
Abstract
USP9X is an X-chromosome gene that escapes X-inactivation. Loss or compromised function of USP9X leads to neurodevelopmental disorders in males and females. While males are impacted primarily by hemizygous partial loss-of-function missense variants, in females de novo heterozygous complete loss-of-function mutations predominate, and give rise to the clinically recognisable USP9X-female syndrome. Here we provide evidence of the contribution of USP9X missense and small in-frame deletion variants in USP9X-female syndrome also. We scrutinise the pathogenicity of eleven such variants, ten of which were novel. Combined application of variant prediction algorithms, protein structure modelling, and assessment under clinically relevant guidelines universally support their pathogenicity. The core phenotype of this cohort overlapped with previous descriptions of USP9X-female syndrome, but exposed heightened variability. Aggregate phenotypic information of 35 currently known females with predicted pathogenic variation in USP9X reaffirms the clinically recognisable USP9X-female syndrome, and highlights major differences when compared to USP9X-male associated neurodevelopmental disorders.
Similar content being viewed by others
Introduction
The deubiquitylating enzyme encoded by USP9X is best known for its roles in brain development and cancer1. It is ranked among the top 5% of evolutionary constrained genes and is highly intolerant to variation (pLI = 1.0; z-score = 6.35)1,2,3,4. It is essential for embryonic viability5. USP9X functions to reverse the effects of protein ubiquitylation, a frequent post-translational modification that often culminates in protein degradation via the proteasome6. USP9X thus protects many of its substrates from degradation, thereby increasing their abundance and hence function1. Many USP9X substrates are encoded by genes involved in brain development and neurodevelopmental disorders (NDDs)1. Furthermore, rare USP9X mutations have been identified to directly cause NDDs7,8,9,10.
USP9X is located on the X-chromosome. The inheritance patterns and clinical presentations of the X-linked disorders often differ between males and females. X-linked disorders predominantly affect hemizygous males while female carriers are generally unaffected. This was the case for the historical family MRX99, with multiple affected males, in which a C-terminal protein truncating variant in USP9X was maternally transmitted across three generations8. An additional 15 likely pathogenic missense variants have since been reported in affected males, with further 26 of uncertain significance (VUS)7,8. Many of these 42 variants were inherited through unaffected mothers, while others arose de novo. We showed that these male USP9X missense variants cause partial, rather than complete loss of USP9X function7,8,11. In particular, these mutations disable brain-specific USP9X functions, while leaving other functions, such as those essential for embryonic viability, intact.
Sparing of heterozygous females in X-linked disorders may involve protective X-inactivation. USP9X is, however, an atypical X-chromosome gene which escapes X-inactivation, and thus the likely mechanisms is also related to its gene expression and as such dosage10. Nonetheless, the USP9X mutations found in the affected females are predominantly complete loss of function (LOF) alleles. In total, 20 females have been reported with a syndromic NDD, also known as MRX99F, caused by de novo heterozygous USP9X null mutations including whole and partial gene deletions, nonsense and early frameshift mutations10,12,13,14. Studies using patient derived cells established that these LOF mutations caused reduction of USP9X mRNA and protein abundance in affected females, suggesting haploinsufficiency as a likely disease mechanism. Such LOF alleles are likely to never be observed in a male as complete loss of USP9X, as would be the case of a hemizygous male, is known to result in early embryonic lethality in at least mouse5.
USP9X is significantly depleted of missense variants, which would indicate that at least some of these variants are likely highly deleterious to USP9X function, e.g. when located in critical domains of the protein. Indeed, a single missense mutation altering a residue in the catalytic domain of USP9X has been identified in a female individual with strong clinical resemblance to others with USP9X-female syndrome10. In this report, we identify and study an additional 10 novel female variants, 8 missense and 2 in-frame single amino acid deletions. In depth comparative in silico predictions and structural modelling provided support of pathogenicity. We also assess and compare the clinical presentations of this cohort against all reported individuals with USP9X-female syndrome. We define an expanded and overlapped phenotypic spectrum of these female cohorts, which collectively revealed similarities and differences in the phenotypic features observed in male versus female USP9X associated NDDs. Collectively, our data expands the mutational mechanisms and phenotypic outcomes relating to USP9X-female syndrome.
Results
Identification of novel USP9X variants in affected females
Following our earlier reports of USP9X variants in male and female cases with NDDs8,9, we have collected, through international clinical, diagnostic and research centres, several additional female ascertained USP9X variants of unknown clinical significance. As part of this study we have further selected and scrutinised eight missense variants and two in-frame single amino acid deletions. These variants were predominantly identified using trio based exome sequencing (Table 1; Supplementary Data). In addition to these 10 novel variants we also analysed one previously reported missense variant (Female 8; ref. 10).
One of the variants (p.Asp1685Asn) was found in two unrelated individuals (Females 26 and 27). In Female 26, this variant was inherited from a mosaic mother (de novo in mother). A second inherited variant in Female 33 (p.Glu1764Lys) was also passed on from the mother, but intriguingly with no evidence of mosaicism, at least in blood (Table 1 and Supplementary Data). This variant was not found in the maternal grandmother, while the maternal grandfather (with no signs of disability) could not be tested. The other nine of eleven variants all arose de novo (Supplementary Data). Applying the guidelines of the American College of Medical Genetics Guidelines (ACMG; ref. 15) we classified all of these 11 variants as likely pathogenic (Table 1 and Supplementary Data). All were also predicted to be deleterious using Polyphen216 and Combined Annotation Dependent Depletion (CADD; ref. 17) algorithms (Table 1 and Supplementary Data). All 11 variants alter highly conserved amino acids, all but one invariable (Fig. 1a). These variants are novel and not present in Genome Aggregation Database (gnomAD V2.1.1;18) consisting of >140,000 exomes or genomes (Table 1). In general, the variants impacted regions of the USP9X protein, which were predicted to be intolerant to variation (Fig. 1b; ref. 19). Five of the variants were located in the ubiquitin C hydrolase (UCH) catalytic domain, while the others were found distributed in the N-terminal extension of the protein of largely unknown structure and function (Table 1 and Fig. 1b). To see if these variants clustered in potential USP9X ‘mutation hotspots’, we compared their location with those of likely pathogenic variants associated with male NDDs (n = 16; ref. 7). Furthermore, as USP9X loss-of-function mutations are also enriched in somatic cancers1, we included variants extracted from the Catalogue of Somatic Mutations in Cancer (COSMIC) database that are predicted to be deleterious (CADD score >30; n = 49). While this analysis did not reveal any striking variant ‘hotspots’, we noted that the catalytic domain was potentially enriched with female variants, and enriched with cancer variants, compared to its flanking regions (p = 0.08 and p = 0.00017 respectively via two-proportion z-test; Fig. 1b).

a Cross species protein alignment of USP9X showing conservation of altered amino acid residues. b Location of female likely pathogenic variants on the USP9X protein structure. USP9X variation tolerance landscape is provided (see Materials and Methods). Locations of male likely pathogenic variants and predicted deleterious missense somatic cancer variants (extracted from COSMIC database with CADD score ≥30) are shown for comparison.
Altogether we identify 10 novel female USP9X missense and single amino acid deletion variants associated with NDDs, which alter generally invariable amino acids and are located in protein regions required for catalytic activity and/or intolerant to variation.
Variant prediction algorithms support pathogenicity of USP9X-female missense variants
To extend our investigations into the functional (or not) effect of these variants, we employed an array of in silico missense variant pathogenicity predictive tools. We compared the outcomes of these predictive algorithms between the female likely pathogenic missense variants and that of common (i.e. assuming to be benign) USP9X missense variants. We defined a common variant as one with an allele frequency >1:100,000 in the gnomAD V2.1.1 database (n = 159)18. The gnomAD derived variants are from individuals devoid of severe paediatric disease, and are found distributed throughout the entire protein (including catalytic domain and regions predicted intolerant to variation). They provide the best, currently available control dataset to further interrogate the pathogenicity of USP9X variation (Supplementary Fig. 1). We submitted all missense variants, including common variants, female and male NDD variants, and all COSMIC variants (n = 358; Supplementary Fig. 1) to the suite of variant prediction tools within the ANNOVAR ensemble20. We previously established several tools with the best predictive power for assessing USP9X missense variation7. Using these same tools, we compared the combined predictive scores of the USP9X common variants with the female, male and cancer variants. For each tool used (CADD, Ployphen2, M-CAP, Mutation Assessor, VEST3, DANN, SIFT and PROVEAN), female missense variants scored significantly higher than common variants, thus supporting pathogenicity (Fig. 2a). The scores of female variants also trended higher than male variants, and that of cancer, suggesting that on average they were more deleterious. We have also reported beforehand that a combined score of CADD and PROVEAN provides a highly stringent predictive rubric for USP9X missense variation7. Applying this approach, we found 100% of these nine female missense variants tested were scored as pathogenic (CADD > 25 and PROVEAN > 0.5665) compared to 75% of male likely pathogenic variants, 45% of cancer variants and only 15% of common variants (Fig. 2b). Thus, several in silico predictive approaches provided congruent support of the pathogenicity of female USP9X likely pathogenic missense variants.

a Aggregate comparison of common variants extracted from gnomAD (allele frequency >1:100000), against females missense variants, likely pathogenic male missense variants, and variants found in cancer (extracted from COSMIC database) using a suite of in silico prediction tools. Box-whisker plots are defined as follows: centre line, median; box limits, upper and lower quartiles; whiskers, min and max values. *significantly different from common variants p < 0.05 by two-tailed equal variance Student’s t-test. b Comparison of CADD and PROVEAN scores reveal clustering of variants all female missense variants in the upper-right quadrant consistent with pathogenicity (CADD > 25, PROVEAN > 0.565). Scores of common variants are significantly correlated (Pearson’s correlation given). Colour scheme as in a. Inset identifies each variant in the ‘pathogenic quadrant’. Graphs show percent of each type of variant, and the overall composition of variant types within the pathogenic quadrant.
Protein structure modelling of the USP9X catalytic domain variants
We employed our recently resolved USP9X UCH catalytic domain structural model7,11 to interrogate the molecular mechanisms underpinning pathogenicity of the five female missense and single amino acid deletion variants found within (Table 1 and Fig. 3). Notably, the predicted effects of missense variation using this structural model thus far have been validated using in vitro recombinant protein binding and deubiquitinating assays11. The p.Tyr1881 residue deleted in Female 29 contributes to a beta-sheet critical for the positioning of the UCH catalytic triad. The p.Tyr1881 deletion is predicted to alter the position of the catalytic residue p.His1879 and likely to have significant effects on catalytic activity. Predicted deleterious cancer variants (i.e. CADD > 30) in close proximity (p.Ser1872Asn, p.Ala1875Val, p.Ser1876Gly, p.Val1870Ile) likely act via similar mechanisms (Fig. 3). The p.Tyr1802 residue altered in Female 28 contributes to the hydrophobic surface involved in ubiquitin binding via interaction with the p.Ile36 residue of ubiquitin. The p.Tyr1802Ser substitution introduces a polar amino acid predicted to disrupt the hydrophobicity and ubiquitin binding. Predicted deleterious cancer variants (p.Lys1798Thr, p.Arg1799Gln, p.Arg1799Leu) are proposed to have similar effect (Fig. 3). The p.Asp1685Asn substitution in Female 27 also results in an amino acid charge reversal, and is predicted to alter the intramolecular charge–charge interaction with p.Gln1796, and as such constrict the ubiquitin binding channel and sterically clash with the backbone amine of the p.Leu73 residue of ubiquitin. This mechanism is similar to that predicted for proximal deleterious cancer variants (p.Glu1688Lys, p.Glu1688Ala; Fig. 3). The p.Leu1693Trp variant in Female 8 introduces a highly bulky tryptophan predicted to disrupt the local hydrophobic core provided by p.Val1643, p.Leu1647, p.Phe1689 and p.Phe1671. The importance of maintaining this fold is highlighted by the presence of predicted deleterious cancer variants acting via similar mechanism (p.Ser1692Leu and p.Val1694Met; Fig. 3). The p.Glu1764Lys variant in Female 33 lies within the zinc finger motif of the catalytic domain, which forms multiple contacts with ubiquitin and is integral to the catalytic activity of several related deubiquitinating enzymes21,22. Indeed, structure based mutations of USP9X which disrupt zinc-binding alter its activity towards specific types of ubiquitin chain linkages23, while the presence of multiple deleterious cancer variants (p.Asp1761Tyr, p.Asp1720Asn and p.Arg1783Cys) in the zinc finger motif provide additional support of its importance. Thus while p.Glu1764Lys is surface exposed and not likely to be involved in stabilizing intramolecular interactions, its close proximity to the zinc-binding site suggests it may alter the positioning or zinc-binding properties of this motif, suggesting profound effect on USP9X ubiquitin chain specificity and activity23. Thus structural modelling of the all likely pathogenic USP9X-female variants located in the catalytic domain provides rationale for disrupted catalytic activity and/or ubiquitin binding, and is supported by analogous mechanisms of several proximal predicted deleterious variants arising in cancer.

a Homology model of USP9X (grey) with catalytic site (magenta), likely pathogenic female variants (red) and location of predicted deleterious cancer variants (blue; extracted from COSMIC database with CADD score >30). Interaction with ubiquitin is shown. Likely pathogenic variants are positioned in regions of well‐ordered secondary structure or flexible regions involved in zinc-binding. b Insets indicate local structural effects of indicated likely pathogenic female USP9X variants. All native amino acid side chains are represented as grey sticks. Variant amino side chains are indicated by red sticks. Side chains of the amino acids forming the core catalytic site are indicated by magenta sticks. Location of cancer variants is highlighted in blue. Zinc ion is represented with a yellow sphere. Hydrophobic van der vaals radii are indicated by dots and charge–charge interactions are shown by dotted lines.
Variable phenotype of females with USP9X missense variants
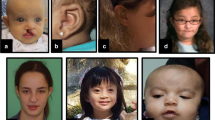
All females in our missense and single amino acid deletion cohort (n = 12) were ascertained primarily on the basis of psychomotor developmental delay (Fig. 4a and Supplementary Data). Intellectual disability (ID) was present in all individuals where assessed, but was variable, ranging from borderline to severe. All individuals displayed problems with speech and language, the severity of which was also across a wide spectrum, ranging from somewhat innocuous delay through to complete absence. There was also variable effects on the development of motor function, ranging from unreported to severe disability, which in two individuals required standing supports or wheel chairs. Motor disability was most frequently related to hypotonia (Fig. 4a). Hearing loss was a prominent feature, and individuals displayed a number of different behavioural disturbances including autism, anxiety and aggression (Fig. 4a and Supplementary Data). All individuals presented with brain malformations, most frequently agenesis of the corpus callosum and ventriculomegaly (Fig. 4a, b and Supplementary Data). Other congenital abnormalities were also observed, involving skeletal defects affecting the spine, feet and hips, and heart defects, the latter of which resulted in neonatal lethality in one case (Female 23; Supplementary Data). In addition, abnormalities affecting skin, gastroenterological, urogenital, metabolic and endocrine systems were observed at lower frequencies (Supplementary Data). Facial dysmorphisms were prevalent in almost all individuals, and was in close alignment with previously reported individuals with USP9X-female syndrome, with common features including deep-set eyes, telecanthus, blepharophimosis, broad nasal tip with wide alae and short collumnella, low set and dysplastic ears, small mouth and micrognathia (Fig. 4a, c, Supplementary Fig. 2 and ref. 10). These data reveal a variable phenotype associated with missense and single amino acid deletion USP9X variants in females, with major features of developmental delay, ID, speech and motor delay, prevalent brain malformations and other congenital aspects particularly affecting the development of the craniofacial structures spine and heart.

a Constellation and frequency of major clinical features. b Magnetic resonance imaging (MRI) of brains of affected individuals. Note prominent extra-axial spaces in all, hypoplastic corpus callosum in Females 25 and 29, optic nerve atrophy and in Female 25, Dandy Walker malformation and Blake’s pouch cyst in Female 26, and ventriculomegaly of the 3rd and 4th ventricles in Female 29. c Images of affected females showing facial dysmorphisms, with common features including deep-set eyes, telecanthus, blepharophimosis, a broad nasal tip with wide alae and short collumnella, and low set and dysplastic ears. Written consent was obtained for the publication of photographs.
Affected females share key clinical presentations, which differ in USP9X males
We also sought to establish if the clinical presentations of our female cohort with missense and single amino acid deletion variants overlapped with those of previously reported individuals with USP9X-female syndrome caused by bona fide complete loss of function alleles. For this purpose we combined the phenotypic information from the 20 reported individuals with heterozygous gene deletion, nonsense and frameshift USP9X alleles (Supplementary Fig. 3; refs. 10,12,13,14). We herein further expand this cohort by reporting an additional three novel cases (Females 30–32; Fig. 5a, b, Supplementary Fig. 2). These individuals presented with phenotypes that also highlight the range of severities associated with USP9X variation (Supplementary Data). Female 30 had a de novo nonsense variant (p.Trp380Ter) and presented with severe ID, absent speech and severe motor disability. In comparison, Female 32 with a de novo frameshift variant (p.Ile535Asnfs*11) had only slight delays in speech, language and motor skills, and is now largely meeting developmental milestones. Furthermore a maternally inherited nonsense variant (p.Arg215Ter) was found following the genetic autopsy of Female 31, a terminated foetus with brain malformation, heart and skeletal defects (Supplementary Data). In this instance, while the mother was assessed as having a history of scoliosis and partial hearing impairment, she was otherwise normal and with no dysmorphic features or additional congenital anomalies. These cases further expand the clinical spectrum associated with bona fide heterozygous loss of function USP9X variants.

a Location of bona fide loss-of-function variants in individuals with USP9X-female syndrome. Three novel variants described in this study are annotated. b Images showing facial dysmorphisms of females affected by novel nonsense and frameshift USP9X variants. Note resemblance to individuals in Fig. 4c. c Comparison of clinical features of females with missense and single amino acid deletions (n = 12) variants with females harbouring null alleles including all previously published cases and three novel cases identified in this study (n = 23). Sankey plot highlights overlap across all features except short stature. d Comparison of clinical features of a combined female cohort (n = 35) with phenotypes of all published males with likely pathogenic variants (n = 16). Sankey plot highlights overlap across neurological features but not in other major female associated congenial phenotypes. Thickness of each stream is proportional to the percentage of cases with each feature, which is also provided numerically at the terminal nodes.
We then combined phenotypical data from all such individuals (n = 23; Supplementary Fig. 3) and compared the frequency of major clinical features to that observed in the female individuals harbouring missense and single amino acid deletion variants (n = 12 cases; Supplementary Data). We found strong concordance in the prevalence of combined neurological and other congenital features between these cohorts (Fig. 5c). These data strongly support the pathogenicity of missense and single amino acid deletion variants in females.
Given this strong overlap, we combined data from both these two female cohorts (n = 23 + 12 = 35 individuals) to best define the clinical presentations in females with deleterious USP9X variants (Fig. 5c). Subsequently, we also used this data to compare females with males (n = 167) with pathogenic or likely pathogenic USP9X variants (Fig. 5d). While the major neurological findings were similar between males and females, males were unlikely to have congenital presentations including skeletal and heart defects, among others (Fig. 5d). These data also support the deleterious effect of the UPS9X missense and single amino acid deletion variants, refine USP9X-female syndrome phenotype from an aggregate of 35 individuals, and point to major differences between males and females with USP9X variants.
Discussion
Here we expand the genotypic and phenotypic spectrum of USP9X-female syndrome. The current state of knowledge suggests that de novo heterozygous complete LOF USP9X variants cause a defined syndrome in heterozygous females10. Twenty-three such females have been now been reported, including the three novel cases herein. These female LOF mutations include whole or partial gene deletions, early nonsense and frameshift mutations10,12,13,14. Males with such LOF mutations are unlikely to survive early stages post fertilisation5. Here we present new evidence that missense and single amino acid deletion variants can also cause a similar female phenotype. Prior to this study, only a single affected female missense variant located in the USP9X catalytic domain had been reported10. We now add 10 additional variants classified as likely pathogenic under the ACMG guidelines, shedding new light into the genetic origin of USP9X disease as well as its clinical presentations.
Eight of these 10 novel variants occurred de novo in these females, which by a traditional view could also have been considered an ‘incidental finding’ of an USP9X unaffected carrier female. One individual had an inherited variant from their mother who were subsequently found to be mosaic, a mode which has been previously reported10. The remaining case of Female 33, with a variant inherited from her unaffected mother, who is unlikely a somatic mosaic, suggests possible modifier(s) of female penetrance, perhaps similar to e.g. the X-chromosome linked PCDH19 clustering epilepsy24. While it is also plausible to question this specific USP9X variant’s pathogenicity, the variant is located within the catalytic domain, it is predicted to be pathogenic universally by several algorithms, and the affected girl bears clear clinical resemblance to USP9X-female syndrome. That includes cardinal clinical presentations such as skin pigmentation, hip dysplasia, heart defects, choanal atresia and hearing loss in addition to other hallmark neurological and craniofacial features. We also identified another similar transmission, in a case of a maternally inherited nonsense variant (Female 31). In this instance the LOF effect of this variant (p.Arg215*) is highly likely. The carrier mother of this child had a history of scoliosis, but was otherwise unaffected. These two cases of variable penetrance of USP9X LOF variants in heterozygous females suggest the existence of a disease modifier. One possible candidate is skewing of X-inactivation, but USP9X is known to escape X-inactivation. That said, the degree to which a gene escapes from X-inactivation is known to be variable, and can be tissue specific25,26,27,28. Complete skewing of X-inactivation may also suggest the existence of another genetic abnormality on one of the X-chromosomes of the affected female, which can be contributing to the phenotype independently of USP9X, e.g. as has been observed in an affected female carrier of Fragile X29. Furthermore, a haploinsufficiency-like mechanism of USP9X-female NDD is not supported by e.g. phenotypes observed in Turner Syndrome with XO sex chromosome karyotype, which generally lack neurological manifestations30. Possible role for X-inactivation in USP9X-female NDDs is suggested by several frequently observed clinical features including mosaic skin pigmentations and asymmetries in brain formation, breast development, limb development and other structures10,12,13. Previous studies have however shown that skewing of X-inactivation in DNA obtained from three out of five patient derived fibroblasts showed no correlation with disease severity10. However, as it is also the case in e.g. PCDH19 clustering epilepsy, where the blood or skin-derived X-inactivation is not informative31, mouse model evidence suggests that it is at play in at least brain32. X-inactivation studies revealed no evidence of skewing in Female 33, whilst studies on the mothers DNA were uninformative across multiple loci (data not shown). We were also unable to conduct further studies on Female 31 and her mother. Other potential modifiers may relate to specific variants, such as interindividual variation in nonsense mediated mRNA decay33 or associated transcriptional compensation34,35. Thus examples and identification of USP9X variants with variable penetrance offer an opportunity to investigate the likely modifiers and their mechanism in a deeper and more systematic manner.
The set of major clinical features of USP9X-female syndrome associated with de novo heterozygous gene deletion, nonsense and frameshift mutations involves developmental delay, ID, brain malformations and other congenital abnormalities impacting craniofacial development, and the heart, skeleton, skin and other organs10. These clinical presentations were also observed in our cohort of females with missense and single amino acid deletion USP9X variants, but which also appeared variable. Variability in female USP9X associated NDDs can also be driven by the underlying mutation type and not just existence of modifiers (as discussed above). All previously reported mutations result in a loss of USP9X dosage10, with likely downstream impact on all USP9X substrates. This uniform molecular mechanism was proposed to underpin the consistency in the phenotypic outcomes shared among different individuals giving rise to the reports of a recognisable USP9X-female syndrome. Furthermore, genetic ablation of Usp9x from the developing mouse brain (loss of dosage) provides a strong recapitulation of the neurological phenotypes of these affected females, including hypoplastic corpus callosum, ventriculomegaly, and learning and memory problems7,11,36,37. The impact of missense mutations is less defined, and may cause differential impact on downstream substrates and as such phenotypic outcome(s). The variants affecting the catalytic domain are most likely similar to complete LOF alleles, whilst those in the N-terminal extensions may disrupt only subsets of USP9X substrates (see below). It is yet to be determined as to whether these missense variants retain residual USP9X function or act as dominant negative alleles.
The affected female missense and single amino acid deletion variants we identified in this study all passed through rigorous in silico testing which further supported pathogenicity. Detailed structural modelling of the five variants impacting the catalytic domain supplied evidence of altered catalytic activity and/or binding to ubiquitin. This was reinforced by the presence of multiple likely deleterious cancer variants (CADD > 30) in close proximity, which were projected to act via overlapping mechanisms. USP9X is a known tumour suppressor38,39,40 and there also exists a significant enrichment of LOF USP9X variants in the COSMIC database1. It is noteworthy that childhood malignancy has been reported in two female individuals with USP9X-female syndrome10, and could potentially be involved in the natural course of the condition. The other variants were all located in the N-terminal region of largely undetermined function1. Male USP9X variants in the N-terminal regions have been shown to disrupt subsets of USP9X substrate interactions, rather than all7. These substrates are, however, critical specifically for the function of neurodevelopmental signalling pathways TGFβ, mTOR, Notch and Wnt7, all of which have been shown to be deregulated in the developing brains of mice lacking Usp9x11,37,41,42,43. It was also striking to see that males shared much of the neurological phenotypical features shared in our female cohorts, but almost none of the other congenital features. At least some of these male variants are inherited from phenotypically normal mothers, and as such likely better tolerated in a heterozygous state. The female missense mutations reported herein the N-terminal region of USP9X are speculated to be more deleterious than their male counterparts, disrupting more critical (but unknown) USP9X functions or overall USP9X protein structural integrity. On aggregate, the outcomes of the in silico predictive tools not only supported the pathogenicity of the female variants, but also showed that they were more deleterious than male variants. This is a preliminary finding and requires larger validation datasets. Highly deleterious variants are unlikely to ever be identified in males due to probable effect on embryonic viability5,37. It was notable that the mosaic mother of Female 26 (p.Asp1685Asn) had a history of five miscarriages.
In conclusion, in this study we identified likely pathogenic missense and single amino acid deletion variants, and additional nonsense and frameshift variants in USP9X in affected heterozygous females. The phenotype of these females affirmed that of previous reports, but also highlighted its considerable variability. Our study thus reveals the complexities in the clinical definition, and genetic aetiology of an emerging USP9X-female syndrome.
Methods
Subjects
This study was approved by the Women’s and Children’s Health Network Human Research Ethics Committee, South Australia, Australia (HREC786–07–2020). All subject information was provided following informed guardian consent. The authors affirm that guardians of human research participants provided written informed consent for publication of images in Figs. 4 and 5 and Supplementary Fig. 2.
Variant analysis
Two-proportion z-test for enrichment of variants in the catalytic domain was conducted using http://www.sthda.com/english/wiki/two-proportions-z-test-in-r where full length USP9X is 2570 amino acids in length and catalytic domain is 396 amino acids in length. Variant predictions were performed using Annovar20 accessed via the webserver http://wannovar.wglab.org/. Box-whisker plots are defined as follows: centre line, median; box limits, upper and lower quartiles; whiskers, min and max values. Statistical significance was assessed using two-tailed equal variance Student’s t-test with p < 0.05. USP9X tolerance to variation landscape was established using methods described in ref. 19 and outputted using the Metadome Version 1.0.1 webserver at https://stuart.radboudumc.nl/metadome/dashboard.
Structural modelling
The USP9X catalytic structural homology model was generated and characterized using Maestro Prime module (Schrodinger, LLC, NY, USA) using the crystal structure 5WCH (http://www.rcsb.org/structure/5WCH) with absent flexible loop regions modelled ab initio through energy minimization7,11,44. Homology model image generation and mutagenesis were performed using PyMol V1.8.2.0 (Schrodinger).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Additional data and materials from this study are available from the authors on reasonable request, subject to compliance with our obligations under human research ethics.
References
Murtaza, M., Jolly, L. A., Gecz, J. & Wood, S. A. La FAM fatale: USP9X in development and disease. Cell Mol. Life Sci. 72, 2075–2089 (2015).
Petrovski, S., Wang, Q., Heinzen, E. L., Allen, A. S. & Goldstein, D. B. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 9, e1003709 (2013).
Cooper, G. M. et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 15, 901–913 (2005).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Pantaleon, M. et al. FAM deubiquitylating enzyme is essential for preimplantation mouse embryo development. Mech. Dev. 109, 151–160 (2001).
Kerscher, O., Felberbaum, R. & Hochstrasser, M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu. Rev. Cell Dev. Biol. 22, 159–180 (2006).
Johnson, B. V. et al. Partial loss of USP9X function leads to a male neurodevelopmental and behavioral disorder converging on transforming growth factor beta signaling. Biol. Psychiatry 87, 100–112 (2020).
Homan, C. C. et al. Mutations in USP9X are associated with X-linked intellectual disability and disrupt neuronal cell migration and growth. Am. J. Hum. Genet. 94, 470–478 (2014).
Paemka, L. et al. Seizures are regulated by ubiquitin-specific peptidase 9 X-linked (USP9X), a de-ubiquitinase. PLoS Genet. 11, e1005022 (2015).
Reijnders, M. R. et al. De novo loss-of-function mutations in USP9X cause a female-specific recognizable syndrome with developmental delay and congenital malformations. Am. J. Hum. Genet. 98, 373–381 (2016).
Yoon, S. et al. Usp9X controls ankyrin-repeat domain protein homeostasis during dendritic spine development. Neuron 105, 506–521. e507 (2020).
Sinthuwiwat, T., Ittiwut, C., Porntaveetus, T. & Shotelersuk, V. Female-restricted syndromic intellectual disability in a patient from Thailand. Am. J. Med. Genet. A 179, 758–761 (2019).
Tsurusaki, Y. et al. Novel USP9X variants in two patients with X-linked intellectual disability. Hum. Genome Var. 6, 49 (2019).
Vianna, E. Q. et al. Understanding the landscape of X-linked variants causing intellectual disability in females through extreme X chromosome inactivation skewing. Mol. Neurobiol. https://doi.org/10.1007/s12035-020-01981-8 (2020).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. https://doi.org/10.1002/0471142905.hg0720s76 (2013).
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894 (2019).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Wiel, L., Venselaar, H., Veltman, J. A., Vriend, G. & Gilissen, C. Aggregation of population-based genetic variation over protein domain homologues and its potential use in genetic diagnostics. Hum. Mutat. 38, 1454–1463 (2017).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Hetfeld, B. K. et al. The zinc finger of the CSN-associated deubiquitinating enzyme USP15 is essential to rescue the E3 ligase Rbx1. Curr. Biol. 15, 1217–1221 (2005).
Tencer, A. H., Liang, Q. & Zhuang, Z. Divergence in ubiquitin interaction and catalysis among the ubiquitin-specific protease family deubiquitinating enzymes. Biochemistry 55, 4708–4719 (2016).
Paudel, P. et al. Crystal structure and activity-based labeling reveal the mechanisms for linkage-specific substrate recognition by deubiquitinase USP9X. Proc. Natl Acad. Sci. USA 116, 7288–7297 (2019).
Kolc, K. L. et al. A systematic review and meta-analysis of 271 PCDH19-variant individuals identifies psychiatric comorbidities, and association of seizure onset and disease severity. Mol. Psychiatry 24, 241–251 (2019).
Garieri, M. et al. Extensive cellular heterogeneity of X inactivation revealed by single-cell allele-specific expression in human fibroblasts. Proc. Natl Acad. Sci. USA 115, 13015–13020 (2018).
Luijk, R. et al. Autosomal genetic variation is associated with DNA methylation in regions variably escaping X-chromosome inactivation. Nat. Commun. 9, 3738 (2018).
Deng, X., Berletch, J. B., Nguyen, D. K. & Disteche, C. M. X chromosome regulation: diverse patterns in development, tissues and disease. Nat. Rev. Genet. 15, 367–378 (2014).
Tukiainen, T. et al. Landscape of X chromosome inactivation across human tissues. Nature 550, 244–248 (2017).
Jorge, P. et al. Classical fragile-X phenotype in a female infant disclosed by comprehensive genomic studies. BMC Med. Genet. 19, 74 (2018).
Sybert, V. P. & McCauley, E. Turner’s syndrome. N. Engl. J. Med. 351, 1227–1238 (2004).
Dibbens, L. M. et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat. Genet. 40, 776–781 (2008).
Pederick, D. T. et al. Abnormal cell sorting underlies the unique X-linked inheritance of PCDH19 epilepsy. Neuron 97, 59–66.e55 (2018).
Nguyen, L. S., Wilkinson, M. F. & Gecz, J. Nonsense-mediated mRNA decay: inter-individual variability and human disease. Neurosci. Biobehav. Rev. 46 Pt 2, 175–186 (2014).
Ma, Z. & Chen, J. Premature termination codon-bearing mRNA mediates genetic compensation response. Zebrafish. https://doi.org/10.1089/zeb.2019.1824 (2020).
Yin, S. et al. Evidence that the nonsense-mediated mRNA decay pathway participates in X chromosome dosage compensation in mammals. Biochem. Biophys. Res. Commun. 383, 378–382 (2009).
Oishi, S. et al. Usp9x-deficiency disrupts the morphological development of the postnatal hippocampal dentate gyrus. Sci. Rep. 6, 25783 (2016).
Stegeman, S. et al. Loss of Usp9x disrupts cortical architecture, hippocampal development and TGFbeta-mediated axonogenesis. PLoS ONE 8, e68287 (2013).
Zhu, C. et al. Deubiquitylase USP9X suppresses tumorigenesis by stabilizing large tumor suppressor kinase 2 (LATS2) in the Hippo pathway. J. Biol. Chem. 293, 1178–1191 (2018).
Toloczko, A. et al. Deubiquitinating enzyme USP9X suppresses tumor growth via LATS kinase and core components of the hippo pathway. Cancer Res. 77, 4921–4933 (2017).
Perez-Mancera, P. A. et al. The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature 486, 266–270 (2012).
Jolly, L. A., Taylor, V. & Wood, S. A. USP9X enhances the polarity and self-renewal of embryonic stem cell-derived neural progenitors. Mol. Biol. Cell 20, 2015–2029 (2009).
Bridges, C. R. et al. USP9X deubiquitylating enzyme maintains RAPTOR protein levels, mTORC1 signalling and proliferation in neural progenitors. Sci. Rep. 7, 391 (2017).
Premarathne, S., Murtaza, M., Matigian, N., Jolly, L. A. & Wood, S. A. Loss of Usp9x disrupts cell adhesion, and components of the Wnt and Notch signaling pathways in neural progenitors. Sci. Rep. 7, 8109 (2017).
Zhang, Q. et al. Crystal structure of a peptidase. http://www.rcsb.org/structure/5WCH (2018).
Acknowledgements
We are grateful for the support and contributions of all families involved in this study. We are thankful for the funding received from Creola Pora with the help of her friends and colleagues. This work was supported by SFARI Explorer Grant 527556 to M.P., S.A.W. and L.A.J. L.A.J. is supported by Australian Research Council ARC DE160100620. J.G. is supported by grants from the National Health and Medical Research Council of Australia: Program Grant (628952) and Research Fellowship (1041920). P.P. is supported by National Institute of Health grant R01MH107182. This research received funding specifically appointed to Department of Medical Sciences from the Italian Ministry for Education, University and Research (Ministero dell’Istruzione, dell’Università e della Ricerca - MIUR) under the programme “Dipartimenti di Eccellenza 2018–2022” Project code D15D18000410001. The whole-exome sequencing of Female 34 was performed as part of the Autism Sequencing Consortium and was supported by the NIMH (MH111661). Sequencing and analysis of Female 29 was provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) and was funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute grant UM1 HG008900 and in part by National Human Genome Research Institute grant R01 HG009141.
Author information
Authors and Affiliations
Contributions
L.A.J. devised and coordinated the study, compiled and analysed all experimental data, wrote and revised the manuscript. E.P., S.Y. and P.P. conducted structural modelling and contributed to the draft manuscript. A.E.G. and M.A.C. contributed to variant prediction and enrichment analysis. L.A.P. contributed to compiling clinical data and manuscript revision. M.S. contributed X-inactivation studies. S.A.W. and M.P. contributed to data interpretation and manuscript revision. G.L., C.K., M.C.S., E.G., F.M., C.K., A.G., K.C., K.R., P.Y., S.W., B.K., J.C., D.B., S.S., S.M.W., M.B.D., T.K., S.K., A.B., E.S. and S.T. contributed to whole-exome sequencing, variant validation and collection of clinical data. J.G. contributed to study design, data interpretation and revision of the manuscript. All authors approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
L.A. P.-J. is scientific advisor and founding partner of qGenomics Laboratory. All other authors have no conflicts of interest or financial disclosures to declare.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jolly, L.A., Parnell, E., Gardner, A.E. et al. Missense variant contribution to USP9X-female syndrome. npj Genom. Med. 5, 53 (2020). https://doi.org/10.1038/s41525-020-00162-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-020-00162-9
This article is cited by
-
BCORL1, POF1B, and USP9X copy number variation in women with idiopathic diminished ovarian reserve
Journal of Assisted Reproduction and Genetics (2024)
-
Molecular diagnosis and novel genes and phenotypes in a pediatric thoracic insufficiency cohort
Scientific Reports (2023)
-
Compound genetic etiology in a patient with a syndrome including diabetes, intellectual deficiency and distichiasis
Orphanet Journal of Rare Diseases (2022)
-
Monogenic causes of pigmentary mosaicism
Human Genetics (2022)
-
Systematic analysis and prediction of genes associated with monogenic disorders on human chromosome X
Nature Communications (2022)
