
Abstract
We introduce three developments within the stochastic many-body perturbation theory: efficient evaluation of off-diagonal self-energy terms, construction of Dyson orbitals, and stochastic constrained random phase approximation. The stochastic approaches readily handle systems with thousands of atoms. We use them to explore the electronic states of twisted bilayer graphene (tBLG) characterized by giant unit cells and correlated electronic states. We document the formation of electron localization under compression; weakly correlated states are merely shifted in energy. We demonstrate how to efficiently downfold the correlated subspace on a model Hamiltonian with a screened frequency-dependent two-body interaction. For the 6° tBLG system, the onsite interactions are between 200 and 300 meV under compression. The Dyson orbitals exhibit spatial distribution similar to the mean-field single-particle states. Under pressure, the electron-electron interactions increase in the localized states; however, the dynamical screening does not fully balance the dominant bare Coulomb interaction.
Similar content being viewed by others

Introduction
First-principles many-body theory provides invaluable insights for predicting and deciphering the behavior of electronic states without relying on empirical parameters. In particular, one encounters the largest need for predictive first principles theory in systems with emergent phenomena. For instance, the coupling between nominally weakly correlated subsystems may lead to new states exhibiting all hallmarks of strong correlations. Describing such phenomena is often hindered by the large system sizes that pose an insurmountable challenge for conventional calculations. Here, by presenting computational developments we expand our previous work that enabled the application of the ab initio many-body theory to giant systems1.
We exemplify the methodology by studying twisted bilayer graphene (tBLG), which is a prototypical moiré superstructure in which the coupling of individual monolayers is controlled primarily by the twist angle, θ2. As θ approaches 1.1° “magic angle”, tBLG transitions from a simple semimetal to a system hosting correlated electronic states2. Under charge carrier doping tBLG at (or near) the magic angle exhibits superconducting, insulating, and magnetic properties2,3,4,5,6,7,8,9,10,11,12. These emergent states are associated with a shallow moiré potential, which localizes electrons in so-called AA stacking regions of the superstructure11,13,14,15,16,17 and is responsible for the formation of flat (i.e., dispersionless) bands near the Fermi level18,19.
Fundamentally, the electron localization is governed by the strong interaction between the monolayers and the states’ hybridization near the respective Dirac points. On the one hand, this is realized at small twists near magic angle, but equally well by the bilayer’s in-plane strain or compression, as was shown by recent experiments and theoretical works4,5,20. In the latter case, the interlayer distance reduction drives electronic localization for angles substantially larger than 1.1°4,5. Unlike the twist angle, the degree of compression can be adjusted even after the deposition of the layers and hence represents a unique control mechanism for realizing correlated states. Yet, it has been so far studied to a lesser degree.
At high pressures, graphene becomes thermodynamically unstable; a suitably chosen combination of pressure transmission media and encapsulation allows reaching pressures up to 37 GPa21,22 for tBLG. Even higher compression may be possible for up to 50 GPa before graphene inevitably transforms to diamond22. Nevertheless, even with the improved experimental setup, the tBLG electronic states cannot be probed in the same detail as at ambient conditions21,22. Further, it is unknown whether the decreasing interlayer spacing affects the entire valence and conduction states or leads to selective hybridization of Dirac point states. Finally, weakly correlated states dynamically screen the many-body interactions within the flat bands and critically affect the emergent many-body phenomena17,23; however, the effect of compression on screening is also unknown.
Previous studies of the tBLG electronic structure were limited to tight-binding and continuum models14,15,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45 that demonstrated the magic angle-induced flat band formation at the Fermi level. The model parameters were usually determined from mean-field (DFT) calculations, which markedly deviate from quasiparticle energies46. The ground state of tBLG at the magic angle was further investigated by atomistic Hartree and Hartree-Fock calculations based on the continuum model45,47,48,49,50,51,52,53,54. These calculations revealed that unscreened Coulomb interactions are responsible for stabilizing the insulating states in tBLG. Recently, exact diagonalization of downfolded many-body Hamiltonians (within a subspace of flat bands) was used to address the superconducting regime of tBLG55. Investigations of the high-pressure behavior remain scarce and limited to the MF or model Hamiltonian treatment56,57,58,59,60,61, which focused on describing the dispersion of states near the Fermi level. Further, the strength of the electron-electron interaction at high pressure was investigated neither. Thus, it remains unclear whether the flat bands’ formation under compression is equivalent to that at (or near) the magic angle. While these questions can be answered by the first principles many-body approaches, they were not applied until now due to their enormous computational cost.
In this work, we overcome practical limitations of ab initio many-body method: we propose a series of developments in the stochastic many-body perturbation theory (MBPT) techniques62,63,64,65,66,67 which can readily elucidate how the electronic structure behaves in giant moiré systems. We investigate tBLG with a large twist angle of θ ≈ 6∘ (with the supercell of size 4 × 7 nm containing 2184 atoms, i.e., 8736 valence electrons), which is weakly correlated at ambient conditions but develops flat bands at high compressions. The pressure-induced coupling of the two monolayers is substantially different and affects only states near the Fermi level. Further, we develop a stochastic constrained random-phase approximation (s-cRPA), which efficiently (i.e., with minimal computational cost) maps the correlated subspace on a Hubbard model with dynamical on-site interactions, U(ω). We find that the electron-electron interactions in the flat bands are more screened under compression. However, the screening does not fully cancel out the bare Coulomb interaction. Thus, the effective interaction increases with pressure. As a result, the strong correlation is not only driven by vanishing band dispersion but also by increased on-site terms. These results are the same for Dyson quasiparticle orbitals and mean field canonical single-particle orbitals. We present our results first, then their significance and implications; the theory is discussed in the Methods at the end of this work.
Results
The simulations employ rectangular supercells of the graphene bilayer with twist angles θ = 0∘ (24 × 12 conventional unitcells with 9216 valence electrons in total) and θ ≈ 6∘ (1 × 3 moiré conventional cells or ~16.5 × 16.5 in terms of conventional unitcells with 8736 valence electrons in total). See Supplementary Note 1 and Supplementary Fig. 2 for more details about the construction of the commensurate tBLG supercells. Within our real-space methodology, the Brillouin zone of the supercell is sampled by the Γ-point. The 1 × 3 moiré supercell makes our real-space grid commensurate with high-symmetry \(\tilde{K}\)-point (coinciding with the Dirac point location), where the electronic localization principally occurs. We extract bandstructures with the projector-based energy-momentum analysis1,68,69,70,71,72. We study undoped tBLG, and thus, the Dirac point in our calculations is aligned with the Fermi level. Further, spin and valley symmetry breaking are not considered here. The ideal bilayer interlayer distance was first optimized with the first-principles DFT calculations with van der Waals corrections. For simplicity and to separate out the effects of electron-electron interactions treated by our methodology, we employed flat geometry, as lattice reconstructions are significant for small twist angles73,74,75. The equation of state is extracted from the total energy calculations for bilayers with variable interlayer distances (the details of the ground state calculations and the pressure estimation are provided in the “Methods” and Supplementary Fig. 1). Our results are in excellent agreement with previous theoretical calculations employing weak interlayer interactions using the random phase approximation76.
Pressure-induced localization
This section provides computational results obtained with the stochastic GW approximation (see “Methods”) for both diagonal and off-diagonal parts and discusses the role of the orbital basis. We will first report results for the ideal Bernal-stacked graphene bilayer and then for the tBLG.
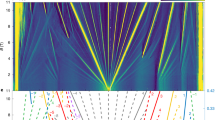
As a first step, we investigate the role of non-local and dynamical correlations in an ideal graphene bilayer. We compare the mean-field DFT and QP energies computed using the diagonal approximation to the self-energy (i.e., Δ = 0 in Eq. (2) of “Methods”). The corresponding bandstructures and the QP densities of states (DOS) are in Fig. 1a–c. As expected77,78,79, MBPT significantly increases the bandwidth (compared to DFT), leading to an excellent agreement with the available experimental data80,81.

Bandstructure of ideal bilayer graphene computed with a DFT and b GW. Due to the rectangular cell, corresponding bands are refolded onto Γ-point. Lines in band structure are guides for the eyes. c Comparison of DFT and GW DOS. Red arrow indicates where should be an experimental peak corresponding to the flattening of the band in ARPES in refs. 80,81. The onset figure schematically shows a comparison of the rectangular and hexagonal first Brillouin zones. d Pressure--interlayer distance (compression) curve for ideal bilayer graphene. Ref. 76. e Charge density of the Dirac point KS states at corresponding pressures of 6° tBLG. The isovalue is the same for all density plots.
Specifically, the experiments show a local minimum of a non-degenerate Dirac band at the M high-symmetry point, which is visible as a peak in the QP DOS at −2.93 eV (red arrow in Fig. 1c). In the bandstructure for the rectangular cell in Fig. 1c (obtained from momentum space projection1,68,69,70) the Γ and M points coincide (due to the Brillouin zone reflection as depicted in the inset figure). The DFT calculation places the peak in DOS incorrectly at −1.94 eV (i.e., 1.0 eV too close to the Fermi level, as shown by the black dashed line and an arrow). This agrees with previous DFT calculations that also underestimated the band dispersion by 10–20% with respect to the experiments11,77,78,79,80,82,83. In contrast, our GW results predict a corresponding feature to appear at −3.11 eV (blue dashed line and an arrow), which is in excellent agreement with the experiment (the peak is placed only 0.18 eV lower than the measured value).
Next, we explore tBLG systems under various pressures ranging from 0 up to 100 GPa (corresponding to maximal compression of 28% of the interlayer distance—see Fig. 1d). The structure is characterized by a hexagonal symmetry with a periodicity of 23.4 Å between the AA stacking regions. Since the twisting angle is high, the coupling between the individual graphene layers is small at ambient conditions, and the system does not differ substantially from the ideal bilayer. First, the electronic states at the Dirac point are fairly delocalized (Fig. 1e), and the distinction between the increase of the orbital density in the AA region is hardly noticeable. Second, (due to the lack of localization) band dispersion is large, and there is no increase in the density of states at the Fermi level (Fig. 2).

a qpDOS of a tBLG θ ≈ 6∘ as a function of pressure. The stochastic error on identifying the quasiparticle energy for the stochastic GW calculation is ~20 meV. b DFT DOS. Both DFT and qpDOS were constructed with Gaussian functions centered at each state (with broadening of 0.35 eV), for more details about qpDOS construction see Supplementary Note 2 and Supplementary Fig. 3 in the Supplementary Information file. (*) In Supplementary Fig. 4 of the Supplementary Information file we show that the DOS eigenenergies are well converged with the supercell size, we note, however, that some k-points are incommensurate with our real-space sampling grids, thus, some DOS features such as van Hove singularities124 are missing.
The situation changes with the compression (for the pressure of 20 GPa and higher). The electronic states become more localized in the AA stacking areas. Already at 20 GPa we clearly observe the spatial redistribution of the orbital density (see illustration in Fig. 1e). In our calculations, we further explore even higher pressures which may be difficult to realize experimentally (though the highest reported pressures achieved for tBLG was P = 37 GPa21,22) With increasing P, localization becomes even more pronounced, and at 100 GPa, roughly 75% of the Dirac point states’ orbital density is localized within 8 Å radius around the AA stacking point. This localization of the Dirac point states translates into the flat-band formation, corresponding to a peak around the Fermi level DOS (Fig. 2). Note that the structure at the pressure of 100 GPa is thermodynamically unstable. Still, the observations are useful as an indicator of the electronic correlation in tBLG: based on experiments5, the same type of localization is expected for lower θ angles at much lower pressures for which graphene is a stable polymorph.
Note that the localization illustrated here lacks contributions beyond those included in the mean-field (DFT) Hamiltonian. In practice, the confinement of orbitals under pressure is driven by the external (ionic) potential and the degree of the localization is impacted by the delocalization error of semilocal DFT functionals. We address this question below and show that the mean field DFT orbitals, however, are remarkably close to the Dyson orbitals computed with MBPT.
Mean-field vs MBPT energy spectrum
At this stage, we compare the first-principles results obtained with the mean-field (DFT) and MBPT (GW) approaches (Fig. 2). While the single particle energies are converged with respect to the supercell (see Supplementary Fig. 4), the DOS curves, however do not have sufficient resolution to capture certain details, such as the van Hove singularities near the Fermi level84. We can extract their position, since it coincides with the energy of the M critical points of the Brillouin zone, which are refolded on the Γ-point. DFT places the singularity at 0.37 eV away from the Fermi level. In contrast, GW positions them at 0.55 eV away from the Fermi level, in excellent agreement with the value of 0.56 eV obtained experimentally84 (see Supplementary Fig. 4). Overall, Fig. 2 shows that MBPT significantly increases the bandwidth and widens the DOS features at 0 GPa compared to the mean-field solution. However, the most apparent changes are for the high pressures at which the DFT DOS significantly contracts and predicts a strong reduction in the width of all bands. While the flat-band formation leads to a peak at the Fermi level, its signature is suppressed by the proximity of the entire set of top valence and bottom conduction bands, which become closer in energy. The DFT results show that occupied and unoccupied states’ behavior is mostly symmetric around the chemical potential (moving up and down in energy, respectively).
The many-body calculations show a different picture. Up to 50 GPa, the entire QP DOS shifts up in energy, i.e., the valence states are moving closer to the Fermi level while the conduction states away from it. The bandwidth of the states away from the Fermi level is mostly unaffected by the increased pressure. Simultaneously, we observe the flat band formation around the chemical potential (indicated by a red arrow in Fig. 2), which does not overlap with the rest of the occupied and unoccupied states. The QP DOS peak comprises eight quasi-degenerate states (corresponding to the Dirac points at K and K′ in the moiré hexagonal Brillouin zone). Increasing the pressure further (i.e., P > 50 GPa) leads to more pronounced structures in the QP DOS, but the peaks’ position remains roughly the same. This difference in the behavior can be understood from the compression curve shown in Fig. 1d: the change of the pressure between 50 and 100 GPa requires only a small decrease in the interlayer distance, i.e., small change of the coupling of the monolayers. For 100 GPa, the flat band is clearly visible in between the conduction and valence bands. The key observation is that the compression-driven flat bands’ formation leaves the rest of the states largely unaffected. Hence, despite the reduced interlayer spacing leads to the electron localization in the vicinity of the Dirac point, the increased coupling between the graphene monolayers is confined to a narrow energy range of the states near the chemical potential.
Role of the off-diagonal self-energy elements
To explore the mutual coupling in the localized moiré states, we investigate the role of off-diagonal self-energy terms \({{{\Sigma }}}_{j\ne k}=\left\langle {\phi }_{j}\right|{{\Sigma }}(\omega )\left|{\phi }_{k}\right\rangle\) in Eq. (2), using our development to the stochastic GW described in the “Methods” section. In practice, we compute the contributions of Δj for the states up to ±0.5 eV away from the highest occupied state. We note that the Σij is computed for a wide frequency range for all off-diagonal terms at once. The cost of the GW calculation is practically identical to the previously developed diagonal implementation (see “Methods” section). Next, we resort to a common procedure in the self-consistent GW85,86 and construct a symmetrized and self-adjoint quasiparticle Hamiltonian H as:
The QP energies of H, however, differ only negligibly from those computed with the diagonal approximation to Eq. (2). We find that the difference for most of the states is less than 1% and only for three states it is roughly 3%. The inclusion of the Δj terms can thus be safely neglected in the QP DOS. This is true also for the flat bands around the Fermi level. Here, the off-diagonal terms are not necessarily small, but they tend to “average-out,” and their net contribution is negligible.
While the QP energies remain unaffected, this does not imply that the QP states, {ψ}, are close to the mean-field DFT orbitals {ϕ}. To investigate this, we diagonalize Hjk (Eq. (1)) for the states in the 0.5 eV vicinity of the Fermi level. Employing the DFT orbitals’ basis, the new eigenstates are ψj = ∑kCkjϕk, with ∑k∣Ckj∣2 = 1. The expansion coefficients are illustrated graphically in Fig. 3. For most states, the Ckj matrix is practically diagonal. On the other hand, the nearly degenerate flat-band states, show substantial mixed character around the Fermi level and ∑k∈{φ}∣Ckj∣2 = 0.995, where {φ} denotes a subspace of eight correlated states at the Fermi level. Nevertheless, each of them has a dominant contribution from a single DFT eigenvector. Indeed, the visual comparison (inset of Fig. 3) shows that {ψ} and {ϕ} are generally similar, e.g., both are localized in the AA stacking regions.

Coefficient matrix obtained from the exact diagonalization of the 51 × 51HQP at 100 GPa pressure. The absolute values of the complex coefficients are plotted. A zero position is set between occupied (negative) and unoccupied (positive) states. With a black circle we indicate 8 Dirac point states that are quasi-degenerate in energy. Inset: We plot a density of state −3 to compare a KS orbital to the QP orbital. State −3 is highlighted with a dotted line in the figure.
We conclude that the stochastic approach efficiently computes both diagonal and off-diagonal terms (at the same cost). Further, our analysis shows that the off-diagonal terms have little impact on the QP density of states and that the pressure-induced coupling is limited to the subset of quasi-degenerate Dirac point states. The differences in the distribution of the single-particle orbitals are visually small, but a more quantitative analysis is provided in the next section.
Pressure dependence of the dynamically screened on-site interaction
In the remainder of the paper, we will demonstrate the stochastic methodology for extracting the dynamically screened on-site interaction U(ω), Eq. (10). With this approach we will explore the role of screening on the orbitals around the Fermi level at various pressures.
We map the strongly correlated states (identified in the previous section and denoted {φ}) onto the Hubbard Hamiltonian (see section “Stochastic Hamiltonian downfolding”), with the effective hopping, t, and on-site interaction, U, terms. The latter contains the information of all other electrons via the non-local and dynamical screening \(\tilde{W}\) in Eq. (10). The electron correlation is commonly characterized by the interplay of the on-site interaction and the kinetic energy38,87,88. In practice, the U ≫ t indicates the regime when the system is strongly correlated. For tBLG close to the magic angle, the strongly correlated regime was driven by the drastic reduction of the hopping (due to localization) to t≤30 meV60. As a result, the U/t ratio becomes very high2,17,87,89, although the screening was predicted to play an important role in reducing the value of the onsite Coulomb term U. Previous calculations suggested that the physics is dominated by the competition between low t and dynamical screening: the dielectric constant was predicted to be 20 times larger at the magic angle than in the ideal bilayer17.
We estimate only the upper bound for the t parameter from the localized states’ bandwidth, extracted17,87,90 from the dispersion of the corresponding QP energies. The hopping term is 1/6 of the bandwidth associated with the flat bands. For the system with the most pronounced localization, i.e., at 100 GPA, we find t ≈ 40 meV, which is in good agreement with the results for the correlated phase at much lower twisting angles. We note that, in contrast, the band dispersion at 0 GPa is very large and t ~ 600 meV. Clearly, the pressure-induced localization is responsible for qualitative changes in the hopping (and the associated t parameter decreases by order of magnitude).
Although the band flattening appears as the primary driver of electron-electron correlation, the on-site Coulomb interaction changes are equally important. Following the approach of refs. 88,91, we provide the mean U values in order to describe the correlation strength of the chosen subspace by a single number rather than comparing U/t ratios for each state. However, we also discuss the variation of U among distinct orbitals. In the text below, we first consider the total screened effective interaction U(ω). Next, we decompose U(ω) into the static bare contribution Ub and dynamically screened counterpart Up(ω). Note, while the U is computed in the basis of correlated states, the idea of the scRPA is to include the dynamical renormalization due to screening from all electronic states in the orthogonal complement of the correlated subspace. The set of localized KS orbitals is a straightforward and convenient choice of basis for scRPA calculations since no additional localization or orthonormalization procedure is required91. The approach to compute U in the KS basis has been previously used in refs. 88,92, but other options (e.g., Wannier basis15) have been proposed. For simplicity, we resort to the first option and compute U in the basis of KS orbitals {ϕ}, next we compare the results to analogous calculations for QP orbital basis {ψ}.
Computed for the Dirac point states, see Eq. (10), the effective interaction U(ω), is screened by all the weakly interacting electrons confined to both monolayers. To account for the dynamical screening, we employ a set of random states which sample the dynamics of all weakly correlated states. The resulting U(ω) converges extremely fast (only 8 random vectors are necessary to yield a negligible stochastic error of 1 meV for a wide range of frequencies—see Fig. 4) with minimal computational requirements (<120 CPU-hours; see section “Stochastic Hamiltonian downfolding”). The frequency dependence of U(ω) is illustrated in Fig. 4. In practice, similar to refs. 91,92, we will discuss the static limit (ω → 0) since there is no elegant mathematical framework to solve the effective Hamiltonian with the frequency-dependent U. For the tBLG at 0 GPa the total screened interaction U(ω → 0) = 202 meV. This is identical to the result for the uncompressed ideal bilayer at 0 GPa where U(ω → 0) = 201 meV. Clearly, our initial assertion holds, and the 6°-tBLG indeed behaves like an ordinary bilayer at ambient conditions.

a Screened and bare U(ω = 0) as a function of pressure for twisted bilayer graphene and screened for the ideal bilayer graphene. Curve labeled with orange triangles is computed in the basis of DFT subspace orbitals, and the curves labeled with violet circles are in the QP basis. The lines connecting the mean values of bare and screened U(ω = 0) are guide for the eyes. Stochastic error in determining the U is smaller than the marker size. The shaded green and orange area provides the standard deviation from a mean value due to the difference of U(ω = 0) for individual states within a correlated subspace. b Frequency dependence of the screened U(ω) computed in the KS basis at 0 (red) and 100 (blue) GPa pressures. Different curves within one color represent individual 8 states within a correlated subspace. The colors of the curves match the triangles' colors from the left panel at 0 and 100 GPa.
Under pressure, the screened on-site interaction in tBLG increases (as much as by ≈ 40% for the highest compression). At 100 GPa, the screened U = 282 meV corresponds to a large ratio U/t ≈ 7 that suggests strongly correlated behavior. This is in striking contrast to the ideal bilayer, for which the U parameter remains practically constant (see Fig. 4). One can intuitively understand the difference in the on-site interaction behavior in tBLG and ideal bilayer from the charge density distributions of the DP states: in tBLG, the electrons in the DP states are trapped in the shallow moiré potential and become confined between the monolayers. The pressure-induced localization leads to increased on-site interaction; the weakly correlated states, on the other hand, remain spread over the entire system. In contrast, the DP electrons in the ideal bilayer experience no localizing potential. Even under compression they remain fully delocalized in the in-plane direction, and their distribution is little affected along the normal to the bilayer. Further, the twist is a necessary prerequisite of coupling between layers since it allows to form energetically degenerate states at K and K′ points of the Brillouin zone. While in the ideal bilayer, K points of two Brillouin zones appear on top of each other forcing the energetic gap between corresponding states, and forbidding them from coupling. Hence, the on-site term in the ideal bilayer is insensitive to pressure.
From the previous analysis (section “Role of the off-diagonal self-energy elements”) we saw that the Dyson orbitals {ψ} are similar to the canonical KS states. Indeed, if we employ the approximate Dyson orbitals instead of the KS states, the U parameter is only insignificantly smaller even at high pressures (cf. Fig. 4): at 100 GPa, the on-site term for the {ψ} states is 260 meV, and the U/t is ≈ 6.5. For this analysis, we use the mean values of the U parameters. However, to emphasize that the individual states of the correlated subspace yield different values of U, we also provide the standard deviation with a shaded area (cf. Fig. 4, left panel). The spread of the individual U values is particularly pronounced for higher pressures (cf. Fig. 4, right panel) and can be explained by lifting the degeneracy within the correlated subspace. In practice, QP and KS orbitals thus produce very similar U interaction. This can be easily understood, given that {ψ} and {ϕ} are very similar.
Finally, the right panel of Fig. 4 shows the frequency dependence of U(ω) for each DP state in tBLG at 0 and 100 GPa. Two main effects of the decreasing interlayer distance can be observed: a vertical shift of the entire U(ω) curve, and an eight-fold increase of the magnitude of the oscillations.
To explore the U(ω) pressure behavior, we now turn to the analysis of the bare and polarization terms, Ub and Up(ω). We find that the (average) values of the bare term are large: when the DFT states are used, Ub = 232 meV at 0 GPa and it increases to Ub = 480 meV at 100 GPa (cf. Fig. 4). The approximate Dyson orbitals yield similar values of 230 meV and 412 meV at 0 and 100 GPa (not shown). Clearly, the difference between the bare terms partially accounts for the discrepancy between the average U values for the {ϕ} and {ψ} states.
The dynamical component of the on-site interaction, Up(ω) contains the effect of the dynamical charge density fluctuations of all the weakly correlated states (outside of the correlated subspace); it is computed via s-cRPA (see the Methods sections). Up(ω) increases under pressure and renormalizes the bare term: the low frequency limit Up(ω → 0) at 0 GPa is −30 meV and at 100 GPa it is −200 meV. Apparently, this screening is not enough to entirely cancel out the Ub contribution. Therefore, the total screened interaction U(ω → 0) is driven by the prevalent bare term and grows with compression. We also see that the magnitude of features in Up(ω) curve increases as the (occupied) states shift closer to the Fermi level. Note that this result is independent of the {ϕ} or {ψ} basis. When computing the screening for the approximate Dyson orbitals, ψ, the weakly correlated subspace remains identical, since ψ is composed of the combination of the quasi-degenerate states.
From the computational prospective we conclude that the stochastic approach is extremely efficient and yields U(ω) for even extremely large systems while requiring only minimal computational resources. This method enabled efficient downfolding and first principles calculations of the renormalized on-site interactions in undoped tBLG supercells.
Discussion
We presented and applied several numerical developments within the stochastic many-body theory which efficiently treat even large systems. We illustrated the method on large scale many-body calculations for twisted bilayer graphene with nearly 9000 valence electrons. We have expanded the stochastic computational toolkit to address the role of off-diagonal self-energy and the basis representation. Further, we have developed stochastic s-cRPA, enabling the downfolding of even giant systems onto model Hamiltonian problems. Our stochastic approaches are applicable to general systems and will find wide application in a wide variety of condensed matter problems.
Our GW results show an excellent agreement with the experimental positions of the van Hove singularities. We also show the formation of electronic localization under compression of the tBLG, which is in agreement with available experimental data and indicate that the compression provides a unique path towards controlled coupling of monolayers and practical realization of moiré states. For systems that are weakly correlated at ambient conditions, the decreased interlayer spacing leads to the formation of flat bands associated with strong correlations. These localized states are found in the vicinity of the Fermi level. In contrast, the majority of the states (delocalized over the individual monolayers) are weakly correlated and remain practically unaffected by the compression.
We compare then effects of pressure to those by varying twist angle on the interplay between screening and electron-electron interaction. The previous theoretical investigations showed that the screening has a crucial role in reducing the on-site interactions, U, as the system approaches the magic twist angle. Thus, the correlations (characterized by large U/t ratios) are primarily driven by the vanishing dispersion (i.e., t → 0) of the states near the Fermi level16,17,23,93,94,95,96,97. In contrast, our stochastic calculations reveal a different scenario: the dynamical screening of the on-site term, U, increases relatively slowly with pressure and the effect of the screening does not fully compensate the bare term. Due to the interlayer coupling, the localized states are strongly affected by compression, and the electronic correlation stems from both small t and large U.
Our calculations indicate that dynamical electronic correlations lead to only small changes in the single-particle orbitals. Consequently, the corresponding U/t ratios in KS and QP basis are practically the same.
By neglecting the graphene layer reconstruction (i.e., in the absence of corrugation) we overestimate the screening effect: structural relaxations in tBLG tend to separate the flat bands from the rest of the spectrum (~20 meV for small angles)44,60,75,98,99 and increased gap would translate to a reduced static limit of the screening. At the same time, the effect of structural relaxations on screening in this system is likely to be small since their effect on bandstructure was shown to be prominent only for twist angles <2°73,74. In addition, tBLG is usually encapsulated with hBN in high pressure experiments, effectively suppressing the out-of-plane relaxation. While further investigation of the role of structural reconstruction is needed, our methodology will likely play a critical role in performing such calculations.
Since the electron-electron interactions in the flat bands appear only mildly screened even at large compression, the electronic structure at high pressures is likely associated with robust insulating states95. However, the absence of internal screening can be efficiently modified, e.g., by encapsulation and by extrinsic adjustable screening23,95.
Our methodology is critical in providing the ab initio information about internal screening effects. It informs future works combining dielectric materials and high pressures. Further, it opens a route to the theoretical understanding of precise control of quasiparticle states.
Methods
Stochastic many-body theory
To compute the QP energies and analyze the MB interactions, we employ a combination of MBPT and mapping of the selected (strongly correlated) subspace on the Hubbard model. Within MBPT, the central quantity is the self-energy, Σ(ω), which is a dynamical and non-local potential acting on a single QP state and incorporates all many-body effects. The QP energies correspond to the poles of the Green’s function, G, representing a QP propagator that is expressed in terms of the Dyson series: \({G}^{-1}(\omega )={G}_{0}^{-1}(\omega )-{{\Sigma }}(\omega )\), where G0 is the reference (non-interacting) Green’s function (GF). Here, the reference GF is taken from DFT calculations with PBE exchange-correlation functional100; Σ is found using a perturbation expansion on top of G0 and is responsible for capturing the dynamical correlation effects.
Here, we employ the basis of single particle states, {ϕj}, obtained from the ground state DFT calculations and the self-energy thus becomes a matrix composed of elements \({{{\Sigma }}}_{j,k}(\omega )\equiv \left\langle {\phi }_{j}\right|{{\Sigma }}(\omega )\left|{\phi }_{k}\right\rangle\). The QP energies, ε, are:
Where \({\varepsilon }_{j}^{0}\) is the Kohn-Sham (KS) eigenvalue, \({v}_{j}^{{{{\rm{xc}}}}}\) is the exchange-correlation potential, and Δj(ω) comprises the coupling due to the off-diagonal elements of Σj,k(ω). The frequency dependent self-energy, Σ(ω), is evaluated at ε. We resort to the GW approximation to the self-energy, which contains exchange and correlation parts; the latter is approximated by the dynamical effects of the charge density fluctuations due to the addition (removal) of an electron to (from) \(\left|{\phi }_{j}\right\rangle\)46,101,102,103. In general, the off-diagonal contributions to the self-energy Δj capture the deviation of the {ϕ}-states from the QP (Dyson) orbitals \(\left|{\psi }_{j}(x)\right\rangle\). The Dyson orbital is defined by an overlap of a many-body wavefunction for the ground state of N particles, \({{{\Psi }}}_{0}^{N}\), and N ± 1 particles state, \({{{\Psi }}}_{j}^{N}\) for the jth excited state. For a hole in the jth state, the Dyson orbital is \({\psi }_{j}(x)\equiv \sqrt{N}{{{\Psi }}}_{j}^{N-1}({\bar{x}}_{1},{\bar{x}}_{2},\ldots ,{\bar{x}}_{N}){{{\Psi }}}_{0}^{N}({\bar{x}}_{1},{\bar{x}}_{2},\ldots ,{\bar{x}}_{N},x)\), where xk is the spin-space coordinate of electron k and one integrates over all coordinates with bar on top. For common weakly correlated systems, the diagonal contributions, Σjj, strongly dominate while the off-diagonal terms are orders of magnitude smaller and can be neglected (i.e., Δj = 0) 85,104.
Our first development efficiently expands the stochastic methodology63,64 and computes both types of contributions using a single-step correction. The expectation values of Σjk are sampled via decomposition of the Green’s function into random vectors ζ spanning the occupied and unoccupied subspace and propagated backward and forward in time, hence representing particle and hole components of the time-ordered Green’s function. The resulting expression is, e.g. for t < 0, \(iG({{{\bf{r}}}},{{{\bf{r}}}}^{\prime} ,t)\equiv \{\zeta ({{{\bf{r}}}}^{\prime} ,t)\zeta ({{{\bf{r}}}})\}\), where {⋯} denotes stochastic averaging and \(\left|\zeta (t)\right\rangle \equiv {e}^{-i\hat{H}t}\left|\zeta \right\rangle\), and \(\hat{H}\) is the system Hamiltonian. For non-interacting Green’s function, G0,(i.e., in the one shot correction scheme), the time evolution is governed by the underlying DFT Hamiltonian \({\hat{H}}_{0}\). The real-time sampling of the induced densities is performed by another set of random vectors η representing the charge density fluctuations, i.e., δn(r, t) ≈ ∣η(r, t)∣2. For nanoscale systems with thousands of atoms, ~ 100 samples suffice to represent the GF, with only ~ 10 needed to represent δn(r, t)62,63,65.
The stochastic methodology capitalizes on the fact that the key quantities (G and W) are determined by collective properties, which are inherently low-rank and captured by the dynamics of a few (random) states within the Hilbert space of single-particle states. This approach leads to a linear scaling algorithm that can treat thousands of atoms63,64. The implementation expands this methodology and efficiently yields also Δj terms (the details are provided in section “Off-diagonal self-energy”). Further, using the QP hamiltonian matrix (represented in the {ϕ}-state basis in the Eq. (1)), we compute the QP orbitals ψ, corresponding to the first step of the self-consistent renormalization loop.
Off-diagonal self-energy
The off-diagonal terms in the polarization self-energy have been implemented in our development version of the stochastic GW code63. In the stochastic GW formalism, the non-interacting Green’s function G0 and the screened Coulomb potential W are sampled with two independent sets of random functions {ζ} and {η} respectively. Additional set of random vectors is used for the sparse stochastic compression in the time-ordering procedure. As a result, the expectation value for the polarization self-energy is a statistical estimator with a statistic error decreasing with number of random vectors as \(1/\sqrt{N}\). A specific off-diagonal term of the self-energy has the following expression:
where ≃ denotes that the expression is exact in the limit of \({N}_{\bar{\zeta }}\to \infty\). The function ζ at time t is defined with a help of the time evolution operator \({U}_{0}(t)\equiv {e}^{-i{H}_{0}t}\) and the projector Pμ(t) that selects the states above or below the chemical potential, μ, depending on the sign of t.:
The ζ vectors in the occupied and unoccupied subspace are propagated backward or forward in time and contribute selectively to the hole and particle non-interacting Green’s functions.
In Eq. (3) the overlap with ϕk is hidden within uζ,k(r, t)—an induced charge density potential:
uζ,k(r, t) represents the time-ordered potential of the response to the charge addition or removal. uζ,k is calculated from the retarded response potential, which is \({\tilde{u}}_{\zeta ,k}=\int {\tilde{W}}_{{{{\rm{P}}}}}({{{\bf{r}}}},{{{\bf{r}}}}^{\prime} ,t)\bar{\zeta }({{{\bf{r}}}}^{\prime} ){\phi }_{k}({{{\bf{r}}}}^{\prime} ){d}^{3}{{{\bf{r}}}}^{\prime}\) with a subsequent time-ordering procedure62,63,105.
Further, the retarded response is related to the time-evolved charge density \(\delta n({{{\bf{r}}}},t)\equiv \frac{1}{\lambda }\left[n({{{\bf{r}}}},t)-n({{{\bf{r}}}},0)\right]\) induced by a scaled perturbing potential \(\delta v=\lambda [\nu ({{{\bf{r}}}},{{{\bf{r}}}}^{\prime} )\bar{\zeta }({{{\bf{r}}}}^{\prime} ){\phi }_{k}({{{\bf{r}}}}^{\prime} )]\). Here λ is selected to be small, i.e., inducing a linear response; in our case we chose λ = 10−4 a.u. The retarded response becomes:
Instead of computing δn(r, t) by a sum over single-particle states, we employ the set of random vectors \(\left\{\eta \right\}\) confined to the occupied subspace. This reduces tremendously a cost of the computation \({\tilde{u}}_{\zeta ,k}\). Time-dependent density n(r, t) is thus62,63,64,106,107,108
where ηl is propagated in time using U0,t
Note, the perturbing potential δv depends explicitly on the state ϕk and the \(\bar{\zeta }\) vector, which samples the whole Hilbert space.
We employ RPA, i.e., performing evolution within the time-dependent Hartree approximation109,110,111, to calculate \({\tilde{u}}_{\zeta ,k}\).
Stochastic Hamiltonian downfolding
The second development presented in this work enables efficient Hamiltonian downfolding and extracting effective parameters for model approaches using first principles. We identify correlated states using the QP orbitals analysis (discussed in the main text) and map the corresponding subspace on a dynamically screened Hubbard model112,113,114:
where \({\hat{c}}_{i\sigma }^{{\dagger} }\), \({\hat{c}}_{i,\sigma }\) are creation and annihilation operators, \({\hat{n}}_{i\sigma }={\hat{c}}_{i\sigma }^{{\dagger} }{\hat{c}}_{i\sigma }\) is a particle number operator.
In practice, we extract the hopping and on-site Coulomb terms, t and U from the first-principles calculations. The latter is91:
Here, {φi} is a set of KS or QP states spanning the correlated subspace (represented by {ϕ} and {ψ} sets, respectively). They are subject to Coulomb interaction \(\tilde{W}\) that contains both bare (instantaneous) and screened (i.e., dynamical) terms. Symbolically, the interaction is \(\tilde{W}=\nu +\nu \tilde{\chi }\nu ,\) where ν is the bare Coulomb kernel and \(\tilde{\chi }\) is the polarizability due to electronic states orthogonal to the {φi}-subspace that contains the DP states.
The cost of the conventional calculation is huge, as it needs to be evaluated by considering all possible transitions between occupied and unoccupied states (see below). Calculations for large systems (such as those studied here) were thus out of reach. In contrast, we propose an efficient approach in which Eq. (10) is evaluated stochastically within the constrained random-phase approximation (cRPA): the real-time formalism samples the dynamics of all occupied states using a new set of random vectors confined to the occupied subspace and orthogonal to {φi}. The separation of the Hilbert space employs our recently developed decomposition technique115. This technique is computationally inexpensive: U(ω) screened by 4364 valence bands require merely <120 CPU ⋅ hrs on a 2.5 GHz processor (the testing configuration was AMD EPYC 7502 with 2.5 GHz frequency using 10 out of 32 physical cores. The total computational time was 9.6 hrs). This methodology thus enables Hamiltonian downfolding even for extremely large systems. The details of the implementation are provided in the next sections on the bare Coulomb interaction (IV D) and its dynamical screening evaluated by s-cRPA (IV E).
The effective bare Coulomb interaction
We calculate the bare effective interaction parameter, the Hubbard Ub, both in a basis of KS or QP wavefunctions (see discussion on the orbital construction in section “Pressure-induced localization”) of a chosen subspace of N = 8 states:
where \(\nu ({{{\bf{r}}}},{{{\bf{r}}}}^{\prime} )\) is a bare Coulomb kernel. The full Hubbard is U(ω) is given by Eq. (10) and contains, besides Ub, also the dynamical screened polarization term. The latter part is computed stochastically as detailed below, in section “Stochastic constrained RPA (s-cRPA)”.
Stochastic constrained RPA (s-cRPA)
Here we discuss the implementation of the dynamical Hubbard term, U(ω) = Ub + Up(ω), where the latter is:
The polarization operator \({\tilde{W}}_{{{{\rm{P}}}}}=\nu \tilde{\chi }\nu\) is computed by the stochastic constrained random-phase approximation (s-cRPA). The key idea is to capture the effect of the entire system on the correlated electrons in states {φ} described by the (downfolded) Hubbard Hamiltonian Eq. (9). In practice, one accounts for the screening through a projection on the subspace, which excludes all correlated states {φ}. In cRPA, \({\tilde{W}}_{{{{\rm{P}}}}}\) thus contains contributions of the induced density fluctuations in the weakly correlated portion of the system.
Conventional techniques evaluate \({\tilde{W}}_{{{{\rm{P}}}}}=\nu \tilde{\chi }\nu\) in frequency domain by the sum over all single-particle transitions outside the correlated subspace (i, j ∉ {φ}), requiring operation on both the entire occupied and unoccupied space91:
Hence, these calculations become expensive for large systems. In contrast, we compute \({\tilde{W}}_{{{{\rm{P}}}}}\) term stochastically in real-time domain:
This expression is computed by time-ordering from the retarded charge density potential:
Where \(\delta \tilde{n}\) is the induced charge density in the weakly correlated subspace perturbed by a potential due to ∣φj(r)∣2. This is formally equivalent to Eq. (5) (representing the action of the self-energy in the GW approximation). In practice, the density is constructed from random vectors \(\{\tilde{\eta }\}\):
where the {η}-states are described in the section “Mean-field vs MBPT energy spectrum” and Pφ is the projection operator on the {φ}-subspace:
Where fk is the occupation of state \(\left|k\right\rangle\). Note that the time evolution of \(\{\tilde{\eta }\}\) vectors follows Eq. (8), which depends on the total density. For details of the time-evolution of subspaces, see ref. 115.
The method is implemented alongside the stochastic GW formalism and both can be evaluated at once. However, in practice, the statistical error in Up(ω) is orders of magnitude smaller since: (i) it stems from one random sampling of W; in contrast, the GW self-energy suffers from larger statistical errors due to the additional random vectors sampling the Green’s function. (ii) it contains only contributions of states orthogonal to those which it is acting on; as a result, the dynamics is “well-behaved” and characterized by only a few dominant (resonant) frequencies which can be efficiently sampled by a small number of random vectors.
Equilibrium geometry and equation of state
The tBLG cells at a specific out-of-plane pressure have been approximated using the interlayer distance of the ideal bilayer graphene in the Bernal stacking at a corresponding pressure. All the calculations for the ideal bilayer graphene have been performed using hexagonal unit cell in QuantumESPRESSO code116 and Tkatchenko-Scheffler’s total energy Van der Waals corrections117 and Effective Screening Medium Method118. Troullier-Martins pseudopotentials119, and the PBE120 functional have been employed. To calculate the pressure–distance curves in the ideal bilayer, we have fitted the total energy E as a function of the volume V with the Murnaghan equation of state121.
where V = S ⋅ z is the volume confined by two graphene layers, \(S={a}_{{{{\rm{lat}}}}}^{2}\) is a surface of the layer, and z is the interlayer distance. S was kept constant using the equilibrium lattice parameter alat = 2.464 Å, while z was varied. The neglect of the pressure-induced in plane expansion is, in part, justified by the large anisotropy of the bulk modulus in the in- and out-of-plane directions5. B0 and \({B}_{0}^{\prime}\) are the bulk modulus and its pressure derivative at the equilibrium volume V0. The resulting fit and fitter parameters are provided in the Supplementary Information file, Supplementary Fig. 1. Using fitted parameters P(z) pressure–distance curves were calculated as a derivative P = dE/dV:
Starting point DFT calculations
The starting-point calculations are performed with the density functional theory (DFT) in a real-space implementation, employing regular grids, Troullier-Martins pseudopotentials119, and the PBE120 functional for exchange and correlation. We investigate tBLG infinite systems using modified periodic boundary conditions with Coulomb interaction cutoffs122. To converge the occupied H0 eigenvalues to <5 meV, we use a kinetic energy cutoff of 26 Eh and 192 × 132 × 66 real-space grid with the step of 0.4 × 0.4 × 0.5 a0, where the z-direction is aligned with the normal of the bilayer plane.
G 0 W 0 calculations
The GW calculations were performed using a development version of the StochasticGW code62,63,64. The calculations employ an additional set of 20,000 random vectors for the sparse stochastic compression used for time-ordering of \({\tilde{u}}_{\zeta }\)63. The sampling of the Green’s function G was performed using Nζ = 500 random vectors. Nη = 8 was used to sample the induced charge density63. The final stochastic error on quasiparticle energies is ≤20 meV. The time propagation of the induced charge density was performed for a maximum propagation time of 50 a.u., with the time-step of 0.05 a.u.
Data availability
All the data supporting the results of this study are available upon reasonable request to the corresponding author
Code availability
The public version of the stochastic GW code is available at www.stochasticGW.com. We used a development version of the stochastic GW to perform calculations, which will be released soon and is available upon reasonable request.
References
Brooks, J., Weng, G., Taylor, S. & Vlcek, V. Stochastic many-body perturbation theory for moiré states in twisted bilayer phosphorene. J. Condens. Matter Phys. 32, 234001 (2020).
Cao, Y. et al. Correlated insulator behaviour at half-filling in magic-angle graphene superlattices. Nature 556, 80 (2018).
Cao, Y. et al. Unconventional superconductivity in magic-angle graphene superlattices. Nature 556, 43 (2018).
Yankowitz, M. et al. Tuning superconductivity in twisted bilayer graphene. Science 363, 1059 (2019).
Yankowitz, M. et al. Dynamic band-structure tuning of graphene moiré superlattices with pressure. Nature 557, 404 (2018).
Sharpe, A. L. et al. Emergent ferromagnetism near three-quarters filling in twisted bilayer graphene. Science 365, 605 (2019).
Lu, X. et al. Superconductors, orbital magnets and correlated states in magic-angle bilayer graphene. Nature 574, 653 (2019).
Saito, Y., Ge, J., Watanabe, K., Taniguchi, T. & Young, A. F. Independent superconductors and correlated insulators in twisted bilayer graphene. Nat. Phys. 16, 926 (2020).
Choi, Y. et al. Electronic correlations in twisted bilayer graphene near the magic angle. Nat. Phys. 15, 1174 (2019).
Xie, Y. et al. Spectroscopic signatures of many-body correlations in magic-angle twisted bilayer graphene. Nature 572, 101 (2019).
Kerelsky, A. et al. Maximized electron interactions at the magic angle in twisted bilayer graphene. Nature 572, 95 (2019).
Jiang, Y. et al. Charge order and broken rotational symmetry in magic-angle twisted bilayer graphene. Nature 573, 91 (2019).
Trambly de Laissardière, G., Mayou, D. & Magaud, L. Localization of dirac electrons in rotated graphene bilayers. Nano Lett. 10, 804 (2010).
Koshino, M. et al. Maximally localized wannier orbitals and the extended hubbard model for twisted bilayer graphene. Phys. Rev. X 8, 031087 (2018).
Kang, J. & Vafek, O. Symmetry, maximally localized wannier states, and a low-energy model for twisted bilayer graphene narrow bands. Phys. Rev. X 8, 031088 (2018).
Calderón, M. & Bascones, E. Interactions in the 8-orbital model for twisted bilayer graphene. Phys. Rev. B 102, 155149 (2020).
Goodwin, Z. A., Corsetti, F., Mostofi, A. A. & Lischner, J. Attractive electron-electron interactions from internal screening in magic-angle twisted bilayer graphene. Phys. Rev. B 100, 235424 (2019).
Bistritzer, R. & MacDonald, A. H. Moiré bands in twisted double-layer graphene. Proc. Natl Acad. Sci. USA 108, 12233 (2011).
Utama, M. I. B. et al. Visualization of the flat electronic band in twisted bilayer graphene near the magic angle twist. Nat. Phys. 17, 184 (2021).
Parker, D. E., Soejima, T., Hauschild, J., Zaletel, M. P. & Bultinck, N. Strain-induced quantum phase transitions in magic-angle graphene. Phys. Rev. Lett. 127, 027601 (2021).
Tao, Z. et al. Raman spectroscopy study of sp2 to sp3 transition in bilayer graphene under high pressures. Appl. Phys. Lett. 116, 133101 (2020).
Clark, S., Jeon, K.-J., Chen, J.-Y. & Yoo, C.-S. Few-layer graphene under high pressure: Raman and x-ray diffraction studies. Solid State Commun. 154, 15 (2013).
Pizarro, J., Rösner, M., Thomale, R., Valentí, R. & Wehling, T. Internal screening and dielectric engineering in magic-angle twisted bilayer graphene. Phys. Rev. B 100, 161102 (2019).
Dos Santos, J. L., Peres, N. & Neto, A. C. Continuum model of the twisted graphene bilayer. Phys. Rev. B 86, 155449 (2012).
Lian, B., Wang, Z. & Bernevig, B. A. Twisted bilayer graphene: a phonon-driven superconductor. Phys. Rev. Lett. 122, 257002 (2019).
Yuan, N. F. & Fu, L. Model for the metal-insulator transition in graphene superlattices and beyond. Phys. Rev. B 98, 045103 (2018).
Yuan, N. F. & Fu, L. Erratum: Model for the metal-insulator transition in graphene superlattices and beyond [phys. rev. b 98, 045103 (2018)]. Phys. Rev. B 98, 079901 (2018).
Po, H. C., Zou, L., Vishwanath, A. & Senthil, T. Origin of Mott insulating behavior and superconductivity in twisted bilayer graphene. Phys. Rev. X 8, 031089 (2018).
Xu, C. & Balents, L. Topological superconductivity in twisted multilayer graphene. Phys. Rev. Lett. 121, 087001 (2018).
Roy, B. & Juričić, V. Unconventional superconductivity in nearly flat bands in twisted bilayer graphene. Phys. Rev. B 99, 121407 (2019).
Volovik, G. E. Graphite, graphene, and the flat band superconductivity. JETP Lett. 107, 516 (2018).
Padhi, B., Setty, C. & Phillips, P. W. Doped twisted bilayer graphene near magic angles: proximity to Wigner crystallization, not Mott insulation. Nano Lett. 18, 6175 (2018).
Dodaro, J. F., Kivelson, S. A., Schattner, Y., Sun, X.-Q. & Wang, C. Phases of a phenomenological model of twisted bilayer graphene. Phys. Rev. B 98, 075154 (2018).
Wu, F., MacDonald, A. & Martin, I. Theory of phonon-mediated superconductivity in twisted bilayer graphene. Phys. Rev. Lett. 121, 257001 (2018).
Isobe, H., Yuan, N. F. & Fu, L. Unconventional superconductivity and density waves in twisted bilayer graphene. Phys. Rev. X 8, 041041 (2018).
Huang, T., Zhang, L. & Ma, T. Antiferromagnetically ordered Mott insulator and d+ id superconductivity in twisted bilayer graphene: a quantum Monte Carlo study. Sci. Bull. 64, 310 (2019).
Zhang, Y.-H., Mao, D., Cao, Y., Jarillo-Herrero, P. & Senthil, T. Nearly flat chern bands in moire superlattices. Phys. Rev. B 99, 075127 (2019).
Kennes, D. M., Lischner, J. & Karrasch, C. Strong correlations and d + id superconductivity in twisted bilayer graphene. Phys. Rev. B 98, 241407 (2018).
Zhang, L. Lowest-energy moiré band formed by dirac zero modes in twisted bilayer graphene. Sci. Bull. 64, 8 (2019).
Guinea, F. & Walet, N. R. Electrostatic effects, band distortions, and superconductivity in twisted graphene bilayers. Proc. Natl Acad. Sci. USA 115, 13174 (2018).
Zou, L., Po, H. C., Vishwanath, A. & Senthil, T. Band structure of twisted bilayer graphene: emergent symmetries, commensurate approximants, and wannier obstructions. Phys. Rev. B 98, 085435 (2018).
Lima, M. P., Padilha, J. E., Pontes, R. B., Fazzio, A. & da Silva, A. J. R. Stacking-dependent transport properties in few-layers graphene. Solid State Commun. 250, 70 (2017).
Rademaker, L. & Mellado, P. Charge-transfer insulation in twisted bilayer graphene. Phys. Rev. B 98, 235158 (2018).
Angeli, M. et al. Emergent d 6 symmetry in fully relaxed magic-angle twisted bilayer graphene. Phys. Rev. B 98, 235137 (2018).
Goodwin, Z. A., Vitale, V., Liang, X., Mostofi, A. A. & Lischner, J. Hartree theory calculations of quasiparticle properties in twisted bilayer graphene. Electron. Struct. 2, 034001 (2020).
Martin, R. M., Reining, L. & Ceperley, D. M. Ceperley. Interacting Electrons (Cambridge University Press, 2016).
Xie, M. & MacDonald, A. H. Nature of the correlated insulator states in twisted bilayer graphene. Phys. Rev. Lett. 124, 097601 (2020).
Cea, T. & Guinea, F. Band structure and insulating states driven by coulomb interaction in twisted bilayer graphene. Phys. Rev. B 102, 045107 (2020).
Zhang, Y., Jiang, K., Wang, Z. & Zhang, F. Correlated insulating phases of twisted bilayer graphene at commensurate filling fractions: a Hartree-Fock study. Phys. Rev. B 102, 035136 (2020).
Bultinck, N. et al. Ground state and hidden symmetry of magic-angle graphene at even integer filling. Phys. Rev. X 10, 031034 (2020).
Liu, S., Khalaf, E., Lee, J. Y. & Vishwanath, A. Nematic topological semimetal and insulator in magic-angle bilayer graphene at charge neutrality. Phys. Rev. Res. 3, 013033 (2021).
Liu, J. & Dai, X. Theories for the correlated insulating states and quantum anomalous Hall effect phenomena in twisted bilayer graphene. Phys. Rev. B 103, 035427 (2021).
González, J. & Stauber, T. Time-reversal symmetry breaking versus chiral symmetry breaking in twisted bilayer graphene. Phys. Rev. B 102, 081118 (2020).
Rademaker, L., Abanin, D. A. & Mellado, P. Charge smoothening and band flattening due to hartree corrections in twisted bilayer graphene. Phys. Rev. B 100, 205114 (2019).
Potasz, P., Xie, M. & MacDonald, A. H. Exact diagonalization for magic-angle twisted bilayer graphene. Phys. Rev. Lett. 127, 147203 (2021).
Munoz, F., Collado, H. O., Usaj, G., Sofo, J. O. & Balseiro, C. Bilayer graphene under pressure: electron-hole symmetry breaking, valley Hall effect, and Landau levels. Phys. Rev. B 93, 235443 (2016).
Chittari, B. L., Leconte, N., Javvaji, S. & Jung, J. Pressure induced compression of flatbands in twisted bilayer graphene. Electron. Struct. 1, 015001 (2018).
Carr, S., Fang, S., Jarillo-Herrero, P. & Kaxiras, E. Pressure dependence of the magic twist angle in graphene superlattices. Phys. Rev. B 98, 085144 (2018).
Padhi, B. & Phillips, P. W. Pressure-induced metal-insulator transition in twisted bilayer graphene. Phys. Rev. B 99, 205141 (2019).
Lin, X., Zhu, H. & Ni, J. Pressure-induced gap modulation and topological transitions in twisted bilayer and twisted double bilayer graphene. Phys. Rev. B 101, 155405 (2020).
Green, B. R. & Sofo, J. O. Landau level phases in bilayer graphene under pressure at charge neutrality. Phys. Rev. B 101, 195432 (2020).
Vlček, V., Rabani, E., Neuhauser, D. & Baer, R. Stochastic GW calculations for molecules. J. Chem. Theory Comput. 13, 4997 (2017).
Vlček, V., Li, W., Baer, R., Rabani, E. & Neuhauser, D. Swift GW beyond 10,000 electrons using sparse stochastic compression. Phys. Rev. B 98, 075107 (2018).
Neuhauser, D. et al. Breaking the theoretical scaling limit for predicting quasiparticle energies: the stochastic GW approach. Phys. Rev. Lett. 113, 076402 (2014).
Vlcek, V. Stochastic vertex corrections: linear scaling methods for accurate quasiparticle energies. J. Chem. Theory Comput. 15, 6254 (2019).
Vlček, V., Baer, R., Rabani, E. & Neuhauser, D. Simple eigenvalue-self-consistent Δ− GW0. J. Chem. Phys. 149, 174107 (2018).
Vlček, V., Rabani, E. & Neuhauser, D. Quasiparticle spectra from molecules to bulk. Phys. Rev. Mater. 2, 030801 (2018).
Popescu, V. & Zunger, A. Extracting E versus k effective band structure from supercell calculations on alloys and impurities. Phys. Rev. B 85, 085201 (2012).
Huang, H. et al. A general group theoretical method to unfold band structures and its application. N. J. Phys. 16, 033034 (2014).
Medeiros, P. V., Stafström, S. & Björk, J. Effects of extrinsic and intrinsic perturbations on the electronic structure of graphene: Retaining an effective primitive cell band structure by band unfolding. Phys. Rev. B 89, 041407 (2014).
Boykin, T. B. & Klimeck, G. Practical application of zone-folding concepts in tight-binding calculations. Phys. Rev. B 71, 115215 (2005).
Boykin, T. B., Kharche, N., Klimeck, G. & Korkusinski, M. Approximate bandstructures of semiconductor alloys from tight-binding supercell calculations. J. Condens. Matter Phys. 19, 036203 (2007).
Yoo, H. et al. Atomic and electronic reconstruction at the van der Waals interface in twisted bilayer graphene. Nat. Mater. 18, 448 (2019).
Liang, X. et al. Effect of bilayer stacking on the atomic and electronic structure of twisted double bilayer graphene. Phys. Rev. B 102, 155146 (2020).
Cantele, G. et al. Structural relaxation and low-energy properties of twisted bilayer graphene. Phys. Rev. Res. 2, 043127 (2020).
Leconte, N., Jung, J., Lebègue, S. & Gould, T. Moiré-pattern interlayer potentials in van der Waals materials in the random-phase approximation. Phys. Rev. B 96, 195431 (2017).
Heske, C. et al. Band widening in graphite. Phys. Rev. B 59, 4680 (1999).
Strocov, V. et al. Photoemission from graphite: Intrinsic and self-energy effects. Phys. Rev. B 64, 075105 (2001).
Grüneis, A. et al. Electron-electron correlation in graphite: a combined angle-resolved photoemission and first-principles study. Phys. Rev. Lett. 100, 037601 (2008).
Ohta, T. et al. Interlayer interaction and electronic screening in multilayer graphene investigated with angle-resolved photoemission spectroscopy. Phys. Rev. Lett. 98, 206802 (2007).
Ohta, T., Bostwick, A., Seyller, T., Horn, K. & Rotenberg, E. Controlling the electronic structure of bilayer graphene. Science 313, 951 (2006).
Zhou, S. et al. Coexistence of sharp quasiparticle dispersions and disorder features in graphite. Phys. Rev. B 71, 161403 (2005).
Ohta, T. et al. Evidence for interlayer coupling and moiré periodic potentials in twisted bilayer graphene. Phys. Rev. Lett. 109, 186807 (2012).
Brihuega, I. et al. Unraveling the intrinsic and robust nature of van Hove singularities in twisted bilayer graphene by scanning tunneling microscopy and theoretical analysis. Phys. Rev. Lett. 109, 196802 (2012).
Faleev, S. V., Van Schilfgaarde, M. & Kotani, T. All-electron self-consistent GW approximation: application to Si, MnO, and NiO. Phys. Rev. Lett. 93, 126406 (2004).
Bruneval, F., Vast, N. & Reining, L. Effect of self-consistency on quasiparticles in solids. Phys. Rev. B 74, 045102 (2006).
Goodwin, Z. A., Corsetti, F., Mostofi, A. A. & Lischner, J. Twist-angle sensitivity of electron correlations in moiré graphene bilayers. Phys. Rev. B 100, 121106 (2019).
Xian, L., Kennes, D. M., Tancogne-Dejean, N., Altarelli, M. & Rubio, A. Multiflat bands and strong correlations in twisted bilayer boron nitride: Doping-induced correlated insulator and superconductor. Nano Lett. 19, 4934 (2019).
Guo, H., Zhu, X., Feng, S. & Scalettar, R. T. Pairing symmetry of interacting fermions on a twisted bilayer graphene superlattice. Phys. Rev. B 97, 235453 (2018).
Neto, A. C., Guinea, F., Peres, N. M., Novoselov, K. S. & Geim, A. K. The electronic properties of graphene. Rev. Mod. Phys. 81, 109 (2009).
Miyake, T., Aryasetiawan, F. & Imada, M. Ab initio procedure for constructing effective models of correlated materials with entangled band structure. Phys. Rev. B 80, 155134 (2009).
Ma, H., Sheng, N., Govoni, M. & Galli, G. Quantum embedding theory for strongly correlated states in materials. J. Chem. Theory Comput. 17, 2116 (2021).
Lu, C.-P. et al. Local, global, and nonlinear screening in twisted double-layer graphene. Proc. Natl Acad. Sci. USA 113, 6623 (2016).
Stauber, T. & Kohler, H. Quasi-flat plasmonic bands in twisted bilayer graphene. Nano Lett. 16, 6844 (2016).
Liu, X. et al. Tuning electron correlation in magic-angle twisted bilayer graphene using coulomb screening. Science 371, 1261 (2021).
Zhu, T., Antezza, M. & Wang, J.-S. Dynamical polarizability of graphene with spatial dispersion. Phys. Rev. B 103, 125421 (2021).
Vanhala, T. I. & Pollet, L. Constrained random phase approximation of the effective coulomb interaction in lattice models of twisted bilayer graphene. Phys. Rev. B 102, 035154 (2020).
Nam, N. N. & Koshino, M. Lattice relaxation and energy band modulation in twisted bilayer graphene. Phys. Rev. B 96, 075311 (2017).
Leconte, N., Javvaji, S., An, J. & Jung, J. Relaxation effects in twisted bilayer graphene: a multi-scale approach. arxiv.Preprint at https://arxiv.org/abs/1910.12805 (2019).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Hedin, L. New method for calculating the one-particle green’s function with application to the electron-gas problem. Phys. Rev. 139, A796 (1965).
Hybertsen, M. S. & Louie, S. G. Electron correlation in semiconductors and insulators: band gaps and quasiparticle energies. Phys. Rev. B 34, 5390 (1986).
Aryasetiawan, F. & Gunnarsson, O. The GW method. Rep. Prog. Phys. 61, 237 (1998).
Kaplan, F., Weigend, F., Evers, F. & van Setten, M. J. Off-diagonal self-energy terms and partially self-consistency in GW calculations for single molecules: efficient implementation and quantitative effects on ionization potentials. J. Chem. Theory Comput. 11, 5152 (2015).
Fetter, A. L. & Walecka, J. D. Quantum Theory of Many-Particle Systems (Dover Publications, 2003).
Gao, Y., Neuhauser, D., Baer, R. & Rabani, E. Sublinear scaling for time-dependent stochastic density functional theory. J. Chem. Phys. 142, 034106 (2015).
Rabani, E., Baer, R. & Neuhauser, D. Time-dependent stochastic Bethe-Salpeter approach. Phys. Rev. B 91, 235302 (2015).
Neuhauser, D., Rabani, E., Cytter, Y. & Baer, R. Stochastic optimally tuned range-separated hybrid density functional theory. J. Phys. Chem. A 120, 3071 (2016).
Baroni, S., de Gironcoli, S. & Dal Corso, A. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 73, 515 (2001).
Baer, R. & Neuhauser, D. Real-time linear response for time-dependent density-functional theory. J. Chem. Phys. 121, 9803 (2004).
Neuhauser, D. & Baer, R. Efficient linear-response method circumventing the exchange-correlation kernel: theory for molecular conductance under finite bias. J. Chem. Phys. 123, 204105 (2005).
Hubbard, J. Electron correlations in narrow energy bands. Proc. R. Soc. Lond., A Math. Phys. Sci. 276, 238 (1963).
Springer, M. & Aryasetiawan, F. Frequency-dependent screened interaction in ni within the random-phase approximation. Phys. Rev. B 57, 4364 (1998).
Kotani, T. Ab initio random-phase-approximation calculation of the frequency-dependent effective interaction between 3d electrons: Ni, Fe, and MnO. J. Condens. Matter Phys. 12, 2413 (2000).
Romanova, M. & Vlček, V. Decomposition and embedding in the stochastic GW self-energy. J. Chem. Phys. 153, 134103 (2020).
Giannozzi, P. et al. Advanced capabilities for materials modelling with quantum espresso. J. Condens. Matter Phys. 29, 465901 (2017).
Tkatchenko, A. & Scheffler, M. Accurate molecular van der waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 102, 073005 (2009).
Otani, M. & Sugino, O. First-principles calculations of charged surfaces and interfaces: a plane-wave nonrepeated slab approach. Phys. Rev. B 73, 115407 (2006).
Troullier, N. & Martins, J. L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 43, 1993 (1991).
Perdew, J. P. & Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 45, 13244 (1992).
Murnaghan, F. The compressibility of media under extreme pressures. Proc. Natl Acad. Sci. USA 30, 244 (1944).
Rozzi, C. A., Varsano, D., Marini, A., Gross, E. K. U. & Rubio, A. Exact coulomb cutoff technique for supercell calculations. Phys. Rev. B 73, 205119 (2006).
Towns, J. et al. Xsede: Accelerating scientific discovery. Comput. Sci. Eng. 16, 62 (2014).
De Laissardiere, G. T., Mayou, D. & Magaud, L. Numerical studies of confined states in rotated bilayers of graphene. Phys. Rev. B 86, 125413 (2012).
Acknowledgements
The development of the off-diagonal self-energy and s-cRPA (VV) was supported by the NSF through NSF CAREER award Grant No. DMR-1945098. The development of the downfolding and the implementation (M.R.) were supported by the Materials Research Science and Engineering Centers (MRSEC) Program through Grant No. DMR-1720256 (Seed Program). M.R.’s work was also supported by the NSF Quantum Foundry through Q-AMASE-i program Award No. DMR-1906325. The calculations were performed as part of the XSEDE123 computational Project No. TG-CHE180051. Use was made of computational facilities purchased with funds from the National Science Foundation (CNS-1725797) and administered by the Center for Scientific Computing (CSC). The CSC is supported by the California NanoSystems Institute and the Materials Research Science and Engineering Center (MRSEC; NSF DMR-1720256) at UC Santa Barbara.
Author information
Authors and Affiliations
Contributions
M.R. conducted the research work under the guidance of V.V. All authors contributed and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Romanova, M., Vlček, V. Stochastic many-body calculations of moiré states in twisted bilayer graphene at high pressures. npj Comput Mater 8, 11 (2022). https://doi.org/10.1038/s41524-022-00697-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-022-00697-8
This article is cited by
-
Dynamical downfolding for localized quantum states
npj Computational Materials (2023)
