
Abstract
The successful discovery and isolation of graphene in 2004, and the subsequent synthesis of layered semiconductors and heterostructures beyond graphene have led to the exploding field of two-dimensional (2D) materials that explore their growth, new atomic-scale physics, and potential device applications. This review aims to provide an overview of theoretical, computational, and machine learning methods and tools at multiple length and time scales, and discuss how they can be utilized to assist/guide the design and synthesis of 2D materials beyond graphene. We focus on three methods at different length and time scales as follows: (i) nanoscale atomistic simulations including density functional theory (DFT) calculations and molecular dynamics simulations employing empirical and reactive interatomic potentials; (ii) mesoscale methods such as phase-field method; and (iii) macroscale continuum approaches by coupling thermal and chemical transport equations. We discuss how machine learning can be combined with computation and experiments to understand the correlations between structures and properties of 2D materials, and to guide the discovery of new 2D materials. We will also provide an outlook for the applications of computational approaches to 2D materials synthesis and growth in general.
Similar content being viewed by others

Introduction
The perfection and physical properties of atomically thin two-dimensional (2D) materials are extremely sensitive to their synthesis and growth process. Achieving desired characteristics such as structural uniformity, high carrier mobility1, strong light–matter interactions, tunable bandgap, and flexibility is the main challenge for the synthesis and growth of next generation, electronics-grade 2D materials. A reliable and optimized growth and manufacturing process is essential for the synthesis of 2D materials with uniform properties at the wafer scale, e.g., for application in flexible and transparent optoelectronics.
Two main approaches have been employed for the synthesis of 2D materials, i.e., (i) top-down approaches such as mechanical2 and liquid-phase exfoliation that allows scalability3, and (ii) bottom-up approaches such as chemical vapor deposition (CVD) and atomic layer deposition techniques4. The former approaches are suitable for mass production of 2D materials but with typically lower quality, whereas the latter approaches can produce high-quality 2D materials but in small amounts. For both types of approaches, the morphology and characteristics of the synthesized 2D materials are very sensitive to the thermodynamic or kinetic conditions5 of the growth processes, e.g., heat transfer and mass transfer of source chemical species, chemical reaction kinetics, adsorption of reaction product species on a substrate surface, and nucleation and growth of the resulting 2D materials. The goal of this review is to provide an overview of the main theoretical and computational methods for understanding the thermodynamics and kinetics of mass transport, reaction, and growth mechanisms during synthesis of 2D materials. We will discuss the possibility of synthesis-by-design of new 2D materials guided by computation to reduce the number of expensive and time-consuming trial-and-error experimentations.
The critical challenge for developing theoretical and computational design tools for the synthesis of 2D materials is the broad range of length and temporal scales involved in their growth process. For example, it may require quantum mechanical and atomistic reactive force-field calculations to determine the activation energies for atomic migration on a surface6 and understand the atomistic surface reaction mechanisms7, and then a finite element method (FEM) to model the mesoscale mass transport phenomena8. Other challenges include incorporating the effects of substrates including the types of substrate defects9, the possible wrinkling of 2D films10, the effect of van der Waals (vdW) interactions at the mesoscale11, and the growth kinetics unique to atomically thin materials12. Also reproducing quadratic dispersion for the flexural acoustic modes of 2D materials using classical or reactive potentials may not be straightforward, it has been already formulated13. Furthermore, a practically useful multiscale model should be computationally efficient, numerically accurate, and, more importantly, able to capture the multi-physical governing relationships among the growth conditions, growth morphology, and materials properties. The eventual goal of developing multiscale computational models is to guide the design of new growth chambers to produce uniform large-area 2D materials.
Atomistic computational methods
First-principles calculations
This section discusses the computational approaches based on the density functional theory (DFT). The primary application of DFT in modeling 2D materials is to determine the relative thermodynamic stabilities of different crystal structures of a 2D material by computing their chemical potentials or to identify kinetic pathways by analyzing the energetics of potential transient structures from one stable equilibrium structure to another as thermodynamic conditions change. Examples of applications of DFT to understanding 2D materials and their growth, as well as the corresponding DFT-based first-principles methodologies and the corresponding experimental observables for validation are summarized in Table 1.
Thermodynamic stability
The thermodynamic stability of a 2D material requires its formation energy to be negative. The formation energies of various 2D materials have been efficiently calculated by DFT in combination with high-throughput screening platforms14. The success of DFT in predicting energies relies on the accuracy of well-tempered approximate exchange-correlation functionals. Standard functionals, e.g., the Perdew-Burke-Ernzerhof parametrization of the generalized gradient approximation exchange-correlation functional (GGA-PBE), accompanied by appropriate corrections15, describe well the formation and atomization energies. However, they are limited by intrinsic delocalization errors when bonds are stretched, resulting in underestimated reaction barriers16. A more realistic criterion for a stable 2D material is a low “above-hull” energy17. Exfoliation energies within 0.2 eV/atom are suggested as a general rule of thumb for the stability in 2D form. A slightly stricter criterion to have low surface energy (<20 meV/Å) is suggested to rule out potential high-surface energy but multi-atom-thick sheets.
Kinetically stabilized 2D Materials
As-grown products may be metastable structures rather than ground-state polytypes18. The well-known phase-stability competition between the 2H and 1T′ types in transition metal dichalcogenides (TMDs) has been studied extensively using DFT. Several possible mechanisms for the 1T′-phase stabilization in the disulfide, including growth-related factors such as the presence of point defects and residual strain, have been elucidated through DFT calculations19. Phase-stability analysis for the entire group -IV metal dichalcogenide family20 revealed that the 1T′ phases are generally more stable for ditellurides. In addition to pure compounds, mixing/segregation behavior and order–disorder transitions in alloys have also been discussed21. Within the W/Mo + S/Se/Te combinations, mixing either chalcogens or metals is thermodynamically favorable22, which can be experimentally quantified using the Warren-Cowley order parameters23. For TMDs, theory predicts that at finite temperatures, the formation of random alloys is favorable. However, kinetically stabilized atomically thin strips of alternating W and Mo in a sulfide alloy have been reported24.
Interlayer/substrate interactions
Accurate description of vdW interactions becomes critical when interlayer sheet–sheet or sheet–substrate interactions are considered, e.g., for identifying the growth orientation. As a non-local effect, vdW forces are not correctly described by DFT with semi-local exchange-correlation functionals and are often corrected by adding pairwise interatomic terms from empirical fittings25, based on charge densities26, or by introducing non-local exchange-correlation functionals, i.e., van der Waals Density Functional27. Thorough testing of these methods has been examined in ref. 28. One common technical problem in this computational approach is that substrates are modeled in a slab geometry where the one surface not in contact with the 2D sheet may host surface states, unless it is passivated. To suppress artificial charge transfer that these may induce, capping using pseudohydrogen29 with fractional charge is often performed. With the advent of new software platforms automating the generation of solid surfaces in combination with stacked 2D sheets and adsorption geometries (e.g., MPInterfaces30), high-throughput screening of possible substrates may lead to a systematic approach to substrate engineering.
Precursor chemistry and kinetics
Transient intermediate states are commonly calculated using transition state theory and the nudged elastic band (NEB) method31 by calculating bond dissociation energies or corresponding activation barriers32. One recently attempted approach to capture precursor reaction kinetics is constrained molecular dynamics (MD)33, where slowly varying coordination constraints are enforced on reacting species to map out free energy barriers, as implemented in VASP34. It is different from NEB calculations in that (1) it calculates free energies at finite temperatures; (2) the final state of the reaction need not to be known; and (3) it has a better numerical stability35. Constrained MD was used to study a sulfur precursor, S2, reacting with a MoO3 surface36, concluding that MoO3 surface vacancies favor the sulfurization process both kinetically and thermodynamically. The downside of constrained MD is that a reaction coordinate is chosen a priori, which may bias the system towards unnatural products with incorrect reaction mechanisms33 and higher reaction barriers34.
Other methods for sampling of rare events such as nucleation of a new structure include umbrella sampling, transition interface sampling, and metadynamics. The umbrella sampling technique was introduced by Torrie and Valleau37, to improve the sampling of systems with energy landscapes containing high energy barriers. The weighted histogram analysis method38 can be used to analyze a series of umbrella sampling methods. In the transition interface sampling39, the transition region is divided into subregions of intermediate states. The rate constant of a reaction in this method is the multiplication of transition probabilities between different intermediate states. The metadynamics technique was introduced in 2002 and is usually used within an atomistic modeling framework40. Selected collective variables of the system not only evolve with time but also periodically leave behind positive Gaussian potentials that are added to the original potential energy of the system, which effectively push the system out of a local minimum and into a neighboring energy well. Informally, metadynamics resembles “filling the free energy wells with computational sand.”41
Growth front advancement
Early studies focused on calculating the surplus energies of edges in the high-symmetry directions and then applying the thermodynamic Wulff construction42. One difficulty in calculating the edge energies is the polar and non-centrosymmetric nature of materials, where the usual ribbon calculation geometry makes the energies of dislike edges inseparable. The usual workaround is by constructing triangular flakes exposing a single type of edge43. Another “energy density method” was introduced in ref. 44 and a recently proposed method42 aimed at finding a general method suitable for high-throughput calculations introduces capping groups to passivate a surface.
One major criticism of further employing thermodynamic Wulff constructions is that growth is by definition out of equilibrium. Therefore, edges that dominate over others should not be the energetically favorable ones, but the slowest growing ones. The step-flow approach was formulated in ref. 45 for graphene growth and was further developed in ref. 46 for polar materials, establishing the use of kinetic Wulff construction. A similar approach involves DFT calculations and fitting to experimental grain morphologies, to construct a kinetic Monte Carlo (KMC) model47.
Defects
The formation of defects may occur within thermodynamic equilibrium such as thermally generated point defects or follow from growth imperfections such as dislocations and grain boundaries (GBs), or reflect the finite size of crystals such as edges and surfaces. Defects can also be deliberately introduced using methods such as electron or ion irradiation and chemical treatment40. The calculation of defect formation energies follows a well-established procedure detailed in ref. 48. 2D materials also host lattice-specific defect types, such as the Stone-Wales defect in graphene49. Single-atom vacancies are another type of defects in 2D materials. A general strategy to ensure correct convergence behavior for charged defects in 2D systems is presented in ref. 50. Defect formation energies in MoS2 have been comprehensively studied in ref. 51; possible strategies of introducing extrinsic dopants have been examined in ref. 52. Defect complexes are also frequently studied to identify likely combinations between simple intrinsic defects and external contamination53, devise possible defect-pairing strategies to neutralize harmful defects54, and investigate their influence on the growth behavior of 2D materials on a different 2D sheet9,55. Multiple vacancies may exist in 2D materials such as double vacancies in graphene, resulting in (i) two pentagons and one octagon—V2 (5-8-5) defect; or (ii) three pentagons and three heptagons—V2(555-777) defect; or (iii) four pentagons, a hexagon, and four heptagons—V2(5555-6-7777) defect56. The formation of defects with an even number of vacancies in graphene is energetically favorable due to the lack of any dangling bonds57, whereas a large number of vacancies may bend and wrap the 2D material58. Table 2 lists the typical values of formation and migration energies of these point defects from atomistic calculations.
Dislocations are line defects in 2D materials that can be of in-plane edge or out-of-plane screw type. The former consists of pentagon–heptagon (5–7) pairs59,60 and the latter makes the structure to become 3D61. In general, dislocations in 2D materials have high gliding energy barriers and are immobile at room temperature. They hinder the plastic deformation of 2D materials62. The in-plane edge dislocations with the smallest Burgers vector, b = (1,0), is typically the energetically most favorable dislocation configuration. The trilayer bi-atomic MoS2 has a complex dislocation structure with two types, i.e., Mo-rich that resembles (5–7) and S-rich that resembles (7–5) pairs. The complex nature of dislocations in MoS2 is a source of its rich chemistry. For example, at specific chemical potentials of S, the (5–7) dislocation reacts with 2S vacancies to form a (4–6) defect.
GBs in 2D materials can be considered as an array of dislocations, which can be divided into low-angle and high-angle GBs depending on the dislocation core density. Atomistic simulations revealed that the mechanical properties of 2D materials, similar to the one-dimensional nanostructures, will be affected by both the density and arrangement of the defects63. Notably, graphene sheets with high-angle tilt GBs can be as strong as the pristine material and much stronger than those with low-angle GBs64. First‐principles calculations also revealed that sinuous GB structures are energetically favorable when the straight GB line cannot bisect the tilt angle65. GB energies of different 2D materials are also reported based on atomistic calculations. For MoS2, the energy of GB with a 20.6° misorientation angle is 0.05 eV/Å62. The GB energy for phosphorene varies between 0.05 and 1.5 eV/Å, depending on its tilt angle66.
Molecular dynamics
The MD approach is one of the main tools that have been used to study the growth of 2D materials at the atomistic scale. The quality of potential function governing the atomic interactions is the key for the accuracy of this technique in predicting the structures and properties of 2D materials. In general, two classes of interatomic potentials are used in MD simulations to study the growth of 2D materials, i.e., empirical and reactive potentials, which we elaborate on here.
Empirical potentials
Empirical potentials are faster and more computationally efficient, but less accurate than reactive potentials. Common empirical potentials for modeling 2D materials are reviewed below.
Lennard-Jones potential
In 1924, Lennard-Jones (LJ) published the 12–6 pairwise potential to describe the vdW interaction between two neutral particles (i.e., rare gases)67,
where r is the distance between two interacting atoms, \({\it{\epsilon }}\) is the potential well depth and σ is the distance at which the potential is zero. The r−12 term captures the strong repulsion occurring when two atoms get closer. The repulsive effect originates from the Pauli exclusion principle, which penalizes the overlap of electron orbitals. The r−6 term describes the vdW interaction, which is due to the coupling between the instantaneous polar charges induced in two neutral molecules and is significantly weaker than the electron overlap effects.
The popularity of the LJ potential is due to its simplicity and it has been widely applied to simulate the interlayer interactions in atomically layered materials. While being used to capture vdW interactions, a limitation of the pairwise interaction is the inability to capture frictional processes, such as the sliding of atomic layers. Different corrections have been proposed to overcome this shortcoming68,69,70. The two potential parameters \({\it{\epsilon }}\) and σ can be determined by using one energy quantity like the binding energy and a structural quantity like the density. The LJ parameters for most elements can be determined systematically, as demonstrated by Rappe et al.71. The LJ parameters for typical 2D materials and substrates in Table 3 are listed in in Table 4 (for σ) and Table 5 (for \({\it{\epsilon }}\)), respectively.
Stillinger-Weber potential
Stillinger and Weber (SW) proposed this potential in 1985, to model bulk silicon72. The SW potential includes both pair (two-body) and three-body interactions,
where V2 describes the pairwise bond-stretching energy and V3 captures the bending energy associated with an angle with initial value θ0.
The cut-offs rmax, rmax12, and rmax13 can be determined by fitting to the average of the first- and second-neighboring bond lengths. The seven unknown potential parameters, including five unknown geometrical parameters, i.e., ρ and B in V2 and ρ1, ρ2, and θ0 in V3, and two energy parameters A and K, can be determined either by a least-squares fit to targeted quantities such as the elastic constants, Poisson’s ratio, and the phonon spectrum, or by analytical derivations from the linear valence force-field potential. It is noteworthy that the nonlinear parameter B could not be determined by the latter approach, which should be determined by nonlinear quantities such as the third-order elastic constants73.
The SW potential can describe nonlinear processes including large deformations, as higher-order terms for the variation of bond length and angle are included in its formulation. Although the SW potential is efficient and suitable for simulating thermal transport and other nonlinear phenomena74, it is not able to describe chemical bond formation or breaking. It should also be noted that the SW potential cannot provide bending energy for atomically layered materials without out-of-plane bonds such as graphene75. Due to its efficiency, the SW potential has been used in the simulation of many atomically layered materials such as 2D nanostructures of Si and their thermal properties76, MoS273,74,76, MoSe276, WS277, and black phosphorus73,78,79. The SW potential has been parametrized for 156 emerging atomic layered materials, along with other available empirical potentials for these atomically layered materials80.
Force-field potential
The force-field (FF) potential describes the variation in the potential energy of a deformed structure with respect to its equilibrium configuration. It is essentially a Taylor expansion of the total energy in terms of the variation in the bond length and angle,
where Δb, Δθ, Δϕ, and Δγ are variations of the bond length, bond angle, twisting angle, and inversion angle, respectively; kb, \(k_b^{(3)}\), \(k_b^{(4)}\), kθ, \(k_\theta ^{(3)}\), \(k_\theta ^{(4)}\), kϕ, kγ, kbb′, kθθ′, and kbθ are force constants. Vb is the bond-stretching energy, Vθ is the bond angle-bending energy, Vϕ is the torsional energy for a bond, Vγ is the inversion energy among four atoms, and Vc is the off-diagonal coupling interaction. It is noteworthy that the cubic and quartic anharmonic terms in the bond-stretching and angle-bending interactions enable the FF model to capture nonlinear phenomena such as thermal expansion and thermal transport.
The FF potential has two significant features: (i) the number of interaction terms and (ii) the force-constant parameters and the equilibrium structural parameters (bond lengths and angles). MM381 and COMPASS82 use all of the interaction terms and can simulate large deformations and nonlinear phenomena. In contrast, the Keating model83 is the simplest model including only the harmonic terms. Parameters of the potential are usually fitted to available data and, thus, the volume and accuracy of the fitted data set determine the accuracy of the FF model. Several generations of FF models have been developed as more data become available, including four versions of the molecular mechanics FF: MM184, MM285, MM381, and MM486. The UFF71 provides the FF parameters for all the elements in the periodic table.
As the energies in the FF model are additive, this potential has been widely used to calculate physical or mechanical properties for various materials via analytic expressions, including many 2D materials. For example, an FF model, with bond stretching and angle-bending terms, has been used to derive the phonon dispersion of graphene layers86. The bond-stretching and angle-bending terms were utilized to extract the bending properties of graphene87. A mapping between the FF model and the elastic beam model has been developed, which is used to derive analytic expressions for Young’s modulus and Poisson’s ratio of graphene and carbon nanotubes88.
Reactive interatomic potentials
The reactive-potential approach has been widely used to explore the synthesis and properties of different materials. The reactive potentials can capture the bond breaking and formation during the classical simulations of 2D materials. They are also computationally less expensive than first-principles methods such as DFT calculations and ab-initio MD. Typical reactive potentials that have been frequently considered are Tersoff Bond Order (Tersoff)89, Reactive Empirical Bond Order (REBO)90, Adaptive Intermolecular Reactive Empirical Bond Order (AIREBO)91, Charge Optimized Many-Body (COMB)92, and Reactive Force Field (ReaxFF)93. These different reactive potentials are reviewed in detail in ref. 94. Performances of several reactive and non-reactive potentials in graphene-based materials are compared in ref. 95. These potentials allow the realization of the thermodynamic stability of graphene-based materials under different conditions, environmental chemistry, substrate, interlayer interactions, and structural defects. However, only a few of them have been employed in the study of computational synthesis of other 2D materials to reveal thermodynamic stability, simulate the process of the growth of layers, model the top-down synthesis of 2D materials such as exfoliation techniques38, and evaluate the properties of 2D nanostructures. This is due to several challenges including the following: (1) the long time required to develop a reactive potential, (2) the non-transferability of potential parameters from one material to another, and (3) the lack of universal functional forms for the reactive potentials.
The Tersoff potentials are mainly used to explore various structures and properties of 2D hexagonal boron nitride (h-BN) materials96. The REBO and AIREBO potentials are used to simulate the CVD process and growth of 2D amorphous carbon on substrates, mechanical properties of MoS2, and even study the thermal stability of C60 2D nanostructures97. The COMB3 potentials have been used to simulate the experimental CVD deposition and growth of graphene and metal on substrates98. The ReaxFF potentials have been used to explore the structures of several 2D materials under intercalation, study the defects and groups on the surfaces of MXene materials, simulate CVD growth of MoS2 layers, and explore the synthesis of 2D polymeric materials in experiments7,99. In general, COMB3 and ReaxFF can further explore 2D materials, as their functional forms can describe heterogeneous systems. It remains crucial to find accelerated methods to develop reactive potentials with high accuracy and transferability for 2D materials.
Tersoff potential
The Tersoff potential was first published in 1986100 and further developed in 1988101. The second version of the Tersoff potential has the following form,
where \(f_R(r_{ij})\) and \(f_A(r_{ij})\) are the repulsive and attractive components, respectively. The cutoff function is:
A distinct feature of the Tersoff potential is the absence of an explicit three-body interaction term. Instead, the concept of bond order is introduced, in which the strength of a given bond depends strongly on its local environment. Bond order is accounted for via the function bij, which has the following form
aij is an alternative parameter to improve the accuracy of the potential, with a similar form
The 13 unknown parameters are typically determined by fitting to data from first-principles calculations or experiments.
The Tersoff potential was used for modeling of graphene, which gives a better description of the phonon spectrum102. It was also used to model germanene103 and silicene103. By introducing one scaling parameter for the quantity bij with atoms i and j of different elements104, the Tersoff potential was generalized to describe the interaction for multicomponent systems including C-Si and Si-Ge104, and boron nitride105. Using the Tersoff potential, the thermal conductivity of silicene was calculated to be 61.7 and 68.5 W mK−1 for the armchair and zigzag configurations, which is reduced when it is doped with heavier silicon isotopes106. This potential has also been used for modeling the misfit dislocations in 2D materials, where a critical thickness was found for the formation of an interface misfit dislocation107.
ReaxFF potential
The ReaxFF93 is a bond-order-based force field, which allows formation and breaking of bonds. It has a general form of
where the energy terms on the right-hand side in the order of appearance are bond, over-coordination penalty, under-coordination stability, lone pair, valence, torsion, non-bonded vdW interactions, and Coulomb energies, respectively. The bond-order parameter determines the bonded interactions between the atoms in the system,
where \({\mathrm {BO}}_{ij}^\sigma ,{\mathrm {BO}}_{ij}^\pi ,\) and \({\mathrm {BO}}_{ij}^{\pi \pi }\) are the partial contributions of associated σ, π, and ππ bonds between the i and j atoms; rij is the distance between the i and j atoms; \(r_o^\sigma\), \(r_o^\pi\), and \(r_o^{\pi \pi }\) are the associated bond radii; and pbo are empirical constants that will be determined from ab-initio simulations or experiments. The energy terms in Eq. (14) are then formulated as a function of the bond-order parameter (see ref. 93 for details).
ReaxFF studies on 2D materials were initiated from studies of various aspects of graphene108 and inspired investigations for a number of other 2D materials, which are elaborated below.
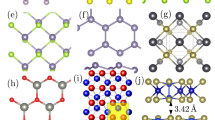
Graphene—Effect of the environment’s chemistry on properties and responses of graphene has been investigated using ReaxFF. For example, effect of oxygen on the mechanical response of graphene109, effect of the addition of fluorine and hydrogen110 on the structural quality of graphene, as well as the Ni-catalyzed growth process of single-layer graphene111, are investigated. The growth mechanism involves atomic deposition and segregation of C atoms on the Ni substrate, forming dome-like carbon nanotube cap and polygonal ring patterns via atoms→chains→graphene-like layer formation (Fig. 1a). The ReaxFF has also been applied to study reactive events at the graphene–water interface (Fig. 1b)112 and selective desalination via the bare and hydrogenated graphene nanopore113.

a Ni-catalyzed growth of single-layer graphene on a Ni(100) surface using MD/UFMC simulations. [Adapted from ref. 111 with permission from The Royal Society of Chemistry]. b Water-mediated proton transfer through O/OH-terminated vacancy defects on a graphene layer. [Adapted from ref. 112 under Creative Commons Attribution 4.0 International License]. c Formation of silicene between graphene bilayers. [Reprinted figure with permission from ref. 114. Copyright (2019) American Chemical Society]. d Atomic structures of ripplocations with different numbers of extra units added on top MoS2 layer. [Adapted with permission from ref. 116. Copyright (2019) American Chemical Society]. e Histograms corresponding to averaged interlayer distance without and with K+ ions [Reprinted from ref. 121. Copyright (2019), with permission from Elsevier].
Silicene and phosphorene—Silicene and black phosphorene (BP) are the silicon and phosphorus analogs of graphene, respectively. A favorable route for silicene formation has been proposed by employing the graphene bilayer as a template (Fig. 1c)114. It was found that vacancy defects reduce the thermal stability of silicene, where the critical temperature reduces by more than 30%115. A P/H parameter set has been developed for monolayer BP114 to examine the mechanical, thermal, and chemical stability of both pristine and defective BP.
Molybdenum disulfide—The ReaxFF potential for 2D MoS2 systems was introduced in ref. 6. This potential could precisely calculate the formation energy of ripplocations in MoS2 (Fig. 1d)116, which result from the slippage of the upper layer against the lower layer to relax the strain without breaking covalent bonds. This potential was also trained over the vacancy formation energies, diffusion barriers, bending rigidity, and kinetics. It has also been applied to design S vacancy defects on MoS2 surface117. Reactive MD simulations have also been used to model the synthesis of MoS2 layers via sulfidation of MoO3 surfaces7. These simulations identified a three-step reaction pathway as follows: (i) evolution of O2 and reduction of MoO3 surface; (ii) S2-assisted reduction and formation of SO/SO2; and (iii) sulfidation of the surface via Mo-S bond formation. The ReaxFF MD simulations have also been employed within a multiscale framework to capture thermal properties of MoS2118. The predicted thermal conductivity of MoS2 structures with infinite length (37 ± 3 W/mK) falls within the range of experimental results (34.5–110 W/mK)119.
MXenes—MD simulations with ReaxFF of MXenes revealed that intercalating with potassium cations drastically improves water stability and homogeneity of MXenes, and reduce the self-diffusion coefficient of water by two orders of magnitude120. The dynamical response of MXenes with different surface terminations to intercalating ions was studied with DFT calculations and ReaxFF MD simulations121. Figure 1e compares the interlayer distance of Ti3C2(OH)2 MXene bilayer with and without K+ ions. Influence of metal ions intercalation on vibrational properties of water molecules trapped between MXene layers was studied using ReaxFF MD simulations122. Widening of the interlayer gap may enable the penetration of molecular reactants such as urea, which decomposes readily and leads to the intercalation of ammonium cations123. ReaxFF simulations were performed for defect formation and homoepitaxial growth of TiC layer on Ti3C2 MXene structures124.
REBO/AIREBO potentials
The REBO potential \(( {E_{ij}^{\mathrm {REBO}}} )\) is a combination of attractive \(( {V_{ij}^A} )\) and repulsive \(( {V_{ij}^R} )\) interactions with certain ratio (bij), \(E_{ij}^{\mathrm {REBO}} = V_{ij}^R + b_{ij}V_{ij}^A\). The bond-order interaction ratio, bij, is dependent on the local coordination in the atomic environment. The REBO potential can define the conjugation of bonds using bij, and therefore the potential has been widely used for modeling hydrocarbon systems125. The repulsive term is expressed by the Brenner equation90,
Here, the Qij, rij, and αij parameters depend on i and j. The bond weighting parameter wij(rij) depends on switching function S(t) as
The attractive interaction is expressed as
the bond-order interaction ratio bij is
Here, \(p_{ij}^{\sigma \pi }\) and \(p_{ji}^{\sigma \pi }\) are not necessarily equal as they depend on the penalty function (gi) of bond angle θjik between the vector rij, and vector rki, i.e.,
The AIREBO potential is the modification over the REBO potential, which also considers the torsional \(( {E_{kijl}^{\mathrm {TORSION}}} )\) and LJ \(( {E_{ij}^{lj}} )\) interactions. The integration of pairwise interactions in AIREBO potential can be represented by the following equation
The REBO potential can describe the breaking, formation, and hybridization of covalent bonds. The REBO and AIREBO potentials have been used to study the growth of 2D amorphous carbon, mechanical properties of MoS2, and even thermal stabilities of C60 2D nanostructures using atomic coordination, internal stress, and mass density analysis126. The subplantation phenomenon during CVD, i.e., penetration of the substrate bulk carbon by carbon atoms, can be explained by this REBO potential. REBO potential is also suitable for modeling and analysis of TMD materials such as MoS2. The potential results in stabilized structure and good agreement of the structural and mechanical properties97,127,128. It was also used for modeling the synthesis of diamane from graphene and its stability under different conditions129.
COMB3 potential
The general formation of the COMB3 potential can be expressed as92
The total potential energy consists of electrostatic energy \(\left( {U^{\mathrm{es}}\left[ {\left\{ q \right\},\left\{ r \right\}} \right]} \right)\), short-range interactions \(\left( {U^{\mathrm {short}}\left[ {\left\{ q \right\},\left\{ r \right\}} \right]} \right)\), long-range vdW interaction \(\left( {U^{\mathrm {vdW}}\left[ {\left\{ q \right\},\left\{ r \right\}} \right]} \right)\), and the correction terms. Here, q and r represent the charge and coordinate array of the elements. The electrostatic energies \(\left( {U^{\mathrm {es}}\left[ {\left\{ q \right\},\left\{ r \right\}} \right]} \right)\) can be represented as
The self-interaction energy \(\left( {U^{\mathrm {self}}\left[ {\left\{ q \right\},\left\{ r \right\}} \right]} \right)\) consists of field-effect and ionization energy, or the affinity energy. Charge-to-charge interaction \(\left( {U^{qq}\left[ {\left\{ {q1s} \right\},\left\{ r \right\}} \right]} \right)\) is associated with the charge density distribution functions. Nuclear-charge interaction \(U^{qZ}\left[ {\left\{ q \right\},\left\{ r \right\}} \right]\) depends on the coulombic interaction between charge density distributions in the system. Polar interaction energy \(U^{\mathrm {polar}}\left[ {\left\{ q \right\},\{ r\} } \right]\) depends on the charge interactions and dipole distributions in response to the external electric field.
The charge-dependent short-range interaction in Eq. (24) is associated with bond distance functions of the atom and the associated charge functions. This energy term can be expressed by
Here, \(V_{ij}^{\mathrm {bond}}\) is the bond energy that is associated with pairwise attraction \(\left( {V^A(r_{ij},q_i,q_j)} \right)\) and pairwise repulsion energy \((V^R(r_{ij},q_i,q_j))\). They exponentially decrease with the interatomic distance rij. Fc(rij) is the cutoff function.
The long-range interaction energy is defined by the vdW interaction, which can be expressed by the LJ interaction formula130
Here, \(\sigma _{ij}^{\mathrm {vdW}}\) and \(\varepsilon _{ij}^{\mathrm {vdW}}\) are the equilibrium distance and interaction energy, respectively; rij is the cutoff radii where the interaction assumes zero.
The correction factor in Eq. (24) adjusts the energy penalties associated with specified angles. It can be calculated by a sixth-order Legendre polynomial and bond bending term.
Here, \(K_{ijk}^\theta\) indicates the specific bond angles and θijk is the bond angle.
The COMB3 potential has been introduced to study the CVD process for various 2D materials. The charge-optimized COMB3 potentials have been used to simulate the experimental CVD deposition and growth of graphene on metal substrates. Especially, the introduction of dihedral interaction terms in the COMB3 potential can capture the delocalized bonding and bond bending during the CVD process on a particular substrate. The wrinkle formation due to the size mismatch of graphene and Cu-substrate has been studied using COMB3 potential, which was comparable to experimental results98. This potential was also used to understand the role of growth parameters such as absorption energy, migration barrier, and temperature in the Cu deposition on ZnO substrate131. COMB3 potential has also been used to investigate the effect of surface hydroxylation of amorphous SiO2 substrate on heat conduction of supported graphene132.
Mesoscale simulations
Phase-field model
At mesoscale, a simple and effective model for 2D materials growth is the classical Burton-Cabrera-Frank (BCF) theory133, where the island growth is realized by the deposition of adatoms from the supersaturated gas atmosphere and the incorporation of diffusing atoms at the step edge. The direct numerical implementation of the BCF model requires tracking the evolving step edges and applying the boundary conditions due to the sharp-interface nature of the model; in contrast, phase-field diffuse-interface models134 avoid the explicit tracking of the interfaces.
The BCF-based phase-field models have been developed since the 1990s for a series of problems related to crystal growth, including collective step motions in a 1-D step train135, spiral surface growth during thin-film epitaxy136, and step flow under different kinetic regimes137. More recently, the combined effects of edge diffusion, the Ehrlich-Schwoebel barrier, deposition, and desorption for epitaxial growth have been investigated in 2-D138. Based on these studies, Meca et al.139,140 applied the model to the growth of 2D materials with anisotropic diffusion on substrates, using the epitaxial graphene growth on copper foil during CVD as an example, with experimental validations. The typical BCF-based phase-field model for 2D materials growth uses a smoothly varying order parameter ϕ to distinguish the phases, e.g., ϕ = 1 in the 2D island, ϕ = −1 in the substrate, and −1 < ϕ < 1 at step edges. The free energy functional ℱ of the system has the general form of:
where the first term f(ϕ) is a double-well (or multi-well, e.g., see refs. 136,141) function with global minima at ϕ = 1 and ϕ = −1; the second term describes the coupling with the reduced saturation field u, with g(ϕ) an interpolation function and λ the coupling coefficient; the third term is the gradient energy due to the inhomogeneous distribution of ϕ at interfaces, where W(θ) is the angle-dependent interface thickness with \(\theta = arctan( {\phi _y/\phi _x})\). u can be directly related to the adatom concentration c through \(u = {\mathrm{\Omega }}( {c - c_{eq}^0} )\), where \(c_{eq}^0\) is the equilibrium concentration for a flat interface and Ω is the atomic area of the solid phase. The evolution of ϕ is governed by
where τϕ is the characteristic time. The evolution of the saturation field u is governed by
where D is the diffusion coefficient, Fd is the effective deposition rate, and τs is the characteristic time scale for atom desorption. The phase-field equations (Eqs. (30) and (31)) can recover the sharp-interface BCF model in the limit of W(θ) → 0140.
The applications of the phase-field models in 2D materials growth can be categorized into the following three aspects. (1) Reproducing and explaining experimentally observed growth morphologies. Examples include the reconstruction of the spiral growth of SnSe2141, the verification of impurity-induced bilayered graphene growth142, and the explanation to island shape change from quasi-hexagons to triangles and dendrite-like morphologies143. (2) Investigating the effect of experimental parameters on growth morphologies. Taking the CVD growth of graphene as an example, the effects of flux of carbon species144, deposition rates145, and equilibrium saturation145, and substrate orientation139 on graphene island morphologies have been investigated using phase-field simulations, which could provide useful guidance to the experimental control of the growth qualities. (3) Further development of the model to include the effect of additional physical/chemical processes during growth. In addition to the deposition, edge diffusion, and edge anisotropies, phase-field simulations have been extended to model multi-island interactions146, tilt GB topology of 2D materials on substrates with topological cones147, and to include the chemical reaction kinetics in the gas phase using a microkinetic model148. These works improve the fidelity and applicable range of the phase-field model for 2D materials growth.
Nevertheless, a primary obstacle to applying phase-field models for computational synthesis of 2D materials is the lack of connection between the model parameters and the experimentally controllable CVD processing parameters, which is largely due to the difference in size scales of interest. Most of the existing phase-field simulations only focus on the growth of a few 2D islands (up to micrometer scale), which can hardly represent the effect of the macroscopic CVD parameters. One possible solution is to enable the “multiscale” modeling approach8, integrating the FEM (see the section on “Macroscale models”) to account for the effect of macroscopic CVD parameters on transport phenomena and the mesoscale phase-field model for 2D crystal growth within selected zones of the whole substrate. In particular, the steady-state velocity, concentration, and temperature profiles from FEM can be incorporated in the phase-field model, as it has been proposed and applied by Momeni et al.8 (see Fig. 2). The incorporation of chemical reaction kinetics in the phase-field model can improve the accuracy of the predictions and expand its applicability. Phase-field models have also been applied to phase changes in 2D materials. Notable examples include the multidomain microstructure during H-T’ structure transformation in MoTe2 and the domain switching behaviors under external stimuli149. A fast Fourier transform algorithm has been implemented to study the domain switching during bending of MoTe2 (see Fig. 3149).

a Initial precursor concentration distribution from FEM. b Phase-field simulation results for MoS2 morphology on substrate at t = 0.25 h, the enlarged images of i–iii boxes are shown in d–f. c Experimental results for deposition of MoS2 after deposition for t = 0.25 h, the enlarged images of 1–3 boxes are shown in g, h. Reprinted from ref. 8 under Creative Commons Attribution 4.0 International License. The scale bar in a, b shows 400 μm and the one in d–f shows 400 μm.

a Phase diagram in \(\left( {\bar \varepsilon _{xx},\bar \varepsilon _{yy}} \right)\) space. Colored (hatched) regions correspond to one (two) phase regions. Solid lines (analytic) and filled data points (simulations) indicate the strain at which H and a given T′ variant have equal energies. Dashed lines and open symbols similarly represent two-phase coexistence bounds. (▲, △) and (●, ○) correspond to simulations with and without bending, respectively, and blue circles (blue solid circle and blue open circle) represent DFT results at 0. b–d Images of the microstructures with increasing time/strain from left to right for strain paths b–d in phase diagram a. Strain values \(\left( {\bar \varepsilon _{xx},\bar \varepsilon _{yy}} \right)\) are given below each panel. e Corresponding maximum principal stress (GPa) maps for microstructures in d. All simulations are for 50 μm × 50 μm simulations. Adapted with permission from ref. 149. Copyright (2017) American Chemical Society.
Phase-field-crystal model
Different from the continuous mesoscale phase-field approach, the phase-field-crystal (PFC) approach150 describes the thermodynamics and dynamics of phase transformations through an atomically varying order parameter related to the atomic density field. With the application of the classical dynamic DFT, PFC can capture the atomistic-scale morphology and evolution dynamics, within diffusional time scales, which is usually challenging for conventional atomistic methods. Recently, PFC has been actively applied to investigate the defect formation and the atomic structure of interfaces, GBs, and triple junctions during the growth of 2D materials151. The unique GB structure and collective domain dynamics in binary 2D materials, e.g., h-BN, was investigated (see Fig. 4152).

a–f Grain coalescence and inversion domain dynamics. c Atomic site number N vs. t, showing two regimes N = −0.065t + 3.045 × 104 and N = −0.085t + 3.612 × 104 via fitting. d, e Domain shrinking in the white boxed region of b. f Transient of three merging heart-shaped defects before their annihilation. g–i Time evolution of collective atomic displacements in the yellow boxed corner of d. Adapted figure with permission from ref. 152. Copyright (2017) by the American Physical Society.
Compared with the continuous phase-field approach, the advantage of PFC lies in resolving atomistic-scale interface structures and growth dynamics of 2D islands; however, due to the limitation of simulation size, interface dynamics under the effect of realistic CVD parameters are difficult to realize. Future directions of PFC simulations for 2D materials growth include the consideration of atomistic-scale island-substrate interactions and the integration with the larger-scale phase-field or FEM simulations.
Kinetic Monte Carlo
KMC is another mesoscale technique for predicting the growth morphology and kinetic mechanisms of 2D materials, where all possible kinetic events are listed in an event catalog, and their stochastic sequence are randomly selected based on their activation energies153. Activation energies of the related kinetic processes are usually obtained from atomistic-scale calculations. KMC identifies the governing kinetic pathways and simulates the kinetic process. Details of its implementation can be found in ref. 154.
KMC has been extensively applied to simulate the growth of graphene, e.g., the evolution of vacancy complexes and formation of vacancy chains155, the formation and kinetic effect of multi-member carbon ring complexes156, etching of graphene GBs due to oxygen migration and reaction157, and GB evolution following the Stone-Wales mechanism158. Growth of nanoscale graphene islands from carbon monomer nuclei or pre-existing growth fronts has been captured, showing catalytic growth behaviors159, anisotropic morphological patterns160, temperature- and deposition-flux-rate-dependent size and shapes161, inhomogeneous and nonlinear growth kinetics due to lattice mismatch162, and geometry-determined growth mechanisms163. The “coastline” graphene morphology during sublimation164 and the step-flow growth of epitaxial graphene have also been reported165. Based on KMC simulations for graphene growth on Cu (111) with and without hydrogen, the growth protocol was designed for bilayer growth and N-doped graphene growth166.
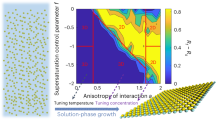
Regarding TMDs, the KMC simulations for the growth of WSe2 monolayer on graphene have been used to develop the phase diagram of domain morphologies as a function of flux and precursor stoichiometry (Fig. 5)154,167. It was found that the fast kink nucleation and propagation, rather than edge attachment and diffusion, could lead to ultrafast growth of monolayer WSe2168. KMC simulations also guided CVD growth of large-scale WSe2 grains by controlling the three-stage adsorption–diffusion–attachment mechanisms169. KMC model is also a key component in a more generalized mechanistic model for growth morphology predictions of 2D materials47.

a Reaction energy diagram of the growth process based on DFT calculations167. The simulation starts with adatoms. They react with each other to form the TMD domains. After bonded into the domain, the in-flake atoms are mobile through edge diffusion and vacancy diffusion. Each state is defined by its state energy, and each process is defined by its transition activation energy. Adapted from ref. 154 under Creative Commons Attribution 4.0 International License. b Domain morphology diagram on the metal flux-C/M ratio plane at 973 K from KMC simulations. Five regions are identified: I: no growth, II: quasi-equilibrium compact domain, III: fractal, IV: dendrite, and V: semi-compact domain. The arrows show a proposed scenario to obtain high-quality domains with high growth rate. Adapted from ref. 167.
In summary, the KMC model can be a useful tool for investigating the kinetic pathways and morphologies during the growth of 2D materials. However, the probability-based nature of KMC makes it most suitable for cases where atomic fluctuations are high, i.e., the atomistic and nanoscale morphology and kinetics of 2D materials. For larger-scale simulations, due to the significantly increased system size and the disparate rates of different KMC events, a full-KMC is computationally expensive. As a compromise, multiscale KMC has been developed163; simplifications should also be made to account for the key events that are most relevant to the large-scale kinetics163. Under such situations, the phase-field approach could be a more efficient option. Meanwhile, the KMC model can provide the governing kinetic mechanisms for phase-field simulations to improve the validity and accuracy of the simulation.
Macroscale models
Although the growth of 2D materials occurs at the nano- and mesoscale, it is controlled by physics and parameters that have a macroscopic nature, e.g., heat and mass transfer, furnace configuration, and gas-phase reactions. Thus, having a thorough understanding of the macroscale physics and processes is essential for controlling the growth of 2D materials and their synthesis by design. We can classify the macroscale models of the growth chamber into four groups: (i) experiment-based models, where rate equations and their constants used to describe the growth170 are determined from experiments; (ii) analytical models, where the governing equations are simplified and solved analytically171; (iii) adaptive models, where a set of experiments are used to train the model172; and (iv) multiphysics models, where the coupled system of governing equations at different length and temporal scales are solved numerically173. Among these methods, the last group of models have key advantages, providing a profound understanding to the growth process, flexibility to apply to different growth conditions, and the ability to optimize the process.
A practical macroscale model of the growth chamber should capture the critical governing physics, e.g., the heat and mass transports and chemical reactions. Setting up these models requires several key information that can be obtained from lower scale simulations7 or experiments174. Identifying the gas-phase reactions, we may approximate some of the reaction parameters using classical theories, e.g., the collision rate to estimate the chemisorption rate of species175 or the group contribution methods to determine the diffusion coefficient176. The other approximation is for low concentrations of reactive species, where the change in pressure and heat of reaction can be neglected. In the latter case, we may decouple the fluid and heat transfer of gaseous materials from the mass transfer and kinetics. In contrast, for high concentrations of reactive species in the gas phase, such as in metalorganic chemical vapor deposition (MOCVD), the coupled system of equations must be solved176. A summary of main equations in macroscale models are presented below.
Gas flow
The Navier-Stokes equation governs the flow rates in the growth chamber,
where u is the velocity field, p is pressure, I is the unit matrix, μ is the dynamic viscosity, and F is the volumetric applied force, i.e., weight. Furthermore, the density ρ is a function of temperature and precursor concentration. In general, the concentration of precursor in the gas phase is low and ρ can be assumed to be only a function of temperature.
Heat transfer
The two main heat transfer mechanisms in the growth chamber are convection and conduction that respectively are
where Q is the heat source, CP is the heat capacity at constant pressure, T is the temperature, q is the heat flux vector, and k is the thermal conductivity coefficient.
Mass transfer
Commonly, the concentration of precursor materials in the growth chamber is negligible. Thus, the equation governing the mass transfer can be formulated with the flow-assisted diffusion of dilute species
where R is the source term for precursor, c is the precursor concentration, D is the diffusion coefficient, and N is the flux of the precursor. Equations (32)–(34) must be solved for a set of initial and boundary conditions, as shown in Table 6.
Chemical reaction
Consider a set of j reactions involving i species of the form \(aA + bB + \ldots \rightleftharpoons xX + yY + \ldots\) with forward and reverse reaction rate constants \(k_j^f\) and \(k_j^r\), respectively. These reaction rates depend on the temperature via an Arrhenius expression, e.g., \(k_j^f = A_j^f\left( {T/T_{{\mathrm{ref}}}} \right)^n{\mathrm{exp}}\left( { - E_j^f/R_gT} \right)\). Here, \(A_j^f\) denotes the frequency factor, n the temperature exponent, \(E_j^f\) the activation energy, and Rg the gas constant. According to the mass action law, the reaction rates rj can be described as,
where ci is the concentration of i-th species and vij is the stoichiometric coefficient (negative for reactants and positive for products). The rate equations provide the information regarding the concentration of different species, which in combination with mass and heat balance equations form the complete system of equations.
Macroscale models provide insight into the growth mechanisms and flow regimes, and play a crucial role in determining proper furnace design and growth parameters. Figure 6 shows the application of a macroscopic model of the flow and concentration diffusion in a vertical furnace with a side inlet to study the effect of the gap between the susceptor and substrate. It shows that the presence of the gap results in the formation of eddy currents close to the substrate edge, which disturbs precursor concentration and hinders the formation of 2D materials that is consistent with the experimental observations. These models have been successfully used in determining the mechanisms governing the growth of 2D materials. For example, it was revealed that the concentration gradient of the precursor is a critical factor in determining the growth morphology of 2D materials, where a low concentration gradient along the substrate leads to the formation of multigrain 2D films. In contrast, moderate planar concentration gradients lead to isolated islands of varying morphologies, and an out-of-plane gradient leads to the formation of standing 2D materials173,177. Furthermore, macroscale models can be combined with lower scale models to form a multiscale platform with high fidelity, e.g., ref. 8.

a Snapshots of the concentration profile within the growth chamber with a heated susceptor where the precursor enters from top left side of the furnace. It indicates the effect of buoyancy forces on inducing instabilities and back flow formation. b, c Magnified views of the flow and concentration profiles close to the susceptor/substrate. Eddy current forms when there is a gap between the susceptor and substrate which hinders formation of 2D materials.
Materials genome approaches
Data-driven methods have been employed to satisfy the urgent need for theoretical development of 2D materials. The improvement of computational power has resulted in extensive collections of data and scientifically organized databases, which can be used by methods such as data mining and machine learning, to accelerate the computational design of new 2D materials.
Database of 2D materials
Compared with the databases of 3D bulk materials such as the Crystallographic Open Database178, the Inorganic Crystal Structure Database179, and the Cambridge Crystallographic Data Centre180, the database focused on 2D materials is still under development. Several 2D materials databases have been attempted primarily based on a high-throughput approach coupled with first-principles and quantum theory to address this shortcoming. Besides, data-mining methods have also been applied to screen those bulk materials databases and identify possible 2D materials from 3D structures. These approaches can provide various information about 2D materials that have been or not been synthesized in experiments. For example, using a criterion to identify 2D materials, a database has been developed on the basis of experimental and DFT data181. This database provides information including exfoliation energy, bandgap, work function, and elastic-constant values of various 2D materials, and also predicted hundreds of new 2D materials for the experimental synthesis. DFT and many-body perturbation theory have been used to generate the Computational 2D Materials Database (C2DB)182. The C2DB contains structural, thermodynamic, elastic, electronic, magnetic, and optical information of about two thousand 2D materials in more than 30 crystal structures. A few hundred new monolayer structures that were previously unknown, but potentially synthesizable were also identified. A data-mining method has also been used to screen the bulk materials from the open database and identified a few hundred 2D weakly bonded solid materials183. The band gaps and point groups that determine piezoelectric and nonlinear optical properties in these materials were also provided. A search in the database of bulk materials was performed and DFT calculations were used to explore these materials for possible exfoliation, where more than one thousand materials have been identified14. Information including electronic, vibrational, magnetic and topological properties of promising materials was also provided. Similarly, the crystal database was screened for layered motifs, and hundreds of stable layered materials were found that can be considered as candidates for the formation of 2D monolayers via exfoliation184.
Machine learning method
Machine learning models trained using 2D material databases can be parameterized, validated, and therefore used for predictive exploration of novel 2D materials with desired properties. Machine learning methods can also aid the understanding of the complex correlations between structures and properties in 2D materials. A combination of machine learning with MD simulations and in-situ high-resolution transmission electron microscopy to explore TMDs has been performed185, where machine learning provided information for structural optimization and evolution of defects to help understand the structural transition in 2D TMDs. A practical method to explore hybrid 2D materials was developed by coupling machine learning with DFT calculations186. The structural and electronic properties of different hybrid 2D materials were provided and various parameters for vdW heterostructures were screened. A machine learning model with force-filed-inspired descriptors in material screening for complex systems has been introduced and used to discover exfoliated 2D-layered materials187. An artificial neural network for titanium dioxide systems was trained based on the a DFT calculated database, where a novel quasi 2D titanium dioxide structure was revealed188. Similarly, new 2D materials with high magnetic moments were found using a machine learning model trained by first-principles data189. Machine learning methods have also been used to aid the development of force fields for classical simulations of materials190. For instance, a force field for classical simulations of stanene was developed using a machine learning method trained by data sets from ab-initio results to calculate the mechanical and thermal properties of stanene190. 2D materials without bulk layered counterparts are also being discovered, using genetic algorithms191,192 or particle swarm optimization, both using energy-based merit criteria and additional biases towards 2D sheets (see refs. 17,193,194).
Outlook
This overview summarizes the state-of-the-art of modeling efforts on the growth of 2D-layered materials and outlines the advantages and limitations of computational models and methods at different length and time scales. The emphasis is on modeling, understanding, and predicting the thermodynamics and kinetics of 2D materials growth. The eventual goal is to develop computational tools and techniques to realize the “synthesis by design” of 2D materials.
An ideal computational model should not only capture accurately the correct physics to allow robust designs but also be computationally efficient to be employed for active control of the growth process. The complex nature of the growth of 2D materials requires applications of a range of models, which operate at multiple length and temporal scales to capture the full range of the growth phenomena from macroscale flow and concentration profiles in the furnace to atomistic reactions leading to formation and growth of 2D materials at mesoscale. Modeling the full spectrum of mechanisms involved in the growth of 2D materials is challenging due to the limitations associated with each of the computational models at different length and temporal scales.
A high-fidelity prediction of 2D materials growth often requires an integration of methods applicable across different scales, ranging from electronic and atomistic, to mesoscale phenomenological, and large-scale continuum models. For example, chemical potentials of different phases and the migration energies of atomic species in different structural and chemical environment from electronic and atomistic models can be employed to parametrize mesoscale phase-field models for predicting the morphology and thus characteristics of as-grown 2D materials with the boundary conditions specified by the information obtained from large-scale continuum models such as temperature distribution in the growth chamber as presented in ref. 8. Furthermore, technical obstacles to in-situ experimental monitoring of growth hinder the validation of computational predictions. The current mathematical and numerical models are computationally expensive and commonly do not capture simultaneously all the physics involved in a growth process of 2D materials, limiting their widespread adoption in active control of 2D materials growth. One promising way to circumvent this challenge is to use machine learning models trained using experimental and computational databases of the 2D materials structure and growth conditions such as the ones available at the 2D Crystal Consortium–Materials Innovation Platform. Finally, the models and approaches applied in understanding the growth of 2D materials can be adapted to the growth of other materials that use the same synthesis techniques, e.g., CVD growth of thin films.
As a future perspective, we can envision two main strategies for the development of computational models and their applications to the design and synthesis of 2D materials, which are long-term challenging goals of the field. First is the open-loop design approach, where the growth chamber design and synthesis conditions are determined by performing a series of simulations mimicking the thermophysical conditions and reaction and growth kinetics that would be expected in experiments. In this approach, the models must have high fidelity and a comprehensive sensitivity analysis should be performed to obtain a robust design. The second approach is the closed-loop design that the developed models will be utilized to determine the states of the system to be passed to a controller which adjusts the growth conditions in real-time. In this approach, the computational efficiency of the models is key to the success of the design.
Data availability
All data that was obtained during this project are available from the authors.
References
Henini, M. Principles of electronic materials and devices (second edition). Microelectron. J. 33, 681 (2002).
Yi, M. & Shen, Z. A review on mechanical exfoliation for the scalable production of graphene. J. Mater. Chem. A 3, 11700–11715 (2015).
Coleman, J. N. et al. Two-dimensional nanosheets produced by liquid exfoliation of layered materials. Science 331, 568–571 (2011).
Lim, Y. R. et al. Roll-to-roll production of layer-controlled molybdenum disulfide: a platform for 2D semiconductor-based industrial applications. Adv. Mater. 30, 1705270 (2018).
McKee, G. S. B. & Vecchio, K. S. Thermogravimetric analysis of synthesis variation effects on CVD generated multiwalled carbon nanotubes. J. Phys. Chem. B 110, 1179–1186 (2006).
Ostadhossein, A. et al. ReaxFF reactive force-field study of molybdenum disulfide (MoS2). J. Phys. Chem. Lett. 8, 631–640 (2017).
Hong, S. et al. Computational synthesis of MoS layers by reactive molecular dynamics simulations: initial sulfidation of MoO surfaces. Nano Lett. 17, 4866–4872 (2017).
Momeni, K., Ji, Y., Zhang, K., Robinson, J. A. & Chen, L.-Q. Multiscale Framew. Simul.-Guide. growth 2D Mater. npj 2D Mater. Appl. 2, 27 (2018).
Zhang, X. et al. Defect-controlled nucleation and orientation of WSe2 on hBN: a route to single-crystal epitaxial monolayers. ACS Nano 13, 3341–3352 (2019).
Chen, W., Gui, X., Yang, L., Zhu, H. & Tang, Z. Wrinkling of two-dimensional materials: methods, properties and applications. Nanoscale Horiz. 4, 291–320 (2019).
Momeni, K. & Levitas, V. I. Propagating phase interface with intermediate interfacial phase: phase field approach. Phys. Rev. B 89 (2014).
Mehdipour, H. & Ostrikov, K. Kinetics of low-pressure, low-temperature graphene growth: toward single-layer, single-crystalline structure. ACS Nano 6, 10276–10286 (2012).
Jiang, J.-W., Wang, B.-S., Wang, J.-S. & Park, H. S. A review on the flexural mode of graphene: lattice dynamics, thermal conduction, thermal expansion, elasticity and nanomechanical resonance. J. Phys. Condens. Matter 27, 83001 (2015).
Mounet, N. et al. Two-dimensional materials from high-throughput computational exfoliation of experimentally known compounds. Nat. Nanotechnol. 13, 246–252 (2018).
Zhuang, H. L. & Hennig, R. G. Stability and magnetism of strongly correlated single-layer VS2. Phys. Rev. B 93, 54429 (2016).
Cohen, A. J., Mori-Sánchez, P. & Yang, W. Insights into current limitations of density functional theory. Science 321, 792–794 (2008).
Paul, J. T. et al. Computational methods for 2D materials: discovery, property characterization, and application design. J. Phys. Condens. Matter 29, 473001 (2017).
Sun, Y. et al. Low-temperature solution synthesis of few-layer 1T′-MoTe2 nanostructures exhibiting lattice compression. Angew. Chem. Int. Ed. 55, 2830–2834 (2016).
Kretschmer, S., Komsa, H.-P., Bøggild, P. & Krasheninnikov, A. V. Structural transformations in two-dimensional transition-metal dichalcogenide MoS2 under an electron beam: insights from first-principles calculations. J. Phys. Chem. Lett. 8, 3061–3067 (2017).
Duerloo, K.-A. N., Li, Y. & Reed, E. J. Structural phase transitions in two-dimensional Mo- and W-dichalcogenide monolayers. Nat. Commun. 5, 4214 (2014).
Gan, L.-Y., Zhang, Q., Zhao, Y.-J., Cheng, Y. & Schwingenschlögl, U. Order-disorder phase transitions in the two-dimensional semiconducting transition metal dichalcogenide alloys Mo1−xWxX2 (X = S, Se, and Te). Sci. Rep. 4, 6691 (2015).
Komsa, H.-P. & Krasheninnikov, A. V. Two-dimensional transition metal dichalcogenide alloys: stability and electronic properties. J. Phys. Chem. Lett. 3, 3652–3656 (2012).
Dumcenco, D. O., Kobayashi, H., Liu, Z., Huang, Y.-S. & Suenaga, K. Visualization and quantification of transition metal atomic mixing in Mo1−xWxS2 single layers. Nat. Commun. 4, 1351 (2013).
Azizi, A. et al. Spontaneous formation of atomically thin stripes in transition metal dichalcogenide monolayers. Nano Lett. 16, 6982–6987 (2016).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Tkatchenko, A. & Scheffler, M. Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 102, 73005 (2009).
Dion, M., Rydberg, H., Schröder, E., Langreth, D. C. & Lundqvist, B. I. Van der Waals density functional for general geometries. Phys. Rev. Lett. 92, 246401 (2004).
He, J., Hummer, K. & Franchini, C. Stacking effects on the electronic and optical properties of bilayer transition metal dichalcogenides MoS2, MoSe2, WS2, and WSe2. Phys. Rev. B 89, 75409 (2014).
Dreyer, C. E., Janotti, A. & Van de Walle, C. G. Absolute surface energies of polar and nonpolar planes of GaN. Phys. Rev. B 89, 81305 (2014).
Mathew, K. et al. MPInterfaces: A Materials Project based Python tool for high-throughput computational screening of interfacial systems. Comput. Mater. Sci. 122, 183–190 (2016).
Henkelman, G., Uberuaga, B. P. & Jonsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Eichfeld, S. M., Colon, V. O., Nie, Y., Cho, K. & Robinson, J. A. Controlling nucleation of monolayer WSe2 during metal-organic chemical vapor deposition growth. 2D Mater. 3, 25015 (2016).
Tuckerman, M. E. Statistical mechanics: theory and molecular simulation (Oxford Univ. Press, 2010).
Bucko, T. Ab initio calculations of free-energy reaction barriers. J. Phys. Condens. Matter 20, 64211 (2008).
Sheppard, D., Terrell, R. & Henkelman, G. Optimization methods for finding minimum energy paths. J. Chem. Phys. 128, 134106 (2008).
Misawa, M. et al. Reactivity of sulfur molecules on MoO3 (010) surface. J. Phys. Chem. Lett. 8, 6206–6210 (2017).
Torrie, G. M. & Valleau, J. P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: umbrella sampling. J. Comput. Phys. 23, 187–199 (1977).
Sun, H. et al. Theory of thin-film-mediated exfoliation of van der Waals bonded layered materials. Phys. Rev. Mater. 2, 94004 (2018).
van Erp, T. S. & Bolhuis, P. G. Elaborating transition interface sampling methods. J. Comput. Phys. 205, 157–181 (2005).
Laio, A. & Parrinello, M. Escaping free-energy minima. Proc. Natl Acad. Sci. USA 99, 12562–12566 (2002).
Darve, E. & Pohorille, A. Calculating free energies using average force. J. Chem. Phys. 115, 9169–9183 (2001).
Kaminski, J. W., Kratzer, P. & Ratsch, C. Towards a standardized setup for surface energy calculations. Phys. Rev. B 95, 85408 (2017).
Liu, Y., Bhowmick, S. & Yakobson, B. I. BN white graphene with ‘colorful’ edges: the energies and morphology. Nano Lett. 11, 3113–3116 (2011).
Chetty, N. & Martin, R. M. First-principles energy density and its applications to selected polar surfaces. Phys. Rev. B 45, 6074–6088 (1992).
Artyukhov, V. I., Liu, Y. & Yakobson, B. I. Equilibrium at the edge and atomistic mechanisms of graphene growth. Proc. Natl Acad. Sci. USA 109, 15136–15140 (2012).
Li, X. et al. Edge-controlled growth and etching of two-dimensional GaSe monolayers. J. Am. Chem. Soc. 139, 482–491 (2017).
Rajan, A. G., Warner, J. H., Blankschtein, D. & Strano, M. S. Generalized mechanistic model for the chemical vapor deposition of 2D transition metal dichalcogenide monolayers. ACS Nano 10, 4330–4344 (2016).
Freysoldt, C. et al. First-principles calculations for point defects in solids. Rev. Mod. Phys. 86, 253–305 (2014).
Stone, A. J. & Wales, D. J. Theoretical studies of icosahedral C60 and some related species. Chem. Phys. Lett. 128, 501–503 (1986).
Komsa, H. P., Berseneva, N., Krasheninnikov, A. V. & Nieminen, R. M. Charged point defects in the flatland: Accurate formation energy calculations in twodimensional materials. Phys. Rev. X 4, 31044 (2014).
Rao, R. et al. Dynamics of cleaning, passivating and doping monolayer MoS2 by controlled laser irradiation. 2D Mater. 6, 45031 (2019).
Rastogi, P., Kumar, S., Bhowmick, S., Agarwal, A. & Chauhan, Y. S. Doping strategies for monolayer MoS2 via surface adsorption: a systematic study. J. Phys. Chem. C. 118, 30309–30314 (2014).
Weston, L., Wickramaratne, D., Mackoit, M., Alkauskas, A. & Van de Walle, C. G. Native point defects and impurities in hexagonal boron nitride. Phys. Rev. B 97, 214104 (2018).
Walsh, A. & Zunger, A. Instilling defect tolerance in new compounds. Nat. Mater. 16, 964–967 (2017).
Zhang, F. et al. Full orientation control of epitaxial MoS2 on hBN assisted by substrate defects. Phys. Rev. B 99, 155430 (2019).
Lee, G.-D. et al. Diffusion, coalescence, and reconstruction of vacancy defects in graphene layers. Phys. Rev. Lett. 95, 205501 (2005).
Kotakoski, J., Krasheninnikov, A. V. & Nordlund, K. Energetics, structure, and long-range interaction of vacancy-type defects in carbon nanotubes: atomistic simulations. Phys. Rev. B 74, 245420 (2006).
Momeni, K., Attariani, H. & Lesar, R. A. Structural transformation in monolayer materials: A 2D to 1D transformation. Phys. Chem. Chem. Phys. 18, 19873–19879 (2016).
Zhou, W. et al. Intrinsic structural defects in monolayer molybdenum disulfide. Nano Lett. 13, 2615–2622 (2013).
Zou, X., Liu, Y. & Yakobson, B. I. Predicting dislocations and grain boundaries in two-dimensional metal-disulfides from the first principles. Nano Lett. 13, 253–258 (2013).
Xu, F., Yu, H., Sadrzadeh, A. & Yakobson, B. I. Riemann surfaces of carbon as graphene nanosolenoids. Nano Lett. 16, 34–39 (2016).
Yu, Z. G., Zhang, Y.-W. & Yakobson, B. I. An anomalous formation pathway for dislocation-sulfur vacancy complexes in polycrystalline monolayer MoS2. Nano Lett. 15, 6855–6861 (2015).
Sha, Z. D. et al. Inverse pseudo Hall-Petch relation in polycrystalline graphene. Sci. Rep. 4, 5991 (2014).
Grantab, R., Shenoy, V. B. & Ruoff, R. S. Anomalous strength characteristics of tilt grain boundaries in graphene. Science 330, 946–948 (2010).
Zhang, Z., Yang, Y., Xu, F., Wang, L. & Yakobson, B. I. Unraveling the sinuous grain boundaries in graphene. Adv. Funct. Mater. 25, 367–373 (2015).
Liu, Y., Xu, F., Zhang, Z., Penev, E. S. & Yakobson, B. I. Two-dimensional mono-elemental semiconductor with electronically inactive defects: the case of phosphorus. Nano Lett. 14, 6782–6786 (2014).
Jones, J. E. On the determination of molecular fields. II. From the equation of state of a gas. Proc. R. Soc. Lond. Ser. A Math. Phys. Eng. Sci. 106, 463–477 (1924).
Wen, M., Carr, S., Fang, S., Kaxiras, E. & Tadmor, E. B. Dihedral-angle-corrected registry-dependent interlayer potential for multilayer graphene structures. Phys. Rev. B 98, 235404 (2018).
Kolmogorov, A. N. & Crespi, V. H. Registry-dependent interlayer potential for graphitic systems. Phys. Rev. B 71, 235415 (2005).
Naik, M. H., Maity, I., Maiti, P. K. & Jain, M. Kolmogorov–Crespi potential formultilayer transition-metal dichalcogenides: capturing structural transformations in Moiré superlattices. J. Phys. Chem. C. 123, 9770–9778 (2019).
Rappe, A. K., Casewit, C. J., Colwell, K. S., Goddard, W. A. & Skiff, W. M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 114, 10024–10035 (1992).
Stillinger, F. H. & Weber, T. A. Computer simulation of local order in condensed phases of silicon. Phys. Rev. B 31, 5262–5271 (1985).
Jiang, J.-W. Parametrization of Stillinger–Weber potential based on valence force field model: application to single-layer MoS2 and black phosphorus. Nanotechnology 26, 315706 (2015).
Jiang, J.-W., Park, H. S. & Rabczuk, T. Molecular dynamics simulations of single-layer molybdenum disulphide (MoS2): Stillinger-Weber parametrization, mechanical properties, and thermal conductivity. J. Appl. Phys. 114, 64307–64311 (2013).
Jiang, J.-W., Qi, Z., Park, H. S. & Rabczuk, T. Elastic bending modulus of single-layer molybdenum disulfide (MoS2): finite thickness effect. Nanotechnology 24, 435705 (2013).
Yapicioglu, H., Kandemir, A., Sevik, C., Çağın, T. & Kinaci, A. Thermal transport properties of MoS2 and MoSe2 monolayers. Nanotechnology 27, 55703 (2016).
Norouzzadeh, P. & Singh, D. J. Thermal conductivity of single-layer WSe2 by a Stillinger–Weber potential. Nanotechnology 28, 75708 (2017).
Jiang, J.-W., Rabczuk, T. & Park, H. S. A Stillinger–Weber potential for single-layered black phosphorus, and the importance of cross-pucker interactions for a negative Poisson’s ratio and edge stress-induced bending. Nanoscale 7, 6059–6068 (2015).
Xu, W., Zhu, L., Cai, Y., Zhang, G. & Li, B. Direction dependent thermal conductivity of monolayer phosphorene: parameterization of Stillinger-Weber potential and molecular dynamics study. J. Appl. Phys. 117, 214308 (2015).
Jiang, J.-W. & Zhou, Y.-P. In Parameterization of Stillinger-Weber Potential for Two-Dimensional Atomic Crystal Chapter 1 (IntechOpen, 2017). https://doi.org/10.5772/intechopen.71929.
Allinger, N. L., Yuh, Y. H. & Lii, J. H. Molecular mechanics. The MM3 force field for hydrocarbons. 1. J. Am. Chem. Soc. 111, 8551–8566 (1989).
Sun, H. COMPASS: an ab initio force-field optimized for condensed-phase applications overview with details on alkane and benzene compounds. J. Phys. Chem. B 102, 7338–7364 (1998).
Keating, P. N. Effect of invariance requirements on the elastic strain energy of crystals with application to the diamond structure. Phys. Rev. 145, 637–645 (1966).
Allinger, N. L. Calculation of molecular structure and energy by force-field methods. Adv. Phys Org. Chem. 13, 1–82 (1976).
Allinger, N. L. Conformational analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms. J. Am. Chem. Soc. 99, 8127–8134 (1977).
Allinger, N. L., Chen, K., Katzenellenbogen, J. A., Wilson, S. R. & Anstead, G. M. Hyperconjugative effects on carbon—carbon bond lengths in molecular mechanics (MM4). J. Comput. Chem. 17, 747–755 (1996).
Tu, Z. & Ou-Yang, Z. Single-walled and multiwalled carbon nanotubes viewed as elastic tubes with the effective Young’s moduli dependent on layer number. Phys. Rev. B 65, 233407 (2002).
Chang, T. & Gao, H. Size-dependent elastic properties of a single-walled carbon nanotube via a molecular mechanics model. J. Mech. Phys. Solids 51, 1059–1074 (2003).
Tersoff, J. Empirical interatomic potential for carbon, with applications to amorphous carbon. Phys. Rev. Lett. 61, 2879–2882 (1988).
Brenner, D. W. et al. A second-generation reactive empirical bond order (REBO) potential energy expression for hydrocarbons. J. Phys. Condens. Matter 14, 783–802 (2002).
Stuart, S. J., Tutein, A. B. & Harrison, J. A. A reactive potential for hydrocarbons with intermolecular interactions. J. Chem. Phys. 112, 6472–6486 (2000).
Liang, T., Devine, B., Phillpot, S. R. & Sinnott, S. B. Variable charge reactive potential for hydrocarbons to simulate organic-copper interactions. J. Phys. Chem. A 116, 7976–7991 (2012).
Van Duin, A. C. T., Dasgupta, S., Lorant, F. & Goddard, W. A. ReaxFF: a reactive force field for hydrocarbons. J. Phys. Chem. A 105, 9396–9409 (2001).
Liang, T. et al. Reactive potentials for advanced atomistic simulations. Annu. Rev. Mater. Res. 43, 109–129 (2013).
Fonseca, A. F. et al. Graphene–titanium interfaces from molecular dynamics simulations. ACS Appl. Mater. Interfaces 9, 33288–33297 (2017).
Mortazavi, B., Pereira, L. F. C., Jiang, J.-W. & Rabczuk, T. Modelling heat conduction in polycrystalline hexagonal boron-nitride films. Sci. Rep. 5, 13228 (2015).
Mukhopadhyay, T., Mahata, A., Adhikari, S. & Asle Zaeem, M. Probing the shear modulus of two-dimensional multiplanar nanostructures and heterostructures. Nanoscale 10, 5280–5294 (2018).
Klaver, T. P. C., Zhu, S.-E., Sluiter, M. H. F. & Janssen, G. C. A. M. Molecular dynamics simulation of graphene on Cu (100) and (111) surfaces. Carbon N. Y. 82, 538–547 (2015).
Hong, S. et al. Chemical vapor deposition synthesis of MoS2 layers from the direct sulfidation of MoO3 surfaces using reactive molecular dynamics simulations. J. Phys. Chem. C. 122, 7494–7503 (2018).
Tersoff, J. New empirical model for the structural properties of silicon. Phys. Rev. Lett. 56, 632–635 (1986).
Tersoff, J. New empirical approach for the structure and energy of covalent systems. Phys. Rev. B 37, 6991–7000 (1988).
Jiang, J.-W. & Wang, J.-S. Manipulation of heat current by the interface between graphene and white graphene. Europhys. Lett. 96, 16003 (2011).
Mahdizadeh, S. J. & Akhlamadi, G. Optimized Tersoff empirical potential for germanene. J. Mol. Graph. Model. 72, 1–5 (2017).
Tersoff, J. Modeling solid-state chemistry: interatomic potentials for multicomponent systems. Phys. Rev. B 39, 5566–5568 (1989).
Jiang, J.-W. & Wang, J.-S. Theoretical study of thermal conductivity in single-walled boron nitride nanotubes. Phys. Rev. B 84, 85439 (2011).
Liu, B. et al. Thermal conductivity of silicene nanosheets and the effect of isotopic doping. J. Phys. D. Appl. Phys. 47, 165301 (2014).
McGuigan, B. C., Pochet, P. & Johnson, H. T. Critical thickness for interface misfit dislocation formation in two-dimensional materials. Phys. Rev. B 93, 214103 (2016).
Raju, M., van Duin, A. & Ihme, M. Phase transitions of ordered ice in graphene nanocapillaries and carbon nanotubes. Sci. Rep. 8, 3851 (2018).
Huang, X., Yang, H., van Duin, A. C. T., Hsia, K. J. & Zhang, S. Chemomechanics control of tearing paths in graphene. Phys. Rev. B 85, 195453 (2012).
Paupitz, R. et al. Graphene to fluorographene and fluorographane: a theoretical study. Nanotechnology 24, 35706 (2012).
Neyts, E. C., van Duin, A. C. T. & Bogaerts, A. Formation of single layer graphene on nickel under far-from-equilibrium high flux conditions. Nanoscale 5, 7250–7255 (2013).
Achtyl, J. L. et al. Aqueous proton transfer across single-layer graphene. Nat. Commun. 6, 6539 (2015).
Raju, M., Govindaraju, P. B., van Duin, A. C. T. & Ihme, M. Atomistic and continuum scale modeling of functionalized graphyne membranes for water desalination. Nanoscale 10, 3969–3980 (2018).
Berdiyorov, G. R., Neek-Amal, M., Peeters, F. M. & van Duin, A. C. T. Stabilized silicene within bilayer graphene: a proposal based on molecular dynamics and density-functional tight-binding calculations. Phys. Rev. B 89, 24107 (2014).
Berdiyorov, G. R. & Peeters, F. M. Influence of vacancy defects on the thermal stability of silicene: a reactive molecular dynamics study. RSC Adv. 4, 1133–1137 (2014).
Kushima, A., Qian, X., Zhao, P., Zhang, S. & Li, J. Ripplocations in van der Waals layers. Nano Lett. 15, 1302–1308 (2015).
Yilmaz, D. E., Lotfi, R., Ashraf, C., Hong, S. & van Duin, A. C. T. Defect design of two-dimensional MoS2 structures by using a graphene layer and potato stamp concept. J. Phys. Chem. C. 122, 11911–11917 (2018).
Mortazavi, B. et al. Strong thermal transport along polycrystalline transition metal dichalcogenides revealed by multiscale modeling for MoS2. Appl. Mater. Today 7, 67–76 (2017).
Yan, R. et al. Thermal conductivity of monolayer molybdenum disulfide obtained from temperature-dependent Raman spectroscopy. ACS Nano 8, 986–993 (2014).
Osti, N. C. et al. Effect of metal ion intercalation on the structure of MXene and water dynamics on its internal surfaces. ACS Appl. Mater. Interfaces 8, 8859–8863 (2016).
Berdiyorov, G. R. & Mahmoud, K. A. Effect of surface termination on ion intercalation selectivity of bilayer Ti3C2T2(T=F, O and OH) MXene. Appl. Surf. Sci. 416, 725–730 (2017).
Osti, N. C. et al. Influence of metal ions intercalation on the vibrational dynamics of water confined between MXene layers. Phys. Rev. Mater. 1, 65406 (2017).
Overbury, S. H. et al. Complexity of intercalation in MXenes: destabilization of urea by two-dimensional titanium carbide. J. Am. Chem. Soc. 140, 10305–10314 (2018).
Sang, X. et al. In situ atomistic insight into the growth mechanisms of single layer 2D transition metal carbides. Nat. Commun. 9, 2266 (2018).
Wang, N. & Komvopoulos, K. The effect of deposition energy of energetic atoms on the growth and structure of ultrathin amorphous carbon films studied by molecular dynamics simulations. J. Phys. D. Appl. Phys. 47, 245303 (2014).
Ma, T., Hu, Y.-Z., Wang, H. & Li, X. Microstructural and stress properties of ultrathin diamondlike carbon films during growth: Molecular dynamics simulations. Phys. Rev. B 75, 35425 (2007).
Wang, X., Tabarraei, A. & Spearot, D. E. Fracture mechanics of monolayer molybdenum disulfide. Nanotechnology 26, 175703 (2015).
Reddy, C. D., Gen Yu, Z. & Zhang, Y.-W. Two-dimensional van der Waals C60 molecular crystal. Sci. Rep. 5, 12221 (2015).
Paul, S. & Momeni, K. Mechanochemistry of stable diamane and atomically thin diamond films synthesis from bi- and multilayer graphene: a computational study. J. Phys. Chem. C. 123, 15751–15760 (2019).
Edward, L.-J. J. & Sydney, C. On the forces between atoms and ions. Proc. R. Soc. A Math. Phys. Eng. Sci. 109, 584–597 (1925).
Cheng, Y.-T. et al. Cu cluster deposition on ZnO(101¯0): morphology and growth mode predicted from molecular dynamics simulations. Surf. Sci. 621, 109–116 (2014).
Cui, L., Shi, S., Li, Z., Wei, G. & Du, X. Manipulating thermal conductance of supported graphene via surface hydroxylation of substrates. J. Phys. Chem. C. 122, 27689–27695 (2018).
Burton, W. K., Cabrera, N., Frank, F. C. & Francis, M. N. The growth of crystals and the equilibrium structure of their surfaces. Philos. Trans. R. Soc. Lond. Ser. A, Math. Phys. Sci. 243, 299–358 (1951).
Steinbach, I. Phase-field model for microstructure evolution at the mesoscopic scale. Annu. Rev. Mater. Res. 43, 89–107 (2013).
Liu, F. & Metiu, H. Stability and kinetics of step motion on crystal surfaces. Phys. Rev. E 49, 2601–2616 (1994).
Karma, A. & Plapp, M. Spiral surface growth without desorption. Phys. Rev. Lett. 81, 4444–4447 (1998).
Pierre-Louis, O. Phase field models for step flow. Phys. Rev. E 68, 21604 (2003).
Hu, Z., Lowengrub, J. S., Wise, S. M. & Voigt, A. Phase-field modeling of epitaxial growth: applications to step trains and island dynamics. Phys. D. Nonlinear Phenom. 241, 77–94 (2012).
Meca, E., Lowengrub, J., Kim, H., Mattevi, C. & Shenoy, V. B. Epitaxial graphene growth and shape dynamics on copper: phase-field modeling and experiments. Nano Lett. 13, 5692–5697 (2013).
Meca, E., Shenoy, V. B. & Lowengrub, J. Phase-field modeling of two-dimensional crystal growth with anisotropic diffusion. Phys. Rev. E 88, 52409 (2013).
Wu, J. et al. Spiral growth of SnSe2 crystals by chemical vapor deposition. Adv. Mater. Interfaces 3, 1600383 (2016).
Li, J. et al. Impurity-induced formation of bilayered graphene on copper by chemical vapor deposition. Nano Res 9, 2803–2810 (2016).
Zhang, K. et al. Manganese doping of monolayer MoS2: the substrate is critical. Nano Lett. 15, 6586–6591 (2015).
Hao, Y. et al. The role of surface oxygen in the growth of large single-crystal graphene on copper. Science 342, 720–723 (2013).
Luo, B. et al. Etching-controlled growth of graphene by chemical vapor deposition. Chem. Mater. 29, 1022–1027 (2017).
Artyukhov, V. I., Hu, Z., Zhang, Z. & Yakobson, B. I. Topochemistry of Bowtie- and star-shaped metal dichalcogenide nanoisland formation. Nano Lett. 16, 3696–3702 (2016).
Yu, H. et al. Tilt grain boundary topology induced by substrate topography. ACS Nano 11, 8612–8618 (2017).
Meca, E., Shenoy, V. B. & Lowengrub, J. H. H2-dependent attachment kinetics and shape evolution in chemical vapor deposition graphene growth. 2D Mater. 4, 31010 (2017).
Berry, J., Zhou, S., Han, J., Srolovitz, D. J. & Haataja, M. P. Dynamic phase engineering of bendable transition metal dichalcogenide monolayers. Nano Lett. 17, 2473–2481 (2017).
Emmerich, H. et al. Phase-field-crystal models for condensed matter dynamics on atomic length and diffusive time scales: an overview. Adv. Phys. 61, 665–743 (2012).
Elder, K. L. M., Seymour, M., Lee, M., Hilke, M. & Provatas, N. Two-component structural phase-field crystal models for graphene symmetries. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 376, 20170211 (2018).
Taha, D., Mkhonta, S. K., Elder, K. R. & Huang, Z.-F. Grain boundary structures and collective dynamics of inversion domains in binary two-dimensional materials. Phys. Rev. Lett. 118, 255501 (2017).
Battaile, C. C. The kinetic Monte Carlo method: foundation, implementation, and application. Comput. Methods Appl. Mech. Eng. 197, 3386–3398 (2008).
Nie, Y. et al. A kinetic Monte Carlo simulation method of van der Waals epitaxy for atomistic nucleation-growth processes of transition metal dichalcogenides. Sci. Rep. 7, 2977 (2017).
Trevethan, T., Latham, C. D., Heggie, M. I., Briddon, P. R. & Rayson, M. J. Vacancy diffusion and coalescence in graphene directed by defect strain fields. Nanoscale 6, 2978–2986 (2014).
Whitesides, R. & Frenklach, M. Effect of reaction kinetics on graphene-edge morphology and composition. Z. fur Physikalische Chem. 229, 597–614 (2015).
Wang, B., Puzyrev, Y. S. & Pantelides, S. T. Enhanced chemical reactions of oxygen at grain boundaries in polycrystalline graphene. Polyhedron 64, 158–162 (2013).
Zhuang, J., Zhao, R., Dong, J., Yan, T. & Ding, F. Evolution of domains and grain boundaries in graphene: a kinetic Monte Carlo simulation. Phys. Chem. Chem. Phys. 18, 2932–2939 (2016).
Taioli, S. Computational study of graphene growth on copper by first-principles and kinetic Monte Carlo calculations. J. Mol. Model. 20, 2260 (2014).
Fan, L. et al. Topology evolution of graphene in chemical vapor deposition, a combined theoretical/experimental approach toward shape control of grapheme domains. Nanotechnology 23, 115605 (2012).
Gaillard, P., Schoenhalz, A. L., Moskovkin, P., Lucas, S. & Henrard, L. Growth of nitrogen-doped graphene on copper: multiscale simulations. Surf. Sci. 644, 102–108 (2016).
Wu, P. et al. Lattice mismatch induced nonlinear growth of graphene. J. Am. Chem. Soc. 134, 6045–6051 (2012).
Jiang, H. & Hou, Z. Large-scale epitaxial growth kinetics of graphene: a kinetic Monte Carlo study. J. Chem. Phys. 143, 84109 (2015).
Huang, J. Y. et al. In situ observation of graphene sublimation and multi-layer edge reconstructions. Proc. Natl Acad. Sci. USA 106, 10103–10108 (2009).
Ming, F. & Zangwill, A. Model and simulations of the epitaxial growth of graphene on non-planar 6H–SiC surfaces. J. Phys. D. Appl. Phys. 45, 154007 (2012).
Qiu, Z., Li, P., Li, Z. & Yang, J. Atomistic simulations of graphene growth: from kinetics to mechanism. Acc. Chem. Res. 51, 728–735 (2018).
Nie, Y. et al. First principles kinetic Monte Carlo study on the growth patterns of WSe2 monolayer. 2D Mater. 3, 25029 (2016).
Chen, S. et al. Origin of ultrafast growth of monolayer WSe2 via chemical vapor deposition. npj Comput. Mater. 5, 28 (2019).
Yue, R. et al. Nucleation and growth of WSe2: enabling large grain transition metal dichalcogenides. 2D Mater. 4, 45019 (2017).
Safron, N. S. & Arnold, M. S. Experimentally determined model of atmospheric pressure CVD of graphene on Cu. J. Mater. Chem. C. 2, 744–755 (2014).
Eversteyn, F. C., Severin, P. J. W., Brekel, C. H. Jvd & Peek, H. L. A stagnant layer model for the epitaxial growth of silicon from silane in a horizontal reactor. J. Electrochem. Soc. 117, 925–931 (1970).
Chen, J., Wong, D. S. H., Jang, S. S. & Yang, S. L. Product and process development using artificial neural‐network model and information analysis. AIChE J. 44, 876–887 (1998).
Vilá, R. A. et al. Bottom-up synthesis of vertically oriented two-dimensional materials. 2D Mater. 3, 41003 (2016).
Lee, P. W., Omstead, T. R., McKenna, D. R. & Jensen, K. F. In situ mass spectroscopy studies of the decomposition of organometallic arsenic compounds in the presence of Ga(CH3)3 and Ga(C2H5)3. J. Cryst. Growth 93, 134–142 (1988).
Theodoropoulos, C. et al. Kinetic and transport modeling of the metallorganic chemical vapor deposition of InP from trimethylindium and phosphine and comparison with experiments. J. Electrochem. Soc. 142, 2086–2094 (1995).
Poling, B. E., Prausnitz, J. M. & O’connell, J. P. The Properties of Gases and Liquids, Vol. 5 (Mcgraw-Hill, New York, 2001).
Zhang, F. et al. Controlled synthesis of 2D transition metal dichalcogenides: from vertical to planar MoS2. 2D Mater. 4, 25029 (2017).
Grazulis, S. et al. Crystallography open database - an open-access collection of crystal structures. J. Appl. Crystallogr 42, 726–729 (2009).
Belsky, A., Hellenbrandt, M., Karen, V. L. & Luksch, P. New developments in the Inorganic Crystal Structure Database (ICSD): accessibility in support of materials research and design. Acta Crystallogr. Sect. B 58, 364–369 (2002).
Allen, F. H. et al. The Cambridge Crystallographic Data Centre: computer-based search, retrieval, analysis and display of information. Acta Crystallogr. Sect. B 35, 2331–2339 (1979).
Choudhary, K., Kalish, I., Beams, R. & Tavazza, F. High-throughput identification and characterization of two-dimensional materials using density functional theory. Sci. Rep. 7, 5179 (2017).
Haastrup, S. et al. The Computational 2D Materials. Database.: high.-throughput modeling Discov. atomically thin Cryst. 2D Mater. 5, 42002 (2018).
Cheon, G. et al. Data mining for new two- and one-dimensional weakly bonded solids and lattice-commensurate heterostructures. Nano Lett. 17, 1915–1923 (2017).
Ashton, M., Paul, J., Sinnott, S. B. & Hennig, R. G. Topology-scaling identification of layered solids and stable exfoliated 2D materials. Phys. Rev. Lett. 118, 106101 (2017).
Patra, T. K. et al. Defect dynamics in 2-D MoS2 probed by using machine learning, atomistic simulations, and high-resolution microscopy. ACS Nano 12, 8006–8016 (2018).
Tawfik, S. A. et al. Efficient prediction of structural and electronic properties of hybrid 2D materials using complementary DFT and machine learning approaches. Adv. Theory Simul. 2, 1800128 (2019).
Choudhary, K., DeCost, B. & Tavazza, F. Machine learning with force-field-inspired descriptors for materials: fast screening and mapping energy landscape. Phys. Rev. Mater. 2, 83801 (2018).
Eivari, H. A. et al. Two-dimensional hexagonal sheet of TiO2. Chem. Mater. 29, 8594–8603 (2017).
Miyazato, I., Tanaka, Y. & Takahashi, K. Accelerating the discovery of hidden two-dimensional magnets using machine learning and first principle calculations. J. Phys. Condens. Matter 30, 06LT01 (2018).
Cherukara, M. J. et al. Ab initio-based bond order potential to investigate low thermal conductivity of stanene nanostructures. J. Phys. Chem. Lett. 7, 3752–3759 (2016).
Revard, B. C., Tipton, W. W., Yesypenko, A. & Hennig, R. G. Grand-canonical evolutionary algorithm for the prediction of two-dimensional materials. Phys. Rev. B 93, 54117 (2016).
Singh, A. K. et al. Genetic algorithm prediction of two-dimensional group-IV dioxides for dielectrics. Phys. Rev. B 95, 155426 (2017).
Chmiela, S. et al. Machine learning of accurate energy-conserving molecular force fields. Sci. Adv. 3, e1603015 (2017).
Botu, V., Batra, R., Chapman, J. & Ramprasad, R. Machine learning force fields: construction, validation, and outlook. J. Phys. Chem. C. 121, 511–522 (2017).
Song, Z., Artyukhov, V. I., Yakobson, B. I. & Xu, Z. Pseudo Hall–Petch strength reduction in polycrystalline graphene. Nano Lett. 13, 1829–1833 (2013).
Okada, M. et al. Direct chemical vapor deposition growth of WS2 atomic layers on hexagonal boron nitride. ACS Nano 8, 8273–8277 (2014).
Kobayashi, Y. et al. Growth and optical properties of high-quality monolayer WS2 on graphite. ACS Nano 9, 4056–4063 (2015).
Huang, J. et al. Large-area synthesis of monolayer WSe2 on a SiO2/Si substrate and its device applications. Nanoscale 7, 4193–4198 (2015).
Chen, L. et al. Step-edge-guided nucleation and growth of aligned WSe2 on sapphire via a layer-over-layer growth mode. ACS Nano 9, 8368–8375 (2015).
Eichfeld, S. M. et al. Highly scalable, atomically thin WSe2 grown via metal–organic chemical vapor deposition. ACS Nano 9, 2080–2087 (2015).
Yanase, T. et al. Chemical vapor deposition of NbS2 from a chloride source with H2 Flow: orientation control of ultrathin crystals directly grown on SiO2/Si substrate and charge density wave transition. Cryst. Growth Des. 16, 4467–4472 (2016).
Friday, M., Angeles, L., Reyes-lillo, S. & Rabe, K. Bulletin of the American Physical Society APS March Meeting 2018. 2018 (2018).
Hotta, T. et al. Molecular beam epitaxy growth of monolayer niobium diselenide flakes. Appl. Phys. Lett. 109, 133101 (2016).
Efetov, D. K. et al. Specular interband Andreev reflections at van der Waals interfaces between graphene and NbSe2. Nat. Phys. 12, 328–332 (2016).
Zhou, L. et al. Large-area synthesis of high-quality uniform few-layer MoTe2. J. Am. Chem. Soc. 137, 11892–11895 (2015).
Farahani, H., Rajabpour, A. & Reyhani, A. Interfacial thermal resistance between few-layer MoS2 and silica substrates: a molecular dynamics study. Comput. Mater. Sci. J. 142, 1–6 (2018).
Hong, Y., Ju, M. G., Zhang, J. & Zeng, X. C. Phonon thermal transport in a graphene/MoSe2 van der Waals heterobilayer. Phys. Chem. Chem. Phys. 20, 2637–2645 (2018).
Filippova, V. P., Kunavin, S. A. & Pugachev, M. S. Calculation of the parameters of the Lennard-Jones potential for pairs of identical atoms based on the properties of solid substances. Inorg. Mater. Appl. Res. 6, 1–4 (2015).
Al-Matar, A. K. & Rockstraw, D. A. A generating equation for mixing rules and two new mixing rules for interatomic potential energy parameters. J. Comp. Chem. 25, 660–668 (2004).
Hiura, H., Miyazaki, T. & Kanayama, T. Formation of metal-encapsulating Si cage clusters. Phys. Rev. Lett. 86, 1733–1736 (2001).
Mundin, K. C. & Ellis, D. E. Stochastic classical molecular dynamics coupled to functional density theory: applications to large molecular systems. Braz. J. Phys. 29, 199–214 (1999).
Morris, G. M. et al. van Der Waals Potential Energy (2003). Available at: http://www.img.bio.uni-goettingen.de/mswww/internal/manuals/autodock/AD3.a.0UserGuide.html (Accessed 23 March 2019).
Lewis, L. J. & Mousseau, N. Tight-binding molecular-dynamics studies of defects and disorder in covalently bonded materials. Comput. Mater. Sci. 12, 210–241 (1998).
Turner, G. W., Chushak, Y. G. & Bartell, L. S. Molecular dynamics investigation of transient nucleation in the freezing of clusters of SeF6. J. Phys. Chem. A 108, 1666–1670 (2004).
Monajjemi, M., Ketabi, S., Hashemian Zadeh, M. & Amiri, A. Simulation of DNA bases in water: comparison of the Monte Carlo algorithm with molecular mechanics force fields. Biochem 71, S1–S8 (2006).
Landron, C. et al. Liquid alumina: detailed atomic coordination determined from neutron diffraction data using empirical potential structure refinement. Phys. Rev. Lett. 86, 4839–4842 (2001).
Bartell, L. S. & Xu, S. Molecular synamics examination of an anomalous phase of TeFe molecules. J. Phys. Chem. 95, 8939–8941 (1991).
Acknowledgements
K.M. and L.Q.C. are partially supported by the Hamer Professorship at Penn State, Louisiana Tech University, the National Science Foundation 2D Crystal Consortium – Material Innovation Platform (2DCC-MIP) under NSF cooperative agreement DMR-1539916, and the NSF-CAREER under NSF cooperative agreement CBET-1943857. Y.Z.J. is supported by the I/UCRC Center for Atomically Thin Multifunctional Coatings (ATOMIC) seed project SP001-17 and the 2DCC-MIP. This project is also partly supported by DoE-ARPA-E OPEN, NASA-EPSCoR, Louisiana EPSCoR-OIA-1541079 (NSF(2018)-CIMMSeed-18 and NSF(2018)-CIMMSeed-19), and LEQSF(2015-18)-LaSPACE. J.W.J. is supported by the Recruitment Program of Global Youth Experts of China, the National Natural Science Foundation of China (NSFC) under Grant Number 11822206, and the Innovation Program of Shanghai Municipal Education Commission under Grant Number 2017-01-07-00-09-E00019. H.S.P. acknowledges the support of the Mechanical Engineering Department at Boston University. A.C.T.v.D., D.Y., and Y.S. acknowledge funding from the Fluid Interface Reactions, Structures and Transport (FIRST) Center, an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Science for MXene force-field development and application work.
Author information
Authors and Affiliations
Contributions
All authors contributed to collecting the references and outlining the review paper. K.M. drafted the review. All authors participated in revising the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Momeni, K., Ji, Y., Wang, Y. et al. Multiscale computational understanding and growth of 2D materials: a review. npj Comput Mater 6, 22 (2020). https://doi.org/10.1038/s41524-020-0280-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-020-0280-2
