
Abstract
A growing number of children born are conceived through in vitro fertilisation (IVF), which has been linked to an increased risk of adverse perinatal outcomes, as well as altered growth profiles and cardiometabolic differences in the resultant individuals. Some of these outcomes have also been shown to be influenced by the use of different IVF culture media and this effect is hypothesised to be mediated epigenetically, e.g. through the methylome. As such, we profiled the umbilical cord blood methylome of IVF neonates that underwent preimplantation embryo development in two different IVF culture media (G5 or HTF), using the Infinium Human Methylation EPIC BeadChip. We found no significant methylation differences between the two groups in terms of: (i) systematic differences at CpG sites or regions, (ii) imprinted sites/genes or birth weight-associated sites, (iii) stochastic differences presenting as DNA methylation outliers or differentially variable sites, and (iv) epigenetic gestational age acceleration.
Similar content being viewed by others

Introduction
Since its first successful implementation in 1978, more than 8 million children1 (~3% of all births in European countries) have been conceived through in vitro fertilisation (IVF)2. Although most of these children are born seemingly healthy, assisted reproductive technology (ART) singletons are at increased risk of adverse perinatal3 and childhood4,5 outcomes as compared to their naturally conceived counterparts. For instance, IVF neonates are at higher risk of preterm birth (<37 weeks, relative risk (RR) 1.4–2.0), low birth weight (<2500 g, RR 1.6–1.7), being small for gestational age (RR 1.5) and perinatal mortality (RR 1.7–2.0)3. Later life outcomes mainly relate to growth and weight, as well as disturbed cardiometabolic function, demonstrated by increased systolic blood pressure, suboptimal diastolic function, lower low-density lipoprotein and higher fasting insulin levels4,5,6.
The IVF process involves 2–6 days of in vitro embryo culture, during which embryos are exposed to an artificial environment that is influenced by the culture medium, atmospheric conditions (oxygen levels) and laboratory plastics. Over the years, a variety of culture media have been used7,8,9,10,11, which have been shown to affect short- and long-term health outcomes of the resultant offspring in both animal and human studies. In human studies culture medium composition has been linked to differences in birth weight12,13,14, postnatal weight15,16 and the childhood developmental profile17. Previously, we conducted a multi-centre randomised controlled trial (RCT) among six Dutch IVF centres to compare the effect of G5 (Vitrolife) and HTF (Lonza) media on pregnancy and neonatal outcomes. Of note is that the G5 medium contains amino acids8,18, while HTF does not. While it was found that G5 led to lower fertilisation rates, it generated more embryos that were suitable for transfer and had a higher implantation rate, leading to a higher cumulative live birth rate14. At birth, G5 neonates were more likely to be born prematurely and with lower birth weights14 even when birth weight was corrected for gestational age, indicating an additional effect of the culture medium on birth weight.
Although no causative mechanism for these differences in outcome has been established, the findings are consistent with the Developmental Origins of Health and Disease (DOHaD) paradigm. This paradigm suggests that adversity during early life, such as during the peri-conception period, makes the resultant offspring more vulnerable to disease in later life19 and this effect may be mediated by the epigenome, and specifically DNA methylation20. In the context of IVF, the handling of gametes and embryos and exposure to the in vitro environment or the hormone-primed uterus represent environmental exposures that could contribute to the observed disease susceptibility21. Further evidence for the involvement of DNA methylation is that epigenetically regulated imprinting disorders, although still rare, are more common after IVF22. Moreover, the period of in vitro embryo culture of IVF procedures coincides with the process of epigenetic reprogramming, during which DNA methylation marks are almost completely erased and re-established23,24. This process has been shown to be responsive to environmental cues24.
Relatively few studies have used molecular assays to assess the effects of different IVF culture media on the resultant embryos and neonates. For instance, the methylome of IVF neonates from a culture medium trial has only been investigated in one prior study. As a follow-up to the aforementioned G5 versus HTF RCT, placental DNA methylation at selected imprinting control regions was compared in resultant singletons finding no significant differences within these regions25. In contrast, most other work so far has focused on comparing the placenta or umbilical cord blood (UCB) methylome of IVF neonates in general to their naturally conceived counterparts26,27,28,29,30,31,32. These studies were recently summarised in a systematic review and meta-analysis30 which described that most sites or regions identified to be differentially methylated were inconsistent or contradictory between studies, likely due to differences in the methylome analysis methods, heterogeneity within the cohorts and due to sample size. The majority of included studies used targeted approaches to look at imprinting genes, and a meta-analysis of such studies conducted on the placenta and UCB samples revealed only significant differential methylation at the PEG1/MEST imprinting gene locus30. Methylation at the imprinted regions KvDMR1, H19 CTCF3 and CTCF6 and SNRPN may also be perturbed in IVF placentas, but these did not reach statistical significance in the meta-analysis30. The epigenetic deregulation in these cases is thought to occur post-fertilisation as it involves both paternally and maternally methylated regions and the methylation levels differ only by a few percent, indicating that the loss or gain of methylation only affects a minority of alleles. The findings from genome-wide methylation studies on these tissues have been contradictory, with some studies identifying differential methylation, predominantly with small differences, and others not29,30,31. Interestingly, some studies report increased variation in DNA methylation in IVF offspring28,29, suggesting a stochastic rather than a systematic universal effect of IVF on the methylome. This is substantiated by the reported increased rate of so-called methylation outliers (i.e. samples with an outlying methylation value at a given site or region) in the IVF group25. The contribution of different culture media to systematic or stochastic methylome differences on a genome-wide scale remains undetermined.
In this study, we investigated the effect of different IVF culture media on the DNA methylation of human IVF neonates on a genome-wide scale. To this end, we profiled the UCB methylome of IVF neonates that underwent embryo culture in G5 or HTF medium as part of a RCT. Additionally, the methylome profiles of this IVF cohort are compared to data from two reference birth cohorts of naturally conceived individuals (Fig. 1a).

a Schematic overview showing sample collection from IVF neonates from the G5 versus HTF RCT as well as the inclusion of naturally conceived neonates, genome-wide DNA methylation data generation, data processing and analyses included in this study. b PCA of all CpG sites passing our QC criteria in data from UCB samples of IVF neonates that underwent embryo culture in G5 (gold) or HTF (blue) medium. c Density plot showing the distribution of beta values from all sites and samples within each group (G5 = gold, HTF = blue).
Results
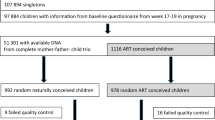
In the present study, we investigated genome-wide DNA methylation patterns of DNA samples derived from UCB collected at birth from 114 IVF neonates that had undergone embryo culture in G5 or HTF medium. 106 of the UCB samples (n = 59 G5, n = 47 HTF) yielded sufficient DNA for DNA methylation profiling using the EPIC array (Fig. 1a). Maternal characteristics, IVF treatment parameters and neonatal outcomes were comparable between the culture medium groups. In the G5 group, although not statistically significant, a higher percentage of pregnancies were complicated by hypertension and pre-eclampsia than in the HTF group (hypertension—14 vs. 6%, pre-eclampsia 7 vs. 2% for G5 and HTF pregnancies respectively). Delivery by caesarean section was lower (12%) in the G5 group compared to the HTF group (23%) (Table 1 and Supplementary Table 1).
All of the 106 samples that underwent DNA methylation analysis by EPIC array met our QC criteria (Methods). One sample from the G5 group was excluded from our analyses based on a mismatch between the recorded and predicted sex (Fig. 2a). Of the approximately 850,000 CpG sites represented on the EPIC array, we retained 696,205 sites for our analyses and 689,139 of these represented complete observations with no missing values in any samples.

a Scatter plot showing the projection of UCB samples (n = 105) into the PC space generated using reference data for sex prediction. The shape of the dots represents the recorded sex of the participants (circles = female, triangle = male), while the colour shows the predicted sex based on results from sEST (blue = male, pink = female, grey = not specified). b Violin plot showing the predicted cellular composition of the UCB samples, split by culture medium group (gold and blue represent G5 and HTF medium respectively). The violin plots are overlaid with boxplots where the horizontal lines represent the 25th percentile, median and 75th percentile respectively while the whiskers extend to the farthest data points that are no more than 1.5 times the IQR from the upper or lower quartile. c Heatmap showing associations between the principal components and biological/technical aspects of the samples. The colour gradient represents the −log10 of the p values. P values < 0.05 are shown. Significance of the correlation between age, maternal age, CD8-T cells, CD4T cells, NK cells, B cells, monocytes, granulocytes and nRBCs and the 8 PCs was tested using a permutation test with 10,000 permutations. The associations of the PCs with variables creating 2 groups (sample plate, sex, culture medium, pregnancy complication) and those creating three or more groups (Sentrix ID, Sentrix position) were tested using two-sided Wilcoxon rank tests and Kruskal–Wallis one-way analysis of variance respectively.
Global analysis of DNA methylation
Principal component analysis (PCA) did not reveal any separation of the culture medium groups within the first eight principal components (PCs) (Figs. 1b, 2c) that explain a total of 46.7% of the variance within our data (Supplementary Table 2), indicating that the culture media are not the main contributors to the variance of our data. Instead, the first eight PCs were significantly associated with sample characteristics including sex (PCs 5 and 8), gestational age (PC7), sample plate (PCs 1, 2, 4 and 6) as well as cellular composition of the samples (PCs 1–7) (Fig. 2b, c). Therefore, we corrected for these technical factors in our subsequent analyses, alongside potential confounders (sex, gestational age, maternal age, treatment centre and pregnancy complications) that were chosen a priori based on literature and expert opinion. The distribution of all beta values (all sites in all samples) was also similar between the culture medium groups (Fig. 1d).
Analysis of DNA methylation at individual CpG sites
Next, we investigated associations between the culture medium and DNA methylation at single CpG sites in an epigenome-wide analysis (EWAS) using linear mixed-effects models (Methods). Less than 0.01% of sites (37 sites in total) had a group mean difference of more than 10%, with the most extreme difference being 23.6%. After correcting for multiple testing, no statistically significant differentially methylated positions (DMPs) were found between the two culture medium groups (Fig. 3a, b). As pregnancy complications, such as gestational diabetes and pre-eclampsia, could affect or be affected by the methylome, we conducted the analyses twice, once with pregnancy complications included as a binary variable (yes/no) and once where all samples from complicated pregnancies (n = 18) were excluded. The results from this analysis were comparable to those of the first analysis (Supplementary Fig. 1).

Volcano plots showing differential methylation between G5 and HTF neonates where the grey dots represent all individual CpG sites (a, b) or multiple CpG sites aggregated into genomic regions, namely genes (c), promoters (d), CpG islands (e). Imprinted genes (c) and sites within them (a) are highlighted in purple while CpG sites associated with birth weight are shown in green (b). No significantly differentially methylation positions or regions (FDR adjusted p value < 0.1) were identified when comparing the two culture medium groups.
To reduce the number of comparisons, we also chose to repeat the analyses with sites of potential interest only, namely sites within imprinted genes33 and sites previously associated with birth weight34. After the data were pre-processed, 8726 sites within imprinted genes were tested. The maximum group mean difference amongst the imprinted sites was 6.9% (Fig. 3a). None of the sites were found to be significantly differentially methylated between the culture medium groups. Of the 914 CpG sites consistently found to be associated with birth weight in the meta-analysis by Küpers et al.34, 749 passed our quality control (QC) criteria and were included in the analysis. Amongst these sites, the maximal group mean difference was 3.0% and we did not find any of them to be statistically differentially methylated (Fig. 3b). Excluding the samples from complicated pregnancies did not change the result of either analysis (Supplementary Fig. 1a, b).
Regional analysis of DNA methylation
After looking at the methylation levels of individual sites, we looked at methylation across larger genomic regions, namely whole genes, promoters, and CpG islands (CGIs). Our analyses included 28,009 genes, of which 207 were imprinted genes, 42,035 promoters and 25,238 CGIs. The maximal group mean difference of any gene was 8.1%. Imprinted genes showed even lower group mean differences than imprinted sites, with a maximal difference of 3.6%. No genes were found to be significantly differentially methylated between the G5 and HTF groups (Fig. 3c). The maximal group mean differences for promoters and CGIs were 10.0% and 12.1%, respectively (Fig. 3d, e) and no promoters or CGIs were found to be significantly differentially methylated between the culture medium groups. Excluding samples from pregnancies with complications did not affect the results (Supplementary Fig. 2a–c).
Differential DNA methylation variance in IVF samples
To assess the contribution of stochastic DNA methylation alterations to the observed phenotypes in our IVF cohorts, we assessed differential variance, using the iEVORA algorithm35, and identified methylation outliers, using previously described thresholds36, in all samples. Applying this threshold, we identified a total of 157,160 outliers within the 105 analysed samples, with a predominance of hypomethylation outliers (114,693 hypomethylation outliers and 42,467 hypermethylation outliers) (Fig. 4). The median number of all, both hypo- and hypermethylation, outliers in each G5 sample was 571 (567.5 IQR) and 536 (269 IQR) in each HTF sample (Fig. 4), which was not found to be significantly different (p = 0.86) between the culture medium groups. Furthermore, when considering hypomethylation and hypermethylation outliers separately, no significant difference was found between the culture medium groups. Outlier burden, the total number of outliers per sample, was not significantly associated with gestational age, birth weight or maternal age. Only technical features of our samples, including sample plate and cell composition, were significantly associated with outlier burden (Supplementary Table 3). An association between pregnancy complications and the total number of outliers was not tested statistically, but amongst the samples with very high numbers of outliers (above the upper quartile), only 1 was born after a pregnancy complicated by pre-eclampsia. The results were comparable when the samples taken from neonates that had experienced pregnancy complications were excluded (Supplementary Fig. 3 and Supplementary Table 4). When applied to the full cohort, the iEVORA algorithm identified 262 CpG sites with significantly different variances between the culture medium groups (Supplementary Table 5, sheet 1). Of these sites, 90% (235 sites) were more variable in the G5 group as compared to the HTF group. 202 of the 262 differentially variable CpG sites were annotated with a gene name and four genes, namely FAM38A, MEF2C, OCA2 and TNNT2, each contained two differentially variable sites. Additionally, three of the differentially variable sites were located within imprinting genes, namely PEX10, MAGI2 and OBSCN. None of the differentially variable sites were amongst the birth weight-associated sites37. We then repeated the analysis excluding all participants who had experienced pregnancy complications which identified 105 differentially variable sites (Supplementary Table 5, sheet 2). Of these sites, 56% (50 sites) were more variable in the G5 group than the HTF group and 65 of the sites were the same as those identified in the analysis where all the participants were included. Seventy-nine of the sites were annotated with a gene name and multiple differentially variable sites were identified in two of the genes, namely two sites within the TNNT2 gene and three sites within the MOV10L1 gene. Furthermore, one site was found to be differentially variable in the imprinted gene PEX10 and none of the identified sites were birth weight-associated sites. GO and KEGG enrichment analyses of the differentially variable sites identified by iEVORA did not identify any significantly enriched ontologies or pathways after multiple testing corrections (Supplementary Table 5, sheets 4–7).

The main panel shows the number of hypomethylation (x-axis) and hypermethylation (y-axis) outliers per UCB sample (G5 = gold, HTF = blue). Distribution summaries, in the form of a density plot and boxplot, are shown for hypomethylation outliers and hypermethylation outliers in the top and right side panels respectively. Lines of the boxplot represent the 25th percentile, median and 75th percentile respectively while the whiskers extend to the farthest data point that is no more than 1.5 times the IQR from the upper or lower quartile. The axes are shown on a log10 scale. The groups were not found to be significantly different (p value > 0.1).
Epigenetic gestational age as a marker of developmental maturity
Gestational age can be predicted from DNA methylation levels at certain CpG sites (epigenetic clock)38,39. Similar to birth weight, these have been used to comment on developmental maturity at birth and gestational age acceleration (GAA), i.e. when epigenetic gestational age (eGA) is more advanced than clinical gestational age (cGA), has been positively correlated with birth weight37,39,40. eGA estimates derived using the Bohlin prediction model38 were more strongly correlated (Pearson correlation coefficient = 0.77) with our data and had a lower root mean squared error (RMSE = 1.29) than the estimates derived with the Knight prediction model39 (Pearson correlation coefficient = 0.55, RMSE = 1.45), therefore only the results from the Bohlin epigenetic clock are shown. However, of note is that both prediction models were trained using data from the HumanMethylation450 (450K) array and of the 96 sites used for the Bohlin eGA prediction model, eight sites with coefficients ranging from −15.5 to 6.1 are no longer present on the EPIC array. We removed these sites from the prediction model. When applying the prediction model to 450K data from the ENVIRONAGE study (n = 159), the omission of these eight CpG sites lead to a mean increase in the predicted gestational age by 0.73 weeks (range 0.17–1.10 weeks) (Supplementary Fig. 4a). Therefore, we cannot thoroughly evaluate absolute epigenetic gestational age, but we assume that all samples will be similarly affected by the missing sites and thus can compare the GAA between culture medium groups. GAA was calculated by regressing eGA on cGA while correcting for cell composition of the samples (Methods). In G5 samples the median GAA was 0.01 (0.64 IQR) and in HTF samples the median GAA was 0.03 (0.92 IQR), which was not significantly different (p = 0.42) (Fig. 5). Additionally, we found no significant correlation between GAA and birth weight (Pearson correlation = −0.17, p = 0.08). The results were comparable when participants who had experienced pregnancy complications were excluded from the analysis (Supplementary Fig. 4b).

Raincloud plot showing the GAA of each UCB sample in each culture medium group. Points represent individual samples of the G5 (gold) and HTF (blue) groups. Above a density plot and boxplot is shown. Horizontal lines of the boxplot represent the 25th percentile, median and 75th percentile respectively while the whiskers extend to the farthest data point that is no more than 1.5 times the IQR from the upper or lower quartile. GAA is represented in weeks. The groups were not found to be significantly different (p value > 0.1).
Comparison of IVF neonates to naturally conceived neonates
Even though the main aim of this study was to investigate the effect of two different culture media on the methylome of IVF neonates, we also sought to compare the methylomes of the IVF neonates to those of naturally conceived neonates using previously published data from the FLEHS and ENVIRONAGE longitudinal cohort studies. However, as these samples were not processed concurrently with the IVF samples it is not possible to correct for technical variation between the studies meaning that any effect of the IVF process cannot be differentiated from technical differences. These findings are demonstrated in the supplementary material (Supplementary Table 6: participant demographics, Supplementary Fig. 5: processing of FLEHS and ENVIRONAGE data, Supplementary Fig. 6: comparison of IVF and naturally conceived neonates).
Discussion
To the best of our knowledge, the genome-wide analysis of the influence of different IVF culture media on the methylome of human IVF neonates presented here is the largest cohort on which such a study has been conducted to date. Despite this, our sample size was insufficient to conduct sub-group analyses looking specifically at sex or treatment type (IVF vs. ICSI), which could reveal clinically relevant differences. We have investigated the impact of two compositionally different media, namely G5 from Vitrolife and HTF from Lonza, which were shown to influence IVF outcomes during the original RCT14. However, these phenotypic differences, e.g. in birth weight, were no longer significant in the sub-group of the original RCT that is presented here. We have found no evidence that these culture media lead to systematic or stochastic methylation differences in the resultant IVF neonates. To facilitate a comparison between different modes of conception, samples from well-matched naturally conceived individuals would have ideally been collected and processed alongside the IVF samples.
In line with findings from previous studies, examining the methylome of IVF children born after embryo culture in different media, we identified no differentially methylated positions or regions and only moderate group mean differences, largely less than 10%. This was also seen when the methylation status of imprinting genes in the placenta of the same individuals was analysed25. Similarly, a comparison of IVF children (aged 7 or 8) born after embryo culture in a global medium (Life Global), or single-step medium (Irvine Scientific) found no evidence of differential methylation between the medium groups at imprinting genes, transposable elements or on a genome-wide scale41,42.
The lack of differential methylation between G5 and HTF neonates may seem surprising given the stark differences in medium composition, which include the complete lack of amino acids in HTF medium, while G5 contains all amino acids except non-essential glutamate, glutamine and glycine8, and the addition of hyaluronan and lipoic acid to G5 medium14. Although the direct interplay between these individual components and DNA methylation has not been investigated in human embryos, it seems plausible that amino acid availability may influence the functional capacity of DNA methylation establishment and maintenance machinery. The sensitivity of embryos to these environmental differences is further supported by the finding that gene expression differences exist between embryos cultured in G5 or HTF medium43,44 and it is known that gene expression can be regulated by DNA methylation. However, the lack of differentially methylated sites or regions could be explained by a number of reasons. Firstly, dysregulated DNA methylation may be transient during in vitro embryogenesis and therefore not be detectable in neonates. Secondly, alternative epigenetic marks, such as histone modifications, may mediate the association between the culture media and the observed gene expression and phenotype differences. Additionally, in the sub-group of participants recruited for this follow-up study, phenotypic differences, such as birth weight, were less than in the full RCT cohort, which may have reduced the magnitude of any culture medium-induced effects. Finally, even though our study is the largest described methylome study after an IVF culture medium trial, we still lack the power to detect methylation differences with a magnitude of less than 10%. Although exact power estimates are challenging without the existence of prior data to establish the expected variance within our study population, simulation studies by Saffari et al.45 and Tsai et al.46 estimates that a sample size of 211 or more participants would be required to achieve 80% power to detect significant methylation differences with an effect size of 7% or less, respectively, using array-based assays such as the EPIC array45,46. However, it remains to be determined whether mean differences of less than 10%, representing methylation loss or gain at any site in just a small proportion of an individual’s cells, represent clinically significant differences47.
An alternative to the theory that peri-conception environmental differences induce systematic methylation differences, relates to the presence of stochastic epimutations that are either induced by the environment48 or provide a survival benefit if selection pressure is applied by certain environmental conditions49. In placenta samples of the same individuals as those described in this study, DNA methylation outliers were also identified in all samples without a difference in outlier burden between the culture medium groups25. Whether the number of outliers identified per individual is comparable between the two studies is difficult to assess due to the different thresholds used to define outliers and the different techniques used to analyse the methylome that differ vastly in their coverage of the genome. In the field of cancer biology, the iEVORA algorithm has been used to identify so-called field defects, which represent stochastic methylation alterations in normal pre-cancerous tissues that later undergo neoplastic transformation35. Frequently, sites identified as differentially variable in pre-cancerous samples become differentially methylated in tumour samples, suggesting that sites of this nature could be interesting biomarkers for disease with a later onset. In this study, such epimutations could be linked to later development of disease phenotypes, such as cardiometabolic diseases, although it should be noted that the differentially variable sites identified were not enriched in pathways relating to cardiovascular or metabolic function and it is not yet known whether there will be a difference in the prevalence of cardiometabolic disease between G5 and HTF offspring. Nonetheless, these sites warrant further clinical and molecular follow-up. Alternatively, differential variability at certain CpG sites could be driven by factors that are only experienced by a few individuals in the study population, such as pregnancy complications. Previously, DNA methylation differences associated with pre-eclampsia50 and gestational diabetes51,52,53 have been described when analysing UCB samples of neonates. According to our findings, there might be an association between culture medium, the number of differentially variable sites and pregnancy complications, but the design of this study does not allow to discuss the direction of causality (culture media, methylation and pregnancy complications).
Although studies comparing naturally conceived and IVF neonates have found some methylation differences, especially at imprinting genes30, this has not been observed in this or other culture medium comparisons25,41,42. This may be due to the fact that the environmental discrepancy between two culture media is less severe than the difference between in vivo and in vitro embryo development, thus leading to a smaller or no effect on the methylome. The concurrent processing of samples from naturally conceived individuals would be required to assess this further.
The lack of difference we observed in GAA may relate to the fact that the eGA prediction tools were trained using the HumanMethylation450K array and eight of the 96 probes required for the prediction model are no longer present on the EPIC array. These probes were therefore excluded from the model leading to a consistent over-estimation of gestational age. The inclusion of these sites may be important to identify a relationship between GAA, birth weight37,39,40 and potentially culture medium.
In conclusion, our genome-wide methylome analysis of IVF neonates that underwent embryo culture in G5 or HTF medium revealed no significant differences between the culture medium groups, suggesting that the use of either culture medium will establish a comparable DNA methylation signature, including at imprinting genes. However, we have observed some differentially variable sites between the culture medium groups, which seem associated with pregnancy complications, but the persistence and clinical significance of these findings should be assessed with further follow-up studies. To assess whether epigenetic reprogramming is transiently affected by differences in culture medium composition, epigenetic studies of embryos cultured in different media are required.
Methods
Ethical approval
This study was approved by the local medical ethical committee, Medische Ethische Commissie academisch ziekenhuis Maastricht/University of Maastricht (METC azM/UM) and registered in the Dutch Trial register (NTR 1979/NL1866). Both parents of all neonates gave written informed consent.
Study population and sample collection
Samples were collected as part of a culture medium comparison study14, which was a multi-centre RCT, involving six IVF centres in the Netherlands. Specifically, couples undergoing IVF treatments were randomised to embryo culture either in HTF medium (Lonza, Verviers, Belgium) or Vitrolife G1TM Version 5 (G5, Göteborg, Sweden), while all other IVF-related procedures and conditions were kept the same. Of the 6 IVF centres, five participated in UCB sampling. In these five centres, the study resulted in 273 singleton live births that occurred after fresh (not frozen) embryo transfers. UCB samples were collected from as many resulting singleton pregnancies as possible, 115 in total, irrespective of birth weight, gestational age at birth and the presence of pregnancy complications. Within 30 min of delivery, UCB was collected by a gynaecologist, nurse or midwife according to a standardised protocol. The samples were sent to the Department of Obstetrics and Gynaecology at Maastricht University Medical Centre (MUMC+) and were stored at −80 °C until they were used.
DNA extraction
DNA was extracted from thawed UCB samples using the Gentra Puregene DNA purification kit (Qiagen Hilden, Germany) according to the manufacturer’s instructions for 3 mL of human whole blood with minor modifications, namely, a smaller volume (8.5 mL) of red blood cell (RBC) lysis solution and longer centrifugation time (4 min where 2 min are indicated and 8 min where 5 min are indicated).
Bisulfite conversion and methylome profiling by EPIC array
One microgram of DNA was bisulfite-treated using the EpiTect® Fast 96 DNA Bisulfite Kit (Qiagen Hilden, Germany) and analysed using the Infinium Human Methylation EPIC BeadChip Kit (Illumina, CA, USA) according to the manufacturer’s protocol.
Data analysis
All data were analysed using R (version 3.6.3)54. The data were visualised using the ggplot255 and ComplexHeatmap56 packages.
Baseline characteristics
Differences in baseline characteristics between the two culture medium groups were compared and evaluated using Student’s t-tests for continuous variables and Pearson’s chi-squared tests for categorical variables.
Quality control and preprocessing
We applied preprocessing functions from the RnBeads package57 to normalise the data using subset-quantile within array normalisation (SWAN)58, and to remove poor quality probes and samples using the greedycut algorithm with a detection p value threshold of 0.05. Subsequently, the following sites were removed: (i) sites on the sex chromosomes, (ii) sites in close proximity to single nucleotide polymorphisms (SNPs), (iii) sites with missing values in more than 5% of the samples, and (iv) sites not in a CpG context. Sites containing missing values in 0–5% of the samples were only used to calculate aggregated beta values for different regions, including genes, promoters and CpG islands (CGI), they were excluded in all analyses looking at individual sites. Unless indicated otherwise, we used methylation beta values, which are calculated for each individual, at each CpG site by dividing the methylated signal intensity by the sum of the methylated and unmethylated signal intensity. The sex of the samples was predicted by comparing the samples’ sex chromosome methylation values and their respective detection p values to reference data using a clustering (PCA)-based approach, as implemented in the sEst package59. Correspondingly, each sample was assigned two predicted sexes based on the X-chromosome and Y-chromosome profiles respectively. If both matched, the sample was labelled as male or female, otherwise, it was labelled as “not specified”. If a mismatch between the recorded and predicted sex was identified, the sample was removed from subsequent analyses (n = 1).
Cellular deconvolution of UCB samples
To estimate the cellular composition of the UCB samples, the reference-based method described by Gervin et al.60 was implemented using the minfi package61. As recommended, the algorithm was applied to data pre-processed with the preprocessNoob method62 and deconvolution was carried out based on the IDOL optimised probes contained within the FlowSorted.CordBloodCombined.450k package63. The algorithm was used to estimate the proportion of natural killer cells, B cells, monocytes, granulocytes, nucleated red blood cells and CD4- and CD8-T cells within each sample.
Comparison of G5 and HTF IVF neonates
All high-quality CpG sites were used to conduct a PCA in which the beta values were centred but not scaled. Associations between the PCs and technical or demographic features of the samples were tested using: (i) permutation tests (with 10,000 permutations) to ascertain the significance of correlations (gestational age, maternal age, predicted cellular sample composition), (ii) a two-sided Wilcoxon rank test for categorical data where there are two groups (sex, culture medium, sample plate, pregnancy complication) or (iii) a Kruskal–Wallis one-way analysis of variance for categorical variables generating 3 or more groups (Sentrix ID, Sentrix position—array number and sample position respectively).
Methylation M-values, representing the log2 ratio of the methylated probe intensity compared to the unmethylated probe intensity64, were used to test for an association between the culture medium and DNA methylation with mixed-effects linear models implemented using the variancePartition package65. The models were corrected for potential confounders, namely gestational age, sex, maternal age, pregnancy complications (included as a binary variable where the presence of gestational diabetes, hypertension and pre-eclampsia were encoded as “yes” and otherwise “no” was recorded) and the predicted cell compositions as fixed effects while the treatment centre and batch effects (sample plate) were included as random effects. As gestational diabetes, hypertension and pre-eclampsia represent pathophysiologically heterogeneous pregnancy complications, the analyses were repeated while excluding participants affected by any of the complications. The models were applied to individual CpG sites or aggregate (mean) values of multiple CpG sites within a region to identify differentially methylated positions (DMPs) or regions (DMRs) respectively. To examine DMRs, the M-values of all probes attributed to a specific gene, promoter or CGI were aggregated by calculating their mean. For the targeted analyses, the models described above were applied to (sites within) imprinted genes33 and probes associated with birth weight34. All analyses were corrected for multiple testing using the Benjamini–Hochberg method66, and an adjusted p value of <0.1 was considered significant.
DNA methylation outliers were defined as described previously36. In short, hypomethylation outliers were defined as beta values lower than three interquartile ranges (IQR) from the 25th percentile, while hypermethylation outliers were defined as beta values greater than three times the IQR above the 75th percentile. The IQR and percentile values were calculated using all UCB samples. Subsequently, an association between the log10 transformed number of outliers and the culture medium was sought using the mixed-effects linear models as described above. To identify CpG sites with differential variance between the culture medium groups, we applied iEVORA35 using the matrixTests package67. At each CpG site, iEVORA applies Bartlett’s test, which is a parametric test for differential variance, as well as a Student’s t-test. Thereafter, sites reaching significance in Bartlett’s test after multiple testing correction (FDR corrected p value < 0.05) and nominal significance in the t-test (p value < 0.05 without multiple testing correction) are considered significant. As such, the output of Bartlett’s test is regularised as it is usually overly sensitive to single outliers. Both DNA methylation outliers and iEVORA analyses were applied to the full cohort as well as the subset of participants that had not experienced pregnancy complications. Gene ontology (GO) and Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathway enrichment analyses were conducted on differentially variable sites using functionality from the missMethyl package68.
Estimation of epigenetic gestational age and gestational age acceleration
Epigenetic gestational age was calculated using methods described by Bohlin et al.38 and Knight et al.39. The accuracy of the respective predictions was evaluated by calculating the Pearson’s correlation and root mean squared error between eGA and cGA. The model described by Bohlin et al. generated more accurate predictions and was therefore used to calculate GAA as previously described38. The Bohlin eGA prediction model was applied exactly as described by Bohlin et al. Firstly, within array normalisation was carried out using the BMIQ method using the RnBeads package57. Subsequently, batch effects attributable to the sample plate were corrected using ComBat from the sva package69. There were no missing values in any samples at the required sites, apart from eight CpG sites of the prediction model that are not present on the EPIC array. These eight sites were therefore excluded from the prediction. GAA represents the residuals from regressing eGA on cGA corrected for sample cell composition. To determine whether there is an association between GAA and culture medium, we applied mixed-effects linear models correcting for sex and maternal age as fixed effects alongside IVF treatment centre as a random effect. Again, the analysis was carried out on the full cohort and repeated excluding those participants who had experienced pregnancy complications.
Comparison of IVF and naturally conceived neonates
Selection and processing of data from naturally conceived individuals
To compare the methylome of our IVF neonates to naturally conceived individuals, we used data from two geographically similar longitudinal birth cohorts, namely the Flemish Environment and Health Study (FLEHS, Flanders Belgium)70,71 and the Environmental Influence on Early Ageing study (ENVIRONAGE)72 that had both undertaken array-based methylome profiling. Samples were considered for inclusion if the neonates were born after at least 36 full weeks of gestation (comparable to the IVF neonates included in this study). A total of 85 individuals from the FLEHS cohort and 502 individuals from the ENIRONAGE cohort were considered for inclusion based on this criterium. Methylation data in these studies were generated either with Illumina’s EPIC or 450K arrays. Preprocessing of the data from the separate studies/arrays was conducted separately but in an identical fashion to the IVF data, with the exception of within-study batch effects, which were corrected using ComBat69 as these could not be corrected for using the mixed-effects models. The sample inclusion and preprocessing steps of these two cohorts is summarised in Supplementary Fig. 5H. After study/data type-specific processing the data were combined, retaining only CpG sites present and passing the QC of all the array types included. Overall, 346,403 CpG sites were common to all platforms and studies.
To select only the data likely to be most similar to our IVF cohort, we generated a matched selection from the ENIRONAGE neonates who had their methylome profiled using the EPIC array. We used nearest neighbour matching (Mahalanobis distance) based on sex, maternal age, birth weight and gestational age to select 105 neonates to compare to the IVF neonates. This matching was carried out using the MatchIt package73.
Comparison of characteristics of matched IVF and naturally conceived individuals
The participant characteristics between IVF and matched naturally conceived individuals were compared and evaluated using Student’s t-tests for continuous variables and Pearson’s chi-squared tests for categorical variables. The p values obtained from this comparison are shown in Supplementary Table 6.
Statistical testing to compare naturally conceived and IVF neonates
Empirical Bayes moderated mixed effect linear models were used to ascertain associations between DNA methylation and mode of conception. These models were corrected for gestational age at birth, cell composition, sex, and maternal age as fixed effects and where relevant, array type as a random effect. Multiple testing correction was applied using the Benjamini–Hochberg method66, and an adjusted p value of <0.05 was considered significant.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The dataset generated during the current study, IVF samples, are available in the Gene Expression Omnibus (GEO) repository74 under the accession number GSE189531. Data included from the FLEHS and ENVIRONAGE cohorts are available from GEO under the accession numbers GSE110128 and GSE151042 respectively.
Code availability
Custom R code used for the processing and analysis of the data described in this article are available at https://github.com/CellularGenomicMedicine/UCB_methylome.
References
Adamson, G. D. et al. ICMART preliminary world report 2015. Hum. Reprod. 34 (2019).
Wyns, C. et al. ART in Europe, 2016: results generated from European registries by ESHRE. Hum. Reprod. Open 2020, hoaa032 (2020).
Bernsten, S. et al. The health of children conceived by ART: ‘the chicken or the egg?’. Hum. Reprod. update 25, 137–158 (2019).
Ceelen, M. et al. Growth during infancy and early childhood in relation to blood pressure and body fat measures at age 8-18 years of IVF children and spontaneously conceived controls born to subfertile parents. Hum. Reprod. 24, 2788–2795 (2009).
Hann, M. et al. The growth of assisted reproductive treatment-conceived children from birth to 5 years: a national cohort study. BMC Med. 16, 224 (2018).
Guo, X. Y. et al. Cardiovascular and metabolic profiles of offspring conceived by assisted reproductive technologies: a systematic review and meta-analysis. Fertil. Steril. 107, 622–631.e5 (2017).
Sunde, A. et al. Time to take human embryo culture seriously. Hum. Reprod. 31, 2174–2182 (2016).
Morbeck, D. E. et al. Composition of commercial media used for human embryo culture. Fertil. Steril. 102, 759–766.e9 (2014).
Morbeck, D. E., Baumann, N. A. & Oglesbee, D. Composition of single-step media used for human embryo culture. Fertil. Steril. 107, 1055–1060.e1 (2017).
Mantikou, E. et al. Embryo culture media and IVF/ICSI success rates: a systematic review. Hum. Reprod. Update 19, 210–220 (2013).
Youssef, M. M. et al. Culture media for human pre-implantation embryos in assisted reproductive technology cycles. Cochrane Database Syst. Rev. 20, Cd007876 (2015).
Dumoulin, J. C. et al. Effect of in vitro culture of human embryos on birthweight of newborns. Hum. Reprod. 25, 605–612 (2010).
Zandstra, H., Van Montfoort, A. P. & Dumoulin, J. C. Does the type of culture medium used influence birthweight of children born after IVF? Hum. Reprod. 30, 530–542 (2015).
Kleijkers, S. H. et al. Influence of embryo culture medium (G5 and HTF) on pregnancy and perinatal outcome after IVF: a multicenter RCT. Hum. Reprod. 31, 2219–2230 (2016).
Kleijkers, S. H. et al. IVF culture medium affects post-natal weight in humans during the first 2 years of life. Hum. Reprod. 29, 661–669 (2014).
Zandstra, H. et al. Association of culture medium with growth, weight and cardiovascular development of IVF children at the age of 9 years. Hum. Reprod. 33, 1645–1656 (2018).
Bouillon, C. et al. Does embryo culture medium influence the health and development of children born after in vitro fertilization? PLoS ONE 11, e0150857 (2016).
Tarahomi, M. et al. The composition of human preimplantation embryo culture media and their stability during storage and culture. Hum. Reprod. 34, 1450–1461 (2019).
Wadhwa, P. D., Buss, C., Entringer, S. & Swanson, J. M. Developmental origins of health and disease: brief history of the approach and current focus on epigenetic mechanisms. Semin Reprod. Med. 27, 358–368 (2009).
Felix, J. F. & Cecil, C. A. M. Population DNA methylation studies in the Developmental Origins of Health and Disease (DOHaD) framework. J. Dev. Orig. Health Dis. 10, 306–313 (2019).
Sullivan-Pyke, C. S., Senapati, S., Mainigi, M. A. & Barnhart, K. T. In Vitro fertilization and adverse obstetric and perinatal outcomes. Semin Perinatol. 41, 345–353 (2017).
DeAngelis, A. M., Martini, A. E. & Owen, C. M. Assisted reproductive technology and epigenetics. Semin Reprod. Med. 36, 221–232 (2018).
Li, L. et al. Single-cell multi-omics sequencing of human early embryos. Nat. Cell Biol. 20, 847–858 (2018).
Hanna, C. W., Demond, H. & Kelsey, G. Epigenetic regulation in development: is the mouse a good model for the human? Hum. Reprod. Update 24, 556–576 (2018).
Mulder, C. L. et al. Comparison of DNA methylation patterns of parentally imprinted genes in placenta derived from IVF conceptions in two different culture media. Hum. Reprod. 35, 516–528 (2020).
Mani, S. & Mainigi, M. Embryo culture conditions and the epigenome. Semin Reprod. Med. 36, 211–220 (2018).
Novakovic, B. et al. Assisted reproductive technologies are associated with limited epigenetic variation at birth that largely resolves by adulthood. Nat. Commun. 10, 3922 (2019).
Turan, N. et al. Inter- and intra-individual variation in allele-specific DNA methylation and gene expression in children conceived using assisted reproductive technology. PLoS Genet. 6, e1001033 (2010).
Melamed, N., Choufani, S., Wilkins-Haug, L. E., Koren, G. & Weksberg, R. Comparison of genome-wide and gene-specific DNA methylation between ART and naturally conceived pregnancies. Epigenetics 10, 474–483 (2015).
Barberet, J. et al. DNA methylation profiles after ART during human lifespan: a systematic review and meta-analysis. Hum. Reprod. Update dmac010 (2022).
El Hajj, N. et al. DNA methylation signatures in cord blood of ICSI children. Hum. Reprod. 32, 1761–1769 (2017).
Tierling, S. et al. Assisted reproductive technologies do not enhance the variability of DNA methylation imprints in human. J. Med. Genet. 47, 371–376 (2010).
Ginjala, V. Gene imprinting gateway. https://genomebiology.biomedcentral.com/articles/10.1186/gb-2001-2-8-reports2009 (2001).
Küpers, L. K. et al. Meta-analysis of epigenome-wide association studies in neonates reveals widespread differential DNA methylation associated with birthweight. Nat. Commun. 10, 1893 (2019).
Teschendorff, A. E., Jones, A. & Widschwendter, M. Stochastic epigenetic outliers can define field defects in cancer. BMC Bioinforma. 17, 178 (2016).
Gentilini, D. et al. Stochastic epigenetic mutations (DNA methylation) increase exponentially in human aging and correlate with X chromosome inactivation skewing in females. Aging 7, 568–578 (2015).
Khouja, J. N. et al. Epigenetic gestational age acceleration: a prospective cohort study investigating associations with familial, sociodemographic and birth characteristics. Clin. Epigenetics 10, 86 (2018).
Bohlin, J. et al. Prediction of gestational age based on genome-wide differentially methylated regions. Genome Biol. 17, 207 (2016).
Knight, A. K. et al. An epigenetic clock for gestational age at birth based on blood methylation data. Genome Biol. 17, 206 (2016).
Bright, H. D. et al. Epigenetic gestational age and trajectories of weight and height during childhood: a prospective cohort study. Clin. Epigenetics 11, 194 (2019).
Barberet, J. et al. Do assisted reproductive technologies and in vitro embryo culture influence the epigenetic control of imprinted genes and transposable elements in children? Hum. Reprod. 36, 479–492 (2021).
Ducreux, B. et al. Genome-wide analysis of DNA methylation in buccal cells of children conceived through IVF and ICSI. Genes 12, 1912 (2021).
Mantikou, E. et al. Factors affecting the gene expression of in vitro cultured human preimplantation embryos. Hum. Reprod. 31, 298–311 (2016).
Kleijkers, S. H. et al. Differences in gene expression profiles between human preimplantation embryos cultured in two different IVF culture media. Hum. Reprod. 30, 2303–2311 (2015).
Saffari, A. et al. Estimation of a significance threshold for epigenome-wide association studies. Genet Epidemiol. 42, 20–33 (2018).
Tsai, P. C. & Bell, J. T. Power and sample size estimation for epigenome-wide association scans to detect differential DNA methylation. Int J. Epidemiol. 44, 1429–1441 (2015).
Breton, C. V. et al. Small-magnitude effect sizes in epigenetic end points are important in children’s environmental health studies: the children’s environmental health and disease prevention research center’s epigenetics working group. Environ. Health Perspect. 125, 511–526 (2017).
Gentilini, D. et al. Multifactorial analysis of the stochastic epigenetic variability in cord blood confirmed an impact of common behavioral and environmental factors but not of in vitro conception. Clin. Epigenetics 10, 77 (2018).
Tobi, E. W. et al. Selective survival of embryos can explain DNA methylation signatures of adverse prenatal environments. Cell Rep. 25, 2660–2667.e4 (2018).
Cirkovic, A. et al. Systematic review supports the role of DNA methylation in the pathophysiology of preeclampsia: a call for analytical and methodological standardization. Biol. Sex. Differ. 11, 36 (2020).
Elliott, H. R., Sharp, G. C., Relton, C. L. & Lawlor, D. A. Epigenetics and gestational diabetes: a review of epigenetic epidemiology studies and their use to explore epigenetic mediation and improve prediction. Diabetologia 62, 2171–2178 (2019).
Awamleh, Z. et al. Exposure to gestational diabetes mellitus (GDM) alters DNA methylation in placenta and fetal cord blood. Diabetes Res. Clin. Pr. 174, 108690 (2021).
Howe, C. G. et al. Maternal gestational diabetes mellitus and newborn DNA methylation: findings from the pregnancy and childhood epigenetics consortium. Diabetes Care 43, 98–105 (2020).
R Core Team. R: a language and environment for statistical computing. R foundation for statistical computing. https://www.R-project.org/ (2021).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer: 2016) .
Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016).
Müller, F. et al. RnBeads 2.0: comprehensive analysis of DNA methylation data. Genome Biol. 20, 55 (2019).
Maksimovic, J., Gordon, L. & Oshlack, A. SWAN: subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol. 13, R44 (2012).
Jung, C. H. et al. sEst: accurate sex-estimation and abnormality detection in methylation microarray data. Int. J. Mol. Sci. 19, 3172 (2018).
Gervin, K. et al. Systematic evaluation and validation of reference and library selection methods for deconvolution of cord blood DNA methylation data. Clin. Epigenetics 11, 125 (2019).
Aryee, M. J. et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 (2014).
Triche, T. J., Weisenberger, D. J., Van Den Berg, D., Laird, P. W. & Siegmund, K. D. Low-level processing of illumina infinium DNA methylation BeadArrays. Nucleic Acids Res. 41, e90 (2013).
Jaffe, A. E. FlowSorted.Blood.450k: illumina HumanMethylation data on sorted blood cell population. https://bioconductor.org/packages/release/data/experiment/html/FlowSorted.Blood.450k.html (2018).
Du, P. et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinforma. 11, 587 (2010).
Hoffman, G. E. & Schadt, E. E. variancePartition: interpreting drivers of variation in complex gene expression studies. BMC Bioinforma. 17, 483 (2016).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. Ser. B57, 289–300 (1995).
Koncevičius, K. matrixTests: fast statistical hypothesis tests on rows and columns of matrices. https://CRAN.R-project.org/package=matrixTests (2020).
Phipson, B., Maksimovic, J. & Oshlack, A. missMethyl: an R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics 32, 286–288 (2016).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883 (2012).
Van Den Heuvel, R. et al. Biobank@VITO: biobanking the general population in Flanders. Front. Med. 7, 37 (2020).
Langie, S. A. S. et al. GLI2 promoter hypermethylation in saliva of children with a respiratory allergy. Clin. Epigenetics 10, 50 (2018).
Janssen, B. G. et al. Cohort profile: the ENVIRonmental influence ON early AGEing (ENVIRONAGE): a birth cohort study. Int J. Epidemiol. 46, 1386–1387m (2017).
Ho, D. E., Imai, K., King, G. & Stuart, E. A. MatchIt: nonparametric preprocessing for parametric causal inference. J. Stat. Softw. 42, 1–28 (2011).
Edgar, R., Domrachev, M. & Lash, A. E. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210 (2002).
Acknowledgements
We thank the IVF couples who have agreed for their children to participate in this study. We thank all the employees of the IVF clinics and obstetrics departments involved in recruiting participants and collecting samples for this study. We thank E. Pishva for his consultation regarding data analysis and for the critical reading of the manuscript. This study was funded by March of Dimes (6-FY13-153). It was further supported by the stichting fertility foundation, EVA (Erfelijkheid Voortplanting & Aanleg) speciality programme (grant no. KP111513) of Maastricht University Medical Centre (MUMC+), and the Horizon 2020 innovation (ERIN) (grant no. EU952516) of the European Commission. We thank the FLEHS Supervisory Board for the provision of data. The FLEHS studies are commissioned, financed, and steered by the Flemish Government (Department of Economy, Science and Innovations, Agency for Care and Health and Department of Environment). The FLEHS dataset has been generated by the Flemish Center of Expertise on Environment and Health (FLEHS 2016–2020), funded by the Environment, Nature and Energy Department of the Flemish government. The views expressed in this manuscript are those of the author(s) and are not necessarily endorsed by the Flemish government.
Author information
Authors and Affiliations
Contributions
R.M.K., A.P.A.v.M. and M.Z.E. study design and conception. D.C., J.v.E.-A., S.M., Y.W., R.v.G., and J.C.M.D. were involved in sample collection for the IVF cohort. A.P.A.v.M. and F.B. carried out the lab work for the IVF samples. S.R., S.L., T.S.N., M.P., R.A. and E.M.B. were involved in sample collection and processing for the naturally conceived cohorts. R.M.K., F.B., J.T., A.P.A.v.M. and M.Z.E. were involved in the data analysis and interpretation. R.M.K. wrote the first draft of the manuscript. M.G., A.P.A.v.M. and M.Z.E. contributed to the writing of the manuscript. All authors provided textual comments and approved the manuscript. H.B., A.P.A.v.M. and M.Z.E. supervised the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Koeck, R.M., Busato, F., Tost, J. et al. Methylome-wide analysis of IVF neonates that underwent embryo culture in different media revealed no significant differences. npj Genom. Med. 7, 39 (2022). https://doi.org/10.1038/s41525-022-00310-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-022-00310-3
