
Abstract
Purpose
Hardikar syndrome (MIM 612726) is a rare multiple congenital anomaly syndrome characterized by facial clefting, pigmentary retinopathy, biliary anomalies, and intestinal malrotation, but with preserved cognition. Only four patients have been reported previously, and none had a molecular diagnosis. Our objective was to identify the genetic basis of Hardikar syndrome (HS) and expand the phenotypic spectrum of this disorder.
Methods
We performed exome sequencing on two previously reported and five unpublished female patients with a clinical diagnosis of HS. X-chromosome inactivation (XCI) studies were also performed.
Results
We report clinical features of HS with previously undescribed phenotypes, including a fatal unprovoked intracranial hemorrhage at age 21. We additionally report the discovery of de novo pathogenic nonsense and frameshift variants in MED12 in these seven individuals and evidence of extremely skewed XCI in all patients with informative testing.
Conclusion
Pathogenic missense variants in the X-chromosome gene MED12 have previously been associated with Opitz–Kaveggia syndrome, Lujan syndrome, Ohdo syndrome, and nonsyndromic intellectual disability, primarily in males. We propose a fifth, female-specific phenotype for MED12, and suggest that nonsense and frameshift loss-of-function MED12 variants in females cause HS. This expands the MED12-associated phenotype in females beyond intellectual disability.
Similar content being viewed by others


INTRODUCTION
MED12 is a member of the preinitiation complex (PIC), a large protein complex that is involved in transcription initiation. Specifically, MED12 is a key component within the PIC Mediator complex, which is comprised of 31 mediator subunits, cyclin-dependent kinase 8 (CDK8), as well as several paralogs (such as MED12L and MED13L).1 The Mediator complex conveys information from gene-specific regulatory proteins to RNA polymerase II (Pol II) to regulate transcriptional initiation, transcriptional termination, and noncoding RNA activation.2,3,4 Furthermore, the mediator complex regulates chromatin looping, higher-order chromatin folding, and messenger RNA (mRNA) processing and export.5,6,7 These functions coordinate a broad range of cellular activities, including cell growth, cell migration, development, and differentiation.8,9,10,11,12,13
MED12 is essential for CDK8 kinase activation, which modulates the interaction between Mediator and Pol II, MED13, and CCNC.2,3,14 Missense variants in MED12 are associated with multiple X-linked intellectual disability syndromes, including Opitz–Kaveggia syndrome (MIM 305450), also known as FG syndrome, Lujan–Fryns syndrome (MIM 309520), and X-linked Ohdo syndrome (MIM 300895).4 Additionally, MED12 missense variants have been reported in individuals with nonsyndromic intellectual disability15,16,17,18 as well as in individuals with intellectual disability and dysmorphic features (summarized in Supplementary Table 1). Generally, heterozgous females are unaffected, but there are a few reports of females with developmental differences, thought to be the result of unfavorable X-inactivation (Supplementary Table 1).
Hardikar syndrome (HS) (MIM 612726) is a well-recognized, multiple congenital anomaly disorder with a specific phenotype of foregut malformations, intestinal malrotation, liver and biliary tract disease, genitourinary abnormalities, cleft lip and palate, and pigmentary retinopathy of unknown genetic basis.19,20,21,22 Since its initial description in 1992, four female patients have been reported, all without a molecular etiology: (1) a female with biliary malformation, unilateral cleft lip and palate, abnormal retinal pigmentation, aortic coarctation, low ureteric obstruction, and intestinal malrotation; (2) a female with biliary malformations, cleft palate, abnormal retinal pigmentation, bilateral hydronephrosis with hydroureters, and intestinal malrotation; (3) a female with bilateral cleft lip and palate, preauricular pits, cat’s paw retinal pigmentation, and ventricular septal defect who died at 21 years due to massive intracerebral hemorrhage; and (4) a female with bilateral cleft lip and palate, intestinal malrotation, cat’s paw retinal pigmentation, unilateral lacrimal duct stenosis, umbilical hernia, and biliary tree malformations resulting in severe cholestasis and fibrosis, necessitating liver transplantation. HS was previously suggested to be X-linked, as all known patients are female, but with small numbers it was unclear if this was chance or biology. Here, we report the clinical findings of seven females with HS, including five previously unreported probands and clinical updates on two previously reported patients. These updates include the fatal unprovoked intracranial hemorrhage at age 21 years in one patient, highlighting the importance of early diagnosis of HS and appropriate clinical management. Exome sequencing identified nonsense or frameshift MED12 variants in all seven individuals, and all five tested patients showed evidence of skewed XCI (Table 1). Our patient cohort expands the phenotypic spectrum of HS and nominates loss-of-function pathogenic MED12 variants as causal for this syndrome.
MATERIALS AND METHODS
Exome sequencing and analysis
Exome sequencing was performed on DNA from probands 1, 3, 4, and 5, and their available relatives. Briefly, all the raw reads were aligned to the reference human genome using the Burrows–Wheeler Aligner (BWA) and single-nucleotide variants and small insertions/deletions were captured using the Genome Analysis Toolkit (GATK). ANNOVAR and SnpEff were used to functionally annotate the variants. Variants generated were then filtered against public variant databases (dbSNP, 1000 Genomes Project, NHLBI ESP6500SI, gnomAD, and Kaviar) and >5000 in-house exomes with a minor allele frequency of 0.01% due to rarity of HS. Subsequent gene prioritization was performed on the basis of biological relevance by referring to the OMIM database, which resulted in heterozygous MED12 variants shared among these four individuals as the most likely disease-causing candidates. Exome sequencing was subsequently performed on patients 2 and 6 and a focused analysis on MED12 revealed two additional loss-of-function variants. Clinical exome sequencing of patient 7 was performed independently. MED12 confirmatory genotyping was performed by Sanger sequencing.
X-chromosome inactivation (XCI) assay
Genomic DNA was isolated from peripheral blood mononuclear cells of patients 1–6. Following the DNA methylation-sensitive restriction digestion with HpaII for each patient, the predigested samples and undigested samples were subsequently amplified by polymerase chain reaction (PCR). Fragment size data for digested and undigested pair for each sample were collected from a capillary electrophoresis instrument to determine XCI pattern.
RESULTS
Clinical presentations of Hardikar syndrome patients
Patient 1
This previously unreported infant is the first child of nonconsanguineous, healthy parents. Pregnancy was complicated by prenatal ultrasounds concerning for unilateral cleft lip and palate and bilateral hydronephrosis. Chromosome and fluorescence in situ hybridization analysis of amniotic fluid cells revealed a normal female karyotype (46, XX) without evidence of 22q11.2 microdeletion. Patient was born at 38 weeks gestation with normal birth parameters. Postnatally, she was diagnosed with unilateral cleft lip and palate, bilateral hydronephrosis and left megaureter, aortic coarctation, and extrahepatic biliary atresia. Clinical examination was notable for a left-sided preauricular pit and a single, transverse palmar crease bilaterally (Fig. 1a). The patient underwent surgical correction of the aortic coarctation at 7 days of life and underwent Kasai portoenterostomy at 27 days of life. Development cannot be accurately assessed due to her young age.

(a) Patient 1; note prominent cleft lip. (b) Patient two; note ear pit. (c) Patient 3; note normal ear. (d) Patient 4 (reprinted with permission [Poley JR, Proud VK. Hardikar syndrome: new features. Am J Med Genet A. 2008;146A:2473–2479]); note repaired cleft lip. (e) Patient 5; note repaired cleft lip. (f) Patient 6; note repaired cleft lip. (g) Patient 7; note V-shaped lower eyelids and left ptosis.
Patient 2
This previously unreported patient presented at 14 months of age with left-sided cleft lip with a paranasal root pit, strabismus, retinal rarefaction, aortic coarctation, diaphragmatic hernia, choledochal cyst, congenital hepatic fibrosis, and severe left-sided hydronephrosis. Physical examination was notable for weight and height in the 50th percentile, head circumference in the 97th percentile, posteriorly rotated ears with underdeveloped helices, preauricular pits on the left side, and a duplicated tragus on the right side with pretragus depressions (Fig. 1b). Development was grossly normal. She was clinically diagnosed with HS at CENTRARE (Centro de Referência em fendas orofaciais, Fundação Benjamin Guimarães–Hospital da Baleia, Belo Horizonte, Brazil), and underwent exome sequencing at Laboratório de Genômica Clínica, Faculdade de Medicina da Universidade Federal de Minas Gerais (UFMG), Belo Horizonte. At 2 years and 8 months of age her development remains appropriate, though she did have early delays in motor development, including crawling at 12 months and walking at 18 months. Her retinal disease has not progressed, her strabismus is improving without surgery, and her vision remains normal.
Patient 3
This previously unreported patient presented at 21 months of age with unilateral cleft palate, a choledochal cyst with absent gall bladder, cloacal anomaly, aortic coarctation, scalp hemangioma, and infantile hypotonia that has now resolved. She had a normal ophthalmology examination at birth. Family history was significant for a maternal half-sister with congenital heart disease. Physical examination of this patient was notable for preauricular pits (Fig. 1c) and a grossly normal neurological examination. She was clinically diagnosed with HS at Seattle Children’s Hospital and referred to ophthalmology for repeat examination, which demonstrated pigmentary retinopathy and strabismus. At 6 years of age there has been no further progression of her retinal disease and her vision remains unaffected. There are no developmental concerns. She was referred for research exome sequencing at the Center for Applied Genomics (CAG) at Children’s Hospital of Philadelphia (CHOP).
Patient 4
This is a previously reported patient with a clinical diagnosis of HS.21,22,23 The patient was delivered at 33 weeks gestational age by Cesarean section. Postnatally, she was diagnosed with symmetric small for gestational age (weight and length at the 5th percentile), bilateral cleft lip and cleft palate, a temporary patent ductus arteriosus, bilateral hydronephrosis with bladder exstrophy, umbilical hernia, and intestinal malrotation (Fig. 1d). The cleft lip and palate were repaired when she was 5 months old, and a Ladd procedure was done to repair her intestinal malrotation. This patient had persistent direct hyperbilirubinemia and severe biliary tree malformations, including choledochal cysts, biliary tree dilation, and “biliary lakes” most prominent on the left side. She required a liver transplant at two years of age for progressive cholestasis and liver failure. At 4 years of age she was noted to have cat’s paw retinal pigmentation. Vision remains normal. At 14 years of age she was diagnosed with aortic coarctation as part of an evaluation for refractory hypertension. Coarctation had not been appreciated previously. Due to the association between aortic coarctation and cerebral aneurysms she underwent magnetic resonance angiograph (MRA) of the head and neck, which identified a carotid artery aneurysm, underwent surgical repair of her aortic coarctation, and her carotid aneurysm was being monitored. Development and cognition have been normal, including graduating from secondary school. She was clinically diagnosed with HS at King’s Daughters Children’s Hospital and enrolled in a research protocol at CAG at CHOP for research-based exome sequencing.
Patient 5
This patient is a previously reported patient with a clinical diagnosis of HS20,23. She presented at birth with symmetric growth restriction (weight 1%ile, height 8%ile, and head circumference 3%ile), cleft lip and palate, preauricular pits, convergent squint, cat’s paw pigmentary retinopathy, patent ductus arteriosus (PDA), and a ventricular septal defect (Fig. 1e). The PDA was repaired on the 7th day of life, and the cleft lip and palate were repaired at 7 months of age. She was additionally found to have low IGF-1 levels during an evaluation for hypopituitarism, and was started on growth hormone due to persistent poor growth. At 9 months of age, she was diagnosed with intestinal malrotation, obstructive liver disease, cholestasis, biliary atresia, and ectopic ureters ending in the urethra. Development and cognition were unaffected. She remained on growth hormone until 18 years of age. Adult height was 166 cm (75%ile). She was clinically diagnosed and reported previously through Universitair Ziekenhuis Leuven, but without molecular testing. Since initial report in the literature20,23, she had a massive, fatal, unprecipitated intracranial hemorrhage involving the frontal lobe with midline shift. Blood was also present in the basal ganglia and subarachnoid space. Computed tomography (CT) did not show any evidence of aneurysm; magnetic resonance imaging (MRI), MRA, and magnetic resonance venography (MRV) were not performed nor was autopsy. She was enrolled into a research study at CAG at CHOP for research-based exome sequencing.
Patient 6
This patient is a 4-year-old female born at 34 weeks’ gestational age. Family history is notable for an unaffected twin brother. Her postnatal course was notable for pigmentary retinopathy detected on ophthalmology examination shortly after birth, strabismus, cleft lip and palate, patent ductus arteriosus, bilateral ectopic ureters, absent gall bladder, cholestasis that self-resolved, Meckel diverticulum, and duodenal stenosis, which was surgically corrected at 6 days of life. Physical examination was notable for preauricular pits. (Fig. 1f) At last evaluation at 4 years of age she is developmentally appropriate. Her retinal findings have remained stable and her vision is unaffected. She was clinically diagnosed with HS and underwent research exome sequencing through the CAG and CHOP.
Patient 7
This patient presented at 19 months of age with a cleft palate, imperforate anus with a perirectal fistula, Stahl’s ear with malformed helices bilaterally, preauricular tags, and bilateral sensorineural hearing loss with cochlear implants (Fig. 1g). Pregnancy was complicated by ultrasound findings concerning for increased nuchal fold thickness and mild aortic dilation with a high arching aorta. Physical examination was notable for normal growth parameters (weight was in the 23rd percentile, height was in the 6th percentile, and her head circumference was in the 25th percentile), V-shaped lower eyelids, left ptosis, normal retina, clinodactyly, hypoplastic toe nails, 2,3 toe syndactyly, and sacral dimple. Postnatal echocardiogram confirmed the prenatal concern for mild aortic dilation. Her development was last assessed at 33 months of age and was normal, though prior to receiving cochlear implants her speech was delayed. She was clinically diagnosed with HS at CHOP, and underwent clinical exome sequencing.
Identification of de novo loss-of-function variants in MED12 in female patients with HS
Four individuals with clinically diagnosed Hardikar syndrome (HS) have been reported in the literature.19,20,22 We performed exome sequencing on two of these reported individuals (patients 4 and 5), and on one additional, previously unpublished patient with a clinical diagnosis of HS (patient 3). Despite multiple contact attempts, we were unable to obtain samples from the other two previously reported individuals with HS. Due to the previously proposed hypothesis that HS represented an X-linked, de novo condition,23 we systematically selected de novo candidate variants based on minor allele frequencies in population genomics resources (1000 Genomes Project, ESP6500SI, and gnomAD), and identified heterozygous, novel nonsense or frameshift variants in MED12 (GenBank: NM_005120.2) in these patients. Sanger sequencing of probands and parents confirmed a de novo variant in patient 3 (c.2663dup; p.[Leu889Profs*11]), a presumed de novo c.4903_4906delinsCCAGCA; p.(Val1635Profs*61) variant in patient 4 (absent in mother, father not available for testing, but reportedly phenotypically normal), and a c.5111G>A; p.(Trp1704*) variant in patient 5 (inheritance undetermined, as parental DNA was not available).
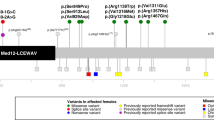
To validate these findings, we performed exome sequencing on three additional individuals with a clinical diagnosis of HS who were not previously reported in the literature, and identified three additional de novo nonsense and frameshift MED12 variants: c.322C>T; p.(Arg108*) (patient 1), c.2207_2210del; p.(Thr736Ilefs*43) (patient 2), and c.5622C>A; p.(Tyr1874*) (patient 6). Interestingly, the variant p.(Arg108*) was suspected to be of somatic origin with a mosaicism level of 15% in leukocytes-derived DNA. We also identified an additional, previously unreported patient with HS, found by clinical exome sequencing to have de novo nonsense variants in MED12 (patient 7: c.6169C>T; p.[Gln2057*]). Interestingly, all seven identified variants localize outside the penultimate exons (Fig. 2), and are predicted to be degraded by nonsense-mediated mRNA decay (NMD) by the exon junction complex (EJC)-dependent model and the 50-bp rule.24

The MED12 gene contains 45 exons and encodes a protein of 2177 amino acids. The MED12 domain (green) is encoded by exons 3–4 and includes amino acids 103–161, the LCEWAV domain (pink) is encoded by exons 7–17 and includes amino acids 286–757, and the PQL domain (blue) is encoded by exons 38–42 and includes amino acids 1616–2051. Pathogenic variants reported in females are indicated above the protein schematic; pathogenic variants reported in males are indicated below the protein schematic. Numbers refer to the accompanying table, which contains variant data and detailed patient phenotype. Black circles denote individuals with nonsyndromic intellectual disability, unfilled circles denote individuals with syndromic intellectual disability, gray squares denote individuals with Ohdo syndrome, blue squares denote individuals with Opitz–Kaveggia syndrome, red squares denote individuals with Lujan syndrome, and unfilled squares denote individuals with limited or prenatal phenotypes. Stars indicate our newly described patient cohort with Hardikar syndrome.
The MED12 gene is extremely intolerant to loss-of-function variation, with a maximum probability of loss-of-function intolerance (pLI) score of 1. We therefore hypothesized that HS patients would have skewed XCI pattern. XCI was analyzed based on the methylation pattern at the androgen receptor (AR) locus in six studied individuals (patients 1–6), four of which showed extremely skewed XCI (97:3–99:1) (Table 1). XCI pattern of patient 3 could not be determined due to lack of polymorphisms at the AR locus. The highest pLI score and the XCI pattern further support the pathogenicity of the nonsense and frameshift variants identified in our patient cohort.
DISCUSSION
Hardikar syndrome (HS) is characterized by the constellation of cleft lip and palate, pigmentary retinopathy, biliary tree anomalies, and intestinal malrotation, with preserved cognition.20,21,22 Despite several articles centered on the disorder, the molecular etiology has not yet been elucidated. Here we present seven female patients with HS found to have nonsense or frameshift variants in MED12 located on the X chromosome. This cohort, which includes five previously unreported patients, expands the clinical spectrum of HS and also suggests that nonsense or frameshift MED12 variants may be the cause of this condition.
MED12 encodes mediator complex subunit-12, a component of the core kinase module of the Mediator complex, which plays a critical role in transcriptional regulation.13,14 Pathogenic MED12 variants are associated with a variety of X-linked intellectual disability syndromes, including Opitz–Kaveggia syndrome, Ohdo syndrome, Lujan syndrome, as well as nonsyndromic intellectual disability25,26,27,28 (summarized in Supplementary Table 1 and Fig. 2). We propose that MED12 variants are additionally associated with a fifth, female-specific phenotype of HS. This is consistent with previous reports proposing X-linked dominant inheritance for HS23. Importantly, HS is the first MED12-associated condition with preserved neurodevelopment and distinct structural anomalies including cleft lip and palate, retinal disease, genitourinary abnormalities, and gastrointestinal malformations.
Previous studies have demonstrated a role for Med12 in developmental patterning, cellular growth, and cellular fate determination. Specifically, knockdown of Med12 in mouse embryonic stem cells led to dysregulated expression of lineage-specific differentiation markers similar to the perturbations seen with knockdown of the pluripotency factor Nanog.8 Forward genetics studies have identified a number of med12 zebrafish mutants whose phenotypes included impairments in neural crest cell formation, craniofacial chondrogenesis, neurogenesis, pancreatic and hepatic development, and heart formation.9,10,11 Moreover, Med12 deficiency in mice is embryonic lethal at E7.5.12 A hypomorphic Med12 variant was identified in mice and was associated with severe defects in neural tube closure and heart formation.12 Taken together, these studies emphasize the critical roles of Med12 in heart, gastrointestinal, and craniofacial development, recapitulating almost all of the characteristic presentations of MED12-associated syndromes, including HS.
Proposed mechanisms of pathogenesis in Hardikar syndrome
The mechanism by which MED12 nonsense or frameshift variants cause HS with preserved intellectual development is unknown. Importantly, the MED12 variants associated with intellectual disability are most frequently missense variants and are found more commonly in males. It is possible that the Hardikar phenotype is caused exclusively by nonsense or frameshift variants in MED12 and that these variant types are not viable in males. However, this is not supported by existing literature, which reports multiple affected females with intellectual disability and missense MED12 variants17,18,29,30 and males and females with frameshift or truncating causative MED12 variants who have severe neurodevelopmental phenotypes, but lack the anomalies associated with HS.15,31,32 It is possible that the nonsense and frameshift variants reported in males and females with severe intellectual disability without HS features are functionally different than those associated with HS.
Another potential explanation relates to XCI. It is possible that females with unfavorably skewed X-inactivation display intellectual disability phenotypes, whereas those with favorably skewed X-inactivation have HS. This is contested by a report of multiple females in which XCI pattern did not predict disease severity.17 This phenomenon is also observed in other X-linked disorders, such as Rett syndrome.33 Importantly, most XCI studies, including ours, are performed on DNA derived from peripheral blood, which does not necessarily reflect XCI patterns across all tissues.15,16,17,18 Indeed, it is widely appreciated that XCI is heterogeneous among tissues with fine spatial and cell type–specific regulation.34 It is therefore still possible that XCI patterns in different tissues dictate the distinct clinical phenotypes of MED12-associated syndromes in females.
An alternate explanation is the destruction of MED12 transcripts via NMD. It is possible that MED12 variants associated with HS undergo NMD, precluding defective protein production, whereas MED12 variants associated with intellectual disability maintain expression of the mutant MED12 allele in brain. A tantalizing hypothesis is that MED12 plays a critical and dose-dependent role in tubulogenesis during embryology, controlling development of the aorta, biliary tree, gastrointestinal tract, and genitourinary tract that can still be performed by MED12 protein with missense variants. In contrast, MED12 haploinsufficiency as a result of a nonsense or frameshift variant that undergoes NMD would preclude proper visceral development, producing the classic phenotypes of HS. The critical role of MED12 in visceral development is supported by animal models of homozygous med12 deficiency, which demonstrate impaired gastrulation, as well as impaired formation of the gastrointestinal tract and endoderm.10,12
In contrast, the neuropathology caused by MED12 missense variants may reflect inappropriate transcriptional pathway activation from variant-bearing MED12 protein expression. Such a model would predict preserved cognition in individuals with HS, as there would be no expression from the variant-bearing allele, due to either XCI or NMD or both, to interfere with proper neurodevelopment, in contrast to what may occur with missense variants with preserved expression. Signaling pathway dysregulation with different MED12 variants has been described previously.35,36
This combined model may explain the male and female individuals reported in the literature with nonsense and frameshift MED12 variants with severe intellectual disability without the systemic features of HS.15,30,31 Specifically, the frameshift and nonsense variants reported in males and females with intellectual disability may escape NMD or be excluded in certain splicing isoforms, allowing for preservation of MED12 expression and appropriate tubulogenesis, but also intellectual disability due to the presence of mutant MED12 protein in the brain and aberrant gene expression and cellular signaling. Further supporting this model, in a family with multiple male and female probands with profound intellectual disability due to a pathogenic frameshift MED12 variant, in vitro studies have demonstrated production of two unique MED12 isoforms in the two males with no evidence of NMD and no correlation between XCI and neurodevelopment in females.15 This suggests that this frameshift MED12 variant associated with intellectual disability is not subject to NMD, as we hypothesize for the HS-causing variants. Further work is required to determine whether NMD and/or skewed XCI are at play to establish the true molecular mechanism of the distinct MED12-associated HS phenotype.
Clinical considerations of Hardikar syndrome
HS has phenotypic overlaps with a variety of other well-known syndromes, most notably CHARGE syndrome (MIM 214800), Alagille syndrome (MIM 118450 and 610205), 22q11.2 deletion syndrome (MIM 611867), and Kabuki syndrome (MIM 147920 and 300867). CHARGE syndrome is an autosomal dominant disorder caused by variants in CHD7, and is characterized by colobomas, heart anomalies, choanal atresia, retardation of growth and development, genitourinary abnormalities, and ear differences. It shares the clefting and cardiac differences seen in HS, but unlike HS, biliary and gastrointestinal tract anomalies are uncommon and developmental delay is seen in over 60% of affected individuals. Alagille syndrome is an autosomal dominant condition caused by variants in either JAG1 or its receptor NOTCH2. It is characterized by bile duct paucity, congenital heart disease (usually pulmonary artery stenosis), butterfly vertebrae, posterior embryotoxon, and a triangular face. Like HS, the patients’ development and cognition are usually unaffected; however, unlike HS, neither facial clefting nor retinal issues are associated with Alagille syndrome. The 22q11.2 deletion syndrome should be considered in the differential of HS, due to the shared features of facial clefting, congenital heart disease, and renal anomalies; however, biliary anomalies are rare. Kabuki syndrome is an autosomal dominant or X-linked dominant condition caused by pathogenic variants in KMT2D and KDM6A, respectively. Kabuki syndrome and HS share many features, including congenital heart disease, genitourinary differences, and biliary disease, though they are unique in many ways including distinct facial features in Kabuki syndrome. There is sufficient overlap between HS and Kabuki syndrome that they were previously postulated to represent a phenotypic spectrum of the same disease process.23,37 Here we demonstrate that HS truly is a distinct syndrome with a distinct genetic basis. Interestingly, the causal genes for Kabuki syndrome also play a role in transcriptional regulation, like MED12. Indeed, MED12, KDM6A, and KMT2D exist in a shared transcriptional network.38,39 The overlapping phenotype of disorders of transcriptional regulation has been well described, most notably for Kabuki syndrome and CHARGE syndrome.40,41 We propose a similar mechanism for the similarities between HS and Kabuki syndrome.
Distinguishing HS from its differential is critical, and we anticipate that the majority of patients with HS will be diagnosed either clinically due to the specificity of the clinical findings, or by the identification of a loss-of-function MED12 variant in an affected female. Our patient cohort emphasizes additional key features of HS including ptosis, preauricular skin tags and pits, external ear malformations, hearing loss, and aortopathy, most commonly aortic coarctation. Blood vessel abnormalities may extend beyond the aorta, as evidenced by the carotid aneurysm identified in patient 4 and the suspected cerebral aneurysm in patient 5. Based on this critical discovery and the novel phenotypes found in our patient cohort, we propose that the standard of care for individuals with HS now include (1) careful examination for evidence of cleft lip and palate and velopharyngeal insufficiency, (2) serial ophthalmology examinations to assess for retinal differences, (3) echocardiogram at diagnosis to evaluate for congenital heart disease and aortic coarctation, (4) annual echocardiograms and carotid ultrasound to assess for development of aortic and carotid dilation, (5) evaluation for liver disease, (6) evaluation for intestinal malrotation, (7) evaluation for genitourinary abnormalities and vesicoureteral reflux, (8) formal audiology evaluation, and (9) MRA of the head and neck at diagnosis to evaluate for vascular malformations and biannually thereafter to scan for development of cerebral aneurysms.
Conclusion
In this study, we report five additional females with Hardikar syndrome, bringing the total in the literature to nine, and identify MED12 nonsense and frameshift variants in seven affected individuals. In addition, all five patients with informative results showed skewed X-inactivation patterns. Limitations of the study include the lack of confirmed de novo inheritance in two patients, and the inability to test two other previously reported HS patients. Despite these limitations, we propose that HS is a unique, female-limited multiple congenital anomaly syndrome caused by loss-of-function variants in MED12. Advances in our understanding of the phenotypic spectrum of HS and the pathophysiology of disease is of immeasurable importance to facilitate counseling of families with MED12 variants and to allow for preventive care for the vascular and other modifiable risks associated with this disorder.
Data availability
All variants have been deposited into ClinVar, VCV000432691, VCV000280361, VCV000620451, VCV000520705.
References
Bourbon, H. M. et al. A unified nomenclature for protein subunits of mediator complexes linking transcriptional regulators to RNA polymerase II. Mol. Cell 14, 553–557 (2004).
Harper, T. M. & Taatjes, D. J. The complex structure and function of mediator. J. Biol. Chem. 293, 13778–13785 (2018).
Fant, C. B. & Taatjes, D. J. Regulatory functions of the mediator kinases CDK8 and CDK19. Transcription 10, 76–90 (2019).
Graham, J. M. & Schwartz, C. E. MED12 related disorders. Am. J. Med. Genet. A 161, 2734–2740 (2013).
Allen, B. L. & Taatjes, D. J. The mediator complex: a central integrator of transcription. Nat. Rev. Mol. Cell Biol. 16, 155–166 (2015).
Hsieh, T. H. S. et al. Mapping nucleosome resolution chromosome folding in yeast by micro-C. Cell 162, 108–119 (2015).
Schneider, M. et al. The nuclear pore-associated TREX-2 complex employs mediator to regulate gene expression. Cell 162, 1016–1028 (2015).
Tutter, A. V. et al. Role for Med12 in regulation of Nanog and Nanog target genes. J. Biol. Chem 284, 3709–3718 (2009).
Hong, S. K. et al. The zebrafish kohtalo/trap230 gene is required for the development of the brain, neural crest, and pronephric kidney. Proc. Natl Acad. Sci. USA 102, 18473–18478 (2005).
Shin, C. H. et al. Multiple roles for Med12 in vertebrate endoderm development. Dev. Biol. 317, 467–479 (2008).
Wu, S. Y., de Borsetti, N. H., Bain, E. J., Bulow, C. R. & Gamse, J. T. Mediator subunit 12 coordinates intrinsic and extrinsic control of epithalamic development. Dev. Biol. 385, 13–22 (2014).
Rocha, P. P., Scholze, M., Bleiß, W. & Schrewe, H. Med12 is essential for early mouse development and for canonical Wnt and Wnt/PCP signaling. Development 137, 2723–2731 (2010).
Yin, J. W. & Wang, G. The mediator complex: a master coordinator of transcription and cell lineage development. Development 141, 977–987 (2014).
Kim, S., Xu, X., Hecht, A. & Boyer, T. G. Mediator is a transducer of Wnt/β-catenin signaling. J. Biol. Chem. 281, 14066–14075 (2006).
Lesca, G. et al. Clinical and neurocognitive characterization of a family with a novel MED12 gene frameshift mutation. Am. J. Med. Genet. A 161, 3063–3071 (2013).
Bouazzi, H., Lesca, G., Trujillo, C., Alwasiyah, M. K. & Munnich, A. Nonsyndromic X-linked intellectual deficiency in three brothers with a novel MED12 missense mutation [c.5922G>T (p.Glu1974His)]. Clin. Case Reports 3, 604–609 (2015).
Charzewska, A. et al. The power of the mediator complex-expanding the genetic architecture and phenotypic spectrum of MED12 -related disorders. Clin. Genet. 94, 450–456 (2018).
Prontera, P. et al. A novel MED12 mutation: evidence for a fourth phenotype. Am. J. Med. Genet. A 170, 2377–2382 (2016).
Hardikar, W., Smith, A. L., Keith, C. G. & Chow, C. W. Multisystem obstruction with cholestasis, pigmentary retinopathy, and cleft palate: a new syndrome? Am. J. Med. Genet. 44, 13–17 (1992).
Cools, F. & Jaeken, J. Hardikar syndrome: A new syndrome with cleft lip/palate, pigmentary retinopathy and cholestasis. Am. J. Med. Genet. 71, 472–474 (1997).
Poley, J. R. & Proud, V. K. Hardikar syndrome: new features. Am. J. Med. Genet. A 146, 2473–2479 (2008).
Ryan, K. M. et al. Aortic coarctation and carotid artery aneurysm in a patient with hardikar syndrome: cardiovascular implications for affected individuals. Am. J. Med. Genet. A 170, 482–486 (2016).
Nydegger, A., Van Dyck, M., Fisher, R. A., Jaeken, J. & Hardikar, W. Hardikar syndrome: long term outcome of a rare genetic disorder. Am. J. Med. Genet. A 146, 2468–2472 (2008).
Kurosaki, T., Popp, M. W. & Maquat, L. E. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat. Rev. Mol. Cell. Biol. 20, 406–420 (2019).
Schwartz, C. E. et al. The original Lujan syndrome family has a novel missense mutation (p.N1007S) in the MED12 gene. J. Med. Genet. 44, 472–477 (2007).
Clark, R. D. et al. FG syndrome, an X-linked multiple congenital anomaly syndrome: the clinical phenotype and an algorithm for diagnostic testing. Genet. Med. 11, 769–775 (2009).
Rump, P. et al. A novel mutation in MED12 causes FG syndrome (Opitz-Kaveggia syndrome). Clin. Genet. 79, 183–188 (2011).
Lyons MJ. MED12-Related Disorders. 2008 Jun 23 [updated 2016 Aug 11]. In GeneReviews® [Internet]. (eds Adam, M. P. et al.) (Seattle (WA): University of Washington, Seattle, 1993–2020).
Murakami, H., Enomoto, Y., Tsurusaki, Y., Sugio, Y. & Kurosawa, K. A female patient with X‐linked Ohdo syndrome of the Maat‐Kievit‐Brunner phenotype caused by a novel variant of MED12. Congenit. Anom. 60, 91–93 (2019).
Fieremans, N. et al. Identification of Intellectual disability genes in female patients with a skewed X-inactivation pattern. Hum. Mutat. 37, 804–811 (2016).
Wang, C. et al. MED12-related disease in a chinese girl: clinical characteristics and underlying mechanism. Front Genet 11, 129 (2020).
Kosmicki, J. A. et al. Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nat. Genet. 49, 504–510 (2017).
Hoffbuhr, K. et al. MeCP2 mutations in children with and without the phenotype of Rett syndrome. Neurology 56, 1486–1495 (2001).
Zito, A. et al. Heritability of skewed X-inactivation in female twins is tissue-specific and associated with age. Nat. Commun. 10, 5339 (2019).
Donnio, L. M. et al. MED12-related XLID disorders are dose-dependent of immediate early genes (IEGs) expression. Hum. Mol. Genet. 26, 2062–2075 (2017).
Srivastava, S. et al. Dysregulations of sonic hedgehog signaling in MED12‐related X‐linked intellectual disability disorders. Mol. Genet. Genomic Med. 7, e00569 (2019).
Ejarque, I. et al. Is Hardikar syndrome distinct from Kabuki (Niikawa—Kuroki) syndrome? Clin. Genet. 80, 493–496 (2011).
Cheng, D. et al. CARM1 methylates MED12 to regulate its RNA-binding ability. Life Sci. Alliance 1, e201800117 (2018).
Aranda-Orgilles, B. et al. MED12 regulates HSC-specific enhancers independently of mediator kinase activity to control hematopoiesis. Cell Stem Cell 19, 784–799 (2016).
Verhagen, J. M. A., Oostdijk, W., Terwisscha van Scheltinga, C. E. J., Schalij-Delfos, N. E. & van Bever, Y. An unusual presentation of Kabuki syndrome: clinical overlap with CHARGE syndrome. Eur. J. Med. Genet. 57, 510–512 (2014).
Schulz Y. et al. CHARGE and Kabuki syndromes: a phenotypic and molecular link. Hum. Mol. Genet. 23, 4396–4405 (2014).
Acknowledgements
We thank all the patients and families who participated in this study. There are no funders to acknowledge.
Author information
Authors and Affiliations
Contributions
Conceptualization: E.B., D.L., H.H.; Data curation: D.L; Formal Analysis: D.L; Investigation: R.L., D.C., M.V., N.D.L., E.R.V, T.W., S.P., J.J., S.V., E.Z., A.H., P.C., A.G., T.S., T.B., D.P.; Resources: H.H.; Visualization: A.S, D.L; Writing – original draft: A.S.,K.S., D.L; Writing – review & editing: E.B., H.H.
Corresponding author
Ethics declarations
COMPETING INTERESTS
The authors declare no competing interests.
Ethics Declaration
All individuals’ families from the different institutions agreed to participate in this study and signed appropriate consent forms. The Institutional Review Board of the Children’s Hospital of Philadelphia approved this study. Permission for clinical photographs was given separately.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Li, D., Strong, A., Shen, K.M. et al. De novo loss-of-function variants in X-linked MED12 are associated with Hardikar syndrome in females. Genet Med 23, 637–644 (2021). https://doi.org/10.1038/s41436-020-01031-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-01031-7
This article is cited by
-
Skewed X-chromosome inactivation in unsolved neurodevelopmental disease cases can guide re-evaluation For X-linked genes
European Journal of Human Genetics (2023)
