
Abstract
PTEN has a strong Mendelian association with autism spectrum disorder (ASD), representing a special case in autism’s complex genetic architecture. Animal modeling for constitutional Pten mutation creates an opportunity to study how disruption of Pten affects neurobiology and glean potential insight into ASD pathogenesis. Subsequently, we comprehensively characterized the neural (phospho)proteome of Ptenm3m4/m3m4 mice, which exhibits cytoplasmic-predominant Pten expression, by applying mass spectrometry technology to their brains at two-weeks- (P14) and six-weeks-of-age (P40). The differentially expressed/phosphorylated proteins were subjected to gene enrichment, pathway, and network analyses to assess the affected biology. We identified numerous differentially expressed/phosphorylated proteins, finding greater dysregulation at P40 consistent with prior transcriptomic data. The affected pathways were largely related to PTEN function or neurological processes, while scant direct overlap was found across datasets. Network analysis pointed to ASD risk genes like Pten and Psd-95 as major regulatory hubs, suggesting they likely contribute to initiation or maintenance of cellular and perhaps organismal phenotypes related to ASD.
Similar content being viewed by others


Introduction
PTEN (Phosphatase and Tensin homolog on chromosome Ten, OMIM #601728), a well-studied, tumor suppressor gene, is known for its canonical role as the major antagonist of PI3K/AKT/mTOR signaling, a crucial growth and survival intracellular cascade, via the lipid modality of PTEN’s dual-specific phosphatase function1,2,3. Germline PTEN mutations are considered a rare cancer predisposition mechanism, while also representing one of the strongest Mendelian associations with autism spectrum disorder (ASD)4,5. Ostensibly, individuals with germline PTEN mutations present with a diverse array of clinical phenotypes, ranging from high risk of benign and malignant neoplasia to ASD6.
While there have been several rigorous efforts to explore the transcriptome of ASD7,8,9,10,11,12,13,14,15, there has been limited exploration of the proteome of ASD16,17,18,19. Little has been done to understand the proteome, let alone characterize the functional effectors of the phosphoproteome in syndromic ASD contexts, such as those arising from Mendelian gene variation, here, germline PTEN mutation. Thus, we believe that animal modeling for PTEN mutation-associated ASD followed by the deployment of proteomic/phosphoproteomic technologies could represent an excellent approach for disentangling the complexity associated with autism pathophysiology.
For insight into potential disease mechanisms of PTEN-ASD (i.e., persons with ASD carrying germline PTEN mutations), we generated the Ptenm3m4 mouse model, which exhibits predominantly cytoplasmic Pten localization20,21,22. The Ptenm3m4 mouse presents with clear neurological phenotypes; homozygous mutants have severe macrocephaly from increased brain mass, subsequent to neuronal hypertrophy, astrogliosis, microgliosis, and white matter overabundance22,23,24. Furthermore, we observed deficits in neuronal differentiation in neural stem cells (NSCs) derived from Ptenm3m4 mice25. Despite these various pathologies, Ptenm3m4/m3m4 mice have fairly normal learning and memory phenotypes. However, Ptenm3m4/m3m4 mice display an aberrant increase in sociability among males, and both sexes have impaired motor coordination22. Most importantly, the neural transcriptome of these mice shares a significant portion of differentially expressed genes with the neural transcriptomes of individuals with idiopathic ASD, namely decreased expression of genes involved in synaptic functions and increased expression of genes related to neuroinflammation26. Moreover, there is a shift toward increased intron retention in the brains of these mice and dysregulation in microexon splicing27, a potential shared hallmark of autism pathophysiology28. Intriguingly, there is growing evidence suggesting mutations in PTEN that decrease nuclear localization (e.g., the M3M4 mutation) associate strongly with autism-like phenotypes29. Thus, the Ptenm3m4 mouse is likely a useful model for exploring the neurobiology that is affected by germline mutations in PTEN.
Given the prior findings from the Ptenm3m4 mouse and the lack of neural (phospho)proteomic data available to interrogate PTEN pathophysiology in the context of ASD, we performed proteomic, phospho-serine/threonine (pS/pT) proteomic, and phospho-tyrosine (pY) proteomic surveys of two-week-old (P14) hemibrains and six-week-old (P40) cortices of Ptenm3m4/m3m4 mice compared to littermate wildtype samples–an experimental design shared by our prior work on the neural transcriptome26. We hypothesized that disruption of Pten, which has poorly characterized but likely promiscuous protein phosphatase function, would likely perturb the phosphoproteome in ways that contribute to pathophysiology that disrupts synaptic functions and promotes stress or neuroinflammatory processes. Furthermore, we anticipated disruption of the proteome given the previously documented changes in the transcriptome and anticipated relationships between the differential expression in the transcriptome and proteome. Ultimately, our transcriptome-proteome-phosphoproteome exploration of the molecular complexity of the Ptenm3m4 brain has illuminated the variation within and between different –omic landscapes, identifying important candidate regulators and effectors of PTEN dysfunction in human ASD.
Results
Differential expression and phosphorylation in Pten m3m4 brain
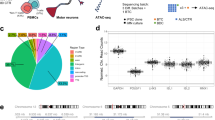
In order to assess how the m3m4 mutation affects the landscape of gene and protein expression and protein phosphorylation, we performed parallel transcriptomic26, proteomic, and phosphoproteomic experiments on Ptenm3m4/m3m4 hemibrains and cortices at two-weeks- (P14) and six-weeks-of-age (P40), respectively (Fig. 1A). At P14, a total of 3207 unique proteins, with an average of 2345 unique proteins per sample, were identified (Supplementary Data 1: Table 1), whereas at P40, a total of 6635 unique proteins, with an average of 5381 unique proteins per sample, were identified (Supplementary Data 1: Table 2). Principal component analysis (PCA) based on the LFQ/NSAF abundance values for a given protein, at both time points revealed completely separate clusters representing homozygous mutant and wild-type samples (Supplementary Information: Fig. 1A, B). At P14, a total of 4080 unique peptides were identified, 97% of which contained phosphorylated residues in the phospho-serine/threonine scan, an average of 2239 unique phosphopeptides per sample (Supplementary Data 1: Table 3). At P40, a total of 8468 unique peptides were identified, 90% of which contained phosphorylated residues, with an average of 5783 unique phosphopeptides per sample (Supplementary Data 1: Table 4). PCA, based on the LFQ abundance values for a given phosphopeptide, at both time points revealed non-overlapping clusters of homozygous mutant and wild-type samples (Supplementary Information: Fig. 1C, D). These findings illustrate robust sampling of the proteome and phosphoproteome, and the captured variability differentiated homozygous mutant and wild-type brain samples well.

A Hemibrain and cortical tissues were extracted from two-week-old (P14) and six-week-old (P40) Ptenm3m4/m3m4 male mice (N = 3) for preparation for RNA sequencing, proteomic scanning, and pS/pT and pY phosphoproteomic scanning. After –omic profiling, these data were analyzed through various bioinformatics approaches. B Volcano plot of differentially expressed protein assessed at P14 (left) and P40 (right) by proteomic scan. C Volcano plot of differentially phosphorylated proteins assessed at P14 (left) and P40 (right) by phosphoproteomic scan.
In assessing differentially expressed proteins, we found 24 over-expressed and 27 under-expressed proteins in the P14 mutants compared to wildtype controls (Table 1; Fig. 1B). At P40, we identified 150 over-expressed and 102 under-expressed proteins in the mutant brains compared to the wild-type brains (Table 1; Fig. 1B). When assessing differential phosphorylation of phospho-serine/threonine phosphopeptides, we found 1 increased and 99 decreased in the P14 mutant hemibrain (Table 1; Fig. 1C). Moreover, we found 113 increased and 185 decreased at P40 in the mutant cortex (Table 1; Fig. 1C). In the special phospho-tyrosine-specific phospho-scan, we identified 39 and 29 relatively increased phosphopeptides at P14 and P40, respectively (Table 1). In the same phospho-tyrosine scan, we found 29 and 26 decreased phosphopeptides at P14 and P40, respectively (Table 1). Overall, the over-expression/under-expression of proteins or enrichment/depletion of phosphopeptides were distributed roughly equally except in the P14 phospho-serine/threonine scan, where phosphopeptide depletion was heavily favored in the mutant by 99-fold (Table 1). Moreover, there is a general increase in differential expression and phosphorylation as the Ptenm3m4/m3m4 mice age, a trend not observed in the phospho-tyrosine scan (Table 1).
Dissimilarity across –omic datasets describing the Pten m3m4 brain
Given the eight different –omic datasets spanning two-time points, we sought to understand what molecules, showing differential expression/phosphorylation in the homozygous mutant brain, were shared among these datasets. Thus, we performed pairwise comparisons of the molecule lists of each separate –omics approach that compared Ptenm3m4/m3m4 to Pten+/+ mice for both P14 and P40 time points. Surprisingly, we found no intersection between the P14 and P40 proteome (0% overlap; Fig. 2A; Supplementary Information: Table 1). For the phospho-serine/threonine scan, we found 21 molecules of all input molecules shared between time points (13% overlap; Fig. 2B; Supplementary Information: Table 1). For the phospho-tyrosine scan, we found 25 molecules of all input molecules shared between time points (48% overlap; Fig. 2C; Supplementary Information: Table 1). Furthermore, 49 molecules were shared between the P14 and P40 transcriptome datasets (7% overlap; Fig. 2D; Supplementary Information: Table 1). These analyses indicate little to marginal overlap within the various –omic scans over developmental time.

A Venn diagram of protein IDs that are shared or not shared between P14 and P40 time points of the MUT vs WT proteomic scan. B Venn diagram of protein IDs that are shared or not shared between P14 and P40 time points of the MUT vs WT pS/pT phosphoproteomic scan. C Venn diagram of protein IDs that are shared or not shared between P14 and P40 time points of the MUT vs WT pY phosphoproteomic scan. D Venn diagram of gene IDs that are shared or not shared between P14 and P40 time points of the MUT vs WT transcriptome. E Venn diagram of protein IDs shared or not shared among the MUT versus WT proteome, pS/pT phosphoproteome, and pY phosphoproteome within time points P14 (left) and P40 (right). F Venn diagram of gene IDs shared or not shared between the transcriptome versus proteome within time points P14 (left) and P40 (right).
Additionally, we assessed overlap among the proteomic and phosphoproteomic datasets within their respective time points. Surprisingly, we found minimal intersection across these datasets; 8% overlap for P14 datasets and 3% overlap for P40 datasets (Fig. 2E; Supplementary Information: Table 1). We proceeded to assess the overlap between the transcriptome and proteome within each time point and found 1 (i.e., 2% overlap) and 30 (i.e., 4% overlap) intersecting molecules at P14 and P40, respectively (Fig. 2F; Supplementary Information: Table 1). Although we expected to find more shared genes between the transcriptome and proteome, these observations suggest that the mechanisms governing changes in protein expression are separate from those governing gene expression in our model. Moreover, the general lack of overlap among the various datasets suggests that in the context of Pten disruption, there may be little redundancy in modes of dysregulation, meaning that molecules are either subject to dysregulation via expression or phosphorylation but not both.
Differentially expressed/phosphorylated molecules associate with major intracellular signaling cascades, neurological processes, and cancer
To understand the biological consequences of the observed changes in protein expression and phosphorylation in the Ptenm3m4/m3m4 cortex, we performed an IPA analysis on the proteomic and phosphoproteomic (pooled phospho-serine/threonine and phospho-tyrosine datasets) findings. The comprehensive summaries for all IPA analyses can be found in the supplemental information (Supplementary Information). The top five most enriched canonical pathways at P14 and P40 for the proteome and phosphoproteome are shown in Fig. 3A, B, respectively. In the Ptenm3m4/m3m4 proteome, the top implicated canonical pathways broadly involve phosphoinositide metabolism (i.e., 3′-phosphoinositide Biosynthesis) or neurological pathways, such as GABA Receptor Signaling and Glutamate Receptor Signaling (Fig. 3A; Supplementary Data 2: Tables S1, S2). In the Ptenm3m4/m3m4 phosphoproteome, the top canonical pathways generally involve neurological and cancer processes or major cellular signaling cascades: Molecular Mechanisms of Cancer, Synaptic Long Term Potentiation, Neuregulin Signaling and cAMP-mediated Signaling (Fig. 3B; Supplementary Data 2: Tables 3, 4). Moreover, we extracted the top two salient disease and bio function networks (i.e., the networks comprised of the largest number of differentially expressed/phosphorylated molecules) from the proteome and phosphoproteome at P40. Cancer and Development of Head are the two largest disease and bio function networks identified by IPA in the Ptenm3m4/m3m4 P40 cortical proteome (Fig. 3C; Supplementary Information), and Synaptic Transmission and Seizure Disorder are two of the largest salient networks from the Ptenm3m4/m3m4 P40 cortical phosphoproteome (Fig. 3D; Supplementary Information). Overall, a broad qualitative appraisal of these enriched pathways and functional networks consistently implicate PTEN signaling, overgrowth or proliferative processes, and neurological process, especially those related to synaptic transmission or neuroinflammation (Supplemental Data 4). These enrichment analyses highlight the broad yet related biological perturbations in the neural (phospho)proteome of Ptenm3m4/m3m4 mice.

A The top five most significant “Canonical Pathways” identified by IPA core analysis in the P40 cortical and P14 hemibrain proteome comparing Ptenm3m4/m3m4 mice to wildtype controls. B The top five most significant “Canonical Pathways” identified by IPA core analysis in the P40 cortical and P14 hemibrain phosphoproteome comparing Ptenm3m4/m3m4 mice to wildtype controls. C The top two “disease and bio function” networks (by size) identified by IPA core analysis from the P40 cortical proteome of Ptenm3m4/m3m4 mice compared to wildtype controls with activity prediction overlaid. Green = decreased expression. Red = increased expression. Color intensity = degree of increased/decreased expression. Hashed line = indirect biological relationship. Arrow = Direction of relationship. Orange = predicted activation. Yellow = observation inconsistent with direction of prediction. Gray = no influence on prediction. D The top two “disease and bio function” networks (by size) identified by IPA core analysis from the P40 cortical phosphoproteome of Ptenm3m4/m3m4 mice compared to wildtype controls with activity prediction overlaid. Green = decreased phosphorylation. Red = increased phosphorylation. Color intensity = degree of increased/decreased phosphorylation. Hashed line = indirect biological relationship. Arrow = Direction of relationship. Orange = predicted activation. Yellow = observation inconsistent with direction of prediction. Gray = no influence on prediction. E The top network identified by IPA from among the differentially expressed proteins with Molecular Activity Predictor (MAP) results overlaid. The network is organized hierarchically, meaning the most important regulatory molecule is placed on top. Green = decreased expression. Red = increased expression. Color intensity = degree of increased/decreased expression. Solid line = direct biological relationship. Hashed line = indirect biological relationship. Arrow = direction of relationship. Orange = predicted activation. Blue = predicted inactivation. Yellow = observation inconsistent with the direction of prediction. Gray = no influence on prediction.
In an effort to understand more about how these biological changes are initiated and maintained, we sought to identify important upstream regulators via network analysis of the differentially expressed proteins. Strikingly, the top protein interaction network identified by IPA from the Ptenm3m4/m3m4 P40 cortical proteome agnostically positions Pten as the pre-eminent regulatory node via hierarchical ordering. The position of Pten at the top of this network’s regulatory architecture establishes its importance and influence as a regulator of the molecules positioned below (Fig. 3E). When IPA’s biological predictions are overlaid on the network using the Molecule Activity Predictor (MAP) tool, the observed decrease in Pten expression predicts an increase in the activity of Pi3k, Akt, Erk, p70 S6, Gfap, and C1qa, while predicting a decrease in P38 Mapk and Creb (Fig. 3E). These predictions are largely consistent with previous observations in the Ptenm3m4/m3m4 cortex22,24,25. The network analysis enables a biological understanding of the changes in protein expression and phosphorylation, which unequivocally point to disrupted Pten function in the brain and highlight its importance in propagating dysfunction to downstream effector molecules.
Pten and Psd-95 are major regulatory nodes in the Pten m3m4/m3m4 cortex
In an effort to expand our biological understanding of the results from the proteomic and phosphoproteomic Ptenm3m4/m3m4 versus Pten+/+ comparisons, we performed STRING analyses on each to identify the relationships among the differentially expressed/phosphorylated molecules. We extracted the largest STRING networks from each –omic dataset at each time points and analyzed the network statistics using Cytoscape (Supplementary Information: Fig. 2). The most striking network findings were observed in the P40 Ptenm3m4/m3m4 cortical proteome and phosphoproteome. In the largest network (83 nodes) constructed from the P40 Ptenm3m4/m3m4 cortical proteome findings, we found that Pten exhibited the greatest degree of connectivity, 12, and betweenness centrality, 0.66, relative to all other nodes (Fig. 4A). These data independently identify Pten as the most connected (i.e., degree connectivity) and most important node for transmitting information across the network (i.e., betweenness centrality), suggesting that Pten is likely to be the dominant regulatory node affecting the network of differentially expressed proteins in the P40 mutant cortex. Additionally, in the largest network (92 nodes) constructed from the P40 Ptenm3m4/m3m4 cortical phospho-serine/threonine scan, we found Psd-95 (also known as Dlg4) exhibited the greatest degree connectivity, 19, and betweenness centrality, 0.68, relative to all other nodes (Fig. 4B). These data identify Psd-95 as the most connected (i.e., highest degree connectivity) and most important node for transmitting information across the network (i.e., highest betweenness centrality), suggesting that Psd-95 is likely to be the dominant regulatory node affecting the network of differentially phosphorylated proteins in the P40 mutant cortex. The Psd-95 finding also implicates Pten given the known and well-described protein-protein relationship between the two30. Ultimately, the STRING network analysis implicates Pten and Psd-95 as the likely perpetrators of the proteomic and phosphoproteomic dysregulation observed in the cortex of the Ptenm3m4/m3m4 model and possibly responsible for some of the pathological cellular, physiological, and behavioral phenotypes.

A Largest network of related molecules constructed from P40 proteomic results (MUT vs WT) using STRING database. Node size trends with degree connectivity. Edge size trends with relationship evidence. Node color (warmth) trends with betweenness centrality. B Largest network of related molecules constructed from P40 pS/pT phosphoproteomic results (MUT vs WT) using STRING database. Node size trends with degree connectivity. Edge size trends with relationship evidence. Node color (warmth) trends with betweenness centrality.
Meaningful overlap between differentially expressed or phosphorylated molecules and known ASD risk genes
We sought to assess whether the differentially expressed/phosphorylated molecules identified by our –omic surveys demonstrate significant overlap with known autism risk genes as curated by the Simons Foundation Autism Research Initiative (SFARI). Accordingly, we compared the SFARI catalog of ASD risk genes with the gene lists of the significant results of –omic surveys (time points and phospho-serine/threonine/tyrosine pooled). We found that 41 molecules intersected between the phosphoproteome and SFARI genes, and 12 molecules intersected between the proteome and SFARI (Fig. 5A). This was modest overlap given the number of genes curated by SFARI, but again, STRING network analysis implicated Pten and Psd-95 as central nodes in association networks derived from the intersecting molecules (Fig. 5B, C). The re-emergence of Pten and Psd-95 in a separate network analysis predicated on known ASD risk genes strengthens the evidence that implicates them as potential drivers of the phenotypes observed in Ptenm3m4/m3m4 mouse model. The overlap that does exist with the known ASD risk genes underscores the importance of Pten biology to ASD pathophysiology overall.

A Venn diagram illustrating the number of shared and not shared targets among autism risk genes (SFARI), the Ptenm3m4/m3m4 CNS proteome, and the Ptenm3m4/m3m4 CNS phosphoproteome. B Proteins differentially expressed in the Ptenm3m4/m3m4 CNS proteome that are known autism risk genes with biological relatedness overlaid via STRING database. C Proteins differentially phosphorylated in the Ptenm3m4/m3m4 CNS phosphoproteome that are known autism risk genes with biological relatedness overlaid via STRING database.
Discussion
This study represents a proteomic and phosphoproteomic exploration of the brain of a Pten model with strong neurological phenotypes. These experiments demonstrate a distinct developmental difference in protein expression and phosphorylation with differences of greater number, magnitude, and significance at P40 versus P14 (Fig. 1). Interestingly, we found little to marginal overlap among the various –omic datasets, but that the affected biological pathways across the different datasets were similar, if not overlapping (Figs. 2 and 3), suggesting potential convergence of non-overlapping, non-redundant regulatory mechanisms. Additionally, the developmental differences and lack of overlap may be a product of the significant decline in Pten expression from P14 to P4024. Network analysis highlighted Pten as a critical regulator of the differential expression observed in the neural proteome (Figs. 3E and 4A), and Psd-95 as a critical regulator of differential phosphorylation observed in the neural phosphoproteome (Fig. 4B). Ultimately, we observed overlap between our scans’ findings and known ASD risk genes, which when that overlap was subject to network analysis, we found Pten and Psd-95 to again be prominent nodes (Fig. 5).
The lack of overlap among the –omic datasets, especially the Ptenm3m4/m3m4 neural proteome and transcriptome, is likely our most surprising finding. As this is the first exploration of a transcriptome and proteome simultaneously in a Pten model, it is difficult to comment whether this finding would be typical. Apparently in mice, the average correlation between the transcriptome and proteome is modest, 27%, which has been shown to be partially explained by alternative splicing31. Our findings still show less overlap than would be expected based on the aforementioned average. However, this overlap approaches expectations if the overlap analysis is repeated with a relaxed significance threshold (p-value < 0.10). Otherwise, these findings may indicate that gene versus protein expression, at least in our model, are regulated by different mechanisms, and perhaps reflecting a lack of redundancy. It is possible that dysregulated transcription and splicing factor expression may be responsible in part for changes in gene expression26,27, whereas translational mechanisms subsequent to changes in the phosphoproteome, including signaling changes downstream of Pten (e.g. dysregulation of translational control from changes in mTOR activity) may be responsible for the changes in protein expression. The possibility of significant changes in translational control in the Ptenm3m4/m3m4 brain are real and tantalizing; disruption of proteins involved in translational control, including many in the PI3K/AKT/mTOR cascade, have been shown to mediate ASD phenotypes32. It is also possible that changes in alternative splicing may affect the differential protein expression that has been observed in the Ptenm3m4/m3m4 brain27. Furthermore, the lack of overlap between the proteome and phosphoproteome suggests differential modes of regulation at these levels with minimal redundancy. Thus, mechanisms that regulate protein expression appear to be distinct from the mechanisms that regulate phosphorylation the Ptenm3m4 brain, and a phenomenon previously observed and described in other models33. This differential regulation seems like a reasonable possibility in our model given that it is probable that a portion of the observed changes in the Ptenm3m4/m3m4 neural phosphoproteome are more proximal to the dysfunction introduced by the m3m4 mutation than the differential gene/protein expression. This is likely due to the primary functional role of Pten as a dual-specificity phosphatase with possibly a large array of diverse protein substrates34,35,36,37,38,39,40,41,42. As mentioned above, the significant developmental decline in Pten expression may explain some of the differences observed between datasets. Moreover, the potential lack of redundancy across different –omic levels appears consistent with the autosomal dominant pattern of PHTS. Additional biochemical experiments and expanded –omic surveys on this model and other various neural Pten models will be required to disentangle the complexity at the various biological levels.
Beyond the lack of overlap between transcriptome and proteomes, we found the continued emergence of Pten and Psd-95 in the proteome and phosphoproteome pathway and network analysis to be a crucial indicator of their importance to the underlying pathology of the Ptenm3m4/m3m4 brain and potentially to ASD in general. This postulate appears to be supported by one of the only two proteomic analyses of post-mortem ASD brains that identified Psd-95 as a major hub protein in the Brodmann area 19 (BA19)19. Although their finding was in the proteome, whereas ours was in the phosphoproteome, the shared finding is striking, and it is also possible that the effect of the perturbations to Psd-95 have similar biological effects in both contexts. There were other hits that our study shared with that of Abraham and colleagues (e.g. MAPT), suggesting proteomic approaches to ASD may uncover shared pathological hallmarks.
The picture of the broader changes in the (phospho)proteome of the Ptenm3m4/m3m4 brain was largely aligned with our hypotheses, and in many ways, consistent with the overall pathophysiological changes we observed previously in the Ptenm3m4/m3m4 neural transcriptome26. Moreover, many of the differentially expressed or phosphorylated proteins were previously identified in prior phenotyping efforts, including many molecules related to microglia and oligodendrocyte functions like C1q and Mbp23,24. Although many of the specific molecules and pathways found to be dysregulated or enriched, respectively, are different across the various datasets, there is a convergence on biological pathways downstream of Pten functions and disease processes that manifest in individuals with germline PTEN mutations, such as neurodevelopmental abnormalities and cancer (Supplementary Information, Supplementary Data 2). Interestingly, the co-occurrence of neurodevelopmental and cancer pathways arising from study of Ptenm3m4/m3m4 brain underscores how closely the biology of these two seemingly disparate disease processes are and that an important nexus between them is PTEN. Our detailed description of the affected pathways and processes at the (phospho)proteomic levels appear to suggest that there may be convergent mechanisms at play in autism and that the apparent complexity might be reduced to a few central molecules and pathways despite a complex phenotype.
The Ptenm3m4 model is excellent for the purposes of this study, but it is quite unique, and our study is limited to this model. Thus, our observations cannot necessarily be extended to all mutations or disruptions of Pten despite some of the evidence here and in the literature that support generalizability. In fact, it is quite likely that at least part of the expression and phosphorylation changes are specific to the m3m4 model due to the homozygosity of the mutation and cytoplasmic predominance of Pten expression–both features that are unique to our model and rarely, if ever, modeled in other studies. Additionally, Pten expression in the Ptenm3m4 brain declines with age, likely through increased proteosomal degradation, and the m3m4 mutant protein is roughly 50% less stable than wildtype24. Our study would have ostensibly been more sensitive to changes in the proteome, phosphoproteome, and transcriptome with a larger sample size. Although the PCA findings cluster Ptenm3m4/m3m4 and Pten+/+ separately, there is still enough variation within each genotype to indicate that our study would likely find more significant hits, if sample size expanded. Subsequently, we have likely only uncovered the most differentially expressed/phosphorylated molecules, but there may remain important subtle changes in major regulators that were missed.
In sum, our study represents an important entry into understanding the proteome and phosphoproteome in a Pten model with well described neurological pathologies and behavioral deficits. Given the current lack of knowledge of what the proteomic and phosphoproteomic landscapes should look like in the context PTEN-ASD or even idiopathic ASD, this study is a step toward filling this critical knowledge gap. The observed changes in protein expression and phosphorylation appear to affect pathways and processes important in the nervous system (e.g. neurodevelopment and synaptic function) and neuroinflammation. Despite the shared biological signature across the various –omic datasets, the implicated molecules appear to be distinct, which suggest differential modes of regulation across the different biological tiers. The molecules that do emerge from our study as major regulatory players (i.e. Pten and Psd-95) are strongly associated with autism biology. Our work suggests that a renewed focus on these molecules, specifically developing an understanding that would allow for rescue of their important functions in the nervous system, is critical if we are to tackle the clinical challenges presented by ASD.
Methods
The Pten m3m4 murine model
Our planned experimental procedures were approved by the Cleveland Clinic’s Institutional Animal Care and Use Committee (IACUC) under protocol numbers 2018-1952 and 2017-1879 and guided by the Principles of Laboratory Animal Care formulated by the National Society for Medical Research. We created the Ptenm3m4 mouse on an outbred CD1 background, which has been studied extensively, shown to have deficits in social behavior, changes in neuron and glia populations, and changes in gene expression in many known autism risk genes22,23,24,26,27,43. The Ptenm3m4 mutation, located within exon 7 of mouse Pten, consists of five nucleotide substitutions that results in four nonsynonymous and one synonymous codon changes. The four missense mutations disrupt the third (m3) and fourth (m4) putative nuclear localization-like sequences of Pten21,22. We performed mouse genotyping on genomic DNA extracted from toe clippings following Jackson Laboratory’s protocol using in-house PCR primers. The wildtype allele primers are mPten-F5, 5′-TGGCAGACTCTTCATTTCTGTGGC-3′, and mPten-R6, 5′-ACTTCTTCACAACCACTTCTTTCAAC-3′, and mutant allele primers are mPten-F3, 5′-TACCCGGTAGAATTTCGACGACCT-3′, and mPten-R6, 5′-ACTTCTTCACAACCACTTCTTTCAAC-3′. Our mice were maintained on a 14:10 light:dark cycle, where access to food and water was ad libitum. We maintained room temperature between 18 and 26 °C. Mice were euthanized via CO2 asphyxiation with additional cervical dislocation. None of the experiments were blinded but all were randomized and conducted under protocols approved by the Institutional Animal Care and Use Committee (IACUC) at Cleveland Clinic. In our experiments, we utilized only male mice because the Ptenm3m4/m3m4 behavioral phenotypes are most pronounced in males, and the previous RNA-Seq experiment used only male mice26,27. A sample size of three biological replicates (N = 3) was used for each genotype and each time point of the study (i.e., P14 and P40). These time points were patterned after the original RNA-sequencing study26. Moreover, we wanted to capture a molecular neurodevelopment signature by using rough “pediatric” versus “adult” time points. The P14 time point represents this younger stage prior to weaning, while the P40 time point represents the oldest stage we can logistically capture (the mice have at least reached sexual maturity). The Ptenm3m4/m3m4 mice exhibit premature mortality from suspected status epilepticus, rarely living beyond 6 weeks-of-age.
Transcriptomic data analysis
The hemibrain and cortical transcriptome of two-week-old and six-week-old Ptenm3m4 mice (GSE59318), respectively, shares a similar differential expression (DE) profile with that of idiopathic autism, as described in detail by Tilot et al.26. We deployed Ingenuity Pathway Analysis (IPA) software (Qiagen, Redwood City, California) to perform a core analysis on the DE genes from the hemibrain and cortex of Pten homozygous mutant and wild-type mice at both time points (i.e. P14 and P40). We also confirmed and extended this analysis with STRING software (version 11.0; https://string-db.org/). We performed STRING analysis on the same list of DE. These comparisons were of the homozygous mutant compared to the wildtype (i.e., MUT versus WT = MUT/WT Fold Change). The transcriptomic DE data are found in Supplementary Data 1.
Proteomic scan
Three wildtype and three homozygous mutant two-week-old mouse hemibrains and three wildtype and three homozygous mutant six-week-old cortices were submitted to the Lerner Research Institute (LRI) Proteomics Core. These samples were homogenized in urea lysis buffer (20 mM HEPES, 9 M urea, 1X HALT phosphatase inhibitor, pH 8.0) with lysis Matrix D beads using FastPrep-24 from MP Biomedical. An aliquot from each tissue lysate was taken out for in-solution tryptic digestion. These samples were solubilized in 50 μL of in-solution digestion buffer, 6M urea, 100 mM Tris pH 8.0, reduced with DTT, alkylated with iodoacetamide, and then diluted to give a final urea concentration of less than 1 M for digestion. All samples were digested in-solution by adding 0.5 μg trypsin and incubating overnight at room temperature. A second aliquot of trypsin was added and digestion was continued for an additional 6 h. After digestion, the peptides were desalted and quantified using Thermo Scientific Pierce Quantitative Colorimetric Peptide Assay kit (Thermo Scientific #23275). A 100 μg peptide aliquot from each sample was dried down and reconstituted in 100 μL 20 mM ammonium formate (pH 10) for offline fractionation. Fractionation was performed by loading 90 μL onto an offline Hp-RP separation column (Waters, XBridge C18 3.5 μm, 2.1 × 100 mm, or Waters XBridge C18 3.5 μm, 2.1 × 150 mm) using an Agilent 1200 microflow pump. The samples were fractionated using a stepwise gradient at 250 μL/min for 40 min. A fraction collector was used for collection with a rate of 1 minute per fraction through the rest of the gradient. One of every four collections were combined and a total of 10 fractions per sample was obtained. Each combined fraction was lyophilized by vacuum centrifugation, and reconstituted in 30 μL 1% acetic acid for LC-MS/MS. The LC-MS system was a Finnigan LTQ-Obitrap Elite hybrid mass spectrometer system. The HPLC column was a Dionex 15 cm × 75 μm id Acclaim Pepmap C18, 2 μm, 100 Å reversed phase capillary chromatography column. Five μL volumes of the extract were injected and the peptides eluted from the column by an acetonitrile/0.1% formic acid gradient at a flow rate of 0.25 μL/min were introduced into the source of the mass spectrometer on-line. The microelectrospray ion source is operated at 2.5 kV. The digest was analyzed using the data dependent multitask capability of the instrument acquiring full scan mass spectra to determine peptide molecular weights and product ion spectra to determine amino acid sequence in successive instrument scans. The data were analyzed by using all CID spectra collected in the experiment to search the mouse UniProtKB databases with the search program Mascot and Sequest. These search results were then uploaded into the program Scaffold. The quantitation was performed by comparing the normalized spectral counts for these samples. The experimental comparison is between homozygous mutant and wildtype (i.e., MUT versus WT = MUT/WT Fold-Change). The proteomic screen data are found in Supplementary Data 1.
Phospho-serine/threonine proteomic scan
Three wildtype and three homozygous mutant two-week-old mouse hemibrain and three wildtype and three homozygous mutant six-week-old cortices were submitted to the Lerner Research Institute (LRI) Proteomics Core. These samples were homogenized in urea lysis buffer (20 mM HEPES, 9M urea, 1X HALT phosphatase inhibitor, pH 8.0) with lysis Matrix D beads using FastPrep-24 from MP Biomedical (Solon, OH). The protein concentration was measured using a Pierce BCA assay kit. The proteins samples were reduced with DTT and alkylated with iodoacetamide. These proteins were then digested with trypsin overnight at room temperature. After digestion, the peptides were purified using Sep-Pak C18 spin columns (Waters Corporation, WAT 051910). Prior to enrichment, an 8 pmol aliquot of phosphopeptide standard mix was spiked into each sample. Phosphopeptide enrichment was performed using Pierce TiO2 Phosphopeptide Enrichment Spin Tips according to the manufacturer’s instructions. The enriched peptide samples were subjected to C18 clean-up prior to LC-MS/MS analysis. The LC-MS system was a ThermoScientific Fusion Lumos mass spectrometry system. The HPLC column was a Dionex 15 cm × 75 μm id Acclaim Pepmap C18, 2 μm, 100 Å reversed-phase capillary chromatography column. Five μL volumes of the extract were injected and the peptides eluted from the column by an acetonitrile/0.1% formic acid gradient at a flow rate of 0.25 μL/min were introduced into the source of the mass spectrometer online. The microelectrospray ion source is operated at 1.9 kV. The digest was analyzed using the data-dependent multitask capability of the instrument acquiring full scan mass spectra to determine peptide molecular weights and product ion spectra to determine amino acid sequence in successive instrument scans. The data were analyzed by using all CID spectra collected in the experiment to search the mouse UniProtKB databases with the programs Proteome Discoverer 2.2. The experimental comparison is between homozygous mutant and wildtype (i.e., MUT versus WT = MUT/WT Fold Change). The phosphoproteomic pS/pT screen data are found in Supplementary Data 1.
Phospho-tyrosine proteomic scan
Three wildtype and three homozygous mutant hemibrain and cortical tissue samples from P14 and P40 mice were delivered to the Proteomics Services Group, Cell Signaling Technology (Danvers, MA). These samples were homogenized and subject to protease digestion yielding peptides. These extracted peptides were loaded directly onto a 50 cm × 100 μm PicoFrit capillary column packed with C18 reversed-phase resin. The column was developed with a 90-minute linear gradient of acetonitrile in 0.125% formic acid delivered at 280 nL/min. After C18 solid-phase extraction, the peptides were enriched for phospho-Y residue by immunoprecipitation using a phosphotyrosine pY-1000 motif antibody (CST #8954) on A/G beads. After immunoprecipitation the resin was washed, the phosphopeptides were eluted, and then analyzed by LC-MS/MS using Orbitrap-Fusion Lumos, ESI-HCD. The MS analysis was performed with the following parameters: MS Run Time 96 min, MS1 Scan Range (300.0–1500.00), Top 20 MS/MS (Min Signal 500, Isolation Width 2.0, Normalized Coll. Energy 35.0, Activation-Q 0.250, Activation Time 20.0, Lock Mass 371.101237, Charge State Rejection Enabled, Charge State 1+ Rejected, Dynamic Exclusion Enabled, Repeat Count 1, Repeat Duration 35.0, Exclusion List Size 500, Exclusion Duration 40.0, Exclusion Mass Width Relative to Mass, Exclusion Mass Width 10ppm). MS/MS spectra were evaluated using SEQUEST. Searches were performed against the most recent update of the Uniprot Mus musculus database with mass accuracy of +/−50 ppm for precursor ions and 0.02 Da for product ions. Results were filtered with mass accuracy of +/−5 ppm on precursor ions and the presence of the intended motif. The experimental comparison is between homozygous mutant and wildtype (i.e., MUT versus WT = MUT/WT Fold Change). The phosphoproteomic pY screen data are found in Supplementary Data 1.
Bioinformatic analysis
In order to assess the biology impacted by the differential expression or differential phosphorylation observed in our various –omic datasets, we employed Ingenuity Pathway Analysis (IPA, www.qiagen.com/ingenuity) to perform core analysis, which provides enrichment scores as p-values for their various outputs (e.g. canonical pathways) determined by one-tailed Fisher’s exact test using Ingenuity Knowledge Base as a reference (restricted to only experimentally observed findings on the mus musculus background). Reported p-values were adjusted for multiple-testing using Benjamini-Hochberg (BH) procedure. We also used STRING software (https://string-db.org/) to obtain gene ontology, Reactome (https://reactome.org), and KEGG results. The STRING analysis served to both confirm the observation in IPA, which are based on a proprietary database, and to obtain the raw data to pass along to network analysis.
Network analysis
We performed network analysis using Cytoscape 3.7.0 (https://cytoscape.org/) on the interaction data generated by STRING software (https://string-db.org/) on all of our –omic datasets. The STRING analysis generates an association network from an input gene list based on their database of biological relationships, which included several types of evidence: co-expression, text-mining, biochemical/genetic data, and previously curated pathway and protein-complex knowledge. Cytoscape 3.7.0 was utilized to generate the network figures and statistics, such as degree connectivity and betweenness centrality.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Any raw data or analyses related to this study are available from the corresponding author upon reasonable request. All proteomic and transcriptomic data are available in Supplementary Data 1. The transcriptomic data are also deposited in the Gene Expression Omnibus (GEO) database, accession code GSE59318. The (phospho)proteomic data are available via ProteomeXchange with identifier PXD025351.
Code availability
All scripts related to this study are available from the corresponding author upon reasonable request.
References
Maehama, T. & Dixon, J. E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 273, 13375–13378 (1998).
Stambolic, V. et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95, 29–39 (1998).
Maehama, T. & Dixon, J. E. PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Biol. 9, 125–128 (1999).
Butler, M. et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J. Med. Genet. 42, 318–321 (2005).
Yehia, L., Ngeow, J. & Eng, C. PTEN-opathies: from biological insights to evidence-based precision medicine. J. Clin. Invest. 129, 452–464 (2019).
Yehia, L., Keel, E. & Eng, C. The clinical spectrum of PTEN mutations. Annu. Rev. Med. 71, 103–116 (2020).
Voineagu, I. et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384 (2011).
Voineagu, I. Gene expression studies in autism: moving from the genome to the transcriptome and beyond. Neurobiol. Dis. 45, 69–75 (2012).
Buxbaum, J. D. et al. The Autism Sequencing Consortium: Large scale, high throughput sequencing in autism spectrum disorders. Neuron 76, 1052–1056 (2012).
Parikshak, N. N. et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 540, 423–427 (2016).
Gandal, M. J. et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362, eaat8127 (2018).
Gandal, M. J. et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 359, 693–697 (2018).
He, Y., Zhou, Y., Ma, W. & Wang, J. An integrated transcriptomic analysis of autism spectrum disorder. Sci. Rep. 9, 1–9 (2019).
Shnier, D., Voineagu, M. A. & Voineagu, I. Persistent homology analysis of brain transcriptome data in autism. J. R. Soc. Interface 16, 20190531 (2019).
Breen, M. S. et al. Transcriptional signatures of participant-derived neural progenitor cells and neurons implicate altered Wnt signaling in Phelan-McDermid syndrome and autism. Mol. Autism 11, 53 (2020).
Junaid, M. A. et al. Proteomic studies identified a single nucleotide polymorphism in glyoxalase I as autism susceptibility factor. Am. J. Med. Genet. A. 131, 11–17 (2004).
Broek, J. A. C. et al. The need for a comprehensive molecular characterization of autism spectrum disorders. Int. J. Neuropsychopharmacol. 17, 651–673 (2014).
Szoko, N., McShane, A. J. & Natowicz, M. R. Proteomic explorations of autism spectrum disorder. J. Int. Soc. Autism Res. 10, 1460–1469 (2017).
Abraham, J. R. et al. Proteomic investigations of autism brain identify known and novel pathogenetic processes. Sci. Rep. 9, 13118 (2019).
Chung, J.-H. & Eng, C. Nuclear-cytoplasmic partitioning of phosphatase and tensin homologue deleted on chromosome 10 (PTEN) differentially regulates the cell cycle and apoptosis. Cancer Res. 65, 8096–8100 (2005).
Chung, J.-H., Ginn-Pease, M. E. & Eng, C. Phosphatase and tensin homologue deleted on chromosome 10 (PTEN) has nuclear localization signal-like sequences for nuclear import mediated by major vault protein. Cancer Res. 65, 4108–4116 (2005).
Tilot, A. K. et al. Germline disruption of Pten localization causes enhanced sex-dependent social motivation and increased glial production. Hum. Mol. Genet. 23, 3212–3227 (2014).
Lee, H., Thacker, S., Sarn, N., Dutta, R. & Eng, C. Constitutional mislocalization of Pten drives precocious maturation in oligodendrocytes and aberrant myelination in model of autism spectrum disorder. Transl. Psychiatry 9, 13 (2019).
Sarn, N. et al. Cytoplasmic-predominant Pten increases microglial activation and synaptic pruning in a murine model with autism-like phenotype. Mol. Psychiatry https://doi.org/10.1038/s41380-020-0681-0 (2020).
Kang, S. C. et al. Decreased nuclear Pten in neural stem cells contributes to deficits in neuronal maturation. Mol. Autism 11, 43 (2020).
Tilot, A. K. et al. Neural transcriptome of constitutional Pten dysfunction in mice and its relevance to human idiopathic autism spectrum disorder. Mol. Psychiatry 21, 118–125 (2016).
Thacker, S., Sefyi, M. & Eng, C. Alternative splicing landscape of the neural transcriptome in a cytoplasmic-predominant Pten expression murine model of autism-like behavior. Transl. Psychiatry 10, 380 (2020).
Irimia, M. et al. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell 159, 1511–1523 (2014).
Fricano-Kugler, C. J. et al. Nuclear excluded autism-associated phosphatase and tensin homolog mutations dysregulate neuronal growth. Biol. Psychiatry 84, 265–277 (2018).
Jurado, S. et al. PTEN is recruited to the postsynaptic terminal for NMDA receptor-dependent long-term depression. EMBO J. 29, 2827–2840 (2010).
Ghazalpour, A. et al. Comparative analysis of proteome and transcriptome variation in mouse. PLoS Genet. 7, e1001393 (2011).
Hooshmandi, M., Wong, C. & Khoutorsky, A. Dysregulation of translational control signaling in autism spectrum disorders. Cell. Signal. 75, 109746 (2020).
Yates, J. R., Ruse, C. I. & Nakorchevsky, A. Proteomics by mass spectrometry: approaches, advances, and applications. Annu. Rev. Biomed. Eng. 11, 49–79 (2009).
Crockett, D. K., Fillmore, G. C., Elenitoba-Johnson, K. S. J. & Lim, M. S. Analysis of phosphatase and tensin homolog tumor suppressor interacting proteins by in vitro and in silico proteomics. Proteomics 5, 1250–1262 (2005).
Herlevsen, M. et al. A novel model to identify interaction partners of the PTEN tumor suppressor gene in human bladder cancer. Biochem. Biophys. Res. Commun. 352, 549–555 (2007).
Ahn, Y., Hwang, C. Y., Lee, S.-R., Kwon, K.-S. & Lee, C. The tumour suppressor PTEN mediates a negative regulation of the E3 ubiquitin-protein ligase Nedd4. Biochem. J. 412, 331–338 (2008).
Gorbenko, O. et al. Identification of novel PTEN-binding partners: PTEN interaction with fatty acid binding protein FABP4. Mol. Cell. Biochem. 337, 299–305 (2010).
Gunaratne, J. et al. Protein interactions of phosphatase and tensin homologue (PTEN) and its cancer-associated G20E mutant compared by using stable isotope labeling by amino acids in cell culture-based parallel affinity purification. J. Biol. Chem. 286, 18093–18103 (2011).
Maddika, S. et al. WWP2 is an E3 ubiquitin ligase for PTEN. Nat. Cell Biol. 13, 728–733 (2011).
Shnitsar, I. et al. PTEN regulates cilia through dishevelled. Nat. Commun. 6, 8388 (2015).
Verrastro, I., Tveen-Jensen, K., Spickett, C. M. & Pitt, A. R. The effect of HOCl-induced modifications on phosphatase and tensin homologue (PTEN) structure and function. Free Radic. Res. 52, 232–247 (2018).
Smith, S. L., Pitt, A. R. & Spickett, C. M. Approaches to Investigating the Protein Interactome of PTEN. J. Proteome Res. https://doi.org/10.1021/acs.jproteome.0c00570 (2020).
Frazier, T. W. et al. Molecular and phenotypic abnormalities in individuals with germline heterozygous PTEN mutations and autism. Mol. Psychiatry 20, 1132–1138 (2015).
Acknowledgements
We are grateful to Qi Yu for helping with mouse breeding and husbandry in the Eng lab, and Peter Bazeley for bioinformatics core support. CE is the Sondra J. and Stephen R. Hardis Endowed Chair of Cancer Genomic Medicine at the Cleveland Clinic, and is an ACS Clinical Research Professor. This work was funded, in part, by the Zacconi Program of PTEN Research Excellence and the Ambrose Monell Foundation.
Author information
Authors and Affiliations
Contributions
S.T. and C.E. conceptualized and designed the project. S.T. performed data analysis S.T. and C.E. interpreted the data. C.E. supervised the project. S.T. and C.E. drafted, critically revised, and gave final approval of the manuscript.
Corresponding author
Ethics declarations
Competing interests
Author Charis Eng is an Associate Editor for npj Genomics Medicine. The remaining authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thacker, S., Eng, C. Transcriptome-(phospho)proteome characterization of brain of a germline model of cytoplasmic-predominant Pten expression with autism-like phenotypes. npj Genom. Med. 6, 42 (2021). https://doi.org/10.1038/s41525-021-00201-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-021-00201-z
This article is cited by
-
Dysregulation of the Wnt/β-catenin signaling pathway via Rnf146 upregulation in a VPA-induced mouse model of autism spectrum disorder
Experimental & Molecular Medicine (2023)
