
Abstract
Heterogeneous catalysts constitute a crucial component of many industrial processes, and to gain an understanding of the atomic-scale features of such catalysts, ab initio density functional theory is widely employed. Recently, growing computational power has permitted the extension of such studies to complex reaction networks involving either high adsorbate coverages or multidentate adsorbates, which bind to the surface through multiple atoms. Describing all possible adsorbate configurations for such systems, however, is often not possible based on chemical intuition alone. To systematically treat such complexities, we present a generalized Python-based graph theory approach to convert atomic scale models into undirected graph representations. These representations, when combined with workflows such as evolutionary algorithms, can systematically generate high coverage adsorbate models and classify unique minimum energy multidentate adsorbate configurations for surfaces of low symmetry, including multi-elemental alloy surfaces, steps, and kinks. Two case studies are presented which demonstrate these capabilities; first, an analysis of a coverage-dependent phase diagram of absorbate NO on the Pt3Sn(111) terrace surface, and second, an investigation of adsorption energies, together with identifying unique minimum energy configurations, for the reaction intermediate propyne (CHCCH3*) adsorbed on a PdIn(021) step surface. The evolutionary algorithm approach reproduces high coverage configurations of NO on Pt3Sn(111) using only 15% of the number of simulations required for a brute force approach. Furthermore, the screening of potentially hundreds of multidentate adsorbates is shown to be possible without human intervention. The strategy presented is quite general and can be applied to a spectrum of complex atomic systems.
Similar content being viewed by others

Introduction
Heterogeneous catalysis plays a pivotal role in the welfare of the society by impacting sectors ranging from sustainable energy generation and storage to synthesis of fine chemicals to food production1,2. These applications have motivated the search for effective and economic heterogeneous catalysts which, in turn, relies on understanding relationships between the underlying material and the chemistry of interest. Elucidation of these structure–property relationships requires the development of fundamental insights into atomic-level phenomena to accelerate the identification of better catalysts for these applications. In obtaining such insights, ab-initio simulation techniques, such as periodic density functional theory (DFT), have played a central role, and their use has both accelerated the discovery of new catalysts and aided the development of fundamental insights into catalytic reaction mechanisms3,4,5.
In the past, DFT studies have largely considered simple reaction chemistries involving monodentate intermediates in the limit of low coverages (one or two adsorbates per eight to nine surface atoms) and have relied on human intuition to generate relevant atomic structures. More recently, however, rapid growth in computational power and resources has enabled the analysis of complex reaction chemistries involving multidentate adsorbates and high coverages (four or more adsorbates per eight to nine surface atoms) of adsorbates6,7,8,9,10,11,12, as well. Nonetheless, it is challenging to tractably model these complex reaction chemistries using chemical intuition alone. To address these problems, workflows and algorithms have been developed to provide initial guesses of adsorbate geometries, which are then refined through DFT calculations, and to efficiently incorporate simulated data into databases for further analysis13,14,15,16,17,18,19,20,21. Various methods, including cluster expansions and genetic algorithms, have been employed to model high coverages of adsorbates on pure metal surfaces22,23,24,25. Although successful advances have been made, analysis of high coverages of adsorbates on low symmetry surfaces, such as steps and multi-elemental alloys, remains challenging, and there exists no generalized procedure to systematically simulate these cases. In the case of multidentate adsorbates, some tools have been developed to provide initial guesses for the geometric structure of adsorbates21, but the challenge of identifying unique configurations after geometric relaxation remains unresolved. These problems motivate the development of a tool that can both efficiently detect unique adsorbate configurations among a dataset of different geometric configurations, and, further, use this information to populate higher coverage configurations for a given adsorbate.
Herein, we present a Python-based algorithm to digitally represent a given heterogeneous catalytic model system by an undirected graphical representation. These representations are multi-molecular, involving a variety of different types of atoms and bonds, with multiple contact points between adsorbate atoms and the underlying surface (mono-, bi-, and tri-dentate). These graphical representations are used to detect unique adsorbate configurations and adsorption sites on catalyst surfaces. Effectively, the graphical representations are compared with one another to identify unique configurations for cases of low or high coverages, as well as monodentate and multidentate geometries. These capabilities are further combined with an evolutionary algorithm-based approach to systematically estimate high coverage models of adsorbates on low symmetry surfaces such as alloys, steps, and kinks. In addition, design rules, such as estimation of the number of nearest neighbor interactions, are chosen to simplify the vast sample space without compromising chemical insights.
The application of these capabilities is illustrated with two examples: construction of a coverage-dependent phase diagram of NO adsorption on the Pt3Sn(111) surface, which reveals the dependence of the adsorption energy on the chemical environment, and identification of unique configurations for the case of bidentate propyne (CHCCH*3) adsorbed on a low symmetry PdIn(021) alloy step surface.
The use of graph-based methods to automate and accelerate the screening of rich combinations of interacting adsorbates with multiple surface contact points constitutes a new use of graph theory in the field of heterogeneous catalysis. Further, the general methods presented herein are flexible in nature with regard to the type of intermediate (multidentate vs. monodentate) and the catalyst models considered (steps, terraces, and alloys) and, therefore, could be extended to a variety of different atomistic modeling applications.
Results and discussion
In the following, a general algorithm to construct undirected graphs from a given atoms object is presented. Next, algorithms utilizing the undirected graph to detect unique adsorbate configurations, as well as adsorption sites, are described. Finally, applications utilizing these algorithms to estimate high coverage models of monodentate adsorbates on an alloy surface and to identify unique configurations and adsorption energies of multidentate adsorbates on step surfaces are demonstrated.
Graph generation algorithm
To represent atomistic models as an undirected graph encoded with chemical bonding information, the NetworkX26 module in Python is employed, and a combination of tools such as edge and node isomorphism, ego networks, and abstract modifications are utilized to analyze complex atomistic models. Further, to accurately represent these models, periodic boundary conditions are accounted for by unfolding the bonds along the cell edge into repetitions of the cell, and nearest neighbor atoms are defined using an atomic radius-based neighbor list generated with the corresponding module in the Atomic Simulation Environment (ASE)27. We note that nearest neighbor atoms can, alternatively, be defined manually or by using methods such as Voronoi coordination from bulk15, and such approaches can be readily incorporated into our approach.
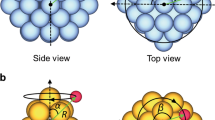
A schematic of the algorithm used to create the undirected graph-based representation is presented in Fig. 1. After reading the atoms object, different kinds of atoms are identified, and a node is defined for each atom. Then, using information regarding the nearest neighbor atoms, edges are created for each node, and finally the full graph is generated. For the specific example presented in Fig. 1, each atom (Fig. 1a) is represented by a node with the same color (Fig. 1c), and edges are created for each node to show bonding with its nearest neighbors. Further, in Fig. 1c, full, dashed, and dotted lines represent edges for bonds between surface–surface (golden atoms), adsorbate–adsorbate, and surface–adsorbate atoms, respectively, and the number of nearest neighbor atoms are reported on each node (without the loss of generality, only catalyst surface atoms are counted as nearest neighbors for the adsorbates).

a Atomic model for a simple nanoparticle with adsorbates, b algorithm to generate graph-based representations, and c generated graph model.
Adsorbate detection and high coverage simulation algorithms
A major bottleneck in simulating heterogeneous catalysts containing either high coverages of adsorbates or multidentate adsorbates is detecting unique adsorbate configurations after full DFT relaxation of the adsorbates has been performed. The full graph representation presented above can be used to solve this problem. First, using the algorithm shown in Fig. 1b, a full graph incorporating all atoms in the system is constructed. Next, adsorbate atoms are detected using the encoded information about the bonding between each atom in the full graph. Subsequently, subgraphs are developed from the full graph with a given adsorbate as the center for each individual subgraph. These individual subgraphs are then used to create an ego graph with a given radius to represent the chemical environment of the adsorbate. Finally, the ego graphs are used to check for isomorphism to find unique configurations for a given adsorbate. A general description of this approach, which is fully automated, is presented in Fig. 2c (blue subset). In addition, as we demonstrate below, encoded in every subgraph is the information related to the chemical environment of the adsorption site which can be leveraged to understand structure–property relationships between the adsorption energy and the chemical identity of the active site.

a, c The site detection and adsorbate detection workflows, respectively. b The overall workflow to utilize the generated graphs to perform atomic scale simulations.
To detect chemically unique adsorption sites on catalyst surfaces, an approach similar to that described above is employed (Fig. 2a—green subset). First, surface atoms are identified, either manually or by checking for asymmetrical bonding in space; the latter strategy involves the calculation of the direction of the normal vector for a given set of atoms (see Supplementary Information, Section A). Different combinations (cycles) for these surface atoms are then analyzed to identify different classes of adsorption sites on the catalyst surface. For example, in the case of fcc(111) type surfaces, cycles of length 1, 2, and 3, representing top, bridge, and hollow sites, respectively, are considered for different sites on the surface. These surface sites are then represented in the full graph by a pseudo atom, and an ego graph is employed to detect unique adsorption sites (Supplementary Information, Section A).
A general workflow to combine the two methods described above is presented in Fig. 2b. The framework to populate unique adsorption sites, along with the capability to detect duplicate adsorption structures, can be fully automated and combined with still more workflows to analyze highly complex adsorption environments. In particular, we have combined these methods with an evolutionary algorithm to predict adsorption configurations of high coverages of adsorbates on model catalyst surfaces. In this approach, sites are first populated with a single adsorbate on the clean surface and subsequently relaxed with DFT using, for example, the Vienna Ab Initio Simulation Package (VASP)28 or GPAW29. Unique configurations are then detected and stored as the set of new starting configurations. The configurations in the new set are scored based on their adsorption energies, and an additional adsorbate is added to the configurations with lowest energies. This process is repeated until the desired total coverage is obtained. This approach has a number of attractive features that should accelerate its adoption in the computational catalysis community. First, it permits efficient analysis of systems that have prohibitively large configurational spaces, such as alloys and other low symmetry surfaces, and the algorithm may be executed in parallel, allowing for scaling as availability of computational resources increases. In addition, the graph representation enables the reduction of sample space by a judicial choice of the local environment of a given adsorbate. Further, the rules for the choice of local environment in terms of the number of atomic shells needed to understand the chemical environment of an adsorbate can be adjusted, depending on the system at hand. For example, in the case of metal surfaces, one can assume that the adsorption site, along with its nearest neighbors, is sufficient to define the chemical environment of a given adsorbate, while more extensive site ensembles might be needed for other types of materials. This flexibility, combined with the scoring strategy of the evolutionary algorithm, reduces the configurational space of adsorbate geometries in high coverage models to tractable amounts (see Supplementary Information, Section B, for an example), even for low symmetry surfaces. We note that the evolutionary algorithm has sufficient flexibility to model high coverages of multidentate adsorbates, as well, although the configurational space of such a problem can be orders of magnitude larger than that of monodentate adsorbates and is currently outside the scope of this work.
Examples demonstrating the utility of the graph-based algorithm
Below, we present two example applications of the graph-based method. In the first case, using the evolutionary algorithm workflow, as well as the adsorbate and site detection algorithms, discussed in the previous section, we construct a coverage-dependent phase diagram of the adsorbate NO on a low symmetry Pt3Sn(111) alloy surface. In the second case, using our adsorbate detection algorithm, we identify unique adsorption configurations of the intermediate propyne (CH3CCH) on a PdIn(021) stepped alloy surface and further estimated a structure–property relationship between the adsorption energy and the chemical environment of the active site.
For many heterogeneous catalytic reactions such as CO oxidation30, NO reduction31, and Fisher–Tropsch chemistry32, it is important to consider high coverages (0.5–0.7 surface monolayers) of adsorbates on the catalyst surface to understand the effects of adsorbate–adsorbate interactions on the underlying chemistry. Herein, we demonstrate the use of our graph-based algorithmic approach to construct a coverage-dependent phase diagram for one such system, NO on a Pt3Sn(111) alloy surface. To illustrate the complexity of the problem, the unique adsorption sites for a single adsorbate on Pt3Sn(111) alloy and pure Pt(111) surfaces are shown in Fig. 3a. A comparison between the two cases shows that the introduction of Sn reduces the symmetry of the surface and increases the number of unique configurations possible by a factor of two. The number of configurations increases by a factor of five when more than one adsorbate is present on the surface. A general analysis of the number of configurations possible on the Pt3Sn(111) surface, compared to Pt(111), in the case of one, two, and three chemically distinct adsorbates, is reported in the Supplementary Information, Section B.

a The unique sites considered for Pt3Sn(111) and Pt(111) model systems. b Coverage-dependent phase diagrams of NO adsorbed on the Pt3Sn(111) surface.
Given the large number of possible configurations, to systematically simulate high coverages of NO (here 2/9, 3/9, 4/9, 5/9, 6/9 ML of NO) on a Pt3Sn(111) surface, we employ the graph-based evolutionary algorithm approach. Unique adsorbate configurations, as well as adsorption sites, are detected for each coverage following the general framework presented in Fig. 2. Initially, all possible sites with one NO molecule on the surface, as shown in Fig. 3a, are considered, and forces are relaxed using DFT. Next, a set of stable configurations are chosen based on rules set in the evolutionary algorithm. For these configurations, a second NO is then placed on the surface, the geometries are relaxed using DFT, and a set of configurations is then chosen to model three NO adsorbates. This procedure is continued to construct the phase diagram shown in Fig. 3b (see also Supplementary Information, Section B). Adsorption energies of the most stable configuration at each coverage are used in the phase diagram (Fig. 3b), and the resulting geometries for the 2/9, 5/9 and 6/9 ML of NO models are shown in the inset. One defining characteristic of these stable configurations is the presence of one or multiple NO atoms adsorbed on a Pt top site, with O atoms pointing towards Sn atoms. This interaction is unique to the Pt3Sn(111) surface and is not observed on Pt(111), where hollow sites are more energetically favorable for NO adsorption at lower adsorbate coverages.
To further validate the accuracy of the graph-based evolutionary method, we simulate another set of high coverage configurations with a brute force approach for testing all possible configurations with adsorbates separated by more than 2.4 Å. The results are nearly identical to what is found using the evolutionary strategy, but the evolutionary algorithm requires only about 15% of the simulations of the brute force method. This significant lowering of the number of simulations can be understood in terms of the strong binding nature of the adsorbate, which leads to small perturbations in the geometries from one coverage to the next and, therefore, permits the use of smaller sets of samples to populate higher coverage configurations (Supplementary Information, Section B). We have also included an analysis of the insights gained from the brute force simulation in the Supplementary Information, Section B. In the future, this approach could be generalized to realize coverage-dependent phase diagrams for even more complicated catalytic systems, including those involving multidentate adsorbates such as propyne (see Supplementary Information, Section B for additional details).
A number of important reactions, including alkane and biomass chemistries, involve the modeling of large multidentate adsorbates on heterogeneous catalytic surfaces. Finding stable configurations of these adsorbates is a formidable task, as the number of possible configurations can be very large. Although tools have been developed to provide initial guesses for geometric structures of such adsorbates21, the challenge of identifying unique configurations after geometric relaxation with DFT remains unresolved. A systematic approach would therefore be desirable to identify the most stable and unique configurations, both before and after relaxation, to elucidate the structure–property relationships between the adsorbate and the catalyst with the greatest possible efficiency. Here, we demonstrate the use of the graph-based method to identify unique configurations, after relaxation, of the adsorbate CH3CCH, which is an important bidentate intermediate in the propane dehydrogenation reaction network, on a characteristic alloy surface. We choose the PdIn(021) stepped surface as a particularly challenging test of the algorithm’s capabilities. Finally, we use the information generated from the graph-based method to understand the adsorption strength of CH3CCH as a function of the chemical environment of the adsorption site.
Figure 4 shows three unique configurations of adsorbed CH3CCH on a PdIn(021) step surface from the simulated dataset, with the step sites of the PdIn(021) surface marked with a green boundary. Below each configuration, using the framework presented in Fig. 1, the corresponding graphs created for each adsorbate at a set radius are reported (here, the radius is reduced for ease of visualization). A comparison of the three graphs shows that the carbon atoms of the adsorbate in Fig. 4a, c are bound to a Pd top site, while carbon atoms in Fig. 4b are bound to a Pd–Pd bridge site. Further, the Pd atoms in Fig. 4a, c have 10 and 8 nearest neighbor atoms, respectively (as reported by the number on the Pd nodes in the graphical representation), making all configurations unique (see Supplementary Information, Section A, for more details on making graphs for multidentate cases). By analyzing the configurations presented in Fig. 4, we see that the binding energy is most favorable when the middle carbon and the terminal carbon atoms, containing only one hydrogen, are both bound to the top sites of the step (Fig. 4c). Further, when one of the carbon atoms is bound to the terrace next to the step sites (Fig. 4b), the adsorption energy is weakened by 0.45 eV, and it is weakened by an additional 0.81 eV when both of the carbon atoms are bound to the Pd atoms on the terrace. As is seen from the corresponding graphs, the Pd atoms encompassing the active site in the three cases (Fig. 4a–c), have (10,10), (10,8), and (8,8) nearest neighbors, respectively. Furthermore, given that in the three cases presented, all the active sites contain only Pd as the element to which the carbon atoms bind, it can be surmised that each undercoordinated Pd atom (coordination of 8) stabilizes the adsorption energy by approximately 0.4 eV. This simple analysis can be extended to gain useful insights for more complicated surface geometries with mixed step terminations, kinks, or even vacancies. Further, similar structure–property relationship-based insights gained for different adsorbates could ultimately be combined to understand the entire reaction network as a function of the chemical environment of the active site by making use of correlations such as scaling and Brønsted-Evans-Polanyi relationships.

a–c show three unique configurations, along with the corresponding graphs generated using the graph-based method, and the relative adsorption energies for the configurations with respect to the most stable configuration.
In conclusion, graph theory-based tools are presented to represent atomic scale models as undirected graphs. These graphs are then combined with workflows to efficiently aid in simulating and understanding complex atomic scale models, including high adsorbate coverages, as well as multidentate adsorbates, present on low symmetry surfaces. Example cases presented show a significant reduction in human effort and in the computational resources needed to model complex reaction geometries. Such analyses can serve as a starting point to more efficiently identify better heterogeneous catalysts for complex reaction chemistries.
Methods
DFT simulation settings
The simulations are performed within the framework of periodic DFT with the VASP code28. The energies and geometries of the most stable configurations on the Pt3Sn(111) surface are obtained through minimization of the total energy with respect to geometry by spin-polarized generalized gradient approximation calculations (GGA–PBE)33. The sensitivity of the reported data was tested with respect to the functional used, and the trends in the adsorption energies do not change with the choice of functional. The projected augmented wave (PAW) method is used to account for the effect of core electrons on the valence electron density34. A Perdew–Burke–Ernzerhof (PBE)-calculated lattice constant of 4.06 Å for Pt3Sn bulk alloy is employed. The Pt3Sn(111) surface is represented by a \(\sqrt {12} \times \sqrt {12}\) unit cell with five layers (total of 60 atoms per unit cell), and the top two layers are relaxed. For the case of PdIn(021), a PBE-calculated lattice constant of 3.3 Å for PdIn bulk alloy is employed. A three atom wide step surface is used to represent the PdIn(021) surface (total of 42 atoms per unit cell), and surface relaxation is allowed in the top two layers perpendicular to the adjacent terraces. A vacuum equivalent to 13 Å is applied between any two successive slabs. A plane wave energy cutoff of 400 eV is used for all the calculations. A minimum k-point grid sampling of 3 × 3 × 1 is employed. The electronic occupancies are determined according to a Methfessel–Paxton scheme with an energy smearing of 0.2 eV. Dipole corrections are used in all cases to decouple the electrostatic interactions between the periodically repeated slabs. Structures are relaxed until the Hellmann–Feynman forces acting on the atoms are smaller than 0.05 eV/Å.
Graph generation settings
The ASE was used to generate neighbor lists for graph generations. Neighbor lists were generated by scaling the covalent radii of elements by a factor of 1.2 and by further considering a skin factor of 0.2 Å. The scaling, as well as the skin factor, are variables that can be tuned for different classes of systems (metals vs. oxides vs. 2D materials) to accurately generate graphs. Additional details are presented in the Supplementary Information, Section A.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Code availability
The code used to generate results in the manuscript is available under the Gitlab repo: https://gitlab.com/jgreeley-group/graph-theory-surfaces.
References
Liu, L. & Corma, A. Metal catalysts for heterogeneous catalysis: from single atoms to nanoclusters and nanoparticles. Chem. Rev. 118, 4981–5079 (2018).
Koval, C. et al. Basic Research Needs for Catalysis Science, 1–164 (2018).
Greeley, J. et al. Alloys of platinum and early transition metals as oxygen reduction electrocatalysts. Nat. Chem. 1, 552–556 (2009).
Nørskov, J. K. et al. Trends in the exchange current for hydrogen evolution. J. Electrochem. Soc. 152, J23–J24 (2005).
Ferrin, P. et al. Modeling ethanol decomposition on transition metals. A combined application of scaling and Brønsted–Evans–Polanyi relations. J. Am. Chem. Soc. 131, 5809–5815 (2009).
Guo, W. & Vlachos, D. G. Effect of local metal microstructure on adsorption on bimetallic surfaces: atomic nitrogen on Ni/Pt(111). J. Chem. Phys. 138, 174702 (2013).
Lausche, A. C. et al. On the effect of coverage-dependent adsorbate–adsorbate interactions for CO methanation on transition metal surfaces. J. Catal. 307, 275–282 (2013).
Frey, K., Schmidt, D. J., Wolverton, C. & Schneider, W. F. Implications of coverage-dependent O adsorption for catalytic NO oxidation on the late transition metals. Catal. Sci. Technol. 4, 4356–4365 (2014).
Hoffmann, M. J., Medford, A. J. & Bligaard, T. Framework for scalable adsorbate–adsorbate interaction models. J. Phys. Chem. C 120, 13087–13094 (2016).
Roling, L. T., Herron, J. A., Budiman, W., Ferrin, P. & Mavrikakis, M. Dimethyl ether electro-oxidation on platinum surfaces. Nano Energy 29, 428–438 (2016).
Tan, L., Huang, L., Liu, Y. & Wang, Q. Augmented pairwise additive interaction model for lateral adsorbate interactions: the NO–CO reaction system on Rh(100) and Rh(111). Langmuir 34, 5174–5183 (2018).
Hansen, M. H., Nørskov, J. K. & Bligaard, T. First principles micro-kinetic model of catalytic non-oxidative dehydrogenation of ethane over close-packed metallic facets. J. Catal. 374, 161–170 (2019).
Medford, A. J. et al. CatMAP: a software package for descriptor-based microkinetic mapping of catalytic trends. Catal. Lett. 145, 794–807 (2015).
Kitchin, J. R. Examples of effective data sharing in scientific publishing. ACS Catal. 5, 3894–3899 (2015).
Montoya, J. H. & Persson, K. A. A high-throughput framework for determining adsorption energies on solid surfaces. npj Comput. Mater. 3, 14 (2017).
Obersteiner, V., Scherbela, M., Hörmann, L., Wegner, D. & Hofmann, O. T. Structure prediction for surface-induced phases of organic monolayers: overcoming the combinatorial bottleneck. Nano Lett. 17, 4453–4460 (2017).
Medford, A. J., Kunz, M. R., Ewing, S. M., Borders, T. & Fushimi, R. Extracting knowledge from data through catalysis informatics. ACS Catal. 8, 7403–7429 (2018).
Tran, K. & Ulissi, Z. W. Active learning across intermetallics to guide discovery of electrocatalysts for CO2 reduction and H2 evolution. Nat. Catal. 1, 696–703 (2018).
Tran, K., Palizhati, A., Back, S. & Ulissi, Z. W. Dynamic workflows for routine materials discovery in surface science. J. Chem. Inf. Model. 58, 2392–2400 (2018).
Margraf, J. T. & Reuter, K. Systematic enumeration of elementary reaction steps in surface catalysis. ACS Omega 4, 3370–3379 (2019).
Boes, J. R., Mamun, O., Winther, K. & Bligaard, T. Graph theory approach to high-throughput surface adsorption structure generation. J. Phys. Chem. A 123, 2281–2285 (2019).
İnoğlu, N. & Kitchin, J. R. Simple model explaining and predicting coverage-dependent atomic adsorption energies on transition metal surfaces. Phys. Rev. B 82, 045414 (2010).
Bajpai, A., Frey, K. & Schneider, W. F. Binary approach to ternary cluster expansions: NO–O–vacancy system on Pt(111). J. Phys. Chem. C 121, 7344–7354 (2017).
Lym, J., Gu, G. H., Jung, Y. & Vlachos, D. G. Lattice convolutional neural network modeling of adsorbate coverage effects. J. Phys. Chem. C 123, 18951–18959 (2019).
Xu, Z. & Kitchin, J. R. Probing the coverage dependence of site and adsorbate configurational correlations on (111) surfaces of late transition metals. J. Phys. Chem. C 118, 25597–25602 (2014).
Hagberg, A. A., Swart, P. J. & Schult, D. A. Exploring Network Structure, Dynamics, and Function Using Networkx. https://doi.org/10.1002/eap.1538 (2008).
Hjorth Larsen, A. et al. The Atomic Simulation Environment—a Python library for working with atoms. J. Phys.: Condens. Matter 29, 273002 (2017).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initiototal-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Mortensen, J. J., Hansen, L. B. & Jacobsen, K. W. Real-space grid implementation of the projector augmented wave method. Phys. Rev. B 71, 035109 (2005).
van Spronsen, M. A., Frenken, J. W. M. & Groot, I. M. N. Surface science under reaction conditions: CO oxidation on Pt and Pd model catalysts. Chem. Soc. Rev. 46, 4347–4374 (2017).
Rodes, A. et al. In situ FTIR spectroscopy characterization of the NO adlayers formed at platinum single crystal electrodes in contact with acidic solutions of nitrite. Langmuir 11, 3549–3553 (1995).
Ojeda, M. et al. CO activation pathways and the mechanism of Fischer–Tropsch synthesis. J. Catal. 272, 287–297 (2010).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Acknowledgements
J.G., S.D., and T.M. acknowledge the United States Department of Energy through the Office of Science, Office of Basic Energy Sciences (BES), Chemical, Biological, and Geosciences Division, Data Science Initiative, Grant DE-SC0020381.
Author information
Authors and Affiliations
Contributions
S.D., T.M., and J.G. conceived the idea. S.D. and T.M. performed the calculations. S.D., T.M., and J.G. co-wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Deshpande, S., Maxson, T. & Greeley, J. Graph theory approach to determine configurations of multidentate and high coverage adsorbates for heterogeneous catalysis. npj Comput Mater 6, 79 (2020). https://doi.org/10.1038/s41524-020-0345-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-020-0345-2
This article is cited by
-
Stability and lifetime of diffusion-trapped oxygen in oxide-derived copper CO2 reduction electrocatalysts
Nature Catalysis (2024)
-
A human-machine interface for automatic exploration of chemical reaction networks
Nature Communications (2024)
-
Python tools for structural tasks in chemistry
Molecular Diversity (2024)
-
Deducing subnanometer cluster size and shape distributions of heterogeneous supported catalysts
Nature Communications (2023)
-
Rapid mapping of alloy surface phase diagrams via Bayesian evolutionary multitasking
npj Computational Materials (2023)
