
Abstract
Copy number (CN) polymorphisms of complement C4 play distinct roles in many conditions, including immune-mediated diseases. We investigated the association of C4 CN with systemic sclerosis (SSc) risk. Imputed total C4, C4A, C4B, and HERV-K CN were analyzed in 26,633 individuals and validated in an independent cohort. Our results showed that higher C4 CN confers protection to SSc, and deviations from CN parity of C4A and C4B augmented risk. The protection contributed per copy of C4A and C4B differed by sex. Stronger protection was afforded by C4A in men and by C4B in women. C4 CN correlated well with its gene expression and serum protein levels, and less C4 was detected for both in SSc patients. Conditioned analysis suggests that C4 genetics strongly contributes to the SSc association within the major histocompatibility complex locus and highlights classical alleles and amino acid variants of HLA-DRB1 and HLA-DPB1 as C4-independent signals.
Similar content being viewed by others


Introduction
Systemic sclerosis (SSc) is a chronic immune-mediated inflammatory disease (IMID) more frequently observed in women (female/male ratio ~3.8–11.5) that affects the connective tissue and is associated with considerable morbidity and mortality1,2,3. The heterogeneous clinical manifestations of SSc are characterized by functional and structural vasculopathy, fibrosis of the skin and internal organs, in addition to inflammatory and immunological alterations like auto-antibody production1. The individual genetic background, together with environmental risk factors and epigenetics factors, play an important role in the pathogenesis of the disease4,5.
A recent genome-wide association study (GWAS) has identified new genes and pathways implicated in the development and progression of SSc6. Similar to other IMIDs, these genetic variations account for a limited portion of estimated heritability7, making clear that additional genetic variants remain to be found with the potential to bring novel insights into disease etiology and pathogenesis. In this sense, structural variants not captured by GWAS, such as copy number (CN) polymorphisms, which have been implicated in the etiology of several diseases8,9, could contribute substantially to the genetic risk of SSc. Several CN variants in immunological genes have been found to be associated with autoimmune diseases10,11,12,13, although technical limitations and the complexity of CN polymorphisms have reduced the impact of their analysis in understanding autoimmunity14,15.
The complement system plays an important role in innate immunity and forms a bridge to the adaptive immune response16,17,18. Functional abnormalities in the complement system have been widely described in rheumatic diseases, such as rheumatoid arthritis (RA) or systemic lupus erythematosus (SLE), and to a lesser extent in SSc19. Furthermore, genetic variability in several complement components may contribute to the development of inflammatory and autoimmune diseases20,21.
Complement component 4 (C4), encoded by two closely linked, highly polymorphic genes C4A and C4B within the major histocompatibility complex (MHC) class III region on chromosome 6, is an important protein in the classical and lectin complement activation pathways, which are major effectors for controlling microbial infections and for promoting clearance of apoptotic cells and soluble immune complexes17,19. C4A and C4B encode proteins with distinct affinities for their molecular targets21,22,23 and present variability in genomic copy number (CN)24 and length. The long form is determined by the presence of a 6.4-kb human endogenous retrovirus K (HERV-K) element in intron 9 of both genes. C4 CN studies in autoimmunity have been mainly focused on SLE providing no definitive results14,22,25 in part due to the complexity of the genetic variation of C4A and C4B and the high linkage disequilibrium (LD) of the MHC locus, which contains the HLA genes, the strongest genetic associations with most autoimmune diseases including SLE, RA, and SSc6,26,27. A recent study, however, managed to attribute most of the genetic association of SLE and Sjögren’s syndrome (SjS) of the MHC locus to C428. Of note, the same study made the complex genetic variation of C4A and C4B accessible through the imputation of SNP data using a large multi-ancestry panel of 2530 reference haplotypes28,29.
CN variations at C4 genes have been implicated as a source of sexual dimorphism in two systemic autoimmune diseases, SLE and SjS28, diseases that have a higher prevalence in women than men. In light of the above, we set out to investigate the contribution of C4 CN to SSc using data from the largest GWAS cohort of SSc, combined with genetic, RNA-sequencing, and C4 serum data from an independent cohort.
Results
Experimental design
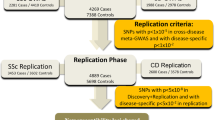
To investigate the role of C4 genetics in SSc, we analyzed two independent cohorts of European descent (see “Methods”, Supplementary Fig. 1 and Supplementary Table 1). We used the first cohort (genetic data, N = 26,633, 34% SSc, 10 countries) to determine the association of C4 CN to SSc. The second cohort (genetic, whole blood expression, and C4 serum data, N = 857, 39% SSc, 9 countries) was analyzed to detect C4 expression quantitative trait loci (eQTL) and for C4 expression- and C4 protein level- modeling. Expression-model eQTLs from the second cohort that explained expression variance were subsequently used in the first cohort as co-factors to determine residual genetic association to SSc unrelated to C4 genetics in the MHC region on chromosome 6. Finally, we utilized classical HLA alleles and amino acids (AAs) to model remnant C4-independent association in the MHC region.
C4 haplotype diversity and its correlation with classical HLA alleles
The complex genetic variation of C4A and C4B, which consists of many haplotypes with different numbers of C4A and C4B genes was recently made accessible for analysis in large cohorts28. We imputed 29 C4 haplotypes independently in both cohorts. Haplotype frequencies were found to be similar in both cohorts and comparable to published results28 but varied substantially across countries (Supplementary Fig. 2 and Supplementary Table 2A). We determined nine C4 haplotypes to have a moderate to strong correlation (r2 > 0.4) to at least one classical HLA allele (Supplementary Dataset 1). Correlation to classical HLA alleles associated to SSc on a genome-wide level was found to be small (r2 < 0.3) for most haplotypes with the exceptions of HLA-B*08:01 and HLA-C*07:01 which had a strong correlation (r2 = 0.82 and 0.67, respectively) with the BS haplotype and HLA-C*16:01 with AL-BS (H2111B) of 0.63 (Supplementary Dataset 1).
The association between C4 copy number variants and SSc is modified by sex and HERV-K
We found a higher C4 CN to be protective for SSc (Fig. 1a and Table 1). Less than four copies of C4 were found in 28.6% of SSc patients, and 2.8% had less than three copies. Interestingly, less than four copies of C4 were found in 28% of female and 32% of male patients. In a simple additive model C4A, C4B and HERV-K CNs exhibited 1.8-fold variation in their relative risk of SSc (95% confidence interval (95% CI), [1.51–2.33]; P = 1.04 × 10−16). Logistic-regression analysis estimated a rather small difference between the protection afforded by each copy of C4A (odds ratio (OR) = 0.73; 95% CI = 0.69–0.78) or C4B (OR = 0.82; 95% CI = 0.77–0.87) (Table 1). We replicated our calculation in the second cohort and performed a meta-analysis, showing consistent results (Table 1).

a depicts relative systemic sclerosis (SSc) risk vs total C4 copy number stratified by C4A CN. The SSc risk score is calculated per individual as the sum of effect sizes (betas) multiplied with the design matrix. Betas of C4A, C4B and C4A:C4B were taken from the most complex model “d” (see “Methods”). Crosses are calculated as average relative risk per rounded C4 CN + /− 2 standard deviations (y axis). Linear regression lines are colored by C4A CN and drawn to visualize the interaction effect of C4A and C4B. The y axis contains a color code to aid a comparison with (b). b depicts the relative SSc risk of combinations of C4A and C4B CNs. Relative risk is calculated as in (a). Outer circles are drawn according to population frequency ranges of each C4A, C4B combination and highlight more common combinations. Diagonal dotted lines help to identify combinations of equal total C4 CN. c depicts relative SSc risk in male individuals vs total C4 CN stratified by C4B CN. Relative risk is calculated like in (a) using effect sizes of C4A, C4B, C4A:C4B, Sex:C4A, and Sex:C4B. Crosses are calculated as average relative risk per rounded C4 CN + /− 2 standard deviations (y axis). Cubic regression lines are colored by C4B CN and drawn to visualize the interaction effect of C4A and C4B. d depicts relative SSc risk vs total C4 CN stratified by HERV-K CN. Relative risk is calculated like in (a) using effect sizes of C4A, C4B, C4A:C4B, HERV-K:C4A, and HERV-K:C4B. Crosses are calculated as average relative risk per rounded C4 CN + /− 2 standard deviations (y axis). Linear regression lines are colored by HERV-K CN.
The number of subjects in the first cohort permits the simple additive model to be expanded to a more complex one investigating the predictors that influence each other. We found evidence for three two-way interactions: an interaction of C4A with C4B, a sexual dimorphism of C4A and C4B, and an interaction of HERV-K CN with C4A and C4B (Supplementary Table 3). The full model log(risk) ~ b1 Sex + b2 C4A + b3 C4B + b4 HERV-K + b5 Sex:C4A + b6 Sex:C4B + b7 HERV-K:C4A + b8 HERV-K:C4A + b9 C4A:C4B predicted 7.7-fold variation in the relative risk of SSc. The following analyses have been derived from this complex model and its calculated coefficients.
The relationship between SSc risk and C4A and C4B gene CNs exhibited consistent, logical patterns across the 18 different CN combinations of C4A and C4B (Fig. 1a, b), which is based on an interaction between C4A and C4B CNs that was suggested by logistic-regression analysis (bc4a:c4b = −0.14, P = 2.1 × 10−5). While higher total C4 CN provide protection, strong deviations from the 1:1 ratio of C4A and C4B are of higher risk like e.g., four copies of C4A and zero copies of C4B (Fig. 1b).
We found evidence for a sexual dimorphism for C4A and C4B but not for HERV-K CN. Stratified analysis showed C4A to be more protective in men (bmale = −0.49, P = 1.7 × 10−9 vs bfemale = −0.27, P = 6.5 × 10−12) while C4B showed statistical evidence to be protective only in women (bfemale = −0.22, P = 1.1 × 10−10 vs bmale = −0.09, P = 0.23) (Table 1). Indeed, C4B CNs of two or higher seem to augment the risk for men (Fig. 1c). Logistic regression with interaction terms confirmed our results for the C4A:sex interaction, while the significance of the C4B:sex interaction was only suggestive (bmale:c4a = −0.17, P = 0.0034, bmale:c4b = 0.13, P = 0.065) (Supplementary Table 3). For total C4 CN, we did not find a significant sex bias (bmale:c4 = −0.1, P = 0.074). We calculated the statistical power for C4B association in males to be 0.41 and for the interaction of C4B:sex to be 0.33.
HERV-K copies generally augment the risk for SSc (Fig. 1d and Table 1). The protection afforded by C4A and C4B CNs are affected by HERV-K copies in a slightly different way. Protection associated with each C4B copy is affected more strongly by HERV-K (bHERV-K:C4B = 0.085, P = 0.053) in comparison to C4A (bHERV-K:C4A = 0.042, P = 0.03). A logistic-regression analysis with separate terms for the long and the short forms of C4A and C4B confirmed that the short forms confer more protection than the long forms (Table 1).
C4 copy number affects C4 expression and C4 protein levels in whole blood
We investigated the effect of C4 and HERV-K CN on whole blood expression levels of C4 (C4 = C4A + C4B, Fig. 2a). Specific analysis for C4A and C4B showed significant positive correlation for both C4A CN (rPearson,C4A = 0.15, P = 1.2 × 10−5) and C4B CN (rPearson,C4B = 0.34, P = 1.7 × 10−24) with C4A or C4B expression. HERV-K CN had a weakening effect on C4 expression at all C4 CN levels (Fig. 2a). C4A or C4B expression models performed better if they contained separate CNs for long (L) and short (S) versions of C4 (AS, AL, BS, BL) instead of C4A plus C4B plus total HERV-K CNs. Interestingly, expression models were best if CNs of C4B were included in the model of C4A and vice versa. About 21% (27%) of expression variance of C4A (C4B) can be attributed to C4A and C4B CNs (Supplementary Table 4B, C).

a depicts residualized total C4 expression levels by total C4 copy number (CN) stratified by HERV-K CN. C4 expression is calculated as the sum of C4A and C4B expression as obtained by RNA-Sequencing. The residualized expression has been calculated by regressing out 20 (18) principal components for controls and cases, respectively. Data has been grouped by rounded C4 and HERV-K CN dosage. b depicts normalized C4 protein levels in plasma by total C4 CN stratified by HERV-K CN. C4 protein levels have been normalized across 10+ laboratory sites. c depicts residualized total C4 expression levels (like in a) for SSc and controls, stratified by sex. Significant comparisons are highlighted by asterisk (*P < 0.05, **P < 0.01, ***P < 0.001). d depicts normalized C4 protein levels (like in b) for SSc and controls, stratified by sex. Significant comparisons are highlighted by asterisk (*P < 0.05, **P < 0.01, ***P< 0.001). e depicts normalized C4 protein levels in blood from 119 adult men (blue) and 447 adult women (red) as a function of age with locally estimated scatterplot smoothing (LOESS). Protein levels are normalized to the number of C4 gene copies in an individual’s genome. All boxplot are drawn with default settings in R 4.0.3: lines are defined as first, second and third quartile (Q1, Q2, Q3), whiskers depict the most extreme data points within Q1–1.5 interquartile range (IQR), and Q3 + 1.5 IQR. Boxplot notches are defined as 95% confidence interval of the median.
Using C4A and C4B eQTLs alone, we were able to explain up to 42% (C4B: 38%) of expression variance with 19 (C4B: 15) SNPs (Supplementary Table 4B, C). This seems to be the upper bound of C4 expression variance explained by C4 genetics as C4A and C4B CN and eQTLs together could not explain more than 40% of expression variance albeit with only 12 (C4B: 13) additional SNPs (Supplementary Tables 4B, C). C4 eQTLs seem to integrate C4 and HERV-K CN information. Indeed, copy numbers of C4AShort, C4ALong, C4BShort, and C4BLong can be predicted well (r2C4AL = 0.5, r2C4AS = 0.58, r2C4BL = 0.54, r2C4BS = 0.67, all P < 2.2 × 10−16) using C4 eQTLs forward selected to explain C4A or C4B expression variance.
Blood serum concentrations of C4 protein were well correlated with C4 CNs (rPearson = 0.25, P = 1.3 × 10−12, Fig. 2b). Regression analysis determined that C4 and HERV-K CNs, sex, age, and SSc explained about 12% of C4 serum concentration variance (P = 1.4 × 10−20) with HERV-K copies again weakening C4 levels (bHERV-K = −0.03, P = 9 × 10−7) (Fig. 2b and Supplementary Table 5). CN-corrected serum C4 levels determined men to have more C4 protein than women (bmale = 0.04, P = 4.6 × 10−4) independent of disease (Fig. 2e and Supplementary Table 5). Individuals with SSc had less CN-corrected C4 protein than healthy subjects independent of sex (bSSc = −0.02, P = 0.012).
Despite that only about a third of SSc patients have less than four copies of C4, we found significant downregulation of C4 expression (pfemale = 0.001, pmale = 0.003) and C4 protein levels (P = 0.004) in SSc patients compared to healthy controls (Fig. 2c, d).
C4 genetics can explain a part of the SSc association to the MHC region
We performed conditional association analysis for genetic markers across the MHC genomic region. Conditioning on C4A, C4B, and HERV-K CN or on a risk score calculated using the complex C4 CN interaction model derived above, showed an impact on residual association levels limited to the vicinity of the C4 gene (Supplementary Fig. 3B, C). In addition, we calculated a C4 risk score recently proposed for SLE and SjS based only on C4A and C4B CN (as: risk = (2.3)C4A CN + C4B CN)28. We applied the SLE/SjS score for conditional association analysis in the first dataset and found almost no effect (Supplementary Fig. 3A).
Given the strong association of C4 CN with SSc and its rather local impact in conditional analysis, we focused on C4 eQTLs as potential modifiers of C4 CN risk as both CN and eQTLs affect C4 expression levels. Our analysis above suggests that the predictive power of C4 CN and C4 eQTLs on C4 expression levels is at least partly redundant. In fact, C4 eQTLs alone can explain more C4 expression variance than C4 CNs and C4 eQTLs are significant predictors for copy numbers of C4. We obtained 10,680 eQTLs of C4A and C4B from the GTEx v8 database and found that 37% of these are associated to SSc with pGWAS < 10−5 using the first dataset (Fig. 3a). This encouraged us to find independent C4 eQTLs using forward selection to explain their contribution to the MHC association to SSc.

Association is calculated in the first dataset (N = 26,633) using logistic regression with cohort, genetic background (PC1-5), and sex as covariates and depicted as position (GRCh38) by significance (Manhattan plot) in gray if no additional covariates were used. The dotted line represents the genome-wide significance cutoff P = 5 × 10−8. a Manhattan plot with marked C4 eQTLs obtained from the GTEx v8 database. b Manhattan plot with additional conditioning on ten independent C4 eQTLs, obtained by forward selection in the first dataset, depicted in blue. The arrow marks the position of HLA-DPB1. c Manhattan plot with additional conditioning on 16 independent C4 eQTLs (obtained by forward selection to explain expression variance in the second dataset (N = 857) depicted in blue. The arrows mark the positions of HLA-DPB1 and HLA-DRB1. d Manhattan plot with additional conditioning on 16 independent C4 eQTLs (obtained by forward selection to explain expression variance in the second dataset) and 8 independent amino acids of DRB1 and DPB1 (obtained by forward selection in the first dataset conditioning on 16 independent C4 eQTLs) depicted in blue.
Conditioned association analysis for genetic markers across the MHC genomic region on ten independent C4 eQTL SNPs rendered most association to SSc nonsignificant (P > 5 × 10−8), except for a peak around HLA-DPB1, the initial association of which was enhanced by conditioning (Fig. 3b). While conditioning on 13 independent C4A exclusive eQTL SNPs had a similar effect on the residual association profile, conditioning on 12 C4B exclusive eQTL SNPs had a smaller effect with residual association (P < 5 × 10−8) to SSc in three regions centered on HLA-DPB1, HLA-DRB1, and HLA-B (Supplementary Fig. 4B, C), which suggests a stronger contribution of C4A to SSc.
While the ten independent C4 eQTL SNPs absorb as much SSc association as possible by design (forward selection), they might have been selected due to the extensive genetic linkage in the MHC region without being implicated in SSc pathogenesis. We therefore asked if SSc association with the MHC could be explained by C4 eQTLs selected to explain C4A and C4B expression variance in the second dataset. Using expression-model eQTL SNPs, selected in the second cohort to explain expression variance, as co-factors in the first cohort to determine residual genetic association with SSc again rendered most MHC association with SSc nonsignificant (P > 5 × 10−8), except for the peaks shown in Fig. 3c centered on HLA-DPB1 and HLA-DRB1.
Remaining MHC signal after conditioning on C4 genetics highlights HLA-DRB1 and HLA-DPB1
Having attributed most SSc association within the MHC to C4 genetics, we investigated which classical HLA alleles and HLA amino acids (AA) demonstrated C4-independent association to SSc. Conditioning on expression derived independent C4 eQTLs results in residual significance (P < 5 × 10−8) for classical alleles and AAs of HLA-DRB1, HLA-DPB1, HLA-DPA1 and HLA-DQA1, HLA-DQB1, and HLA-B (Supplementary Dataset 2A, B). Forward selection to derive independent residual signals marked HLA classical alleles for HLA-DRB1 (*07:01, *11:03, *11:04, *13:01), HLA-DPB1 (*13:01, *26:01, *40:01, *06:01), HLA-DQA1 (*04:01), and HLA-DQB1*05:01.
We tested if classical alleles and AAs of HLA-DRB1 and HLA-DPB1 alone could explain residual C4-independent association. We found that 9 HLA classical alleles (HLA-DRB1: *11:04, *08:01, *07:01, *13:01, *11:03 and HLA-DBP1: *13:01, *26:01, *40:01, *06:01) together with 16 expression-model-derived independent C4 eQTLs can explain almost all associations of SSc with the MHC region (Supplementary Fig. 5B). The same is true for AAs of HLA-DRB1 and HLA-DPB1, which can also explain residual C4-independent association with eight independent AAs (HLA-DRB1: 37SY, 58A, 74RL, 96E, and HLA-DPB1: 8, 76I, 91H, 96x) (Fig. 3d).
To complement our analysis, we repeated our search for residual C4-independent association to SSc, this time conditioning on the C4 genetic signal which was not derived from the expression dataset but from the first dataset as ten independent C4 eQTL signals as described. Repeating the forward selection of AAs or classical HLA alleles conditioning on the ten first dataset-derived independent C4 eQTLs signals resulted in four independent AAs from HLA-DPB1 (9F, 76I, 91H, 96x) or five independent alleles (HLA-DRB1: *11:04 and HLA-DBP1: *13:01, *26:01, *28:01, *30:01) (Supplementary Fig. 4D, E) supporting the role of HLA-DRB1 and HLA-DPB1 as a C4-independent association.
Discussion
In this study, we found a strong association of low C4 and high HERV-K CN with SSc in two independent SSc datasets and their meta-analysis, supporting the protective role of C4 copies in IMIDs. C4A gene copies were slightly more protective than C4B as has been shown in SLE and SjS28 but our data suggest a complex interaction of C4A and C4B CNs that has to be evaluated in the context of HERV-K copies and sex. We found that in SSc, an equal number of C4A and C4B gene copies grants more protection than (strongly) imbalanced numbers which we found to be a risk for SSc (Fig. 1b). Our results might differ from recent observations in SLE and SjS where C4A and C4B copies have been described to act in an additive way, but the authors did not describe the interaction with HERV-K copies in detail28.
Our results showed a sexual dimorphism with respect to the protection afforded by C4A and C4B. While in female individuals, C4A copies grant slightly more protection than C4B copies, our data suggest that in male individuals only C4A confers protection while we did not observe a strong effect for C4B. In male individuals, C4B might therefore function like a null allele with respect to protection from SSc as higher CNs of C4B are associated to higher SSc risk (Fig. 1c). However, as the power of our study to detect significant C4B signals in males was limited, and the sex:C4B interaction was only of suggestive significance, we cannot rule out that C4B has a protective effect in males. While C4 alleles have been described to act more strongly in men, no distinction was made between C4A and C4B activity in SLE or SjS in a recent study of similar size28. It has been described that activated C4A bonds preferably with protein antigens, such as immune complexes, while activated C4B reacts rapidly with carbohydrate antigens, such as bacterial cell walls30. This could partly explain the greater susceptibility to and severity of infections reported in men and the higher incidence of autoimmune diseases in women31,32. In addition, low C4B CNs have been associated to cardiovascular disease33 where the incidence in women is usually lower than in men34. Interestingly, in a recent paper studying the female-biased expression in human skin, several genes from the complement activation pathway were identified as a molecular signature and in genome-wide co-expression networks35. Sexual dimorphism has been extensively reported in vascular physiology and pathophysiology36, where women more commonly develop microvascular dysfunction, and in autoimmune-related interstitial lung diseases, where young women are most commonly affected37. All of these clinical manifestations, for which the role of C4 is yet to be elucidated, are hallmarks of SSc.
Our data confirm that C4 and HERV-K CNs are strong predictors of C4 expression levels in blood and other tissues29. While the major site of C4 expression is the liver38, it has been shown that whole blood can be used with some caution as a surrogate tissue for quantitative trait analysis39,40,41. In addition, local complement production by bone-marrow-derived monocytes and macrophages can restore humoral response in c4 deficient mice42. Interestingly C4A and C4B expression models both profit strongly from the other gene’s CN as a predictor, which supports the genetic interaction between them suggested in this study. Furthermore, the distinction between the long and the short forms of C4: AS, AL, BS, and BL as expression predictors instead of C4A, C4B, and HERV-K CNs alone, greatly favors the accuracy of the expression model. This suggests that HERV-K acts specifically on the gene where it is located, to suppress its expression, which is supported by studies in brain and serum22,29,43. C4A and C4B CNs were able to explain about 20% of C4A and C4B expression variance, which is clearly lower than the ability of C4 eQTLs, which could explain ~40%. Although we most likely over-fitted the expression data, SNPs seem to be the superior instrument in predicting C4 expression as they can integrate CN as well as classical eQTL signals. This finding might help to bring C4 genetics to the clinic in the form of simple genetic tests. The interconnectedness of C4 CN and eQTLs is further supported by C4 eQTLs being able to predict C4 CNs (AS, AL, BS, BL) with coefficients of determination of r2 > 0.5.
C4 and HERV-K CNs are strong predictors of C4 protein levels in blood serum43. Interestingly, it was reported that men had on average 27% more C4 protein per C4 CN than women and that this bias is stronger during reproductive years28. While we found the difference between men and women to be smaller; men had on average 17% more C4 protein per C4 CN than women; we think our dataset confirms this finding and its timeframe (Fig. 2f) reinforcing the role of C4 in the differential susceptibility between men and women observed in SSc. The deficiency of C4 may trigger an inappropriate clearance of apoptotic debris and stimulate chronic activation of myeloid cells. Also, it results in a defect to eliminate autoreactive B-cell clones, and a higher tendency to form self-reactive germinal centers23 and has been previously associated with more severe SLE with earlier disease onset44. We also observed that patients with SSc have lower C4 serum levels than unaffected individuals even after correcting for C4 gene CN, suggesting that hypocomplementemia in SSc is not simply due to C4 genetics but also reflects disease effects on background complement levels45.
C4 expression was clearly down-regulated in SSc patients compared to healthy individuals, as were C4 protein levels, although to a lesser extent (Fig. 2c, d). This might be explained by the difficulty in standardizing C4 protein assays across 10+ laboratory sites but might also point to differential mRNA stability adding another layer of complexity yet to be analyzed. Indeed, while we observed clear disease-independent differences of C4 protein levels between men and women, there was no significant differential expression of C4 between healthy men and women, which suggests post-translational effects to play a role. In this line it has been proposed that IFN-gamma increases the stability of C3 and C4 mRNA46 and a recent expression analysis in SSc detected a strong IFN signature in a subset of patients47.
More than a third of more than 10,000 C4 eQTLs from the GTEx v8 database are associated with SSc (pGWAS < 10−5) and C4 eQTLs alone can be used to explain most of the association of SSc within the MHC region, further supporting their importance in SSc. Interestingly, C4A-specific eQTLs can explain more SSc association than C4B-specific eQTLs (Supplementary Fig. 4B, C), which supports a stronger role for C4A in SSc. While C4 eQTLs could in principle be associated with SSc by the strong linkage structure present in the MHC, our data suggest that C4 eQTLs, forward selected to explain expression variance in blood, can also explain most genetic association with the MHC. Both analyses raise the possibility that C4 genetics is indeed the main signal on chromosome 6 for SSc, as has been suggested for SLE and SjS28, both rheumatic diseases that can co-occur with SSc.
C4-independent genetic association with SSc centers on two peaks (Fig. 3c), most of which can be explained by four AAs each of HLA-DPB1 and HLA-DRB1 (Fig. 3d). Interestingly, the AA positions for HLA-DPB1 and HLA-DRB1 overlap and all 8 AAs positions can be associated with four of five binding pockets described for class II HLA molecules48 likely interfering with (auto-)antigen binding. In addition, three of the HLA-DRB1 AA positions (37, 58, and 74) are close to sites (30, 60, and 74) which have been described to play a role in binding the consensus antigenic peptide of the topoisomerase I epitope, auto-antibodies to which define the ATA+ subgroup of SSc patients49. Furthermore, we found that the C4-independent genetic association with SSc can be explained by 10 independent classical HLA-Alleles instead of AAs, seven of which overlap with a model of nine independent HLA-Alleles recently described50, which supports the independence of C4 and HLA associations with SSc.
Our study has some limitations. First, the number of samples in the second dataset is very low in terms of GWAS. While we were able to replicate the association of C4A, C4B, and HERV-K CNs, replication of the most complex model was out of reach and needs to be the subject of further study. Second, we did not stratify the patients by clinical or serological subgroups. While our results on HLA-DRB1 AAs, being associated with SSc independently of C4 genetics, point towards anti-topoisomerase auto-antibodies and probably diffuse cutaneous SSc, the topic is too vast to explore in this study. Third, unfortunately, we could not distinguish C4A and C4B protein levels in serum, which would have been very useful, to further investigate the sexual dimorphism described. Fourth, the exact amino acid positions and classical alleles from the models calculated in our study might change in the future. New imputation reference panels might provide new associations that could influence the models as forward selection is sensitive to statistical fluctuations. Last, C4 forms a genetic module termed RCCX with three genes: serine/threonine nuclear protein kinase RP, steroid 21-hydroxylase CYP21, and extracellular matrix protein tenascin TNX51. Although we only assessed C4 CNs associated with SSc, we cannot discard the possibility that this module plays a role in disease susceptibility. Specifically, TNX is involved in the maintenance of collagen networks and tissue integrity52, as well as in TGFB activation and signaling, typical for fibrotic conditions such as SSc53,54.
Many rheumatic diseases, including SSc, could benefit from therapies directed toward the complement system. These are currently under active development and are not only focused on inhibitory mechanisms, but on activators or downstream activation fragments23. The inhibition of the complement pathway has proven challenging. Eculizumab, a C5 inhibitor, is a complement-targeting approved drug for a variety of vascular disorders and has recently been approved in kidney diseases55. Moreover, it has been studied in idiopathic inflammatory myopathies and SSc renal crisis, with promising results56,57. Our data suggest that C4 genetics in SSc, by affecting expression and C4 protein levels, plays an important role in mediating the genetic association in the MHC locus and might also be involved in the epidemiological sex bias of SSc. This highlights the contribution of the complement to the development of SSc and to autoimmune disorders in general, which could benefit from therapies directed towards the complement system. Our findings might help to bring C4 genetics to the clinic in the form of simple genetic tests.
Methods
Patients
All patients fulfilled the classification criteria of the 2013 American College of Rheumatology (ACR) or The European League Against Rheumatism (EULAR) or the criteria proposed by LeRoy and Medsger for early SSc58,59. CSIC’s Ethics Committee approved the study and written informed consent was obtained in accordance with the Declaration of Helsinki.
Cohorts and datasets
We (re-)analyzed two independent cohorts of European descent:
First cohort: genome-wide genotyped data from 14 independent epidemiological cohorts comprising a total of 28,179 unrelated individuals (9846 SSc patients and 18,333 healthy subjects) from 10 European countries6. To identify ancestry outliers ~100,000 quality-filtered independent SNPs were selected from each case–control GWAS cohort. Principal component (PC) analysis was performed using PLINK v1.07. Samples showing >4 standard deviations from the cluster centroids of each cohort were considered outliers and removed from further analyses. The presence of relatives and/or duplicates was assessed by computing identity-by-descent (IBD) estimation using PLINK v1.07. An individual from each pair of relatives (Pi_Hat > 0.45) or duplicates (Pi_Hat > 0.99) was removed. After exclusion of non-European samples, we recalculated genetic PCs using the merge of all imputed datasets, selecting ~100,000 independent markers using PLINK v1.9. Missing data values due to the different platforms used for genotyping were corrected by PLINK v1.9 (parameter –correct_for_missingness). We obtained informative principal components as the visualization of the first two PCs can be interpreted as a “map” of the European continent (Supplementary Fig. 1). Second cohort: this cohort included genome-wide genotyped data, whole blood expression data and blood serum C4 protein concentrations from 333 SSc patients and 524 healthy individuals from 9 European countries39. This second cohort is a subset of a larger cohort of seven immune-mediated diseases plus controls described here60. Individuals were excluded on the basis of incorrect sex assignment, high missingness (>10%), non-European ancestry (<55% using Frappe61 and high relatedness (PLINK v1.9 Pi_Hat >0.5). In addition, population stratification was also analyzed by PC analysis selecting ~100,000 independent markers using PLINK v1.9. We obtained informative principal components as the visualization of the first two PCs can be interpreted as a “map” of the European continent (Supplementary Fig. 1).
Basic clinical epidemiological information by cohort can be found in Supplementary Table 1.
Expression data
Whole blood expression data was obtained from alpha and beta globin depleted (globinCLEAR, Ambion) total RNA. Single end 50 bp stranded sequencing was performed on a HiSeq2500 Illumina within the PRECISESADS consortium60 and processed with bcl2fastq (Illumina), Cutadapt62, STAR v2.5.2 (2-pass default mapping to GRCh1963, and RSEM v1.2.3164 to obtain estimated counts per gene. Raw count data were normalized for quantitative trait analyzes39. Briefly: Three genetic principal components (PCs) were regressed out from VSN-normalized65 raw read count data. Potential non-genetic influences were regressed out for SSc and controls separately by 20 (SSc:18) PCs calculated from inter-sample expression correlation matrices.
C4 protein data
Human complement C4 serum data was obtained from the PRECISESADS consortium60 from a turbidimetric immunoassay method according to the manufacturer’s recommendations (SPAPLUS analyzer)66. A corrective factor was calculated in order to normalize the data between the centers as described60.
Imputation
SNPs
For both cohorts, we imputed SNPs from chromosome 6 using the TOPMed reference panel with default settings at https://imputation.biodatacatalyst.nhlbi.nih.gov/67. Stringent QC measures were applied to both cohort’s pre-imputation as follows: SNPs with call rates < 0.98; minor allele frequencies (MAFs) <0.01; and those that deviated from Hardy–Weinberg equilibrium (HWE; P < 0.001 in both case and control subjects) were filtered out from further analysis; samples with call rates <0.95 were removed. Relatives and/or duplicated samples were removed. Post-imputation quality control included filtered for imputation quality (r2 > 0.3), MAF > 0.05, and HWE, which 6,39 resulted in 9,068 SSc patients and 17,565 healthy individuals for C4 haplotype imputation for the first cohort.
C4 haplotypes
A set of 7021 SNPs TOPMed imputed SNPs were selected as they were (a) imputed in all individuals in both cohorts and (b) overlapped the C4 CN reference panel. C4 haplotype imputation was carried separately for both cohorts using the software imputec429 and https://github.com/freeseek/imputec4 and the reference panel downloaded from the dbGaP study accession: phs001992.v1.p128. Weighted imputation accuracy was calculated by multiplying r2Allele by Allele frequency in Supplementary Table 2B.
C4 copy numbers
Each C4 haplotype carries a specific number of C4 isotypes (C4A, C4B) and HERV-K elements (Supplementary Table 2C). We calculated total C4, C4A, C4B, and HERV-K CN dosages by multiplying the allele dosages of the structural haplotype by the number of copies of each C4 isotype and HERV-K that the haplotype contains. For instance, the haplotype AL-BL contains one C4A gene and one C4B gene and two HERV-K copies. The numbers of short and long forms of C4A and C4B (AL, AS, BL, BS) per haplotype are self-evident for 17 of 29 imputed haplotypes. For the remaining, long and short forms were inferred by the consensus that ~95% of C4A is present in the long form43,68,69,70. The haplotype AL-BS for instance can be coded as 0.95 AL, 0.05 AS, 0.05 BL, and 0.95 BS. CNs per haplotype can be found in Supplementary Table 2C.
Classical HLA alleles and HLA amino acids (AA)
Data for the classical HLA alleles and AA variants were obtained from the first cohort by imputation using SNP2HLA71 and the reference panel from the Type 1 Diabetes Genetic Consortium72, described in ref. 50. After genotyping QC, all variants were imputed for each case–control dataset separately in the extended MHC region in chromosome 6. Imputed data were also filtered for 95% success call rate for alleles and amino acids, deviation from HWE considering a P value of <0.001 for SNPs in controls and 95% total call rate for individuals50.
Pearson correlation of C4 haplotypes and classical HLA Alleles
Was calculated among the C4 haplotype dosages and the allele dosages from the HLA imputation.
C4 copy number association analysis
Logistic-regression models from simple to complex were calculated (using the function glm in R 4.0.3) to assess the association of total C4 dosage and its isotype dosages with the disease. We included cohort, five genome-wide principal components (PCs) and sex as covariates, assuming their effects were not collinear:
(a) SSc ~ C4 + HERV-K + PC1-5 + cohort + sex
(b) SSc ~ C4A + C4B + HERV-K + PC1-5 + cohort + sex
(c) SSc ~ C4Ashort + C4Along + C4Bshort + C4Blong + PC1-5 + cohort + sex
The number of subjects in our first cohort permits us to expand the simple additive model to a more complex one investigating the predictors that influence each other. We included three two-way interaction terms in the logistic-regression model:
(d) SSc ~ C4A + C4B + HERV-K + PC1-5 + cohort + sex + C4A:C4B + C4A: HERV-K + C4B: HERV-K + C4A:sex + C4B:sex
Meta-analysis was conducted with Metasoft73 using data from model a, b, and c from both datasets.
Power calculation
Power calculations in CNV studies are problematic because effect sizes and models of the association are based on approximations that may be unrealistic74.
C4
Power calculations for C4B in males was carried out using the GAS Power Calculator [https://csg.sph.umich.edu/abecasis/gas_power_calculator/]. Here, we calculated the disease allele frequency as (sum(CN C4B < 2)/N) = 0.26 and the genotype relative risk as e0.09 = 1.09. Using an additive disease model and 1278 male cases and 6875 male controls, results in a power of 0.406 to detect an association with P < 0.05.
C4:sex interaction
We calculated the power to detect C4B:sex interactions using “powerGWASinteraction”75 in R 4.0.3 which can treat sex as environmental variable. We used: prevalence = 0.00034, pEnv = 0.306, betaC4B = −0.04, beta.sex = −1.35, beta.c4b:sex = 0.13, caseControlRatio = 0.34, ORgeneEnvironment = 1.03, alpha = 0.05 and alpha1 = 1. pGene probability was calculated as (sum(CN C4B > = 2)/N) = 0.73. This results in a power to detect a C4B:sex interaction with P values <0.05 of 0.33.
Calculation of composite C4 risk score for SSc
For each individual (i) a composite C4 risk score can be calculated as the sum of betas “Sb,i” from the (model-specific) effect sizes multiplied by the design matrix (of CN dosages, sex, interactions.. etc.) of the predictors. An individual relative risk score was then calculated as riski = eSb,i/(1+eSb,i).
To visualize the interaction effect of C4A and C4B CNs on relative risk, one multiplies the effect sizes (betas) of the most complex model “d” of C4A, C4B, and C4A:C4B with the design matrix and calculates the relative risk score as above. To visualize the effect of HERV-K on risk we calculated a composite score with model “d” betas and C4A, C4B, HERV-K, C4A:HERV-K, C4B:HERV-K, and C4A:C4B. To visualize the effect of C4B CN in males subjects, we used effect sizes from model “d” for sex, C4A, C4B, sex:C4A, sex:C4B, and C4A:C4B.
Pearson correlation of C4 CN and C4 expression and C4 serum levels
Was calculated with C4 CN dosages, the PC residualized expression data (see above) and the center corrected C4 protein serum concentrations. For visualization, CN dosages were rounded to integers.
C4 expression modeling
Total C4 expression was calculated as the sum of C4A and C4B expression. We used the linear model function “lm” in R 4.0.3 to calculate the adjusted coefficient of determination (r2) for each model with C4 CN, C4 CN + C4 eQTLs and C4 eQTLs alone as predictors. Model evolution is noted in Supplementary Table 4A–C. To add C4 eQTLs to the C4 CN model as expression modifiers, we used forward selection. In a stepwise manner, we selected the SNP to add to the model which had the most significant P value conditioning on all predictors already in the model until no one more SNP was found with P < 0.01. In the same way, SNPs were selected for the eQTLs only model until no more SNPs were found with P < 0.01. To select SNPs in the expression (=second) dataset which were to be used for conditioned analysis of the MHC SNPs in the first dataset, forward selection was applied with SNPs which had pGWAS < 10−5 until no more SNP was found with P < 0.01.
Modeling of C4 copy numbers using eQTLs
We tested if the eQTLs found to explain C4A or C4B expression variance (see C4 expression modeling) can predict copy number dosages of the long and short forms of C4A and C4B: AS, AL, BS and BL. We used the linear model function “lm” in R 4.0.3 to calculate the adjusted coefficient of determination (r2) for each model with either C4A eQTLs or C4B eQTLs as predictors.
C4 gene expression analysis in whole blood
Using raw count data, we included disease, blood cell composition, and effective library size (calculated by EdgeR in R 4.0.3) in the final model. While cell type-specific expression changes between SSc and controls were found significant at a nominal level for most cell types, the direction of expression change coincided for all cell types. We decided to report only whole blood expression changes controlling for blood cell composition.
C4 protein blood serum analysis
We included disease, sex, age, AS, AL, BS, and BL in the final model. The significance for the difference between SSc and controls in men and women was calculated with both the Mann–Whitney test and a t test.
Residual association of genetic variants across the MHC region to SSc
We performed conditional association analysis for genetic markers across the MHC genomic region. The first dataset was analyzed. In all models, we included cohort, five genome-wide PCs and sex as basic covariates. Association analysis of MHC region variants was conditioned on the basic covariates plus:
(1) nothing;
(2) a risk score: 2.3 × C4A CN + C4B CN as proposed;28
(3) covariates from model “b”: C4A CN + C4B CN + HERV-K CN;
(4) covariates from the most complex model “d” described above;
(5) C4 (C4A or C4B or both) eQTLs from GTEx v8 (obtained by forward selection in the first dataset until no SNP had pSNP < 10−5, see Supplementary Tables 4B, 6 and 4C, 6);
(6) C4A -specific eQTLs from GTEx v8 (obtained by forward selection in the first dataset until no SNP had pSNP < 10−5). EQTLs were called C4A-specific if no C4B eQTL was reported in GTEx v8 with P < 0.01 for each SNP;
(7) C4B-specific eQTLs from GTEx v8 (obtained by forward selection in the first dataset until no SNP had pSNP < 10−5);
(8) expression-model SNPs (with pGWAS < 10−5) (obtained by forward selection in the second dataset as described above, see Supplementary Tables 4B, 7 and 4C, 7).
Residual, C4-independent, the association of the MHC region with SSc
After accounting for the contribution of C4 genetics with models “5” or “8” above, we sought to model residual, C4-independent, association of MHC SNPs with (a) forward selection of classical HLA alleles; (b) forward selection of classical HLA alleles of HLA-DRB1 and HLA-DPB1; (c) forward selection of AAs of HLA genes; (d) forward selection of AAs of HLA-DRB1 and HLA-DPB1. Forward selection was carried out until no more HLA allele or AA was found with P < 5 × 10−8.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Summary statistics of the SSc meta-GWAS are available through the NHGRI-EBI GWAS Catalog (https://www.ebi.ac.uk/gwas/downloads/summary-statistics): GCST009131. Data from the PRECISESADS consortium are hosted by ELIXIR Luxembourg https://elixir-luxembourg.org/ and are available upon request. The access procedure is described on the data landing page (https://doi.org/10.17881/th9v-xt85). All other data are contained in the article file and its Supplementary Information.
Code availability
All analysis has been performed with either the software described in “Methods” or within R 4.0.3. For logistic-regression analysis, the glm function of R was used.
References
Denton, C. P. et al. Systemic sclerosis. Lancet 390, 1685–1699 (2017).
Elhai, M. et al. Mapping and predicting mortality from systemic sclerosis. Ann. Rheum. Dis. 76, 1897–1905 (2017).
Bergamasco, A. et al. Epidemiology of systemic sclerosis and systemic sclerosis-associated interstitial lung disease. Clin. Epidemiol. 11, 257–273 (2019).
Angiolilli, C. et al. New insights into the genetics and epigenetics of systemic sclerosis. Nat. Rev. Rheumatol. 14, 657–673 (2018).
Truchetet, M. E. et al. Current concepts on the pathogenesis of systemic sclerosis. Clin. Rev. Allergy Immunol. https://doi.org/10.1007/s12016-021-08889-8 (2021).
Lopez-Isac, E. et al. GWAS for systemic sclerosis identifies multiple risk loci and highlights fibrotic and vasculopathy pathways. Nat. Commun. 10, 4955 (2019).
Bossini-Castillo, L. et al. Immunogenetics of systemic sclerosis: defining heritability, functional variants and shared-autoimmunity pathways. J. Autoimmun. 64, 53–65 (2015).
Zhang, F. et al. Copy number variation in human health, disease, and evolution. Annu Rev. Genomics Hum. Genet. 10, 451–481 (2009).
Henrichsen, C. N. et al. Copy number variants, diseases and gene expression. Hum. Mol. Genet. 18, R1–R8 (2009).
Fanciulli, M. et al. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat. Genet. 39, 721–723 (2007).
Yang, Y. et al. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am. J. Hum. Genet. 80, 1037–1054 (2007).
de Cid, R. et al. Deletion of the late cornified envelope LCE3B and LCE3C genes as a susceptibility factor for psoriasis. Nat. Genet. 41, 211–215 (2009).
McKinney, C. et al. Association of variation in Fcgamma receptor 3B gene copy number with rheumatoid arthritis in Caucasian samples. Ann. Rheum. Dis. 69, 1711–1716 (2010).
Olsson, L. M. et al. Copy number variation in autoimmunity—importance hidden in complexity? Eur. J. Immunol. 42, 1969–1976 (2012).
Usher, C. L. et al. Complex and multi-allelic copy number variation in human disease. Brief. Funct. Genomics 14, 329–338 (2015).
Carroll, M. C. The role of complement and complement receptors in induction and regulation of immunity. Annu. Rev. Immunol. 16, 545–568 (1998).
Walport, M. J. Complement. First of two parts. N. Engl. J. Med. 344, 1058–1066 (2001).
West, E. E. et al. Complement and the regulation of T cell responses. Annu. Rev. Immunol. 36, 309–338 (2018).
Chen, M. et al. The complement system in systemic autoimmune disease. J. Autoimmun. 34, J276–J286 (2010).
Holers, V. M. Complement and its receptors: new insights into human disease. Annu. Rev. Immunol. 32, 433–459 (2014).
Goicoechea de Jorge, E. et al. Common and rare genetic variants of complement components in human disease. Mol. Immunol. 102, 42–57 (2018).
Yang, Y. et al. Diversity in intrinsic strengths of the human complement system: serum C4 protein concentrations correlate with C4 gene size and polygenic variations, hemolytic activities, and body mass index. J. Immunol. 171, 2734–2745 (2003).
Wang, H. et al. Complement C4, infections, and autoimmune diseases. Front. Immunol. 12, 694928 (2021).
Banlaki, Z. et al. Fine-tuned characterization of RCCX copy number variants and their relationship with extended MHC haplotypes. Genes Immun. 13, 530–535 (2012).
Wu, Z. et al. Association between complement 4 copy number variation and systemic lupus erythematosus: a meta-analysis. Clin. Exp. Med. 20, 627–634 (2020).
Okada, Y. et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506, 376–381 (2014).
Langefeld, C. D. et al. Transancestral mapping and genetic load in systemic lupus erythematosus. Nat. Commun. 8, 16021 (2017).
Kamitaki, N. et al. Complement genes contribute sex-biased vulnerability in diverse disorders. Nature 582, 577–581 (2020).
Sekar, A. et al. Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183 (2016).
Yu, C. Y. et al. Dancing with complement C4 and the RP-C4-CYP21-TNX (RCCX) modules of the major histocompatibility complex. Prog. Nucleic Acid Res. Mol. Biol. 75, 217–292 (2003).
Klein, M. et al. Contribution of CD8+ T cells to inflammatory cytokine production in systemic sclerosis (SSc). Autoimmunity 49, 532–546 (2016).
Ingersoll, M. A. Sex differences shape the response to infectious diseases. PLoS Pathog. 13, e1006688 (2017).
Blasko, B. et al. Low complement C4B gene copy number predicts short-term mortality after acute myocardial infarction. Int. Immunol. 20, 31–37 (2008).
Walli-Attaei, M. et al. Variations between women and men in risk factors, treatments, cardiovascular disease incidence, and death in 27 high-income, middle-income, and low-income countries (PURE): a prospective cohort study. Lancet 396, 97–109 (2020).
Liang, Y. et al. A gene network regulated by the transcription factor VGLL3 as a promoter of sex-biased autoimmune diseases. Nat. Immunol. 18, 152–160 (2017).
Boese, A. C. et al. Sex differences in vascular physiology and pathophysiology: estrogen and androgen signaling in health and disease. Am. J. Physiol. Heart Circ. Physiol. 313, H524–H545 (2017).
Han, M. K. et al. Female sex and gender in lung/sleep health and disease. increased understanding of basic biological, pathophysiological, and behavioral mechanisms leading to better health for female patients with lung disease. Am. J. Respir. Crit. Care Med. 198, 850–858 (2018).
Blanchong, C. A. et al. Genetic, structural and functional diversities of human complement components C4A and C4B and their mouse homologues, Slp and C4. Int. Immunopharmacol. 1, 365–392 (2001).
Kerick, M. et al. Expression quantitative trait locus analysis in systemic sclerosis identifies new candidate genes associated with multiple aspects of disease pathology. Arthritis Rheumatol. 73, 1288–1300 (2021).
Basu, M. et al. Predicting tissue-specific gene expression from whole blood transcriptome. Sci. Adv. 7, eabd6991 (2021).
Mu, Z. et al. The impact of cell type and context-dependent regulatory variants on human immune traits. Genome Biol. 22, 122 (2021).
Gadjeva, M. et al. Macrophage-derived complement component C4 can restore humoral immunity in C4-deficient mice. J. Immunol. 169, 5489–5495 (2002).
Wouters, D. et al. High-throughput analysis of the C4 polymorphism by a combination of MLPA and isotype-specific ELISA’s. Mol. Immunol. 46, 592–600 (2009).
Juptner, M. et al. Low copy numbers of complement C4 and homozygous deficiency of C4A may predispose to severe disease and earlier disease onset in patients with systemic lupus erythematosus. Lupus 27, 600–609 (2018).
Esposito, J. et al. The association of low complement with disease activity in systemic sclerosis: a prospective cohort study. Arthritis Res. Ther. 18, 246 (2016).
Mitchell, T. J. et al. IFN-gamma up-regulates expression of the complement components C3 and C4 by stabilization of mRNA. J. Immunol. 156, 4429–4434 (1996).
Beretta, L. et al. Genome-wide whole blood transcriptome profiling in a large European cohort of systemic sclerosis patients. Ann. Rheum. Dis. 79, 1218–1226 (2020).
Agudelo, W. A. et al. Quantum chemical analysis of MHC-peptide interactions for vaccine design. Mini Rev. Med. Chem. 10, 746–758 (2010).
Kongkaew, S. et al. Interactions of HLA-DR and topoisomerase I epitope modulated genetic risk for systemic sclerosis. Sci. Rep. 9, 745 (2019).
Acosta-Herrera, M. et al. Comprehensive analysis of the major histocompatibility complex in systemic sclerosis identifies differential HLA associations by clinical and serological subtypes. Ann. Rheum. Dis. 80, 1040–1047 (2021).
Blanchong, C. A. et al. Deficiencies of human complement component C4A and C4B and heterozygosity in length variants of RP-C4-CYP21-TNX (RCCX) modules in caucasians. The load of RCCX genetic diversity on major histocompatibility complex-associated disease. J. Exp. Med. 191, 2183–2196 (2000).
Matsumoto, K. I. et al. The roles of tenascins in cardiovascular, inflammatory, and heritable connective tissue diseases. Front. Immunol. 11, 609752 (2020).
Kasprzycka, M. et al. Tenascins in fibrotic disorders-from bench to bedside. Cell Adh. Migr. 9, 83–89 (2015).
Valcourt, U. et al. Tenascin-X: beyond the architectural function. Cell Adh. Migr. 9, 154–165 (2015).
Ricklin, D. et al. The renaissance of complement therapeutics. Nat. Rev. Nephrol. 14, 26–47 (2018).
Gouin, A. et al. Role of C5 inhibition in idiopathic inflammatory myopathies and scleroderma renal crisis-induced thrombotic microangiopathies. Kidney Int. Rep. 6, 1015–1021 (2021).
Devresse, A. et al. Complement activation and effect of eculizumab in scleroderma renal crisis. Medicine 95, e4459 (2016).
van den Hoogen, F. et al. 2013 classification criteria for systemic sclerosis: an American college of rheumatology/European league against rheumatism collaborative initiative. Ann. Rheum. Dis. 72, 1747–1755 (2013).
LeRoy, E. C. et al. Criteria for the classification of early systemic sclerosis. J. Rheumatol. 28, 1573–1576 (2001).
Barturen, G. et al. Integrative analysis reveals a molecular stratification of systemic autoimmune diseases. Arthritis Rheumatol. 73, 1073–1085 (2021).
Tang, H. et al. Estimation of individual admixture: analytical and study design considerations. Genet Epidemiol. 28, 289–301 (2005).
Martin, M. Cutadapt remove adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Li, B. et al. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinforma. 12, 323 (2011).
Huber, W. et al. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 18, S96–S104 (2002).
Capaldo, C. et al. The active immunological profile in patients with primary Sjogren’s syndrome is restricted to typically encountered autoantibodies. Clin. Exp. Rheumatol. 34, 722 (2016).
Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299 (2021).
Mason, M. J. et al. Low HERV-K(C4) copy number is associated with type 1 diabetes. Diabetes 63, 1789–1795 (2014).
Zai, C. C. et al. Association study of the complement component C4 gene in tardive dyskinesia. Front. Pharm. 10, 1339 (2019).
Mariaselvam, C. M. et al. The complement C4 genetic diversity in first episode psychosis of the OPTiMiSE cohort. Schizophr Bull Open 2, sgab003 (2021).
Jia, X. et al. Imputing amino acid polymorphisms in human leukocyte antigens. PLoS ONE 8, e64683 (2013).
Brown, W. M. et al. Overview of the MHC fine mapping data. Diabetes Obes. Metab. 11, 2–7 (2009).
Han, B. et al. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am. J. Hum. Genet. 88, 586–598 (2011).
Rucker, J. J. et al. Phenotypic association analyses with copy number variation in recurrent depressive disorder. Biol. Psychiatry 79, 329–336 (2016).
Dai, J. Y. et al. Two-stage testing procedures with independent filtering for genome-wide gene-environment interaction. Biometrika 99, 929–944 (2012).
Acknowledgements
We would like to thank Guillermo Barturen Briñas and Elena Carnero-Montoro for fruitful discussions and Sofia Vargas and Gema Robledo for their excellent technical assistance. We would like to thank Elena López-Isac for organizing all SSc GWAS datasets and all members of the PRECISESADS consortium, especially Ralf Lesche, Sepideh Babaei, Anne Buttgereit, Suzana Makowska and Martina Runge for preparing the RNA Seq data and Johan Frostegård and Jacques-Olivier Pers for preparing and normalizing the serum C4 data. We greatly appreciate the patients and healthy donors for their essential participation in these studies. This work was supported by grant RTI2018101332-B-100 funded by MCIN/AEI/10.13039/501100011033 by “ERDF A way of making Europe”, Red de Investigación en Inflamación y Enfermedades Reumáticas (RIER) from Instituto de Salud Carlos III (RD16/0012/0013). This work has received funding from the Innovative Medicines Initiative 1 & 2 Joint Undertaking (JU) under grant agreements No 115565 (PRECISESADS) and No 831434 (3TR). The JU receives support from the European Union’s FP7 and Horizon 2020 research and innovation programs and EFPIA. MAH was supported by the Juan de la Cierva Incorporacion program, grant IJC2018-035131-I funded by MCIN/AEI/10.13039/501100011033. This work is dedicated to the memory of Annette Kerick (1945-2020).
Author information
Authors and Affiliations
Consortia
Contributions
M.K., M.A.H., and J.M. contributed to the conception and study design. M.K. and M.A.H. contributed to data collection, QC, imputation, and data analysis. C.P.S-A., J.L.C., S.A., S.M.P., M.N., N.H., G.M., J.K.V-B., G.O., A.B., A.H., C.T., Y.A., C.F., M.E.A-R., T.R.D.J.R., L.B., C.P.D., and M.D.M. contributed to GWAS and RNA-Sequencing data collection. All co-authors made substantial contributions to data acquisition, data interpretation, and revised the work critically for important intellectual content.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kerick, M., Acosta-Herrera, M., Simeón-Aznar, C.P. et al. Complement component C4 structural variation and quantitative traits contribute to sex-biased vulnerability in systemic sclerosis. npj Genom. Med. 7, 57 (2022). https://doi.org/10.1038/s41525-022-00327-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-022-00327-8
