
Abstract
Genetic disorders are a leading cause of morbidity and mortality in infants in neonatal and pediatric intensive care units (NICU/PICU). While genomic sequencing is useful for genetic disease diagnosis, results are usually reported too late to guide inpatient management. We performed an investigator-initiated, partially blinded, pragmatic, randomized, controlled trial to test the hypothesis that rapid whole-genome sequencing (rWGS) increased the proportion of NICU/PICU infants receiving a genetic diagnosis within 28 days. The participants were families with infants aged <4 months in a regional NICU and PICU, with illnesses of unknown etiology. The intervention was trio rWGS. Enrollment from October 2014 to June 2016, and follow-up until November 2016. Of all, 26 female infants, 37 male infants, and 2 infants of undetermined sex were randomized to receive rWGS plus standard genetic tests (n = 32, cases) or standard genetic tests alone (n = 33, controls). The study was terminated early due to loss of equipoise: 73% (24) controls received genomic sequencing as standard tests, and 15% (five) controls underwent compassionate cross-over to receive rWGS. Nevertheless, intention to treat analysis showed the rate of genetic diagnosis within 28 days of enrollment (the primary end-point) to be higher in cases (31%, 10 of 32) than controls (3%, 1 of 33; difference, 28% [95% CI, 10–46%]; p = 0.003). Among infants enrolled in the first 25 days of life, the rate of neonatal diagnosis was higher in cases (32%, 7 of 22) than controls (0%, 0 of 23; difference, 32% [95% CI, 11–53%];p = 0.004). Median age at diagnosis (25 days [range 14–90] in cases vs. 130 days [range 37–451] in controls) and median time to diagnosis (13 days [range 1–84] in cases, vs. 107 days [range 21–429] in controls) were significantly less in cases than controls (p = 0.04). In conclusion, rWGS increased the proportion of NICU/PICU infants who received timely diagnoses of genetic diseases.
Similar content being viewed by others

Introduction
A premise of pediatric precision medicine is that outcomes are improved by replacement of clinical diagnosis and empiric management with genetic diagnosis and genotype-differentiated treatment.1,2,3,4,5,6,7,8,9 The evidence base for pediatric precision medicine is still underdeveloped.10, 11 Ill infants are especially in need of precision medicine since genetic diseases are a leading cause of mortality, particularly in neonatal intensive care units (NICU) and pediatric intensive care units (PICU).5,6,7, 12,13,14,15,16 Among high-cost health care, NICU treatment is one of the most cost-effective.17,18,19 Since disease progression can be very rapid in infants, genetic diagnoses must be made quickly to permit consideration of precision interventions in time to decrease morbidity and mortality.5,6, 20,21,22,23 For a few genetic diseases, newborn screening has shown early neonatal diagnosis and rapid, precise intervention to dramatically improve outcomes.24, 25 The potential expansion to newborn diagnosis for symptomatic infants for all 5000 genetic diseases26 has been made technically possible by the advent of clinical genomic sequencing (whole-genome sequencing (WGS) or whole-exome sequencing (WES), and next-generation sequencing gene panel tests (NGS). In particular, rapid WGS (rWGS) can allow genetic diagnosis in 2 days.20, 27
There is substantial evidence that a higher proportion of symptomatic children with likely genetic disease receive etiologic diagnoses by WGS and WES than other genetic tests.3,4,5,6,7, 28,29,30,31,32,33,34,35 Published NICU or PICU experience with rWGS, however, is limited to case reports and one retrospective study.5,6, 20,21,22,23 In the latter, 57% of infants received genetic diagnoses in a median of 23 days (day of life 49).6 However, it has not yet been unequivocally demonstrated whether rWGS improves timeliness of genetic diagnosis relative to standard genetic tests. Here we report results of newborn sequencing in genomic medicine and public health randomized controlled trial (RCT) 1 (NSIGHT1), an RCT of genomic testing in patients (ClinicalTrials.gov Identifier: NCT02225522).24 Specifically, NSIGHT1 compared rates of genetic diagnosis in NICU and PICU infants with possible genetic diseases at 28 days from enrollment by standard tests alone vs. standard tests plus trio rWGS.
Results
Patients
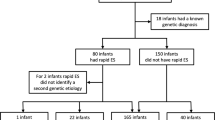
Of 129 nominated infants, 65 (50%) completed the NSIGTH1 study (Figs. 1 and 2). Sixty four enrollees were NICU infants. The infants nominated represented 7% of NICU and PICU admissions during this interval. Thirty-two infants randomized to rWGS plus standard genetic tests (cases) and 33 to standard tests alone (controls, Figs. 1 and 2). The baseline characteristics of the infants were similar in the two arms and similar to those of a previous retrospective case series of infants receiving rWGS in this NICU and PICU (Table 1).6 Detailed (deep) phenotypes of infants were extracted from the electronic medical record in 42 infants receiving genomic sequencing, since this was a prerequisite for interpretation. On average, infants receiving rWGS had 5.9 phenotypic features (range 1–17; Table S1). Phenotypes were highly diverse and typically present at birth (Table 1, S1). The most common indications for nomination were congenital anomalies (35%) and neurological disorders (25%; Table 1). Fewer control infants had cardiovascular findings (6 vs. 28%; difference, −22% [95% CI, −40 to −4%]; p = 0.02) than cases, which may have affected likelihood for genetic disease (Table 1).

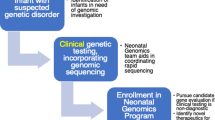
Design of “Newborn Sequencing In Genomic medicine and public HealTh” study 1 (NSIGHT1; ClinicalTrials.gov accession NCT02225522). Time (t) is in days. WGS whole-genome sequencing, EHR Electronic Health Record

CONSORT flow diagram of NSIGHT1 enrollment and randomization. Major reasons for non-enrollment were family refusal (13%), the infant had a diagnosis that explained the phenotype (9%), and incomplete nominations (9%). At unblinding of clinicians (by 10 days after enrollment), a provision was made whereby clinicians could request compassionate cross-over to the rWGS group if the infant was critically ill. Cross-over was requested for 7 (21%) of 33 infants who randomized to standard tests alone, of which 5 met these criteria and were granted
Standard diagnostic tests
Standard diagnostic tests for genetic diseases were performed as clinically indicated in 63 of the 65 infants (Table 1). They included all postnatal diagnostic tests that could be ordered through the electronic medical record. The proportion of infants receiving standard genetic tests and age at first standard test order were similar in both arms (Table 1). In addition to newborn screening infants received an average of 3.1 (range 0–10) standard genetic tests (Table 1, S3), which was similar to a previous retrospective case series of infants receiving rWGS from the same NICU and PICU.6 During the study, non-expedited WGS became available as a standard diagnostic test. Of 33 control infants, 24 (73%) received non-expedited clinical NGS panel tests, WES or WGS standard tests, compared with seventeen (53%) of 32 cases (Table 1, S3). Other than newborn screening, the average age at first standard test order was 14 days (range 0–120 days). Standard tests yielded fifteen (24%) genetic diagnoses in the 63 subjects tested, seven (23%) in 30 cases, and eight (24%) in 33 controls (Table 2, S4). The rates of diagnosis by individual standard clinical tests were: chromosomal microarray 6% (three of 48 tests); Clinical NGS panel test 18% (nine of 49 tests); Clinical WES 33% (one of three tests); Methylation 13% (one of eight). Of note, five (33%) of 15 diagnoses by standard tests would not have been detected by rWGS at the time of study: four were copy number or structural variants and one was a change in DNA methylation. The median time from first standard test order to diagnosis was 64 days (range 16–450 days). The average age at diagnosis by standard genetic tests was 113 days (range 16–451 days). Six (10%) of 63 infants received a diagnosis by standard tests prior to hospital discharge (Table S5).
Rapid whole-genome sequencing
rWGS was performed on infant-parent trios with Illumina HiSeq instruments, with paired reads to an average depth of 40-fold, detecting an average of 5.0 million nucleotide variants per genome (standard deviation 0.3 million variants; Table S2, Figure S1).
Ten of 32 cases (31%) received diagnoses by rWGS (Table 2, Table S4). Upon un-blinding of clinicians to randomization at day 10 after enrollment, compassionate cross-over to rWGS was requested for seven (21%) of the 33 controls. Cross-over to rWGS was declined in two infants who were not acutely ill; both were about to be discharged to home, with follow-up of their medical conditions as outpatients. Five cross-over requests were granted, yielding two diagnoses. In both, diagnosis by rWGS occurred first but was recapitulated by standard tests (Table 2). Including five crossovers, 12 (32%) of 37 infants received rWGS diagnoses (Table 2, S5). On average, enrollment occurred on DOL 22 (range 1–101; Table 1), which was earlier than in our previous report of rWGS (DOL 26; Table 1),6 but an average of 8 days later than standard tests. The median time to rWGS diagnosis, including clinical confirmatory testing, was 14 days (range 8–35 days; Table S5), which was also faster than our previous report of rWGS (23 days; Table S5).6 The median age at diagnosis in patients randomized to rWGS was 28.5 days (range 14–90 days). Among crossovers, the median age at rWGS diagnosis was 94.5 days.
The research protocol required confirmation of rWGS results by another method prior to clinical reporting except in cases where life-threatening progression was imminently likely. There were no such cases, and no provisional diagnostic reports of rWGS results were returned prior to confirmatory testing. Sanger sequencing confirmed all rWGS results.
Diagnoses
Twenty-two genetic diagnoses were reported in 21 (32%) of 65 infants (Table 2). Thirteen cases (41%) received diagnoses by rWGS or standard tests. Eight controls (24%) received diagnoses (Table 1). One individual received two diagnoses. Only one diagnosis was recurrent (X-linked myotubular myopathy in two infants), reflecting substantial genetic heterogeneity among NICU disease presentations6 (Table 2). The most common mechanism was de novo variant occurrence (12 of 21 (57%) diagnoses; Table 2). Seventeen (65%) causative variants were reported as pathogenic, and nine (35%) as likely pathogenic. Eight variants (31%) were predicted to result in amino acid substitutions, six (23%) were indels, five (19%) were predicted to result in stop codon loss or gain, four (15%) were structural variations, two (8%) were predicted to alter splicing, and one (4%) impacted methylation. The most common inheritance pattern was autosomal dominant (14 of 19 (74%) diagnoses), followed by autosomal recessive (three, 16%) and X-linked recessive (two, 11%). In 19 of 21 (90%) diagnosed probands, the diagnosis explained all of the clinical features (Table 2). Two possible diagnoses provoked discussion regarding inclusion. Control infant 5053 had altered mental status, decreased deep tendon reflexes, hypotonia, and cryptorchidism (Table S1). He had a duplication of Chr 1p36.32 as well as X-linked Myotubular Myopathy. Chr 1p36.32 duplication syndrome was not included as a diagnosis. The second, paternally inherited SCN5A c.6010 T > C (p.F2004L) in Case infant 5033 with persistent, symptomatic atrial fibrillation/flutter (Table S1), was not included as a diagnosis. While several case studies had reported this variant as pathogenic and two functional studies showed it to be deleterious, the allele frequency (0.3%) was considered too high to be likely pathogenic, and a diagnosis of familial atrial fibrillation type 10 was not reported.36,37,38,39,40
Clinical utility of molecular diagnoses
The short-term clinical impact of diagnoses was assessed by chart reviews and surveys with referring physicians (Table S4). Thus, clinical utility reflected actual practice, rather than an ideal or maximal personalization of treatment. Clinical utility did not include the impact of negative test results on management. 20 (31%) of the 65 infants enrolled (95% of those receiving diagnoses) had a consequent change in management. Ten (15%) infants (48% of those receiving diagnoses) had a change in management other than genetic or reproductive counseling of parents. Diagnoses were not associated with any harms.
Early study termination
The study was terminated after 21 months due to growing availability of targeted NGS panels, WES and WGS as standard tests, which shifted the baseline of comparison over the course of the study. These were associated with high rates of cross-over requests and higher utilization of targeted NGS panels, WES or WGS as standard clinical genetic tests among controls (73%) than cases (53%; Table S3).
End-point testing
End-points were analyzed on the basis of intention to treat (Figs. 1 and 2). Thus all patients were analyzed in the groups to which they were randomized. The primary end point, rate of genetic diagnosis within 28 days of enrollment, was higher in cases (31%, ten of 32) than controls (3%, one of 33; difference, 28% [95% CI, 10–46%]; p = 0.003 Table 3). Kaplan–Meier curves supported the conclusion that there was a significantly higher probability of receiving a diagnosis by rWGS until DOL 99 or 67 days after test order (Fig. 3). For neonates enrolled within the first 25 days of life, the rate of diagnosis by DOL 28, a secondary end-point, was higher in cases (32%, seven of 22) than controls (0%, zero of 23; difference, 32% [95% CI, 11–53%]; p < 0.01; Table 3). In practice, crossovers did not materially affect these end-points, since the two diagnoses made by rWGS among five cross-over infants occurred later than DOL 28 and 28 days after enrollment (Table S3).

Kaplan–Meier curves of time to diagnosis in cases and controls. The cumulative probability of a diagnosis (Dx) in cases (infants randomized to receive rWGS plus standard genetic tests; shown in red; n = 32) and controls (infants randomized to standard genetic tests alone; shown in blue; n = 33). Differences in probability of receiving a diagnosis were significant between the two arms from day 12–67 after enrollment (a asterisks) and DOL 19 - 99 (b asterisks)
Age at diagnosis and time to diagnosis differed significantly between arms, after accounting for non-proportional rates of diagnosis (Table 4, Table S5): The median age at diagnosis in cases was 25 days (range 14–90 days) vs. median in controls was 130 days (range 37–451). The median time to diagnosis in cases was 13 days (range 1–84 days) vs. median in controls 107 days (range 21–429 days). Two diagnoses explained part of the infant’s disorder rather than the entire presentation. In a sensitivity analysis, when patients adjudged to have a partial diagnosis were considered undiagnosed, age at diagnosis and time to diagnosis were no longer significant (Table 4).
Six other secondary end-points did not differ significantly between arms in an intention to treat analysis (Tables 1, 3, 4, S4). They were the proportion of infants receiving diagnoses of genetic diseases (41% of cases vs. 24% of controls; difference, 16% [95% CI, −6 to 39%])), proportion in whom diagnoses had clinical utility (41% of cases vs. 21% of controls; difference, 19% [95% CI, −3 to 42%]), proportion of infants with a change in medical management (clinical utility, 22% of cases vs. 9% of controls; difference, 13% [95% CI, −5 to 30%]), proportion of patients who received diagnoses prior to hospital discharge (28% of cases vs. 9% of controls; difference, 19% [95% CI, 0–38%]), average length of NICU/PICU stay (average 67 days), 6-month mortality (12%, 8 of 65), and age at death.
Discussion
NICU and PICU infants receiving trio rWGS plus standard clinical testing had a higher rate of genetic diagnosis and shorter time to diagnosis than infants receiving standard tests alone. In intention to treat analysis, rWGS was associated with significantly more genetic diagnoses within 28 days of enrollment (31%, 10 of 32) than standard tests alone (3%, 1 of 33; difference, 28% [95% CI, 10–46%]; p = 0.003). The rate of neonatal (DOL 28) diagnosis was higher in cases (32%, 7 of 22) than controls (0%, 0 of 23; difference, 32% [95% CI, 11–53%]; p = 0.004). Of note, standard genetic testing was ordered an average of 8 days before enrollment, which benefitted the control arm over rWGS cases for these analyses. Nevertheless, age at diagnosis and time to diagnosis were significantly shorter in rWGS cases, after accounting for non-proportional rates of diagnosis.
The rate of genetic diagnosis by rWGS in a NICU or PICU was reported previously in one cohort.6 Enrollment in that study was at average DOL 26 (vs. DOL 22 herein). The rate of diagnosis by rWGS therein was 14% (5 of 35) by DOL 28, and 34% (12 of 35) within 28 days of enrollment, which were similar to herein (32% and 31%, respectively). The total rate of genetic diagnosis by rWGS herein (32%) was within the range reported for WGS and WES studies.3,4,5,6,7, 28,29,30,31,32,33,34,35
Timely return of rWGS diagnoses was limited by two research factors that may not be part of routine clinical practice: firstly, confirmatory testing by “the clinically accepted standard” was required for research rWGS diagnoses—but was not necessarily required for laboratory developed, clinical WGS, WES, and targeted NGS panel tests—which lengthened the time to rWGS diagnosis by 7–10 days. Indeed, all diagnostic rWGS findings in the current study were concordant with orthologous methods. For well covered, pathogenic and likely pathogenic, single nucleotide variants in regions of high WGS quality, a median time-to-result of 5 days is anticipated.6,20, 27 Secondly, enrollment occurred relatively late during the NICU or PICU stay (DOL 22). While parents are interested in receipt of genomic sequencing at birth, an enrollment rate of 6% was reported for WES in NICU infants in another cohort.41, 42 Delay in enrollment herein reflected two logistical factors. First, since a criterion for enrollment was suspicion by the provider of an underlying genetic disease, nomination was often delayed until a genetic test or consult had been ordered. In such cases, the time of enrollment delayed the study test, rWGS, compared to standard testing; nevertheless, there was still a decreased time to diagnosis with rWGS. Secondly, NSIGHT1 required informed consent from both parents; the logistics and complexity of obtaining informed consent in a NICU or PICU setting are arduous. A follow-on study, NSIGHT2, has started in which enrollment occurs close to the day of NICU or PICU admission (ClinicalTrials.gov Identifier: NCT03211039). This was facilitated by simpler enrollment criteria, requirement of informed consent from a single parent, and limiting eligibility for enrollment to within several days of admission.
Since the current study, clinical rWGS has improved with respect to rate of genetic diagnosis and time to diagnosis.27 In particular, the diagnostic rate has increased through ongoing identification of novel disease genes, improved reference genome sequences, and better identification of disease-causing copy number, repeat expansion, regulatory, splicing and structural variations.32, 43,44,45,46,47,48,49,50 These recent advances were not reflected in the current study. Indeed, in three cases herein, causative chromosomal deletions were detected by microarray but not by rWGS. rWGS has recently also become much more feasible in clinical laboratories due to improved throughput of rapid sequencing instruments (Illumina NovaSeq 6000), and the availability of robust commercial interpretation software. Rapid trio exome sequencing (rWES) has also become feasible, and has demonstrated similar performance to rWGS51: In a recent study, rWES revealed a molecular diagnosis in 51% of infants at an average of 33 days of life, and with a mean turnaround time of 13.0 days.51 Randomized, controlled studies are needed that compare the diagnostic and clinical utility and cost effectiveness of rWES and rWGS in NICU and PICU infants
NSIGHT1 was terminated early, primarily due to loss of equipoise noted by some nominating clinicians during the study. Some practitioners grew to regard randomization to standard tests alone to be an inferior intervention than standard tests plus trio rWGS. This was associated with seven (21% of controls) requests to cross-over control infants to the rWGS arm following clinician un-blinding, five of which were granted. It was also associated with a higher rate of order of targeted NGS panels, WES or WGS standard genetic tests in controls (43 tests in 24 controls) than cases (22 tests in 17 cases). Standard genomic sequencing tests accounted for 63% (5) of the eight genetic diagnoses in controls. As a result, there was not a significant difference between arms in the total number of genetic diagnoses, a secondary end-point (41% [13] diagnoses among 32 infants in the rWGS arm, 24% (8) of 33 in controls; difference, 16% [95% CI, −6 to 39%]; p = 0.19). Future pragmatic RCT designs in genomic medicine will require careful attention to the principle of equipoise and to the rapid evolution of clinical NGS-based testing.52, 53 The more widespread use of gene panel testing in the NICU during the course of this study was a significant departure from our experience at study conception. Our study was not intended to evaluate the relative diagnostic yield of panel testing over rWGS. Consequently, the study was not powered to evaluate the non-inferiority of panels over rWGS.
The rationale for rWGS in NICU infants is to enable consideration of acute precision interventions in time to decrease morbidity and mortality.5,6, 21,22,23,24 In two prior studies of genomic sequencing in infants, genetic diagnoses led to precision medicine that was considered life-saving in 5%, and that avoided major morbidity in 6%.6, 7 In those studies, early diagnosis (DOL 49) led to greater implementation of precision medicine (65%) than later diagnosis (DOL 374, 39%), particularly with regard to palliative care guidance. As in the current study, assessments of clinical utility were based on actual changes in management, which were limited by clinician experience with genomic medicine and rare genetic diseases. This is a major challenge for NICU and PICU implementation of genomic medicine for rare genetic diseases.54, 55 Unfortunately, early termination of the current study resulted in loss in power for the secondary end-points: there were not significant differences in the overall rate of clinical utility of diagnoses, length of admission, rate of diagnosis before discharge, mortality and age at death. The clinical utility of diagnoses and rate of diagnosis before hospital discharge trended towards being higher in the rWGS arm (difference, 19% [95% CI, −3 to 42%], p = 0.11, and 19% [95% CI, 0–38%], p = 0.06, respectively). Additional studies are needed to clarify whether shorter time to diagnosis is associated with changes in clinical utility of diagnoses, outcomes, or healthcare utilization.
Conclusions
Among infants with suspected genetic diseases in a regional NICU or PICU, the addition of rWGS decreased the time to diagnosis. Since genetic diseases are among the leading cause of death in the NICU and PICU, as well as overall infant mortality, implementation of rWGS is likely to have broad implications for the practice of neonatology.
Methods
Trial design
NSIGHT1 tested the a priori hypothesis that rWGS increases the proportion of infants receiving a genetic diagnosis within 28 days in a partially blinded, randomized controlled study in a regional NICU and PICU in a tertiary referral children’s hospital (Children’s Mercy—Kansas City, CM-KC)(Fig. 1). Infants were born at CM-KC or transferred from outside birthing hospitals to CM-KC for intensive care at various ages. Inclusion criteria were infants in the NICU or PICU of age less than four months with illnesses of unknown etiology and one of the following: 1. A genetic test order or genetic consult; 2. A major structural congenital anomaly or at least three minor anomalies; 3. An abnormal laboratory test suggested a genetic disease; or 4. An abnormal response to standard therapy for a major underlying condition. Exclusion criteria were a previously confirmed genetic diagnosis that explained the clinical condition, or features pathognomonic for a chromosomal aberration. The NICU census was reviewed daily for eligible infants by enrollment coordinators. The eligibility criteria did not change after trial commencement. NICU clinicians were notified of eligible infants, who were nominated through a standard form. NICU and PICU clinicians notified families of eligible infants about the study, and enrollment coordinators then approached parents for informed consent. Enrolled infants were randomly assigned in a 1:1 ratio to receive standard, clinically determined tests (controls) or standard clinical tests plus trio (infants and parents where available) rWGS for etiologic diagnosis of infants’ underlying conditions (cases; Fig. 1). Randomization was performed automatically by the RAND function in Microsoft Excel at enrollment. Parents and clinicians were initially blinded. However, by day ten they were notified of randomization assignment, to minimize parental anxiety and allow consideration of crossover to rWGS. The study design was adaptive, with modification of enrollment prospectively planned following interim data analysis after approximately 2 years of accrual.
Sample size determination
The study proposed a sample size of 500 in each group (1000 total), with 82% power to detect a difference of 0.05 in the proportion of molecular diagnoses (using a case diagnosis proportion of 0.1 and a control diagnosis proportion of 0.05; two group, continuity-corrected χ2 test with a 0.05 two-sided significance level). We assumed that both primary outcome groups were independent, and contained 500 subjects. We are also interested in comparing the mean time to molecular diagnosis among the two independent study arms. Assuming that 5% of control subjects and 10% of those receiving rWGS receive a molecular diagnosis, the study had more than 98% power to detect the difference in mean time to molecular diagnosis between the cases and controls (96 ± 24 h in rapid whole-genome sequencing cases vs. 240 ± 72 h in controls; two group Satterthwaite t-test with a 0.05 two-sided significance).
Ascertainment of clinical features and study measures
The clinical features of affected infants receiving rWGS were ascertained comprehensively by review of the electronic medical record and discussion with physicians and entered in a study REDCap database.56 Phenotypic features were translated into Human Phenotype Ontology terms and mapped to ~5000 monogenic diseases with the clinicopathologic correlation tools SSAGA, Phenomizer and Phenolyzer, generating rank-ordered, deep differential diagnosis lists27,57, 58 (Table S1). Baseline demographics including age, gender, gestational age, birth weight, APGAR scores, and family history were collected. Other study measures were entered into the REDCap database, including diagnostic tests ordered during hospitalization, changes in clinical management following diagnostic test reporting, length of hospitalization, and mortality. Enrollment was from October 2014 to June 2016, and data collection continued until November 2016.
Trial oversight
The investigators designed the trial in consultation with NICU and PICU staff and program managers of the funding agencies, the National Human Genome Research Institute (NHGRI) and Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD).24 The investigators received a pre-submission opinion from the Food and Drug Administration (FDA), Center for Devices and Radiological Health (CDRH), Office of In Vitro Diagnostics and Radiological Health (OIR), that the study posed a nonsignificant risk for enrollees, and did not need to be performed under an Investigational Device Exemption (FDA/CDRH/OIR submission Q140271, May 8, 2014). The study was approved by the Institutional Review Boards at Children’s Mercy—Kansas City (CM-KC) and Rady Children’s Hospital, San Diego, and conducted in accordance with the Declaration of Helsinki. Data were collected and analyzed by the investigators. All authors participated in the writing of the manuscript and approved the draft that was submitted for publication. The funding sources were not involved in the collection, analysis, or interpretation of the data, or the writing of the report. The first draft of the manuscript was written by the corresponding author. The authors vouch for the accuracy and completeness of the data and data analyses and for the fidelity of the trial to the protocol (ClinicalTrials.gov accession NCT02225522).
Rapid whole-genome sequencing
rWGS was performed under a research protocol and employed 26-h–7-day methods, guided by acuity of illness of the proband as described.5,6,20, 27 When possible, rWGS was performed on specimens from both biological parents and affected infants simultaneously. Of 37 infants receiving rWGS, 31 were analyzed as trios, 3 as mother-infant duos, 2 as singletons, and 1 as a quad with two affected siblings. Genomic DNA was prepared for rWGS using either TruSeq PCR Free (Illumina, San Diego) or KAPA HYPER (KAPA Biosystems), and resultant libraries were quantified by real-time PCR. WGS was with the Illumina HiSeq 2500 (v4 chemistry, 2 × 125 or 2 × 101 nucleotides, nt) in rapid run or high output mode, or HiSeq 4000 (2 × 125 nt). rWGS was to a minimum depth of 90 Gb per genome, and the average genome coverage was 40-fold (Table S2, Figure S1). All samples met established quality metrics.
Rapid WGS analysis and diagnostic interpretation
rWGS were generated with Illumina RTA 1.12.4.2 and CASAVA-1.8.2, and aligned to the human reference genome GRCh37.p5 using GSNAP and bwa-mem v0.7.12. Nucleotide (nt) variants were detected and genotyped with the Genome Analysis Tool Kit (v1.6-13. and v3.2-2).5,6,20, 27 Copy number variants and structural variants were not detected in WGS. Nucleotide variants were annotated with the Rapid Understanding of Nucleotide variant Effect Software (RUNES).5,6, 20,21,22,23,24,25,26,27 Variants were interpreted by board certified molecular geneticists using American College of Medical Genetics guidelines for pathogenic and likely pathogenic classifications.59 Causative variants were identified primarily with Variant Integration and Knowledge INterpretation in Genomes (VIKING) software.5, 6 Inputs for VIKING were the annotated genomic variant file produced by RUNES and a Phenomizer file of the genes on the comprehensive differential diagnosis.5, 6 Alternatively, diagnostic searches utilized pre-calculated candidate gene lists, such as genes with OMIM records or genes associated with mitochondrial disorders. Interpretation considered multiple sources of evidence, including variant pathogenicity, inheritance pattern, strength of disease–gene association, and match of the clinical features of the disease with a deep patient phenotype. All inheritance patterns were examined. Analysis was performed sequentially by two experts. Where a single likely causative variant for a recessive disorder was identified, the locus was manually inspected using the Integrated Genome Viewer in the trio for uncalled variants.60 Expert interpretation and literature curation were performed for likely causative variants with regard to evidence for pathogenicity. The FDA and IRB approved return of verbal, provisional rWGS diagnosis to the treating physician in exceptional cases, where the results were actionable and the infant was imminently likely to die or have worsening morbidity. Familial relationships were confirmed by segregation analysis of variants. All diagnostic genotypes were confirmed by Sanger sequencing prior to final reporting. rWGS and Sanger sequencing were performed in a laboratory licensed by the Clinical Laboratory Improvement Amendments and accredited by the College of American Pathologists. Additional expert consultation and functional confirmation were performed in selected cases when the subject’s phenotype differed from previous mutation reports for that disease gene or when the pathogenicity of variants was uncertain. In the absence of a diagnosis, a research note was placed in the medical record to indicate that testing was complete. At time of study performance, clinical grade detection of copy number and structural variants was not performed. Secondary and incidental findings were not reported.
Standard genetic testing
Standard clinical testing for genetic disease etiologies was performed in infants based on physician clinical judgment, assisted by subspecialist recommendations. Specimens for all standard tests were collected and transported as quickly as possible, and all standard tests were performed as expeditiously as possible. The set of genetic tests considered to be standard was developed by three pediatrician subspecialists. Standard genetic tests were those ordered through the electronic medical record, and included biochemical and immunologic testing for genetic diseases, array comparative genomic hybridization, fluorescence in situ hybridization, high resolution chromosomes analysis, Sanger sequencing of genes, non-expedited proband targeted next-generation sequencing (NGS) gene panels, non-expedited proband whole-exome sequencing (WES), non-expedited proband WGS, methylation studies, and gene deletion/duplication assays (Table S3), as well as Kansas or Missouri state newborn screening (including five lysosomal storage diseases).
Trial end points
The pre-specified primary end point was a comparison of the proportion of patients receiving a molecular diagnosis within 28 days of enrollment. The pre-specified secondary end points were the proportion of patients receiving a molecular diagnosis within the neonatal period (28 days of life), total diagnostic rate, time-to-diagnosis, and percentage of patients with a change in management related to test results in the two arms. Other pre-specified end-points were the length of hospitalization and short term mortality rate. Data related to these end-points were entered into the REDCap database and audited by two study investigators. The end-points were not changed after the trial commenced. Change in management was determined by a survey of nominating clinicians and review of the electronic health record by at least two pediatrician subspecialists with substantial expertise in genomic medicine to identify change in treatments and procedures, canceled tests, new focused tests, recommendation for specific follow-up related to the diagnosis, and changes in consultation related to the diagnosis.61 A modified Delphi method was used to determine inclusion of change in management where there was disagreement.
Statistical analysis
Statistical analyses were based on the intention-to-treat principle to avoid confounding due to the crossover of controls to the rWGS group. Variables representing whether patients received a diagnosis and whether they received a change in care were treated as dichotomous (y/n). Controls were considered diagnosed only if they received a molecular diagnosis from standard tests regardless of whether they were crossed over and received a diagnosis from rapid WGS. Cases were classified as diagnosed if they received at least one diagnosis from either rapid WGS or standard tests.
Primary analyses comparing 28-day diagnostic rates between study arms were conducted using Fisher’s exact test. Differences in secondary endpoints, including total diagnoses, clinical utility of diagnoses, and diagnoses before discharge, were also assessed with Fisher’s exact test. A two-sample t-test was performed to evaluate the null hypothesis of no difference in mean age at hospital discharge for rapid WGS cases and controls.
Kaplan–Meier analyses were used to compare time to diagnosis for each study arm. We generated plots of the cumulative diagnostic rates, estimated as one minus the Kaplan–Meier function, and 95% confidence intervals. For the analysis of age at diagnosis, time was measured from date of birth to date of diagnosis. For the analysis of time to diagnosis from first test, time to diagnosis was measured from the date of enrollment. Patients that did not receive a diagnosis by the end of the study had their data censored at the final date of data collection (November 2016). In general, the log-rank test is most powerful in the presence of proportional hazards. Thus for age at death, for which there was no evidence of non-proportional hazards, the log-rank test was performed.62 When there was evidence of a non-constant hazard ratio, between-group differences were evaluated with the Peto-Peto test.63, 64 The latter was used as an alternative to the log-rank test when comparing time to diagnosis between WGS plus standard tests and standard tests alone because of its increased power, especially when differences are hypothesized to occur early in time.
As a sensitivity analysis, we reclassified patients who received a partial diagnosis as undiagnosed and repeated analyses for relevant primary and secondary end-points. All reported p-values are two-sided and were considered statistically significant if less than 0.05. Statistical analyses were performed in R version 3.3.0.65
Data and material availability
Data are available at LPDR (https://www.nbstrn.org/research-tools/longitudinal-pediatric-data-resource).
Key points
Question: What proportion of acutely ill inpatient infants receive a diagnosis of a genetic disease within 28 days with rapid whole-genome sequencing?
Findings: In a RCT of 65 infants, the diagnostic sensitivity of rapid whole-genome sequencing within 28 days was 31 vs. 3% with standard genetic testing, a significant difference.
Meaning: In NICU and PICU infants with diseases of unknown etiology, rapid whole-genome sequencing may be warranted as a first-line diagnostic test.
References
Green, E. D. & Guyer, M. S. National Human Genome Research I. Charting a course for genomic medicine from base pairs to bedside. Nature 470, 204–213 (2011).
Worthey, E. A. et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet. Med. 13, 255–262 (2011).
Bainbridge, M. N. et al. Whole-genome sequencing for optimized patient management. Sci. Transl. Med. 3, 87re3 (2011).
Dixon-Salazar, T. J. et al. Exome sequencing can improve diagnosis and alter patient management. Sci. Transl. Med. 4, 138ra78 (2012).
Soden, S. et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci. Transl. Med. 6, 265ra168 (2014).
Willig, L. K. et al. Whole-genome sequencing for identification of Mendelian disorders in critically ill infants: a retrospective analysis of diagnostic and clinical findings. Lancet Respir. Med. 3, 377–387 (2015).
Stark, Z. et al. A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet. Med. 18, 1090–1096 (2016).
Manolio, T. A. et al. Global implementation of genomic medicine: We are not alone. Sci. Transl. Med. 7, 290ps13 (2015).
Collins, F. S. & Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 372, 793–795 (2015).
Phillips, K. A. et al. Making genomic medicine evidence-based and patient-centered: a structured review and landscape analysis of comparative effectiveness research. Genet. Med. 19, 1081–1091 (2017).
Roberts, M. C. et al. The current state of implementation science in genomic medicine: opportunities for improvement. Genet. Med. 19, 858–863 (2017).
Xu, J., Murphy, S. L., Kochanek, K. D. & Arias, E. Mortality in the United States, 2015. NCHS Data Brief. 267, 1–8 (2016).
Wilkinson, D. J. et al. Death in the neonatal intensive care unit: changing patterns of end of life care over two decades. Arch. Dis. Child. Fetal Neonatal Ed. 91, F268–F271 (2006).
Hagen, C. M. & Hansen, T. W. Deaths in a neonatal intensive care unit: a 10-year perspective. Pediatr. Crit. Care. Med. 5, 463–468 (2004).
O’Malley, M. & Hutcheon, R. G. Genetic disorders and congenital malformations in pediatric long-term care. J. Am. Med. Dir. Assoc. 8, 332–334 (2007).
Stevenson, D. A. & Carey, J. C. Contribution of malformations and genetic disorders to mortality in a children’s hospital. Am. J. Med. Genet. A 126A, 393–397 (2004).
Lantos, J. D. & Meadow, W. L. Costs and end-of-life care in the NICU: lessons for the MICU? J. Law Med. Ethics 39, 194–200 (2011).
Couce, M. L. et al. Inborn errors of metabolism in a neonatology unit: impact and long-term results. Pediatr. Int. 53, 13–17 (2011).
Weiner, J., Sharma, J., Lantos, J. & Kilbride, H. How infants die in the neonatal intensive care unit: trends from 1999 through 2008. Arch. Pediatr. Adolesc. Med. 165, 630–634 (2011).
Saunders, C. J. et al. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci. Transl. Med. 4, 154ra135 (2012).
Priest, J. R. et al. Molecular diagnosis of long-QT syndrome at 10 days of life by rapid whole genome sequencing. Heart Rhythm 11, 1707–1713 (2014).
Farnaes, L. et al. Rapid whole genome sequencing identifies a novel GABRA1 variant associated with West syndrome. Cold Spring Harb. Mol. Case Stud. 3, a001776 (2017).
Hildreth, A. et al. Rapid whole genome sequencing identifies a novel homozygous NPC1 variant associated with Niemann-Pick Type C1 Disease in a 7 week old male with cholestasis. Cold Spring Harb. Mol. Case Stud. 3, a001966 (2017).
Berg, J. S. et al. Newborn sequencing in genomic medicine and public health. Pediatrics 139, e20162252 (2017).
Downing, G. J., Zuckerman, A. E., Coon, C. & Lloyd-Puryear, M. A. Enhancing the quality and efficiency of newborn screening programs through the use of health information technology. Semin. Perinatol. 34, 156–162 (2010).
OMIM. Online mendelian inheritance in man, OMIM. http://www.omim.org/ (2017).
Miller, N. A. et al. A 26-hour system of highly sensitive whole genome sequencing for emergency management of genetic diseases. Genome Med. 7, 100 (2015).
Vissers, L. E. et al. A clinical utility study of exome sequencing versus conventional genetic testing in pediatric neurology. Genet. Med. 19, 1055–1063 (2017).
Stavropoulos, D. J. et al. Whole-genome sequencing expands diagnostic utility and improves clinical management in paediatric medicine. npj Genom. Med. 1, 15012 (2016).
Tarailo-Graovac, M. et al. Exome sequencing and the management of neurometabolic disorders. N. Engl. J. Med. 374, 2246–2255 (2016).
Baldridge, D. et al. The exome clinic and the role of medical genetics expertise in the interpretation of exome sequencing results. Genet. Med. 19, 1040–1048 (2017).
Ellingford, J. M. et al. Whole genome sequencing increases molecular diagnostic yield compared with current diagnostic testing for inherited retinal disease. Ophthalmology 123, 1143–1150 (2016).
Farwell, K. D. et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet. Med. 17, 578–586 (2015).
Taylor, J. C. et al. Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat. Genet. 47, 717–726 (2015).
Valencia, C. A. et al. Clinical impact and cost-effectiveness of whole exome sequencing as a diagnostic tool: a pediatric center’s experience. Front. Pediatr. 3, 67 (2015).
Olesen, M. S. et al. High prevalence of long QT syndrome-associated SCN5A variants in patients with early-onset lone atrial fibrillation. Circ. Cardiovasc. Genet. 5, 450–459 (2012).
Albert, C. M. et al. Cardiac sodium channel gene variants and sudden cardiac death in women. Circulation 117, 16–23 (2008).
Arnestad, M. et al. Prevalence of long-QT syndrome gene variants in sudden infant death syndrome. Circulation 115, 361–367 (2007).
Wang, D. W. et al. Cardiac sodium channel dysfunction in sudden infant death syndrome. Circulation 115, 368–376 (2007).
Bébarová, M. et al. Subepicardial phase 0 block and discontinuous transmural conduction underlie right precordial ST-segment elevation by a SCN5A loss-of-function mutation. Am. J. Physiol. Heart Circ. Physiol. 295, H48–H58 (2008).
Green, R. C. et al. The BabySeq Project: Preliminary findings from a randomized trial of exome sequencing in newborns. Presented at the American Society of Human Genetics 2016 Annual Meeting. Vancouver, B.C. https://ep70.eventpilot.us/web/page.php?page=IntHtml&project=ASHG16&id=160122602 (2016).
Waisbren, S. E. et al. Parents are interested in newborn genomic testing during the early postpartum period. Genet. Med. 17, 501–504 (2015).
Eldomery, M. K. et al. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 9, 26 (2017).
Känsäkoski, J. et al. Complete androgen insensitivity syndrome caused by deep intronic pseudoexon-activating mutation in the androgen receptor gene. Sci. Rep. 6, 32819 (2016).
Hartmannová, H. et al. Acadian variant of Fanconi syndrome is caused by mitochondrial respiratory chain complex I deficiency due to a non-coding mutation in complex I assembly factor NDUFAF6. Hum. Mol. Genet. 25, 4062–4079 (2016).
Smedley, D. et al. A whole-genome analysis framework for effective identification of pathogenic regulatory variants in Mendelian disease. Am. J. Hum. Genet. 99, 595–606 (2016).
Noll, A. C. et al. Clinical detection of deletion structural variants in whole-genome sequences. npj Genom. Med. 1, 16026 (2016).
Schneider, V. A. et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 27, 849–864 (2017).
Kojima, K. et al. STR-realigner: a realignment method for short tandem repeat regions. BMC Genom. 17, 991 (2016).
Dolzhenko, E. et al. Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res. 27, 1895–1903 (2017).
Meng, L. et al. Use of exome sequencing for for infants in intensive care units: ascertainment of severe single-gene disorders and effect on medical management. JAMA Pediatr. 171, e173438 (2017).
London, A. J. Equipoise in research: integrating ethics and science in human research. JAMA 317, 525–526 (2017).
Lilford, R. J. & Jackson, J. Equipoise and the ethics of randomization. J. R. Soc. Med. 88, 552–559 (1995).
Bowdin, S. et al. Recommendations for the integration of genomics into clinical practice. Genet. Med. 18, 1075–1084 (2016).
Gaff, C. L. et al. Preparing for genomic medicine: a real world demonstration of health system change. npj Genom. Med. 2, 16 (2017).
Harris, P. A. et al. Research electronic data capture (REDCap)—a metadata-driven methodology and workflow process for providing translational research informatics support. J. Biomed. Inform. 42, 377–381 (2009).
Köhler, S. et al. Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am. J. Hum. Genet. 85, 457–464 (2009).
Yang, H., Robinson, P. N. & Wang, K. Phenolyzer: phenotype-based prioritization of candidate genes for human diseases. Nat. Methods 12, 841–843 (2015).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Thorvaldsdottir, H., Robinson, J. T. & Mesirov, J. P. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 14, 178–192 (2013).
ACMG, Board of Directors. Clinical utility of genetic and genomic services: a position statement of the American College of Medical Genetics and Genomics. Genet. Med. 17, 505–507 (2015).
Grambsch, P. M. & Therneau, T. M. Proportional hazards tests and diagnostics based on weighted residuals. Biometrika 81, 515–526 (1994).
Peto, R. & Peto, J. Asymptotically efficient rank invariant test procedures. J. R. Stat. Soc. Ser. A 135, 185–207 (1972).
Leton, E. & Zuluaga, P. Equivalence between score and weighted tests for survival curves. Commun. Stat. Theory Methods 30, 591–608 (2001).
R Core Team. R: A language and environment for statistical computing. (R Foundation for Statistical Computing, Vienna, 2016).
Acknowledgements
We thank Drs. John Lantos, Sarah Soden and Howard Kilbride for advice and assistance. A Deo lumen, ab amicis auxilium. Grant U19HD077693 from NICHD and NHGRI. NICHD and NHGRI programmatic staff assisted investigators during performance of the research activities. Funders had no role in the collection, management, analysis, or interpretation of the data; preparation, review, or approval of the manuscript; or decision to submit the manuscript for publication
Author information
Authors and Affiliations
Contributions
Concept and design: S.F.K., L.K.W., J.E.P., J.A.C. Acquisition, analysis or interpretation of data: all authors. Drafting of manuscript: S.F.K., J.E.P., M.M.C., N.M.S., J.A.C. Critical revision of the manuscript for important intellectual content: L.K.W., E.G.F., L.D.S., D.P.D., C.J.S., I.T., N.A.M., L.Z., S.M.H., A.M.H., S.B., N.V., J.S.L. Statistical analysis: M.M.C., SB. Obtained funding: S.F.K., L.K.W., J.S.L. Administrative, technical or material support: S.F.K., J.A.C. Study supervision: J.E.P., S.F.K., J.A.C.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Petrikin, J.E., Cakici, J.A., Clark, M.M. et al. The NSIGHT1-randomized controlled trial: rapid whole-genome sequencing for accelerated etiologic diagnosis in critically ill infants. npj Genomic Med 3, 6 (2018). https://doi.org/10.1038/s41525-018-0045-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-018-0045-8
This article is cited by
-
Evidence review and considerations for use of first line genome sequencing to diagnose rare genetic disorders
npj Genomic Medicine (2024)
-
Role of next generation sequencing in diagnosis and management of critically ill children with suspected monogenic disorder
European Journal of Human Genetics (2024)
-
Rapid genomic sequencing for genetic disease diagnosis and therapy in intensive care units: a review
npj Genomic Medicine (2024)
-
Whole genome sequencing diagnostic yield for paediatric patients with suspected genetic disorders: systematic review, meta-analysis, and GRADE assessment
Archives of Public Health (2023)
-
Challenges and recommendations to increasing the use of exome sequencing and whole genome sequencing for diagnosing rare diseases in Brazil: an expert perspective
International Journal for Equity in Health (2023)
