
Abstract
The search for new materials requires effective methods for scanning the space of atomic configurations, in which the number is infinite. Here we present an extensive application of a topological network model of solid-state transformations, which enables one to reduce this infinite number to a countable number of the regions corresponding to topologically different crystalline phases. We have used this model to successfully generate carbon allotropes starting from a very restricted set of initial structures; the generation procedure has required only three steps to scan the configuration space around the parents. As a result, we have obtained all known carbon structures within the specified set of restrictions and discovered 224 allotropes with lattice energy ranging in 0.16–1.76 eV atom−1 above diamond including a phase, which is denser and probably harder than diamond. We have shown that this phase has a quite different topological structure compared to the hard allotropes from the diamond polytypic series. We have applied the tiling approach to explore the topology of the generated phases in more detail and found that many phases possessing high hardness are built from the tiles confined by six-membered rings. We have computed the mechanical properties for the generated allotropes and found simple dependences between their density, bulk, and shear moduli.
Similar content being viewed by others

Introduction
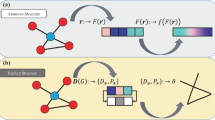
Modeling methods are more and more often used in materials science for predicting substances and polymorphic modifications. We know many fascinating examples of successful experimental synthesis of theoretically foreseen crystal structures. However, the correct and comprehensive prediction of all stable and metastable phases of a particular compound remains challenging. The main problem is that the number of geometrical embeddings (configurations) of a particular crystal structure in the Euclidean space is infinite and impossible to be completely explored. Thus, the search for an effective method of scanning the configuration space (CS) of a crystalline chemical system is a crucial task in modern materials science. A solution was proposed within the evolutionary approach1, in which the target structures are selected in the CS after modifying the initial random set of configurations by their mutation or combination. This approach showed its efficiency in the prediction of many crystalline phases and molecular species2. However, it has at least two issues: (i) the initial generation should be chosen according to some rational criteria, not randomly, and (ii) the set of modification operations should be extended to make the scanning steps more accurate. One of the promising ways to address these issues consists of the analysis of the connectivity of the crystal structure, i.e., the topological properties of the corresponding atomic system. Recently3, a topological generator was proposed to derive the initial set of parent structures for the evolutionary algorithm from a database of atomic topological patterns, i.e., to address the first issue. At the same time4, we developed a theoretical background for the topological description of the CS, the so-called topological network model of solid-state transformations (TNMST), and showed that this model can be used for a high-throughput search for possible new phases.
In this paper, we propose an effective algorithm of CS exploration, which is based on the TNMST and described in the “Methods” section, and apply it to the CS of the carbon system. Crystal structures of carbon allotropes have attracted great attention in the last 15 years after publishing a paper5 on the experimental investigation of superhard graphites under high pressure and especially after awarding a Nobel Prize for discovering graphene. However, at the ambient pressure, only the hexagonal graphite allotrope can be obtained, while many other carbon allotropes, besides diamond, were synthesized and predicted at high pressures. The number of hypothetical not-yet-synthesized carbon allotropes exceeds 500 according to the SACADA database6, but no systematic way of enumerating all possible low-energy structures has been proposed so far. With its simple chemical composition, but extremely high structural diversity, the carbon system seems a proper object for the application of the TNMST. Some examples of deriving carbon allotropes with this model were presented in [4]; however, the carbon CS was not systematically explored. The topological approach is especially important for carbon allotropes because many of those generated in recent years are actually more or less distorted representations of already-known topologies. For example, most of the structures obtained in the latest global DFT search for low-energy and high-density carbon allotropes7,8 have the same topologies that are already deposited in SACADA. Some other approaches, though appealing to SACADA for checking the topology of the resulted carbon networks, do not consider the topology during generation9. The known methods, which account for the structure topology, are based on a systematic10 or random11 generation of the carbon networks and do not provide cover even a part of the CS since they consider no structural relations to already-known allotropes.
Results
Generating procedure
Strictly speaking, the final result of scanning CS with the proposed algorithm should be independent of the initial set of structures; this set should influence only the number of the generation steps, i.e., the speed of the generation convergence. Indeed, since the topological regions cover the whole CS, starting from even one region (net), one can explore the whole CS sooner or later. To check this corollary of the TNMST, we have included into the initial set, not the most stable and experimentally proved structures, but metastable hypothetical allotropes that have no more than three independent atoms in the unit cell and the lattice energy not higher than 0.2 eV atom−1 compared to diamond. The same energy level has been chosen to select the most stable (low-energy) structures at step (v); thus, it plays the role of convergence limit of the generation procedure; the procedure stops when all offsprings have the relative lattice energies higher than this level. From these considerations, we have picked out six qualified carbon allotropes from SACADA, whose topologies are cfc, cfe, cdp, mtn, unj, and 43T129, as the initial set of structures. These structures were randomly selected to demonstrate the universality of the approach, although we deliberately avoided the most stable diamond and lonsdaleite structures.
The whole scanning has been finished in three steps (Table 1). Using a high-throughput topological algorithm, we have generated plenty of topological nets from just a few known allotropes. From these nets, we screen out the geometrically robust structures and check their stability. As a result, we have found 29 experimental and hypothetical low-energy carbon allotropes collected in the SACADA database, besides the 6 parent structures, while 224 new, compared to SACADA, stable hypothetical carbon allotropes have been predicted (Supplementary Tables 1–4). Among these 224 allotropes, 184 have topologies, which have never been found in crystal structures. When naming these topologies, we use the suffix “-CA” (“Carbon Allotrope”) in addition to the standard ToposPro nDk name12. For example, the name 4,4,4T16-CA = 43T16-CA means that this hypothetical carbon allotrope has three inequivalent 4-coordinated atoms, is three-periodic, and is 16th in the set of other three-periodic hypothetical carbon allotropes with the same number of inequivalent four-coordinated atoms. All these allotropes are now included in SACADA as well as in our online service TopCryst13, which enables one to check if a given allotrope topology is novel or it has been already found in real or hypothetical structures. Note that 66 generated allotropes have topologies of silicon frameworks in hypothetical zeolites (Supplementary Table 5); these topologies were recently deposited into TopCryst with the suffix “-HZ”14.
First generation
Starting from the initial set of the 6 parent structures, we have obtained 43 stable hypothetical carbon allotropes, 9 of which are known in SACADA (Supplementary Table 1), while cfd is indicated as carbon 6H in the RCSR database15. In this generation, five allotropes (cfd, crb, dia, lon, and sie) have relative lattice energy less than 0.2 eV atom−1 and have been used as parents for the next generation. Diamond (dia) and lonsdaleite (lon) are two most stable carbon allotropes, while cfd represents a 6-layered SiC polymorph, whose structure belongs to the same polytypic series as lon, dia, cfc, and cfe, which are topologically equivalent to 2-, 3-, 4-, and 9-layered SiC polymorphs, respectively. Thus, in the first generation, we have already obtained the most stable allotropes. All the polytypes are built from the diamondoid building blocks t-kah, t-hes, or t-afi. Importantly, the mtn allotrope has not provided any unstrained offsprings that reflect the isolation of its topological region from the regions of other parents. This allotrope was generated from the zeolite MTN framework16, which contains large cages of icosahedral symmetry. Its topological isolation indicates that it cannot be easily obtained from known carbon allotropes; an essential reconstruction of the structure is required after breaking many bonds.
Second generation
We could expect that the most stable allotropes would provide many low-energy offsprings, and indeed this is the case (Supplementary Table 2). Among 182 offsprings, 20 allotropes were already reported, while the other 162 are unknown. In this generation, we have obtained eight allotropes, whose relative energy is lower than 0.2 eV atom−1; they were used as parents for the third generation. Out of these eight allotropes, only two are known, while the other six have new topologies. All eight parents contain t-kah, t-hes, or t-afi building blocks, hence being locally similar to the allotropes of the diamond series, however, being inferior to them by mechanical properties (Supplementary Table 4). The hardest allotrope among all 224 generated allotropes is 43T16-CA, which, however, has much higher lattice energy compared to diamond (Supplementary Table 2). This allotrope is denser than a diamond with density ρ = 3.614 g cm−3 compared to ρ = 3.535 g cm−3 for diamond. Vicker’s hardness of 43T16-CA is comparable with diamond being a bit higher or lower according to models (1) or (2), respectively (see Methods and Supplementary Table 4). The structure of this allotrope is also unique: it is built from equivalent building units [614] confined by 14 six-membered rings (Fig. 1); such units have never been detected in crystal structures. The energy gap between 43T16-CA and diamond decreases with increasing pressure but remains significant at experimentally achievable conditions (Supplementary Fig. 1).

Transitions from M carbon (cbn) to a diamond (dia) and b lonsdaleite (lon) through common 4,4,5,5-coordinated supernets by creating red bonds in cbn and breaking blue bonds. The green bonds are preserved during the transition and correspond to topologically different common 3,3,4,4-coordinated subnets. Breaking these bonds results in the decomposition of the 3D structure into graphite sheets. The 43T16-CA carbon allotrope: c network and the corresponding tiling and d the tile [614].
Third generation
The eight parents obtained in the second generation produced 28 allotropes with unknown topologies (Supplementary Table 3). Since all of them had the relative lattice energy higher than 0.2 eV atom−1, no new parents were chosen, and the generation procedure was stopped at this step.
Discussion
We see that the generation procedure has rapidly converged at the chosen limit of 0.2 eV atom−1. This does not mean that we have walked around the whole CS of the carbon system, but this does mean that we have scanned the region around the diamond-like structures with the chosen level of topological complexity, i.e., at a given maximal number of inequivalent atoms. In this region, we have found many known carbon allotropes including the most stable structures, but also revealed a lot of unknown topological motifs. We believe that no new low-energy allotropes with one, two, or three inequivalent carbon atoms can be found in this region. However, the following questions emerge when assessing the universality of the approach:
-
(i)
Can we find topologically more complicated allotropes within this region? This is possible if we increase the limit for the number of inequivalent atoms in the structure. For example, considering the offsprings with four inequivalent atoms, we have found well-known superhard M carbon17, which has the cbn topology. The analysis of the supernet–subnet relations shows that M carbon has only two different minimal 4,4,5,5-coordinated supernets, which represent transition states for the diamond (dia)—M carbon (cbn) and lonsdaleite (lon)—M carbon (cbn) transformations. The corresponding common maximal subnets for these transitions are 3,3,4,4-coordinated, but also topologically different, while the common subnet for both transformations is the three-coordinated graphite layer (Fig. 1). This confirms the conclusions17 that M carbon, similar to diamond and lonsdaleite, can be obtained from graphite. However, this enables one to consider M carbon as an intermediate phase between cubic (dia) and hexagonal (lon) diamond, thus demonstrating that the transitions between these three phases do not require graphitization. However, the increase in the number of inequivalent atoms leads to a sharp increase in the number of topologically different subnets and supernets, while the number of unstrained offsprings remains small (Table 1). This means that additional criteria should be applied while generating subnets and supernets; the search for such criteria is a way to developing the TNMST.
-
(ii)
Can we scan the whole CS with this method? For this purpose, we should increase the convergence limit for the parent structures and the number of inequivalent atoms in the offsprings to let the generator overpass the topological regions with high relative lattice energy. The whole analysis can be time-consuming; a more effective way seems to explore the vicinity of a known topology as was done in this work. In this case, one can find more phases, which are related to the structures with important properties, like the superhard M carbon, which exists in the topological vicinity of the diamond structure.
The mechanical properties of the generated structures cover a broad range of values, from 130 to 425 GPa for the bulk modulus and from 55 to 507 GPa for the shear modulus, while the density varies from 2.0 to 3.6 g cm−3 (Fig. 2 and the Supplementary Information). This good deal of data enables us to explore the general dependences in mechanical properties. To make our consideration complete, we have extended our sample with 45 four-coordinated allotropes from the SACADA dataset, for which both bulk and shear moduli are known. Since the density reflects the average distance between carbon atoms, which in turn strongly depends on the bond strength and, hence, should correlate with the mechanical properties, we have explored the “elastic moduli–density” dependences, which indeed show a good correlation (Fig. 2). The BH–GH dependence is also practical since in many cases only one of the moduli is known in the literature: it can be expressed as GH = 0.0203BH1.66 with the correlation coefficient of 0.89 (Supplementary Fig. 2). Another important correlation concerns the topological tiling structure of the allotrope that was discussed above for the 43T16-CA allotrope. Note that most of the hard allotropes possess the same topological feature: their tiles are confined by only 6 rings thus being represented by symbols [6n] (n > 2); this can be called “[6n] criterion of hardness”. The simplest tiles of this type confined by three, four, or five 6 rings (n = 3–5) exist in diamond and lonsdaleite (Fig. 3); the role of these tiles in the allotrope stability and hardness was recently discussed18,19. However, our results indicate the importance of more complicated tiles with n > 5; moreover, they seem at least not less important than the simplest tiles. Thus, the hardest allotrope 43T16-CA is built from only [614] tiles, which do not exist in any other allotropes. Other generated allotropes composed of [610], [68], [67], or [66] tiles occupy the first place by hardness (Fig. 4, Supplementary Table 6); moreover, the hardest 43T16-CA, as well as the stc-4,4-Fddd and 42T39 allotropes, are isohedral, i.e., composed of only one type of tile. However, high hardness does not intend high stability: the most stable allotrope of this type, 43T107-CA, has a relative energy of 0.377 eV atom−1. Moreover, the [6n] criterion does not rigorously predetermine high hardness of the allotrope; the hardness of the last allotrope in Supplementary Table 6 (42T20-CA) is much lower than that of the others. Obviously, some other descriptors should also be responsible for hardness; we plan to search for them in further study.

The dependences of the bulk modulus BH (a) and the shear modulus GH (b) on the density ρ for the 269 four-coordinated carbon allotropes. Two trend lines show the dependences for the data from this work (‘new structures only’; BH = 40.661ρ1.86 with R2 = 0.86 and GH = 8.136ρ3.21 with R2 = 0.84) and the data merged with the information from the SACADA database (BH = 62.382ρ1.50 with R2 = 0.72 and GH = 11.034ρ2.97 with R2 = 0.80).

Natural tilings and tiles (t-kah [63], t-hes [64], and t-afi [65]) in the diamond (dia) and lonsdaleite (lon) nets.

Natural tilings and tiles [68] and [610] in the carbon allotropes with a stc-4,4-Fddd and b 42T39 topologies; c [66] and [67] tiles.
We have also indicated the structures with unusual local geometry of the sp3 carbon atoms, in particular with C–C bonds longer than 1.70 Å; there are totally of 40 such structures among the 224 generated polymorphs (Supplementary Tables 1–3) and all of them have relative energies above 0.45 eV atom−1. Such structures can hardly be realizable from a chemical point of view but can serve as intermediates between stable configurations. Note that usually, the authors do not analyze the C–C distances, thus considering only the positions of atoms in space, although the robust structures should fit both local and overall stability criteria and should be chemically reasonable at all levels of its organization. For example, the large set of 281 hypothetical sp3 carbon allotropes11 contains 125 structures with C–C bonds longer than 1.70 Å.
The presented study is the first extensive application of the TNMST to the exploration of the crystal CS. We see that the topologically different configurations can be effectively enumerated, thus enabling one to obtain all possible phases around the parent structures at a small number of generation steps. The topological approach enables one to exclude topological duplicates, i.e., the structures with the same network connectivity, and hence, to sharply decrease the number of configurations to be considered. Another advantage is that TNMST enables one to search for promising structures around the parents that possess useful properties, thus tuning the property by design. This opens a prospective way of discovering crystalline materials as the approach is universal and can be applied to the crystalline substances of any chemical nature.
Methods
Topological network model of solid-state transformations
The TNMST4 is based on the representation of a crystal structure as an atomic net, i.e., periodic graph, whose nodes and edges correspond to atoms and interatomic contacts (bonds). This net rigorously determines the structure topology, i.e., the connectivity of the atomic system. A keystone of the TNMST is the statement that (i) a particular topology determines a region in the CS, (ii) the regions do not intersect each other, and (iii) each region contains no more than one local energy minimum that coincides with the most stable geometrical embedding (configuration) of the corresponding atomic net. This means that the search for new phases requires the exploration of a discrete and countable set of topologically different atomic nets, not the continuous CS. The transformations of neighboring topologies A and B in CS are performed through common minimal supernets, i.e., the nets of higher connectivity that can be reduced to A or B by breaking a minimal number of edges. Equivalently, the transition can pass through common maximal subnets, i.e., the nets of lower connectivity that can be transformed into A or B by creating a minimal number of edges. The common minimal/maximal supernets/subnets form conjugated pairs, which describe the routes of solid-state reconstructive phase transitions.
To determine the topology of an atomic net, we have used the set of topological invariants (indices), which includes coordination sequences and point symbols20 of all symmetry- inequivalent atoms. If the symmetry-inequivalent atoms had equal topological indices, they are considered topologically equivalent; this means that the maximal symmetry of the network is higher than the embedding under consideration. Each topological region of the CS is characterized by the net A in the highest symmetry embedding; different points of the region corresponding to geometrical distortions of A. The names of the net topologies are given according to conventional nomenclatures12, the most widespread of which is the RCSR nomenclature of bold three-letter symbols (e.g., dia designates the diamond topology). This enables us to avoid slightly distorted copies of the same topology, which are often derived in the DFT generation.
The network representation gives rise to another method for the description of the crystal topology, the tiling method. Tiling is a partition of the crystal space by polyhedral building units confined by rings of the atomic net. These building units (tiles) can be unambiguously chosen according to a rigorous algorithm21 and characterize the way of the structure assembly. If two structures are built from the same or similar tiles, they can be considered topologically similar. For example, carbon structures can be identified as constructed from fragments of cubic or hexagonal diamond if they contain the tiles, from which the diamondoid structures are assembled, i.e., t-kah, t-hes, or t-afi consisting of three, four, or five six-membered rings (Fig. 3)18. The Topological Types of Tiles database (TTT collection)12 was used to determine the topological types of tiles.
We have implemented the description of the net topology by sets of topological indices and tiles, the procedure of generating subnets and supernets, as well as establishing relations between them into the ToposPro program package22, which was used at all stages of the following algorithm.
Algorithm of topological scanning of CS
The features of the topological description of CS enable one to use the TNMST for a fast screening of the candidate structures. The following algorithm can be proposed (Fig. 5):
-
(i)
Forming an initial generation from already-known topologies for a particular chemical system. For example, for carbon allotropes, the generation can include both experimentally obtained phases, like graphite or diamond, and low-energy hypothetical phases. Since the topological complexity, i.e., the number of topologically inequivalent atoms, of the nets influences the speed of the topological analysis, merely rather simple nets should be chosen at this stage. We have restricted ourselves to only four-coordinated carbons, which contain no more than three inequivalent atoms in the unit cell, that fit the complexity of the most stable 3D-framework carbon allotropes (e.g., diamond and lonsdaleite have one inequivalent atom). The carbon allotropes with a lower dimensionality and connectivity (e.g., 2D-layered graphite with two inequivalent atoms) can be derived from the 3D-framework nets as their subnets4.
-
(ii)
Generating all minimal supernets for the initial set of nets. The first step of the generation procedure consists of decreasing the symmetry of the initial net A by consideration of all (translation-equivalent, class-equivalent, and general) subgroups of its space group up to a particular index that provides the specified number of inequivalent atoms in the unit cell. In our study, we have limited this number to three atoms. At the second step, all possible minimal supernets have been generated by adding one independent bond to the net in such a way that the coordination number of the inequivalent atoms would increase no more than by unity. This means that for the carbon system, no more than five-coordinated supernets are considered. All atoms within three-coordination spheres of a given inequivalent atom have been considered as possible additional neighbors to form the extra bond. All the nets have been checked for topological equivalence by comparing their invariants and only topologically different nets have been used at the next step.
-
(iii)
Generating all maximal subnets of the minimal supernets, i.e., all their subnets where only one inequivalent bond is broken. In our study, we have selected only three-periodic four-coordinated subnets, which can correspond to sp3-hybridized carbon allotropes. Obviously, all the initial nets were among these subnets, and all other subnets could be considered as topological offsprings of the initial nets. To extract the most stable allotropes, we have discarded those subnets that contained 3-membered rings of carbon atoms.
-
(iv)
Searching for the best geometrical embeddings for the offsprings. It is important to keep the topology of the net after optimization, so all atoms should gain optimal distances to the adjacent atoms even if some bonds were very long after the supernet–subnet transformations. We have used the Gavrog Systre software23, which provides the most symmetrical barycentric embedding for the net, i.e., the embedding where all atoms occupy barycenters of their neighborhood. Then we have discarded those Systre-optimized nets that contained nonbonded atoms allocated at distances less than 1.4 × 1.54 Å = 2.31 Å to avoid too strained carbon structures.
-
(v)
Searching for the most energetically favorite embeddings of the offsprings. For this purpose, the geometrically unstrained structures obtained at the previous step have been optimized and characterized by DFT modeling. Importantly, the topology of the net can be changed after the DFT optimization; this means that the corresponding network region of the CS is topologically unstable and contains no local energy minimum. The structures from the topologically stable regions have formed the parent pool to generate the next set of topological offsprings. We have discarded from the pool those structures, which were topologically equivalent to the structures from the previous generations, or had the lattice energy higher than a specified level.

The multistep procedure of scanning a topological configuration space applied to the carbon system. The number of parent structures used and the number of offstrings generated, as well as examples of the networks of new carbon allotropes, are given for each step.
This procedure has been repeated until no parent structures have been obtained at step (v).
DFT calculations
We have applied density functional theory (DFT) calculations24,25 for the structure relaxations and for the evaluation of stability and mechanical properties of the generated carbon allotropes. The Vienna Ab initio Simulation Package26 with the projector augmented-wave method and generalized gradient approximation27 has been used. The Perdew–Burke–Ernzerhof28 exchange-correlation functional has been adopted for calculations. The plane-wave kinetic energy cutoff has been set at 600 eV and uniform k-point meshes for sampling the Brillouin zone with a reciprocal-space resolution of 2π × 0.025 Å−1 have been used. The convergence thresholds have been set at 10−6 eV for energy and 10−5 eV Å−1 for ionic forces. The mechanical stability of the allotropes was treated by the calculation of single-crystal elastic constants as well as the total energy versus volume relation according to the equation of states. For the most intriguing lowest-energy and hardest allotropes, we have also checked their dynamic stability through the calculation of the phonon dispersion curves (Supplementary Fig. 3) calculated using the Phonopy package29. Polycrystalline elastic moduli—the bulk modulus BH, the shear modulus GH, Young’s modulus EH, and the Poisson’s ratio ν—have been calculated within the Voigt–Reuss–Hill30 approximation. The Vicker’s hardness HV has been estimated according to two models31,32
and
where χ(ν) = (1–8.5ν + 19.5ν2)(1–7.5ν + 12.2ν2 + 19.6ν3)−1.
Data availability
The data that support the findings of this study are available from the corresponding authors (V.A.B. and Q.Z.) upon reasonable request.
Code availability
ToposPro is available for free at https://topospro.com.
References
Oganov, A. R. & Glass, C. W. Crystal structure prediction using ab initio evolutionary techniques: principles and applications. J. Chem. Phys. 124, 244704 (2006).
Oganov, A. R., Pickard, C. J., Zhu, Q. & Needs, R. J. Structure prediction drives materials discovery. Nat. Rev. Mater. 4, 331–348 (2019).
Bushlanov, P. V., Blatov, V. A. & Oganov, A. R. Topology-based crystal structure generator. Comp. Phys. Commun. 236, 1–7 (2019).
Blatov, V. A., Golov, A. A., Yang, C., Zeng, Q. & Kabanov, A. A. Network topological model of reconstructive solid-state transformations. Sci. Rep. 9, 6007 (2019).
Mao, W. et al. Bonding changes in compressed superhard graphite. Science 302, 425–427 (2003).
Hoffmann, R., Kabanov, A. A., Golov, A. A. & Proserpio, D. M. Homo citans and carbon allotropes: for an ethics of citation. Angew. Chem. Int. Ed. 55, 10962–10977 (2016).
Takagi, M. et al. Global search for low-lying crystal structures using the artificial force induced reaction method: a case study on carbon. Phys. Rev. B 95, 184110 (2017).
Takagi, M. & Maeda, S. Global search for crystal structures of carbon under high pressure. ACS Omega 5, 18142–18147 (2020).
Deringer, V. L., Csányi, G. & Proserpio, D. M. Extracting crystal chemistry from amorphous carbon structures. ChemPhysChem 18, 873–877 (2017).
Strong, R. T., Pickard, C. J., Milman, V., Thimm, G. & Winkler, B. Systematic prediction of crystal structures: an application to sp3-hybridized carbon polymorphs. Phys. Rev. B 70, 045101 (2004).
Shi, X., He, C., Pickard, C. J., Tang, C. & Zhong, J. Stochastic generation of complex crystal structures combining group and graph theory with application to carbon. Phys. Rev. B 97, 014104 (2018).
Blatov, V. A., Alexandrov, E. V., Shevchenko, A. P. Topology: ToposPro. In: ‘Reference Module in Chemistry, Molecular Sciences and Chemical Engineering’. https://doi.org/10.1016/b978-0-12-409547-2.14576-7 (Elsevier, 2019).
Alexandrov, E. V., Shevchenko, A. P. & Blatov, V. A. Topological databases: why do we need them for design of coordination polymers? Cryst. Growth Des. 19, 2604–2614 (2019).
Blatov, V. A., Blatova, O. A., Daeyaert, F. & Deem, M. W. Nanoporous materials with predicted zeolite topologies. RSC Adv. 10, 17760–17767 (2020).
O’Keeffe, M., Peskov, M. A., Ramsden, S. J. & Yaghi, O. M. The reticular chemistry structure resource (RCSR) database of, and symbols for, crystal nets. Acc. Chem. Res. 41, 1782–1789 (2008).
Zeolite framework database. http://www.iza-structure.org/ (2020).
Boulfelfel, S. E., Oganov, A. R. & Leoni, S. Understanding the nature of “superhard graphite”. Sci. Rep. 2, 471 (2012).
Baburin, I. A., Proserpio, D. M., Saleev, V. A. & Shipilova, A. V. From zeolite nets to sp3 carbon allotropes: a topology-based multiscale theoretical study. Phys. Chem. Chem. Phys. 17, 1332–1338 (2015).
Avery, P. et al. Predicting superhard materials via a machine learning informed evolutionary structure search. npj Comput. Mater. 5, 89 (2019).
Blatov, V. A., O’Keeffe, M. & Proserpio, D. M. Vertex-, Face-, Point-, Schlafli-, and Delaney-symbols in nets, polyhedra and tilings: recommended terminology. CrystEngComm 12, 44–48 (2010).
Blatov, V. A., Delgado-Friedrichs, O., O’Keeffe, M. & Proserpio, D. M. Three-periodic nets and tilings: natural tilings for nets. Acta Cryst. A63, 418–425 (2007).
Blatov, V. A., Shevchenko, A. P. & Proserpio, D. M. Applied topological analysis of crystal structures with the program package. ToposPro. Cryst. Growth Des. 14, 3576–3586 (2014).
Delgado-Friedrichs, O. & O’Keeffe, M. Identification of and symmetry computation for crystal nets. Acta Cryst. A59, 351–360 (2003).
Hohenberg, P. & Kohn, W. Inhomogeneous electron gas. Phys. Rev. 136, B864 (1964).
Kohn, W. & Sham, L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133 (1965).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Langreth, D. C. & Mehl, M. J. Beyond the local-density approximation in calculations of ground-state electronic properties. Phys. Rev. B 28, 1809–1834 (1983).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Togo, A. & Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 108, 1–5 (2015).
Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. A 65, 349–354 (1952).
Chen, H., Yang, L. & Long, J. First-principles investigation of the elastic, Vickers hardness and thermodynamic properties of Al–Cu intermetallic compounds. Superlattices Microstruct. 79, 156–165 (2015).
Mazhnik, E. & Oganov, A. R. A model of hardness and fracture toughness of solids. J. Appl. Phys. 126, 125109 (2019).
Acknowledgements
V.A.B., A.A.G., and A.A.K. thank the Russian Science Foundation (grant No. 16-13-10158) for support of developing the network topological model. C.Y., D.T., and Q.Z. thank the National Key R&D Program of China (grant No. 2017YFB0703200), National Natural Science Foundation of China (grant Nos. 51972268, 51372203, and 51761135032) for the financial support. We also acknowledge the high-performance computing center of NWPU (China).
Author information
Authors and Affiliations
Contributions
V.A.B. proposed the network topological model, implemented it into the ToposPro code, and wrote the main text, A.A.G. and C.Y. performed the topological analysis for the carbon phases, Q.Z. contributed to discussions of the model and to the main text, and D.T. and A.A.K. performed the DFT computation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Blatov, V.A., Yang, C., Tang, D. et al. High-throughput systematic topological generation of low-energy carbon allotropes. npj Comput Mater 7, 15 (2021). https://doi.org/10.1038/s41524-021-00491-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-021-00491-y
