A. et al. Microglia activation in Niemann-Pick disease, type C1 is amendable to therapeutic intervention. Hum. Mol. Genet. 27, 2076–2089 (2018).
Colombo, A. et al. Loss of NPC1 enhances phagocytic uptake and impairs lipid trafficking in microglia. Nat. Commun. 12, 1158 (2021).
Lopez, M. E., Klein, A. D. & Scott, M. P. Complement is dispensable for neurodegeneration in Niemann-Pick disease type C. J. Neuroinflammation 9, 216 (2012).
Anthony, K. & Gallo, J. M. Aberrant RNA processing events in neurological disorders. Brain Res. 1338, 67–77 (2010).
Schieweck, R., Ninkovic, J. & Kiebler, M. A. RNA-binding proteins balance brain function in health and disease. Physiol. Rev. 101, 1309–1370 (2021).
Paron, F., Dardis, A. & Buratti, E. Pre-mRNA splicing defects and RNA binding protein involvement in Niemann Pick type C disease. J. Biotechnol. 318, 20–30 (2020).
Macías-Vidal, J., Gort, L., Lluch, M., Pineda, M. & Coll, M. J. Nonsense-mediated mRNA decay process in nine alleles of Niemann-Pick type C patients from Spain. Mol. Genet. Metab. 97, 60–64 (2009).
Tamura, H. et al. Niemann-Pick type C disease: novel NPC1 mutations and characterization of the concomitant acid sphingomyelinase deficiency. Mol. Genet. Metab. 87, 113–121 (2006).
Bychkov, I. et al. Additive effect of frequent polymorphism and rare synonymous variant alters splicing in twin patients with Niemann-Pick disease type C. Eur. J. Hum. Genet. 30, 133–136 (2022).
Goina, E., Musco, L., Dardis, A. & Buratti, E. Assessment of the functional impact on the pre-mRNA splicing process of 28 nucleotide variants associated with Pompe disease in GAA exon 2 and their recovery using antisense technology. Hum. Mutat. 40, 2121–2130 (2019).
Zampieri, S. et al. Splicing mutations in glycogen-storage disease type II: evaluation of the full spectrum of mutations and their relation to patients’ phenotypes. Eur. J. Hum. Genet. 19, 422–431 (2011).
Pagani, F., Buratti, E., Stuani, C. & Baralle, F. E. Missense, nonsense, and neutral mutations define juxtaposed regulatory elements of splicing in cystic fibrosis transmembrane regulator exon 9. J. Biol. Chem. 278, 26580–26588 (2003).
Reunert, J. et al. Rapid diagnosis of 83 patients with Niemann Pick type C disease and related cholesterol transport disorders by cholestantriol screening. EBioMedicine 4, 170–175 (2015).
Dardis, A. & Buratti, E. Impact, characterization, and rescue of pre-mRNA splicing mutations in lysosomal storage disorders. Genes 9, 73 (2018).
Santos, J. I. et al. Splicing modulation as a promising therapeutic strategy for lysosomal storage disorders: the mucopolysaccharidoses example. Life 12, 608 (2022).
Ferri, L. et al. Double-target antisense U1snRNAs correct mis-splicing due to c.639+861C>T and c.639+919G>A GLA deep intronic mutations. Mol. Ther. Nucleic Acids 5, e380 (2016).
Matos, L. et al. Therapeutic strategies based on modified U1 snRNAs and chaperones for Sanfilippo C splicing mutations. Orphanet J. Rare Dis. 9, 180 (2014).
Dufner-Almeida, L. G., do Carmo, R. T., Masotti, C. & Haddad, L. A. Understanding human DNA variants affecting pre-mRNA splicing in the NGS era. Adv. Genet. 103, 39–90 (2019).
Broeders, M. et al. A generic assay to detect aberrant ARSB splicing and mRNA degradation for the molecular diagnosis of MPS VI. Mol. Ther. Methods Clin. Dev. 19, 174–185 (2020).
Acknowledgements
This work was supported by ANID-CHILE: Fondecyt grant No 1230317 (A.D.K.) (2023–2027), 1230337 (S.Z.) (2023–2027), NPSuisse (A.D.K.) (2022–2023), and by the International Centre for Genetic Engineering and Biotechnology (ICGEB) grant CRP/CHL22–02 (A.D., E.B., and A.D.K.) (2023–2026).
Author information
Authors and Affiliations
Contributions
M.L.H. and A.D.K. conceptualized and wrote the article. B.S., R.A.B., E.B., S.Z., and A.D. also wrote the manuscript. M.L.H., B.S., and A.D.K. made the figures.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Las Heras, M., Szenfeld, B., Ballout, R.A. et al. Understanding the phenotypic variability in Niemann-Pick disease type C (NPC): a need for precision medicine. npj Genom. Med. 8, 21 (2023). https://doi.org/10.1038/s41525-023-00365-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-023-00365-w
This article is cited by
-
An overview of the role of Niemann-pick C1 (NPC1) in viral infections and inhibition of viral infections through NPC1 inhibitor
Cell Communication and Signaling (2023)
