
Abstract
Understanding causes of infant mortality shapes public health policy and prioritizes diseases for investments in surveillance, intervention and medical research. Rapid genomic sequencing has created a novel opportunity to decrease infant mortality associated with treatable genetic diseases. Herein, we sought to measure the contribution of genetic diseases to mortality among infants by secondary analysis of babies enrolled in two clinical studies and a systematic literature review. Among 312 infants who had been admitted to an ICU at Rady Children’s Hospital between November 2015 and September 2018 and received rapid genomic sequencing, 30 (10%) died in infancy. Ten (33%) of the infants who died were diagnosed with 11 genetic diseases. The San Diego Study of Outcomes in Mothers and Infants platform identified differences between in-hospital and out-of-hospital causes of infant death. Similarly, in six published studies, 195 (21%) of 918 infant deaths were associated with genetic diseases by genomic sequencing. In 195 infant deaths associated with genetic diseases, locus heterogeneity was 70%. Treatment guidelines existed for 70% of the genetic diseases diagnosed, suggesting that rapid genomic sequencing has substantial potential to decrease infant mortality among infants in ICUs. Further studies are needed in larger, comprehensive, unbiased patient sets to determine the generalizability of these findings.
Similar content being viewed by others

Introduction
Infant mortality has been tracked annually worldwide for many years (http://www.geoba.se/population.php?pc=world&type=019&year=2017&st=rank&asde=d&page=1, https://data.worldbank.org/indicator/SP.DYN.IMRT.IN?locations=US). Knowledge of causes of infant mortality shapes public health policy and prioritizes diseases for investments in surveillance, intervention and medical research. As a result of such prioritization, programs such as Back to Sleep/Safe to Sleep® have significantly reduced rates of many causes of infant mortality over the past 50 years in the United States (https://safetosleep.nichd.nih.gov/, https://www.cdc.gov/reproductivehealth/maternalinfanthealth/infantmortality.htm.)1,2,3,4. However, knowledge of the underlying causes of infant mortality is based on death certificates, which are estimated to be inaccurate or incomplete in 33–53% of cases5,6,7,8,9,10. In addition, the vast majority of death certificates are not informed by molecular diagnoses.
Congenital malformations, deformations and chromosomal abnormalities (International Classification of Diseases (ICD-10) codes Q00–Q99) have been reported to be the leading causes of infant death in the United States for the past 50 years1,2,3,4. Many, but not all, of these are associated with genetic diseases. Several of the other nine leading causes of infant mortality are also associated with genetic diseases, including low birth weight and prematurity, sudden infant death syndrome, newborn sepsis, diseases of the circulatory system and neonatal hemorrhage1,2,3,4. Molecular autopsies have transformed the forensic evaluation of sudden unexplained death in the young, leading to new guidelines and treatments and a decreased number of deaths9,10. Despite this success, and while the molecular basis of 14,000 genetic diseases has been determined11,12, it is not known which genetic diseases are leading contributors to infant mortality other than for sudden unexplained death and chromosomal aneuploidies.
Knowledge of the contribution of genetic diseases to infant mortality has grown indirectly. Many infant deaths occur in hospitals, particularly in neonatal, pediatric and cardiovascular intensive care units (ICUs)13,14. Rapid genomic or precision medicine has created a new opportunity to decrease infant mortality associated with genetic diseases in ICUs15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45. Many genetic diseases have specific, effective, evidence-based treatments. However, when diagnosis of a genetic disease is delayed or absent, treatments must be chosen empirically, with suboptimal outcomes, including unnecessary infant mortality. In response, rapid and ultra-rapid clinical genomic sequencing methods have been developed for prompt diagnosis in infants and children in ICUs who may have a genetic disorder for which effective treatment exists15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45. It is estimated that genetic diseases occur in about 16% of infants in regional ICUs24,46. Over the past 5 years, rapid genomic sequencing has demonstrated consistent utility in this population. In 873 children in ICUs described in 13 studies, 36% received a genetic disease diagnosis by rapid genomic sequencing17,20,21,23,24,25,26,28,30,33,34,36,38,39,40,41,42,43,44,45. The average clinical or therapeutic utility of those diagnoses was 71%. Changes in patient outcomes were reported in 27%. The latter included both avoidance of infant mortality by therapeutic interventions and palliative care decisions. These data suggest that clinical-grade genomic sequencing has the analytic and diagnostic performance to ascertain the contribution of genetic diseases to infant mortality. Furthermore, determination of the leading genetic diseases associated with infant mortality may provide information to direct clinical and public health interventions to reduce infant mortality and decrease suffering. The current manuscript reports results of a scoping study and literature review to assess whether genetic diseases in toto are a leading cause of infant mortality.
Results
Cohort Studies
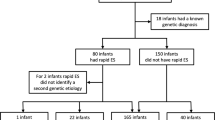
Among 312 probands who had received rapid genomic sequencing in two research datasets, we examined the characteristics of infants who died prior to their first birthday. Both studies enrolled inpatient infants at Rady Children’s Hospital in San Diego between 11/23/2015 and 9/7/2018, primarily from a level IV neonatal intensive care unit. Of these, 205 infants were enrolled in the NSIGHT2 randomized controlled trial24 and 107 infants were enrolled under a biorepository protocol17,18,23,30. All infants had an acute illness at the time of enrollment, with a disease of unknown, possibly genetic, etiology. At one year follow up, 30 (9.5%) of 312 enrollees had died in infancy. They represented approximately 7% of infant deaths in San Diego County during this period (https://www.sandiegocounty.gov/content/dam/sdc/hhsa/programs/phs/documents/MchSt-InfMort.pdf).
We compared the demographic characteristics of infants who died with those who survived in the two cohorts, and with those of all infant deaths in San Diego County during the same period. Among participants in the two research cohorts, infant mortality did not differ significantly between males (9.5%; 16 of 169) and females (12.3%; 14 of 114; P = 0.45). In contrast, in San Diego County from 2015–2019, infant mortality was lower among females (0.35%, 282 of 81,612) than males (0.42%, 359 of 85,710, p < 0.02) (https://www.sandiegocounty.gov/content/dam/sdc/hhsa/programs/phs/documents/MchSt-InfMort.pdf). Excluding 23 infants of unknown race, mortality in the two studies did not differ significantly between white infants (10.3%; 19 of 185 infants) and those of other races (9.6%; 10 of 104, P = 0.86). In contrast, in San Diego County from 2015–2019, white infants had a lower mortality (0.34%, 199 of 58,224) than non-white infants (0.41%, 444 of 109,100; p < 0.05) (https://www.sandiegocounty.gov/content/dam/sdc/hhsa/programs/phs/documents/MchSt-InfMort.pdf). Excluding 8 infants of unknown ethnicity, mortality did not differ significantly between Hispanic infants (11.7%; 16 of 137 infants) and non-Hispanic infants (8.4%; 14 of 167; P = 0.34) in the two studies. In San Diego County from 2015–2019, Hispanic infants had a higher mortality rate (0.45%, 304 of 66,928) than non-Hispanic infants (0.34%, 339 of 100,396; P < 0.0002) (https://www.sandiegocounty.gov/content/dam/sdc/hhsa/programs/phs/documents/MchSt-InfMort.pdf).
Among the 30 infants in the research studies who died, we evaluated whether the likelihood of death had been anticipated or palliative care discussed with parents by EHR review. Eleven (37%) of 30 deceased infants had received a palliative care consult, and 18 (60%) of 30 deceased infants had a modified “Code Blue” resuscitation status in the event of cardiorespiratory arrest. Of these, 16 (53%) had “allow natural death” as their Code Blue status at time of death and 2 (7%) had a partial resuscitation Code Blue status. Of the infants who had received a genetic diagnosis, 4 (40%) out of 10 had a palliative care consult, and 7 (70%) out of 10 had a modified “Code Blue” resuscitation status. Of the 20 infants who did not receive a genetic diagnosis, 7 (35%) had a palliative care consultation and 11 (55%) had a modified “Code Blue” status.
We examined phenotypic characteristics of the infants who died and survived in the two studies. Using methods that had previously been validated for identification of patients with orphan diseases17, we extracted HPO terms corresponding to the clinical features of the 312 infants from unstructured text in the Rady Children’s Hospital EHR at time of enrollment (prior to death) using clinical natural language processing. Of 2,313 HPO terms extracted from Epic only four terms differed in frequency significantly between enrollees who died in infancy and those who survived (Table 1)—abnormal urinary system, respiratory failure, hypotension and cardiac arrest. All occurred in a higher proportion of records of infants who died.
We examined phenotypic characteristics of all infants who died in San Diego County during the same period, and compared in-hospital deaths with out-of-hospital deaths. Using the SOMI platform47, we identified ICD-10 (International Statistical Classification of Diseases and Related Health Problems, 10th Revision) codes in 1,179 infant deaths in San Diego County between 2005 and 2011 (Table 2). Four hundred and thirty-five (37%) infant deaths occurred in a hospital. Among 656 infant deaths, associated with an ICD10 code, the most frequent conditions were gestation <32 weeks (46%), perinatal conditions (45%), congenital malformations and chromosomal anomalies (29%), and sudden infant death syndrome (13%). In-hospital compared to out-of-hospital deaths were enriched for perinatal conditions (63% vs 2.7%; p-value < 0.001), congenital malformations (34% vs 5.9%; p-value: < 0.001), and circulatory system diseases (3% vs 0.4%; p-value: < 0.001). Sudden Infant Death Syndrome was under-represented in-hospital deaths (1.8% vs 9.9%; p-value < 0.001).
In the two RCHSD studies, 81 (26%) of the 312 infants were diagnosed with a genetic disease by rapid genomic sequencing. Of the 30 infants who died, 10 (33%) were diagnosed with 11 genetic diseases (Table 3). The mortality rate, although higher among those with genetic diagnoses, did not differ significantly between infants with or without genetic diseases (12.3%; 10 of 81 versus 8.7%; 20 of 231; P = 0.85), respectively. Nor did the average age at time of death differ significantly between infants with or without genetic diseases (104 days versus 89 days, P = 0.72). All of the genetic diseases diagnosed in infants who died had previously been associated with infant or childhood mortality, with the exception of Chr 12q21.33q22 del, which has only been described in six children48,49,50,51,52,53,54,55,56,57,58,59,60,61,62. In addition, the symptoms and signs observed in these infants were consistent with causal association of genetic diseases and infant death (Table 3). No genetic disease was recurrent. Of the genetic diseases identified in infants who died, only CHARGE syndrome was diagnosed in at least one of the 282 infants who survived.
Systematic literature review
To evaluate whether the findings reported herein were generalizable, we searched the literature for genomic sequencing studies in acutely ill infants in which mortality was recorded. Six additional studies were identified that utilized current genetic disease databases and current variant detection and interpretation methods (Table 4)25,28,45,63,64,65,66. Three studies had similar experimental designs as herein–diagnostic genomic sequencing was performed in infant inpatients in United States hospitals who had acute diseases of unknown etiology and in whom genetic diseases were suspected25,28,45. In two studies, genome sequencing was performed post mortem63,64. One study had both ante and post mortem genomic sequencing65. While all included inpatient and community deaths, the inclusion criteria differed in each study (Table 4). In toto, the studies examined 918 infant deaths. The overall contribution of genetic disease to infant mortality was 21% (weighted average, median 31%, range 6 to 86%). Locus heterogeneity (number of affected loci divided by number of infant deaths) was 70% (137 genetic diseases in 195 infant deaths). The most common genetic causes of death were trisomy 21 (9%), spinal muscular atrophy (5%), 22q11 deletion syndrome (4%), trisomy 18 (4%), CHARGE syndrome (3%), and trisomy 13 (2%). It should be noted that most studies excluded infants with known chromosomal anomalies and those who died prior to enrollment, resulting in under-reporting of these. In two of the studies, a further 8% of infant deaths were associated with variants of uncertain significance (VUS) in established disease loci63,64. Infant mortality attributable to genetic diseases was not restricted to congenital malformations, nor were all infant deaths associated with congenital malformations attributable to genetic diseases. A literature review identified treatment guidelines for 70% of the genetic diseases diagnosed in the 195 infant deaths. These include case series and case reports, as standard of care practices have yet to be fully established for many exceedingly rare genetic conditions. Furthermore, effective, standard of care treatments were available for 18% (Table 5). Many of these treatments are aimed at surveillance and management of symptoms, although some of them are curative, such as bone marrow transplant in severe cases of autoimmune lymphoproliferative disorder67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97. Remaining treatments were supportive or ameliorative. Additionally, genetic evaluation of family members is recommended for all the conditions summarized in Table 5, in particular for autosomal dominant conditions like Alagille Syndrome. Some patients may not be suitable for treatment, and in other cases treatments may not prove effective, in which case palliative and supportive care may be offered. The parents of 31 (35%) of 88 infants who received diagnoses of untreatable genetic diseases before death in our cohort elected palliative care as a result.
Discussion
Genomic sequencing has greatly increased the number of infants in ICUs who are diagnosed with genetic diseases and holds promise to decrease infant mortality in this setting15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45. Little, however, has yet been published with regard to the contribution of genetic diseases to infant mortality using this technology. In six published studies and the cohort reported herein a weighted average of 21% of infant deaths was attributable to single locus genetic diseases25,28,45,63,64,65. However, the range of attribution varied widely between studies (6%-86%), reflecting different experimental designs and inclusion and exclusion criteria. This estimate excluded VUS in disease genes known to be associated with infant mortality. In two studies, the contribution of genetic disease to infant mortality increased 8% upon inclusion of VUS63,64. These data also excluded genes of uncertain significance that are known to be loss of function intolerant or associated with perinatal death in animal models. While the observed between-study heterogeneity precludes a precise estimate the contribution of single locus genetic disease to infant mortality, these data suggest that it is considerable. This is important since it informs estimates of potential reduction in infant mortality achievable by genomic medicine and infant suffering through genome-informed palliative care.
All but two of the studies reviewed herein evaluated mortality in infants who had been admitted to a regional or level IV neonatal, pediatric and cardiovascular ICU during the first year of life. Since only ~4% of infants are admitted to regional ICUs, there is concern that findings from these studies may not be generalizable. This concern was offset in part by two observations. Firstly, 37% of infant mortality in San Diego County occurred in hospitals (https://data.chhs.ca.gov/dataset/licensed-bed-classification-and-designations-trends). Secondly, two studies that were not restricted to infants who had been in an ICU had the same weighted average (22%) of infant mortality attributable to genetic diseases as the others. From a public health standpoint, however, it would be of greater value to know the proportion of all infant mortality that is associated with genetic diseases. Many of the most common genetic diseases that contribute to infant mortality, including trisomies 21, 18, and 13 (15% of total deaths) and 22q11 deletions (4% of total deaths), are well-characterized and are often detectable prenatally using maternal cell-free DNA screening. Rapid, accurate identification of these more common genetic conditions in the prenatal period allows for more targeted use of genomic sequencing and more efficient use of this technology for patients with a possible genetic etiology of their illness but without a prenatal diagnosis.
Comparison of causes of in-hospital and out-of-hospital infant mortality in San Diego County revealed significant differences. Mortality associated with perinatal conditions, congenital malformations, and circulatory system diseases was greater in hospitalized infants. Significantly more sudden infant death syndrome occurred in out-of-hospital deaths. Thus the incidence of specific genetic diseases associated with infant deaths in hospital are likely to differ from those that occur out-of-hospital.
More than 14,000 single locus genetic diseases have been described11,12, and locus heterogeneity is high among infants diagnosed with genetic diseases. In a cohort of 504 patients diagnosed with genetic diseases by exome sequencing, for example, locus heterogeneity was 72%98. A striking finding herein was that locus heterogeneity was similar among infant deaths (70% in 195 infant deaths). Thus, strong negative selection occurs broadly across the genome in human populations. From a practical standpoint, locus heterogeneity implies that effective molecular diagnosis in infants requires comprehensive genomic sequencing, rather than panel tests.
A surprising finding was that published treatment guidelines existed for 70% of infant deaths associated with genetic diseases. Although some patients may not be suitable for treatment even if guidelines exist, it is possible that if substantiated in a larger, unbiased cohort this would imply that there exists a considerable opportunity to decrease infant mortality and morbidity associated with genetic diseases by early neonatal diagnosis. It should be noted that it is now possible to diagnose a genetic disease in 19 h by ultra-rapid whole genome sequencing17, albeit such testing is not yet reimbursed by most healthcare payers. There is now a growing literature of infants whose lives have been saved by early diagnosis of genetic diseases and timely implementation of targeted treatments15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45.
Another interesting finding herein was that palliative care counseling was provided to 40% of parents of infants who died of genetic diseases. Palliative care counseling was often associated with changes in parental decisions with regard to treatment intensity, such as modified “Code Blue” resuscitation status in the event of cardiorespiratory arrest. If substantiated in a larger, unbiased cohort this would imply that there exists a potential opportunity to decrease suffering in infants with genetic diseases that have fatal prognosis by early diagnosis, and to facilitate a healthy grieving process among parents of deceased infants99.
In summary, this study demonstrates that it is possible to identify the contribution of genetic diseases to infant mortality by genomic sequencing. Larger studies using the methods described herein have potential for molecular redefinition of the leading causes of infant death. By analysis of the specific diseases that cause infant mortality it is possible to identify primary and secondary strategies that have the potential to reduce infant mortality and decrease suffering. There was notable heterogeneity in the studies included here, particularly regarding inclusion criteria, geography and initial selection of the cohort. Future studies are therefore warranted that examine large, sequential cohorts of individuals from a single geography over several years in order to more accurately quantify the contribution of genetic disease to overall infant mortality. A larger study could also provide an estimate of the potential impact of genomic medicine on reducing infant mortality. Finally, this study has demonstrated the potential for the SOMI platform to determine whether spatial, environmental, sociodemographic and health-related factors differ among those with and without a known genetic contribution to infant mortality. As with previous population studies of San Diego County, many of the results will have generalizable implications100,101,102,103.
Methods
Cohort studies
We examined infant mortality among 312 children who had received rapid genomic sequencing in two research datasets. The first was NSIGHT2, a prospective, blinded, randomized controlled trial in infants in the neonatal, pediatric and cardiovascular ICUs at Rady Children’s Hospital, San Diego (RCHSD). NSIGHT2 compared the effectiveness and outcomes of rapid whole genome sequencing, rapid whole exome sequencing and ultra-rapid whole genome sequencing (ClinicalTrials.gov NCT03211039, registered July 7, 2017)24. NSIGHT2 was approved by the institutional review board (IRB) at RCHSD, was designated non-significant risk by the Food and Drug Administration (FDA) in an Investigational Device Exemption presubmission, and was performed in accordance with the Declaration of Helsinki. Informed consent was obtained from at least one parent or guardian for each infant. The inclusion criteria were age <4 months and time from admission or time from development of a feature suggestive of a genetic condition of <96 h. The clinical inclusion criteria were broad in order to include all acutely ill infants with diseases of unknown etiology, as well as those with presentations highly suspicious for a genetic cause. Infants in whom there was a very low likelihood that a genetic disease diagnosis would change management were excluded. Specific exclusion criteria included infection or sepsis with normal response to therapy, isolated prematurity, and previously confirmed genetic diagnosis that explained their clinical condition. Infants with a confirmed molecular diagnosis of certain common genetic conditions that are detected by prenatal screening were therefore exlcuded. Full inclusion and exclusion criteria have been previously published24. Genome interpretation was performed as singleton probands. Infants undiagnosed as singletons were re-analyzed as familial trios.
The second dataset was from a biorepository cohort17,18,23,30. Inpatient infants at RCHSD without etiologic diagnoses, in whom a genetic disorder was possible, were enrolled between 11/23/2015 and 9/7/2018. Retrospective comparison of outcomes and healthcare utilization was approved by the IRB at RCHSD and the FDA (ClinicalTrials.gov NCT02917460, registered September 28, 2016). Informed consent was obtained from at least one parent or guardian. Inclusion criteria were symptomatic inpatients less than 1 year of age who were affected by an illness potentially attributable to a genetic disorder. There were no specific exclusion criteria. All patients enrolled underwent whole genome sequencing.
Clinical, rapid whole genome and exome sequencing, analysis and interpretation
Clinical rapid whole genome sequencing, rapid whole exome sequencing and ultra-rapid whole genome sequencing were performed in laboratories accredited by the College of American Pathologists and certified through the Clinical Laboratory Improvement Amendments24. Experts selected a few clinical features representative of each child’s illness from the Electronic Health Record (EHR, Epic) and mapped them to simple genetic diseases with VAAST (Fabric Genomics). Trio EDTA-blood samples were obtained where possible. Genomic DNA was isolated with an EZ1 Advanced XL robot and the EZ1 DSP DNA Blood kit (Qiagen). DNA quality was assessed with the Quant-iT Picogreen dsDNA assay kit (ThermoFisher Scientific) using the Gemini EM Microplate Reader (Molecular Devices). Genomic DNA was fragmented by sonication (Covaris) and bar-coded, paired-end, PCR-free libraries were prepared for rapid whole genome sequencing with TruSeq DNA LT kits (Illumina) or Hyper kits (KAPA Biosystems). Sequencing libraries were analyzed with a Library Quantification Kit (KAPA Biosystems) and High Sensitivity NGS Fragment Analysis Kit (Advanced Analytical), respectively. 2 ×101 nt rapid and ultra-rapid whole genome sequencing was performed to at least 40-fold coverage with Illumina HiSeq 2500 (rapid run mode), HiSeq 4000, or NovaSeq 6000 (S1 or S2 flow cell) instruments, as described24.
Sample preparation and sequencing for rapid whole exome sequencing was performed by an external clinical laboratory (GeneDx). Exome enrichment was with the xGen Exome Research Panel v1.0 (Integrated DNA Technologies), and amplification used the Herculase II Fusion polymerase (Agilent)24. FASTQ files for rapid whole exome sequencing were transferred to Rady Children’s Institute for Genomic Medicine (RCIGM) for analysis and interpretation24.
Rapid whole genome, rapid whole exome and ultra-rapid whole genome sequences were aligned to human genome assembly GRCh37 (hg19), and variants were identified with the DRAGEN Platform (v.3.5, Illumina, San Diego)24. Structural variants were identified with Manta and CNVnator (using DNAnexus)24. Structural variants were filtered to retain those affecting coding regions of known disease genes and with allele frequencies <2% in the RCIGM database. Nucleotide and structural variants were annotated, analyzed, and interpreted by clinical molecular geneticists using Opal Clinical (Fabric Genomics), according to standard guidelines24. Opal annotated variants with respect to pathogenicity, generated a rank-ordered differential diagnosis based on the disease gene algorithm VAAST, a gene burden test, and the algorithm PHEVOR (Phenotype Driven Variant Ontological Re-ranking), which combined the observed Human Phenotype Ontology (HPO) phenotype terms from patients, and re-ranked disease genes based on the phenotypic match and the gene score24. Automatically generated, ranked results were manually interpreted through iterative Opal searches. Initially, variants were filtered to retain those with allele frequencies of <1% in the Exome Variant Server, 1000 Genomes Samples, and Exome Aggregation Consortium database24. Variants were further filtered for de novo, recessive and dominant inheritance patterns. The evidence supporting a diagnosis was then manually evaluated by comparison with the published literature. Analysis, interpretation and reporting required an average of six hours of expert effort. If genomic sequencing established a provisional diagnosis for which a specific treatment was available to prevent morbidity or mortality, this was immediately conveyed to the clinical team, as described24. All causative variants were confirmed by Sanger sequencing or chromosomal microarray, as appropriate. Secondary findings were not reported, but medically actionable incidental findings were reported if families consented to receive this information.
Infant mortality data collection
Characteristics of infant mortality (up to one year of age) were assessed among the 312 infants by structured EHR queries and manual EHR review. Characteristics included sex, race, ethnicity, clinical presentation, age at time of enrollment for rapid genomic sequencing, molecular diagnosis, age at time of death, “Code Blue” resuscitation status in the event of cardiorespiratory arrest, and whether a palliative care consultation had been performed. HPO terms at time of enrollment were extracted from the Epic EHR using clinical natural language processing as previously described17.
The San Diego study of outcomes in mothers and infants (SOMI)
SOMI is a community-level, spatially referenced research platform with data derived from a complete collection of birth and death records and hospital discharge summaries for all births in San Diego County47. Integrated into the platform are spatially referenced data on a wide variety of environmental measures, including census, air and water quality, climate, economic, crime, the built environment, transportation and social system data. The SOMI dataset represents the entire population of ~1 million mothers and their children born between 2005 and 2024. These data can also be linked to banked maternal serum samples from up to 70% of mothers who have received prenatal care in San Diego County, and archived newborn blood spots for virtually all live-born infants in the county. The SOMI dataset can be queried with a unique data visualization platform that incorporates many sources and types of data into one interactive mapping tool. The SOMI platform enables exploration of multi-level data to identify relationships between infant mortality and many pre- and post-natal environmental, genetic and epigenetic factors. The IRBs at the University of California San Diego and San Francisco have approved a waiver of consent for queries of the de-identified SOMI dataset.
Systematic literature review
A literature review to compare published evidence with findings from the two cohort studies and from data about overall infant deaths occurring in San Diego was conducted according to the MOOSE and PRISMA guidelines.
Data sources and record identification
We searched PubMed from January 1, 2010, to February 22, 2020 with the terms “infant mortality” and “genetic disease”. There were no language restrictions. There were no study design restrictions.
Study screening and eligibility
Studies that evaluated the etiologic contribution of single locus genetic diseases to infant mortality using comprehensive genomic methods (genome or exome sequencing with or without chromosomal microarray) were eligible. We limited eligibility to studies of cohorts with a broad range of genetic diseases, rather than one or a few disease types or clinical presentations, and in which the majority of probands were less than 18 years old.
Inclusion criteria and data extraction
Data extraction was manual. Data was reviewed for completeness and accuracy by at least two expert investigators and disparities were reconciled by consensus. Data extraction was limited to infant deaths which occurred at an age less than 1 year. Inclusion criteria for data abstraction included the use of genome or exome sequencing with or without chromosomal microarray, and cases were considered positive if they had a confirmed diagnosis of a single locus genetic disease detected by sequencing. The primary outcome considered was infant mortality associated with a molecular diagnosis of a genetic disease. Molecular diagnoses were defined as pathogenic or likely pathogenic diplotypes affecting single genes or loci with definitive, strong or moderate associations with infant mortality causation. Variants of uncertain significance and secondary findings were not extracted. Where more than one publication reported results from a cohort, we included the most recent value for diagnostic utility.
Testing was performed clinically in hospital laboratories and reference laboratories, and experimentally in research laboratories. Hospital and reference laboratory clinical tests were defined primarily by the site of testing and, as disclosed in the methods, and, secondarily, by the affiliations of the authors. Clinical testing was defined as testing under fixed protocols that were attested to comply with state or national regulatory guidelines for in vitro diagnostic testing. Experimental research tests were those that explored the utility of novel or bespoke methods of testing or analysis.
Statistical analysis
For continuous outcomes, such as age at death, P-values were determined with a two-tailed t-test for independent samples. Counts of outcomes were compared between groups with chi-square. A z-test was used to evaluate the association of proportions between populations, using the binomial approximation of the normal distribution. A significance cutoff of 0.05 was used for all comparisons.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All data associated with this study are present in the paper or are available at the Longitudinal Pediatric Data Resource under a data use agreement and subject to the limitations of the informed consent documents for each subject (Accession Number nbs000003.v1.p, https://www.nbstrn.org/research-tools/longitudinal-pediatric-data-resource).
References
Murphy, S. L., Xu, J., Kochanek, K. D. & Arias, E. Mortality in the United States, 2017. NCHS Data Brief. 1–8 (2018).
Xu, J., Kochanek, K. D., Murphy, S. L. & Tejada-Vera, B. Deaths: final data for 2007. Natl. Vital Stat. Rep. 58, 1–19 (2010).
Hoyert, D. L., Kochanek, K. D. & Murphy, S. L. Deaths: final data for 1997. Natl Vital Stat. Rep. 47, 1–104 (1999).
MacDorman, M. F. & Rosenberg, H. M. Trends in infant mortality by cause of death and other characteristics, 1960-88. National Center for Health Statistics. Vital Health Stat. 20 (1993).
Adeyinka, A. & Bailey, K. Death, Certification. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing (2019).
McGivern, L., Shulman, L., Carney, J. K., Shapiro, S. & Bundock, E. Death certification errors and the effect on mortality statistics. Public Health Rep. 132, 669–675 (2017).
Mieno, M. N. et al. Accuracy of death certificates and assessment of factors for misclassification of underlying cause of death. J. Epidemiol. 26, 191–198 (2016).
Kircher, T., Nelson, J. & Burdo, H. The autopsy as a measure of accuracy of the death certificate. N. Engl. J. Med 313, 1263–1269 (1985). Nov 14.
Ackerman, M. J., Tester, D. J. & Driscoll, D. J. Molecular autopsy of sudden unexplained death in the young. Am. J. Forensic Med. Pathol. 22, 105–11. (2001).
Loporcaro, C. G., Tester, D. J., Maleszewski, J. J., Kruisselbrink, T. & Ackerman, M. J. Confirmation of cause and manner of death via a comprehensive cardiac autopsy including whole exome next-generation sequencing. Arch. Pathol. Lab. Med. 138, 1083–1089 (2014).
OMIM Entry Statistics (Johns Hopkins University, Baltimore, MD, 2018; https://www.omim.org/statistics/geneMap).
National Center for Biotechnology Information, National Library of Medicine, Database of Structural Variants (2018; https://www.ncbi.nlm.nih.gov/dbvar?term=(%22clin%20pathogenic%22%5BFilter%5D)%20AND%20homo%20sapiens%5BOrganism%5D).
Weiner, J., Sharma, J., Lantos, J. & Kilbride, H. How infants die in the neonatal intensive care unit: trends from 1999 through 2008. Arch. Pediatr. Adolesc. Med. 165, 630–634 (2011).
Jacob, J., Kamitsuka, M., Clark, R. H., Kelleher, A. S. & Spitzer, A. R. Etiologies of NICU Deaths. Pediatrics. 2015.
Briggs, B. et al. Novel Factor XIII variant identified through whole-genome sequencing in a child with intracranial hemorrhage. Cold Spring Harb. Mol. Case Stud. 4, a003525 (2018).
Chen, D. Y. et al. Rapid diagnosis of KCNQ2-associated early infantile epileptic encephalopathy improved outcome. Pediatr. Neurol. 86, 69–70 (2018).
Clark, M. M. et al. Diagnosis of genetic diseases in seriously ill children by rapid whole-genome sequencing and automated phenotyping and interpretation. Sci. Transl. Med. 11, eaat6177 (2019).
Farnaes, L. et al. Rapid whole-genome sequencing decreases infant morbidity and cost of hospitalization. npj Genomic Med. 3, 10 (2018).
Farnaes, L. et al. Rapid whole-genome sequencing identifies a novel GABRA1 variant associated with West syndrome. Cold Spring Harb. Mol. Case Stud. 3, a001776 (2017).
French, C. E. et al. Whole genome sequencing reveals that genetic conditions are frequent in intensively ill children. Intensive Care Med. 45, 627–636 (2019).
Gubbels, C. S. et al. Prospective, phenotype-driven selection of critically ill neonates for rapid exome sequencing is associated with high diagnostic yield. Genet. Med. 22, 736–744 (2019).
Hildreth, A. et al. Rapid whole-genome sequencing identifies a novel homozygous NPC1 variant associated with Niemann-Pick type C1 disease in a 7-week-old male with cholestasis. Cold Spring Harb. Mol. Case Stud. 3, a001966 (2017).
James, K. N. et al. Partially automated whole-genome sequencing reanalysis of previously undiagnosed pediatric patients can efficiently yield new diagnoses. NPJ Genom. Med. 11, 5–33 (2020).
Kingsmore, S. F. et al. A Randomized, Controlled Trial of the Analytic and Diagnostic Performance of Singleton and Trio, Rapid Genome and Exome Sequencing in Ill Infants. Am. J. Hum. Genet. 105, 719–733 (2019).
Meng, M. et al. Use of exome sequencing for infants in intensive care units: ascertainment of severe single-gene disorders and effect on medical management. JAMA Pediatr. 171, e173438 (2017).
Mestek-Boukhibar, L. et al. Rapid paediatric sequencing (RaPS): comprehensive real-life workflow for rapid diagnosis of critically ill children. J. Med. Genet. 55, 721–28. (2018).
Miller, N. A. et al. A 26-hour system of highly sensitive whole genome sequencing for emergency management of genetic diseases. Genome Med. 7, 100 (2015).
Petrikin, J. E. et al. The NSIGHT1-randomized controlled trial: rapid whole-genome sequencing for accelerated etiologic diagnosis in critically ill infants. NPJ Genom. Med. 3, 6 (2018).
Petrikin, J. E., Willig, L. K., Smith, L. D. & Kingsmore, S. F. Rapid whole genome sequencing and precision neonatology. Semin. Perinatol. 39, 623–631 (2015).
Sanford, E. F. et al. Rapid whole genome sequencing has clinical utility in children in the pediatric intensive care unit. In Press.
Sanford, E. et al. Concomitant diagnosis of immune deficiency and Pseudomonas sepsis in a 19 month old with ecthyma gangrenosum by host whole-genome sequencing. Cold Spring Harb. Mol. Case Stud. 4, a003244 (2018).
Sanford, E. et al. Rapid whole genome sequencing identifies a novel AIRE variant associated with Autoimmune Polyendocrine Syndrome Type 1. Cold Spring Harb. Mol. Case Stud. 4, a002485 (2018).
Saunders, C. J. et al. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci. Transl. Med. 4, 154ra135 (2012).
Schofield, D., Rynehart, L., Shresthra, R., White, S. M. & Stark, Z. Long-term economic impacts of exome sequencing for suspected monogenic disorders: diagnosis, management, and reproductive outcomes. Genet. Med. 21, 2586–2593 (2019).
Smith, L. D., Willig, L. K. & Kingsmore, S. F. Whole-exome sequencing and whole-genome sequencing in critically ill neonates suspected to have single-gene disorders. Cold Spring Harb. Perspect. Med. 6, a023168 (2015).
Soden, S. E. et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci. Transl. Med. 6, 265ra168 (2014).
Stark, Z. et al. Meeting the challenges of implementing rapid genomic testing in acute pediatric care. Genet. Med. 20, 1554–1563 (2018).
Stark, Z. et al. Does genomic sequencing early in the diagnostic trajectory make a difference? A follow-up study of clinical outcomes and cost-effectiveness. Genet. Med. 21, 173–180 (2019).
Stark, Z. et al. Prospective comparison of the cost-effectiveness of clinical whole-exome sequencing with that of usual care overwhelmingly supports early use and reimbursement. Genet. Med. 19, 867–874 (2017).
Stark, Z. et al. A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet. Med. 18, 1090–1096 (2016).
Tan, T. Y. et al. Diagnostic impact and cost-effectiveness of whole-exome sequencing for ambulant children with suspected monogenic conditions. JAMA Pediatr. 171, 855–862 (2017). Sep 1.
Tan, N. B. et al. Diagnostic and service impact of genomic testing technologies in a neonatal intensive care unit. J. Paediatr. Child Health. 55, 1309–1314 (2019).
van Diemen, C. C. et al. Rapid targeted genomics in critically ill newborns. Pediatrics 140, pii: e20162854 (2017).
Wang, H. et al. Optimized trio genome sequencing (OGTS) as a first-tier genetic test in critically ill infants: practice in China. Hum. Genet. 139, 473–482 (2020).
Willig, L. K. et al. Whole-genome sequencing for identification of Mendelian disorders in critically ill infants: a retrospective analysis of diagnostic and clinical findings. Lancet Respir. Med. 3, 377–387 (2015).
Ceyhan-Birsoy, O. et al. Interpretation of genomic sequencing results in healthy and ill newborns: results from the BabySeq Project. Am. J. Hum. Genet. 104, 76–93 (2019).
Araneta, M. R. G. et al. Health advantages and disparities in preterm birth among immigrants despite disparate sociodemographic, behavioral, and maternal risk factors in San Diego, California. Matern. Child Health J. 24, 153–164 (2020).
van der Werf, I. M. et al. Novel microdeletions on chromosome 14q32.2 suggest a potential role for non-coding RNAs in Kagami-Ogata syndrome. Eur. J. Hum. Genet. 24, 1724–1729 (2016).
Schlade-Bartusiak, K., Ardinger, H. & Cox, D. W. A child with terminal 14q deletion syndrome: consideration of genotype-phenotype correlations. Am. J. Med. Genet. A. 149A, 1012–1018 (2009).
Issekutz, K. A., Graham, J. M. Jr., Prasad, C., Smith, I. M. & Blake, K. D. An epidemiological analysis of CHARGE syndrome: preliminary results from a Canadian study. Am. J. Med. Genet. A. 133A, 309–17 (2005).
Corsten-Janssen, N., van Ravenswaaij-Arts, C. M. A. & Kapusta, L. Congenital arch vessel anomalies in CHARGE syndrome: a frequent feature with risk for co-morbidity. Int. J. Cardiol. Heart Vasc. 12, 21–25 (2016). May 25.
Hendrix, N. W., Clemens, M., Canavan, T. P., Surti, U. & Rajkovic, A. Prenatally diagnosed 17q12 microdeletion syndrome with a novel association with congenital diaphragmatic hernia. Fetal Diagn. Ther. 31, 129–133 (2012).
Haeri, S. et al. Deletion of hepatocyte nuclear factor-1-beta in an infant with prune belly syndrome. Am. J. Perinatol. 27, 559–63. (2010).
Kang, S. L., Forsey, J., Dudley, D., Steward, C. G. & Tsai-Goodman, B. Clinical characteristics and outcomes of cardiomyopathy in Barth syndrome: the UK experience. Pediatr. Cardiol. 37, 167–76. (2016).
Rigaud, C. et al. Natural history of Barth syndrome: a national cohort study of 22 patients. Orphanet. J. Rare Dis. 8, 70 (2013). May 8.
Stoeva, R. et al. A novel SOX9 nonsense mutation, q401x, in a case of campomelic dysplasia with XY sex reversal. Genet. Couns. 22, 49–53 (2011).
Suzumura, H., Sakurai, K., Kano, K. & Ichimura, T. Coffin-Siris syndrome: a case of an extremely low birthweight infant with severe kyphoscoliosis. Acta Paediatr. Jpn. 38, 537–40. (1996).
Al-Maawali, A. et al. Clinical characteristics in patients with interstitial deletions of chromosome region 12q21-q22 and identification of a critical region associated with keratosis pilaris. Am. J. Med. Genet. A. 164A, 796–800 (2014).
Jackson, C. B. et al. Mutations in SDHD lead to autosomal recessive encephalomyopathy and isolated mitochondrial complex II deficiency. J. Med. Genet. 51, 170–175 (2014).
Wiseman, D. H. et al. A novel syndrome of congenital sideroblastic anemia, B-cell immunodeficiency, periodic fevers, and developmental delay (SIFD). Blood 122, 112–23. (2013). Jul 4.
Tan, H. L., Hofman, N., van Langen, I. M., van der Wal, A. C. & Wilde, A. A. Sudden unexplained death: heritability and diagnostic yield of cardiological and genetic examination in surviving relatives. Circulation 112, 207–13. (2005). Jul 12.
Procaccio, V. et al. Nuclear DNA origin of mitochondrial complex I deficiency in fatal infantile lactic acidosis evidenced by transnuclear complementation of cultured fibroblasts. J. Clin. Invest. 104, 83–92 (1999).
Yang, L. et al. Genetic etiology of early infant deaths in a neonatal intensive care unit. J. Med. Genet. 57, 169–177 (2020).
Armes, J. E. et al. Application of whole genome sequencing technology in the investigation of genetic causes of fetal, perinatal, and early infant death. Pediatr. Dev. Pathol. 21, 54–67 (2018).
Wojcik, M. H. et al. Infant Mortality: the contribution of genetic disorders. J. Perinatol. 39, 1611–1619 (2019).
Alagille Syndrome. Spinner NB, et al. 2000 May 19 [Updated 2019 Dec 12]. GeneReviews® [Internet]. Adam MP, Ardinger HH, Pagon RA, et al., editors. (University of Washington, Seattle, 1993–2020). www.ncbi.nlm.nih.gov/books/NBK1273.
Autoimmune Lymphoproliferative Syndrome. Bleesing JJH, Nagaraj CB, Zhang K. 2006 Sep 14 [Updated 2017 Aug 24]. GeneReviews® [Internet]. Adam MP, Ardinger HH, Pagon RA, et al., editors. Seattle (WA): University of Washington, Seattle; 1993-2020. www.ncbi.nlm.nih.gov/books/NBK1108.
Polycystic Kidney Disease, Autosomal Recessive. Sweeney WE, Avner ED. 2001 Jul 19 [Updated 2019 Feb 14]. GeneReviews® [Internet]. Adam MP, Ardinger HH, Pagon RA, et al., editors. Seattle (WA): University of Washington, Seattle; 1993-2020. www.ncbi.nlm.nih.gov/books/NBK1326.
Barth Syndrome. Ferreira C, Thompson R, Vernon H. 2014 Oct 9. GeneReviews® [Internet]. Adam MP, Ardinger HH, Pagon RA, et al., editors. Seattle (WA): University of Washington, Seattle; 1993–2020. www.ncbi.nlm.nih.gov/books/NBK247162
Brugada Syndrome. Brugada R, Campuzano O, Sarquella-Brugada G, et al. 2005 Mar 31 [Updated 2016 Nov 17]. GeneReviews® [Internet]. Adam MP, Ardinger HH, Pagon RA, et al., editors. Seattle (WA): University of Washington, Seattle; 1993-2020. www.ncbi.nlm.nih.gov/books/NBK1517.
Wilde, A. A. M. & Amin, A. S. Clinical spectrum of SCN5A mutations: long QT syndrome, Brugada syndrome, and cardiomyopathy. JACC Clin. Electrophysiol. 4, 569–579 (2018).
Hershberger R. E., Morales A. LMNA-Related Dilated Cardiomyopathy. 2008 Jun 12 [Updated 2016 Jul 7]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1674/.
Hasselberg, N. E. et al. Lamin A/C cardiomyopathy: young onset, high penetrance, and frequent need for heart transplantation. Eur. Heart J. 39, 853–860 (2018).
Vatanavicharn, N. et al. Carnitine-acylcarnitine translocase deficiency: two neonatal cases with common splicing mutation and in vitro bezafibrate response. Brain Dev. 37, 698–703 (2015).
Yan, H. M. et al. Carnitine-acylcarnitine translocase deficiency with c.199-10 T>G and novel c.1A>G mutation: two case reports and brief literature review. Med. (Baltim.) 96, e8549 (2017).
Chronic Granulomatous Disease. Leiding JW, Holland SM. 2012 Aug 9 [Updated 2016 Feb 11]. GeneReviews® [Internet]. Adam MP, Ardinger HH, Pagon RA, et al., editors. Seattle (WA): University of Washington, Seattle; 1993-2020. www.ncbi.nlm.nih.gov/books/NBK99496.
X-Linked Myotubular Myopathy. Dowling JJ, Lawlor MW, Das S. 2002 Feb 25 [Updated 2018 Aug 23]. GeneReviews® [Internet]. Adam MP, Ardinger HH, Pagon RA, et al., editors. Seattle (WA): University of Washington, Seattle; 1993–2020. www.ncbi.nlm.nih.gov/books/NBK1432.
Carnitine Palmitoyltransferase II Deficiency. Wieser T. 2004 Aug 27 [Updated 2019 Jan 3]. GeneReviews® [Internet]. Adam MP, Ardinger HH, Pagon RA, et al., editors. Seattle (WA): University of Washington, Seattle; 1993–2020. www.ncbi.nlm.nih.gov/books/NBK1253.
Sanders, S. J. et al. Progress in understanding and treating SCN2A-mediated disorders. Trends Neurosci. 41, 442–456 (2018).
Turkdogan, D., Thomas, G. & Demirel, B. Ketogenic diet as a successful early treatment modality for SCN2A mutation. Brain Dev. 41, 389–391 (2019).
Flor-Hirsch, H. et al. Lacosamide for SCN2A-related intractable neonatal and infantile seizures. Epileptic Disord. 20, 440–446 (2018).
Wang, J., Wu, J. C., Yu, X. E., Han, Y. Z. & Yang, R. M. Long-term outcomes of a patient with late-onset multiple acyl-CoA dehydrogenase deficiency caused by novel mutations in ETFDH: A case report. Med. (Baltim.) 97, e13153 (2018).
Missaglia, S., Tavian, D., Moro, L. & Angelini, C. Characterization of two ETFDH mutations in a novel case of riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Lipids Health Dis. 17, 254 (2018).
Goh, L. L. et al. Patient with multiple acyl-CoA dehydrogenase deficiency disease and ETFDH mutations benefits from riboflavin therapy: a case report. BMC Med. Genomics 11, 37 (2018).
Lymphoproliferative Disease, X-Linked. Zhang K., Wakefield E., Marsh R. 2004 Feb 27 [Updated 2016 Jun 30]. GeneReviews® [Internet]. Adam MP, Ardinger HH, Pagon RA, et al., editors. Seattle (WA): University of Washington, Seattle; 1993–2020. www.ncbi.nlm.nih.gov/books/NBK1406.
Chediak-Higashi Syndrome. Toro C, et al. 2009 Mar 3 [Updated 2018 Jul 5]. GeneReviews® [Internet]. Adam MP, Ardinger HH, Pagon RA, et al., editors. Seattle (WA): University of Washington, Seattle; 1993-2020. www.ncbi.nlm.nih.gov/books/NBK5188.
Peng, K. et al. Umbilical cord blood transplantation corrects very early-onset inflammatory bowel disease in Chinese patients with IL10RA-associated immune deficiency. Inflamm. Bowel Dis. 24, 1416–1427 (2018).
Bartsakoulia, M. et al. Cysteine supplementation may be beneficial in a subgroup of mitochondrial translation deficiencies. J. Neuromuscul. Dis. 3, 363–379 (2016). Aug 30.
Isolated Methylmalonic Acidemia. Manoli I, Sloan JL, Venditti CP. 2005 Aug 16 [Updated 2016 Dec 1]. GeneReviews® [Internet]. Adam MP, Ardinger HH, Pagon RA, et al., editors. Seattle (WA): University of Washington, Seattle; 1993–2020. www.ncbi.nlm.nih.gov/books/NBK1231.
Ornithine Transcarbamylase Deficiency. Lichter-Konecki U, et. al. 2013 Aug 29 [Updated 2016 Apr 14]. GeneReviews® [Internet]. Adam MP, Ardinger HH, Pagon RA, et al., editors. Seattle (WA): University of Washington, Seattle; 1993–2020. www.ncbi.nlm.nih.gov/books/NBK154378.
Sobacchi, C., Schulz, A., Coxon, F. P., Villa, A. & Helfrich, M. H. Osteopetrosis: genetics, treatment and new insights into osteoclast function. Nat. Rev. Endocrinol. 9, 522–36. (2013).
Pompe Disease. Leslie N, Bailey L. 2007 Aug 31 [Updated 2017 May 11]. GeneReviews® [Internet]. Adam MP, Ardinger HH, Pagon RA, et al., editors. Seattle (WA): University of Washington, Seattle; 1993-2020. www.ncbi.nlm.nih.gov/books/NBK1261.
Sofou, K. et al. Ketogenic diet in pyruvate dehydrogenase complex deficiency: short- and long-term outcomes. J. Inherit. Metab. Dis. 40, 237–245 (2017).
Chida, R., Shimura, M., Nishimata, S., Kashiwagi, Y. & Kawashima, H. Efficacy of ketogenic diet for pyruvate dehydrogenase complex deficiency. Pediatr. Int. 60, 1041–1042 (2018).
Spinal Muscular Atrophy. Prior TW, Leach M. E., Finanger E. 2000 Feb 24 [Updated 2019 Nov 14]. GeneReviews® [Internet]. Adam MP, Ardinger HH, Pagon RA, et al., editors. Seattle (WA): University of Washington, Seattle; 1993–2020. www.ncbi.nlm.nih.gov/books/NBK1352.
Timothy Syndrome. Napolitano C, et al. 2006 Feb 15 [Updated 2015 Jul 16]. GeneReviews® [Internet]. Adam MP, Ardinger HH, Pagon RA, et al., editors. Seattle (WA): University of Washington, Seattle; 1993-2020. www.ncbi.nlm.nih.gov/books/NBK1403.
Tuberous Sclerosis Complex. Northrup H, et. al. 1999 Jul 13 [Updated 2018 Jul 12]. GeneReviews® [Internet]. Adam MP, Ardinger HH, Pagon RA, et al., editors. Seattle (WA): University of Washington, Seattle; 1993-2020. www.ncbi.nlm.nih.gov/books/NBK1220.
Yang, Y. et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 312, 1870–1879 (2014).
Smith, E., du Souich, C., Dragojlovic, N. & Elliott, A. M. CAUSES Study; RAPIDOMICS Study. Genetic counseling considerations with rapid genome‐wide sequencing in a neonatal intensive care unit. J. Genet Couns. 28, 263–272 (2019).
Ratigan, A. R. et al. Community BMI surveillance using an existing immunization registry in San Diego, California. J. Community Health 42, 558–564 (2017).
West, J. H. et al. The role of parenting in alcohol and tobacco use among Latino adolescents. J. Child Adolesc. Subst. Abus. 22, 120–132 (2013). Apr 1.
Hughes, S. C. et al. Inconsistent report of pre-pregnancy-recognition alcohol use by Latinas. Matern. Child Health J. 13, 857–64. (2009).
Chambers, C. D. et al. Alcohol consumption among low-income pregnant Latinas. Alcohol Clin. Exp. Res. 29, 2022–2028 (2005).
Acknowledgements
This study was supported by grant U19HD077693 from NICHD and NHGRI to S.F.K., grant UL1TR002550 from NCATS to E.J. Topol, grant R01HD101540 from NICHD to S.F.K. and C.C., and gifts from the Liguori Family, John Motter and Effie Simanikas, Ernest and Evelyn Rady and RCHSD.
Author information
Authors and Affiliations
Contributions
S.F.K. led the project, obtained funding, and wrote the manuscript. M.O. revised the manuscript. A.H., M.M.C. and C.H. performed data collection and analysis. C.D.C. provided oversight of part of the project and obtained funding. L.L.J.-P. extracted SOMI data. C.H. provided oversight of part of the project. All authors reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
D.D. received funding from Biomarin (consultant for Pegvaliase trials), Audentes Therapeutics (Scientific Advisory Board), and Ichorion Therapeutics (consultant for mitochondrial disease drugs). The remaining authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kingsmore, S.F., Henderson, A., Owen, M.J. et al. Measurement of genetic diseases as a cause of mortality in infants receiving whole genome sequencing. npj Genom. Med. 5, 49 (2020). https://doi.org/10.1038/s41525-020-00155-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-020-00155-8
This article is cited by
-
Whole-exome sequencing as the first-tier test for patients in neonatal intensive care unit: a Chinese single-center study
BMC Pediatrics (2024)
-
Scalable, high quality, whole genome sequencing from archived, newborn, dried blood spots
npj Genomic Medicine (2023)
-
An automated 13.5 hour system for scalable diagnosis and acute management guidance for genetic diseases
Nature Communications (2022)
