
Abstract
Transparent conducting materials (TCMs) are required in many applications from solar cells to transparent electronics. Developing high performance materials combining the antagonistic properties of transparency and conductivity has been challenging especially for p-type materials. Recently, high-throughput ab initio computational screening has emerged as a formidable tool for accelerating materials discovery. In this review, we discuss how this approach has been applied for identifying TCMs. We provide a brief overview of the different materials properties of importance for TCMs (e.g., dopability, effective mass, and transparency) and present the ab initio techniques available to assess them. We focus on the accuracy of the methodologies as well as their suitability for high-throughput computing. Finally, we review the different high-throughput computational studies searching for new TCMs and discuss their differences in terms of methodologies and main findings.
Similar content being viewed by others
Introduction
Transparent conducting materials (TCMs) display simultaneously high electrical conductivity and optical transparency. This peculiar combination makes these materials very interesting both from a fundamental standpoint and for a large variety of applications such as solar cells, flat panel displays, touchscreens, light emitting diodes, and smart-windows (low emissivity and electrochromic windows). The most studied and used TCMs are oxides, referred to as TCOs. TCOs are usually made of high band gap (>3 eV) oxides which are intrinsically or extrinsically doped to reach a very low resistivity (10−4 Ω cm). The first TCO, cadmium oxide (CdO), was discovered more than a century ago. The breakthrough in the field of TCOs and associated applications came in the 1960s with the development of two compounds: indium oxide (In2O3) usually doped with tin (ITO) and tin dioxide (SnO2) usually doped with fluorine (FTO). ZnO has also been widely studied recently in part due to concerns about the cost of indium. For extensive reviews about these typical TCOs, the reader may refer to refs. 1,2,3,4,5 More recently, new n-type TCOs have also emerged such as Ga2O3 which offers a very wide band gap and is considered for power electronics,6,7 or BaSnO3 which shows one of the highest mobility ever measured in a TCO (>120 cm2 V−1 s−1 at room temperature).8,9
The traditional and widely used TCOs are all n-type doped. In fact, one of the main challenges in the field is to develop novel p-type TCOs with performances on a par with their n-type counterparts. While some p-type materials have been suggested (e.g., CuAlO2, SnO, NiO, and LaCrO3)10,11 their materials properties remain poor compared to n-type TCOs. This is especially the case for the carrier mobility, with values typically lower than 5 cm2 V−1 s−1 for p-type oxides. A high mobility/conductivity p-type TCO could be used in combination with known n-type TCOs to develop p–n heterojunctions and enable the development of many new devices opening new avenues in transparent electronics. In addition, p-type TCOs would find applications in thin-film transistors,12 as hole-transport layers in solar cell architectures, as efficient photocathodes in water-splitting solar cells,13 or in dye sensitized solar cells.14,15
So far, the identification and development of efficient TCMs has been driven mainly by chemical intuition, trial-and-error methods, or experimental serendipity. The quality of the current first-principles or ab initio computational approaches combined with the large computational resources available nowadays offer an alternative avenue to materials discovery in general. Starting from a large set of known or hypothetical crystal structures, their physical properties are calculated automatically and stored in databases. Promising candidates are then searched for in this vast data set. The available data also offers a unique playground to search for structure–chemistry–property relationships. This high-throughput (HT) computational approach is emerging in many fields16 together with the development of large online databases of material properties such as AFLOWLIB,17 the Materials Project (MP),18 or the open quantum materials database (OQMD).19 The aim of this review is to present the HT efforts dedicated to the search of new TCMs. We will first review the materials physics behind the properties of interest (electrical conductivity and transparency to visible light) and the different levels of theory that can be used to compute these properties. Figure 1 summarizes how materials physics and chemistry (e.g., band structure and point defects) drive the properties of interest for TCMs. We separate the properties used on a large scale (in green in Fig. 1) from those that are still only available for lower throughput (in red in Fig. 1). Using these different techniques several studies have used HT computing to search for novel TCMs and the last section of this paper will report on their different approaches and main results.

The transparency of a material is described by its transparency T, determined by the absorption coefficient α and the reflectivity R. The energy onset above which light is absorbed is determined by the band gap Eg of the semiconductor and the oscillator strengths 〈ψnk|r|ψnk〉 between states nk and mk. The reflectivity onset is called the plasma frequency ωp. The conductivity σ of a material can be computed using the BTE. The most used approximation is to consider a constant relaxation time τnk = τ. In the CRTA, the effective mass m* is used to describe the transport capabilities of a material. This effective mass depends on the band structure εnk and on the Fermi level εF, or the carrier concentration n, at a temperature T. Quantities in green (red) are usually (not) used in HT studies
Electrical conductivity and mobility
Materials physics
The textbook expression linking the electrical conductivity to materials properties is σ = eμn = ne2τ/m* where μ is the mobility of the carriers which depends on m* their effective mass and τ their scattering time. The effective mass is related to how the band energy (ε) varies with the wavevector (k) by (m*)−1 = ℏ−2∂2ε/∂k2. This simple expression captures much of the physics of carrier transport in TCOs but is not applicable to the many materials deviating from a single parabolic band picture. Indeed, many materials present multiple bands, different valleys in the conduction or valence bands (CB and VB, respectively) and nonparabolicity. Moreover, the conductivity and the effective mass are in general direction dependent and are therefore tensorial properties. It is possible to define more strictly the effective mass taking into account all those effects by resorting to the Boltzmann transport equation (BTE), as illustrated in Fig. 1. The BTE is a semiclassical theory linking materials properties to transport properties (conductivity, Seebeck coefficient, etc.). Adopting the linearized Boltzmann formalism and the relaxation time approximation, the conductivity tensor is given by20,21,22,23,24
where α and β are Cartesian coordinates, e is the elementary charge, f(εnk; εF,T) is the Fermi-Dirac distribution function, εF is the Fermi level, εnk is the electron energy of the band n at the wavevector k, vnk,α is the α-component of its velocity defined as ∂εnk/∂k, τnk is its lifetime or relaxation time, and T is the temperature. The computation of the conductivity tensor requires the knowledge of the band structure εnk and the relaxation time τnk. The relaxation time accounts for the different scattering mechanisms occurring in a material:3,25 scattering by defects of the crystal, by impurities, by grains boundaries, by acoustic and optical phonons, etc. Often, the constant relaxation time approximation (CRTA) is used, where τnk is considered to be constant (see Fig. 1). This can be justified assuming that there is no large variation of the relaxation time in an energy range of kBT, with kB the Boltzmann constant.26 This assumption is usually valid in metals and degeneratly doped semiconductors.22 In the CRTA, the computed conductivity depends on the band structure only, and on τ as a multiplicative parameter:23,24,27
Using an integration by parts, the CRTA conductivity tensor becomes
introducing the effective mass tensor
In the case of a single isotropic parabolic band, Eq. (3) becomes the usual textbook expression for the conductivity mentioned in the beginning of this section. The effective mass tensor is an intrinsic property depending only on the band structure of the material. However, it varies in the Brillouin zone depending on k and n. It is possible to define an average effective mass tensor taking into account all bands and the full Brillouin zone. This average effective mass tensor can be obtained by dividing the conductivity tensor at a given carrier concentration and temperature obtained within the CRTA by the carrier concentration and relaxation time
This is equivalent to evaluating23,24,27,28
We note that this definition is dependent on the temperature and carrier concentration used. The effective mass is a symmetric tensor for which quantities invariant with the chosen axes (e.g., 3 eigenvalues or the trace) are often used. Other methods have been used in the literature based on the fitting of a parabola to the VB maximum (VBM) or CB minimum (CBM) along certain symmetry lines.29,30,31,32,33,34 This approach has been adopted in screening studies but it has strong limitations as it fails to take into account the nonparabolicity, multiple bands, multiple pockets nature of many band structures. It is problematic for the less symmetric space groups where the symmetry lines might not lie along the eigendirections of the effective mass tensor. A density of state (DOS) effective mass (i.e., obtained by fitting the DOS of a parabolic band to the actual one) has also been used in the literature.35 This DOS effective mass solves the problem of nonparabolicity or multiple bands but inherently does not take into account the directional aspect of transport. For the sake of completeness, we note that other types of effective masses can be defined in a similar way such as the Seebeck effective mass widely used in thermoelectrics.27,36 For these reasons, Eq. (6) is, in our opinion, the most rigorous and general way of defining an effective mass to compare materials in terms of their conductivity (or mobility) based solely on their band structure.
Computational approach for the conductivity and mobility
The conductivity effective mass (Eq. (6)) described above depends solely on the band structure. The latter can be obtained from density functional theory (DFT) with a relatively low computational budget. While standard DFT (i.e., based on semilocal functionals such as the local density approximation or generalized-gradient approximation (GGA)) tends to underestimate band gaps dramatically, the effective mass agree reasonably well with experiments.23 The band gap underestimation can, however, be problematic when the semiconducting material is metallic in DFT. We note that in the case of transition metals, Hubbard U corrections can be used to open the gap at no additional computational cost. For transport computations, the density of k-points has to be large enough to capture all the details of the band structure. Interpolation techniques implemented in softwares such as Boltztrap have been effective in providing satisfying effective masses,20,21,23,24 but other approaches have been suggested as well.37
Scattering processes are usually not taken into account in HT studies and effective masses are used as a proxy to mobility and conductivity. A low effective mass is necessary for high mobility/conductivity. In fact, all the best n-type TCOs (SnO2, ZnO, and In2O3) show among the lowest computed electron effective masses in oxides.24 Finally, we stress that all the described approaches assume that band transport is occurring and that small polarons are not forming. Small-polaron formation energies as well as their energy barriers for hopping are assessable through ab initio techniques.38,39,40,41 In general, small-polaron computations require a good amount of human intervention and more costly techniques beyond standard DFT such as hybrid functionals. When they are included in screening studies it is only at the very end of the screening process.35,42 An interesting future avenue would be to develop efficient descriptors for small-polaron formation. It is already possible to relate small-polaron formation energies and the coordination environments of certain transition metals.38 If this approach could be generalized, the recent development of algorithms to automatically identify coordination environments could provide interesting tools to help in the screening of small-polaron formation.43,44
Dopability of a material
The conductivity depends on the mobility (i.e., effective mass and scattering) but also on the amount of carriers, as illustrated in Fig. 1. TCMs being typically doped semiconductors, their carrier concentration n is set by the amount and type of defects in the crystal. The intrinsic (or native) point defects consist of vacancies, self-interstitials and antisites (cations on sites of anions, for example). The extrinsic point defects (impurities or dopants) are substitutional and interstitial foreign atoms.
Point defects strongly influence the carrier concentration and hence the conductivity of semiconductors and TCMs. Indeed, charged defects in semiconductors can generate free electrons or holes. The ability to intrinsically or extrinsically dope a material can be assessed by computing defect formation energies as functions of the Fermi level. This defect formation energy (Ef) can be written as45
where the defect D is in the charge state q, E[Dq] and E[bulk] are the total energies of the crystal with the defect Dq and without any defects, respectively, ni is the number of atoms of type i that have been removed (ni < 0) or added (ni > 0) by the formation of the defect and μi is their chemical potential which is set by growth conditions and phase stability, EF is the Fermi level referenced with respect to the VBM, and Ecorr[Dq] is a correction term introduced to take care of the spurious image charge interactions and the alignment of the potential for charged defects (see the Computational approach for defects studies section). Figure 2 shows a typical defect formation energy versus Fermi level plot. In Fig. 2a, each defect (D1–D4) has charged (positive or negative slope depending on the sign of the charge) and uncharged (horizontal lines) states. The defect D1 has a rather low formation energy and easily exists in charge state −1, making it a shallow acceptor ((0/n−) charge-state transition state close to the VBM, with n a positive number). The defect D2 is on the contrary a shallow donor ((n+/0) charge-state transition close to the CBM). The defect D3 is another shallow donor but it is less stable as it has a larger formation energy. The defect D4 is a deep defect because it displays charge-state transitions (0/−1) and (−1/−2) in the middle of the gap. Different defects always compete with each other in a material and it is the result of these competitions that sets the Fermi level. For instance, Fig. 2b presents an n-type tendency: the donor defect D2 has a much lower formation energy than the others and therefore easily exists in charge-sate +1 around the CBM and releases a free electron into the crystal. In the same way, Fig. 2c shows a p-type tendency. The case of undopability (neither n- or p-type) is shown in Fig. 2d. The “hole-killers” defects D2–D4, in charge-state +1, are very active around the VBM, completely dominating the acceptor defect D1. Around the CBM, D1 and D4 have lower formation energies than the donor D2 and are working as “electron-killers”. Of course, this picture might be different in different chemical potential conditions. It is in practice not always very clear how certain chemical potential conditions could be reached experimentally. Chemical potential of elements and thus synthesis conditions will affect the defect formation energies and therefore their behavior in the material. Certain materials are intrinsically n-type and will be very difficult to form as p-type, especially in equilibrium conditions. For instance, many good n-type oxides have an energetically favorable shallow oxygen vacancy intrinsically generating electrons (e.g., SnO2 and In2O3).46 This oxygen vacancy acts also as a hole-killer as it compensates any attempt to dope the material p-type. This hole-killing process has been demonstrated computationally for many n-type oxides such as SnO2, ZnO, or In2O3.47,48 Some materials cannot be doped neither n- nor p-type and will behave as insulators. Finally, some rare materials can be ambipolar such as silicon but also SnO.49,50

a Schematic illustration of defect formation energies Ef as a function of the Fermi level EF of four defects Di in different charge states. b–d Defect formation energies for n-, p-type dopings and the case of undopability, respectively. In all cases, D1 is a shallow acceptor, D2 a shallow donor, D3 a less stable shallow donor, and D4 a deep defect. In a, c and d, D1 and D4 are electron-killers. In d, D2–D4 are hole-killers
A complete defect study is an expensive task. Alternative methods to determine the dopability at a lower cost have been greatly sought for. Band edges versus vacuum are known to correlate with dopability.48,51,52,53 For instance, p-type oxides tend to have a valence band higher in energy than n-type or insulating oxides. This has provided chemical understanding for intrinsic p-type doping in certain oxides. A case in point is the p-type behavior of copper based oxides which can be related to the copper mixing with oxygen character of their valence band pushing the valence band to higher energies than the purely oxygen band present in many oxides. However, accurate band edges computations with respect to a common reference such as vacuum can be computationally challenging as periodic boundary codes require to construct slabs. Moreover, the accuracy of DFT is here questionable but more appropriate hybrid or GW computations are more expensive. A very cheap alternative, the branch-point energy (BPE) method is more attractive in terms of computational time. Originally suggested by Tersoff,54 this approach has been applied with modern electronic structures calculations by Schleife et al.55 and used in many studies afterwards.30,56,57 The BPE is computed using only the band structure as
where Nk is the number of k-points used to sample the Brillouin zone, NCB and NVB are the number of conduction and valence bands considered, with \(\varepsilon _{c_{n{\mathbf{k}}}}\) and \(\varepsilon _{v_{n{\mathbf{k}}}}\) their energies. Below (above) the BPE, defect states will be mainly donor- (acceptor-) like. Therefore, if the BPE of a material is close to the CBM or inside the CB, the material will likely be easily n-type dopable. Similarly, if the BPE is close to the VBM or inside the VB, the material will likely be easily p-type dopable. If the BPE is in the middle of the gap, nothing can be said about the dopability.
The use of the BPE is controversial. A priori, the formula requires to choose adequately the bands to use through NCB and NVB, giving a sense of arbitrariness. Moreover, the fact that complex defect physics could be captured by band shape only is surprising. Recently, stricter assessment of the BPE was performed by Woods-Robinson et al.58 as they analyzed the computed BPE of 74 known TCMs and compared their results with the known dopability of these materials. Part of their results are reported in Figure 3a. Their analysis showed that the BPE is able to predict the n-type dopability, but completely fails to predict the p-type dopability: for most of the tested p-type materials, the BPE is in the middle of the gap. However, as pointed out already by the authors, a selection bias might be at play here as most n-type TCOs have lower electron effective mass moving the BPE in the conduction band to the contrary of the higher effective mass of p-type TCOs. Recently, Yim et al. clearly questioned the prediction power of the BPE providing evidence that it cannot efficiently discriminate between n- and p-type TCOs.37 Instead of the BPE, Yim et al. suggested the formation energy of the hydrogen (FEH) interstitial defect, with the Fermi level at the VBM, to be the descriptor for how strong the p-type dopability is.37 The rationale behind this descriptor is the fact that the positively charged hydrogen atom has transferred an electron to the VBM (at the Fermi level), which therefore correlates with the vacuum level. Using this FEH, they find a clear distinction between experimentally n- and p-type materials, as reproduced in Figure 3b. We should however note that work from Varley et al. showed that the BPE correlated very well with the interstitial hydrogen transition level, for II-VI and III-V zinc-blend compounds. Moreover, Shapera et al.57 showed that the BPE is able to predict the band offsets in heterojunctions.

a HSE06 band structure calculations aligned to BPE levels (dotted line at 0 eV), for representative TCMs. Blue (red) compounds are n- (p-) type TCMs. Calculations have been performed with NVB = 4 and NCB = 2 and error bars indicate how the computed BPE varies with different selections of NVB and NCB. Adapted with permission from Woods-Robinson et al.58 (Copyright (2018) American Chemical Society.) b FEH versus hole effective mass of known p-type and n-type oxides. n- (p-) type TCOs have a FEH <(>) −1 eV. Reprinted from Yim et al.37
Computational approach for defect studies
There are two natural ways to compute charged defects in solids: the use of clusters and supercells. In these approaches, ionized states of defects in semiconductors are investigated by using clusters or supercells containing defects in the corresponding charge states. While using clusters leads to problems with the dangling bonds on their surfaces that are difficult to eliminate, using supercells with periodic boundary conditions induces spurious Coulomb interactions between image charges (Ecorr[Dq] in Eq. (7)).59,60,61 Nowadays, thanks to the advances of DFT-based calculations and hardware, large supercells can easily be treated for which the image charge interactions are minimized. Furthermore, corrections methods have been developed (see below) making supercell calculations a tool of choice.
Supercell computations are used to evaluate Eq. (7). DFT with semilocal functionals provides fairly good defect formation energies and general doping tendencies.62 However, it is well-known that DFT relying on semilocal exchange-correlation functional drastically underestimates the band gap. Empirical corrections to the band edges to fit to experimental data (scissor operator) have been used to correct this underestimation with much controversies in the literature on the right method to perform this. When working with 3d transition metals, a Hubbard U correction has also been used. The current method of choice is to use hybrid functionals which add a fraction of exact exchange to the DFT functional and provide good energetics in addition to much improved band gaps. The Heyd–Scuseria–Ernzherof (HSE) hybrid functional is especially powerful in solids.63,64,65 Another way to obtain a correct evaluation of the band gap is to use many-body perturbation theory within the GW approximation,66,67,68 implemented in many first-principles codes.69 This approach is, however, problematic as it does not provide forces and cannot be used for atomic relaxation around the defect (contrary to hybrid functionals).
Charged defects computations are intrinsically difficult to perform in periodic boundary condition codes. Indeed, potential alignment (due to the background charge always present in charged defect computations) as well as spurious image charge Coulomb interactions have been known to affect the accuracy of the supercell computations. Different techniques have been suggested to correct these errors with a recent popularity for the methods developed by Freysoldt and Kumagai.70,71,72,73 Although defects calculations with supercells containing around 200 atoms are doable, they are challenging to perform HT both for computational and automation reasons. Recent tools such as PyCDT, PyDEF, or other Python frameworks have been developed to handle such calculations as automatically as possible.74,75,76
While the increase in carrier concentration provided by shallow defects is expected to directly increase the conductivity, we should note that the point defects providing the doping can lower the mobility for instance through ionized impurity scattering. Additionally, higher carrier and defect concentrations also affect transparency as explained in Section “Transparency to visible light”. This leads in practice to some material-dependent optimal carrier concentration maximizing both transport and optical properties.
Transparency to visible light
The transparency of a material (T(ω)) of thickness t, at a given light frequency ω depends on its reflectivity R(ω) and its absorption coefficient α(ω) (see Fig. 1) through
An increase of the reflectivity or the absorption coefficient naturally leads to a decrease of the transparency. In a semiconductor with a direct band gap, a strong absorption onset at the direct band gap energy Eg due to interband transitions sets the higher bound of the light frequency above which the transparency drastically decreases, see Figs. 1 and 4.2,3,77 In an indirect gap material, the interband transitions at the fundamental (electronic) gap have a smaller effect on the absorption because phonons are needed to activate these transitions. A more pronounced absorption onset starts at the direct (optical) gap. The minimal value of the optical band gap of a TCM is usually recommended to be around 3 eV, to avoid absorption in the visible spectrum.77 For applications based on thin films (~100 nm), the indirect absorption can be so weak that in practice an indirect gap lower than 3 eV is acceptable. A few known TCOs rely on this, for instance SnO which shows a direct band gap of 2.7 eV and an indirect band gap of 0.7 eV.78 In this material, the absorption coefficient due to indirect transitions is negligible (~104 cm−1) compared to direct transitions (~105 cm−1), and the material has a transparency of 70–80% for a 100 nm film.78,79

Schematic of the ideal transmittance window of a TCM, where the free carrier reflection (plasma) edge is below 1.75 eV, and the onset of absorption due to the strong interband optical transitions is above 3.25 eV, opening a transparent window over the entire visible spectrum. Reprinted by permission from Springer Nature (Zhang et al.77)
Absorption can also come from additional effects. For example, deep defect levels can absorb a part of the visible light which can be detrimental to the transparency, see Fig. 5a.45,80 In addition, since in doped TCMs some of the CB (VB) states are occupied by electrons (holes), new transitions to higher (lower) empty states become possible, as sketched in Fig. 5b. This so-called second band gap effect has been recently studied in a series of typical TCOs.81 This effect can be slightly detrimental for materials with a second band gap that is significantly lower than the optical band gap. The effect has been shown to be not strong enough to prevent materials with smaller second band gaps to be of interest. In certain circumstances, the second band gap can be larger than the band gap and the filling of the states improves transmittance. This effect is called the Moss–Burstein effect82,83 and is responsible for the good transparency of CdO for instance.84,85

a (Left) Formation energy as a function of Fermi level EF (referenced to the VBM) for N subsitutional for O atoms in ZnO under Zn-rich conditions. The position of the (0, −) acceptor level is indicated. (Right) Configuration coordinate diagram indicating optical absorption energy for exchanging an electron with the CB. Reprinted from Lyons et al.,80 with the permission of AIP Publishing. b New transitions in highly doped TCMs. In the left panel, n-type TCMs, electronic transitions from the CB to higher energy states. In the right panel, p-type TCMs, electronic transitions from lower energy states to the VB. If the second band gap is large enough, the absorption edge can shift to blue wavelengths. Reprinted from Ha et al.,81 with the permission of AIP Publishing
The addition of free carriers has an additional effect. At lower light frequencies, the excess carriers collectively oscillate and increase the reflectivity and absorption of the material. These effects can be quantified by phonon absorption or emission processes.86 More phenomenologically, the Drude model is generally and successfully used to describe the reflection edge: light with a frequency lower than the plasma frequency is reflected by the free carrier gas, defining the lower bound of the transparency window, see Figs. 1 and 4.2,3,77,87 Following the Drude model, the plasma frequency ωp is defined by \(\omega _p^2 = e^2n/(m^ \ast \varepsilon _r\varepsilon _0)\), with εr and ε0 the relative and vacuum permittivity, respectively. In a TCM, the maximum value of the plasma edge is around 1.75 eV, to avoid reflection in the visible spectrum.77
The existence of the plasma edge imposes a limit on the carrier concentration in a TCM:88 the ratio n/m* has to be small enough not to reflect part of the visible spectrum.2,77,87 The electrical conductivity σ is limited by this effect, giving rise to the well-known transparency-conductivity compromise that has to be achieved in TCMs.89 To put it simply, while increasing conductivity naturally pushes to increase the carrier concentration of a TCM, a too large concentration will transform the material in a metal that will reflect most of the light and compromise its transparency. Finally, the optical transparency window mentioned here is suitable for transparent electronics applications, as it corresponds to the visible spectrum. However, the required transparency window depends on the application. For example, a TCM used as a contact in solar cells should also be infrared transparent. This further decreases the maximum carrier concentration allowed through the plasma frequency.
Computational approach
The computation of the onset of absorption due to the interband transitions relies on the accurate computation of the band gap of the material, as shown in Fig. 1. We have already mentioned some of the different methods used to compute the band gap of materials (see Section “Electrical conductivity and mobility”). DFT with semilocal functionals will underestimate the absorption onset sometimes dramatically as in ZnO (0.7 eV in GGA and ~3.3 eV experimentally).24 Hybrid functionals such as HSE, or the GW method, both yield better estimates of the absorption onset.24,81 For example, the G0W0 method (single-shot GW) underestimates the band gap by 0.18 eV on average,69 and HSE06 has an average error of 0.26 eV for typical semiconductors and 0.41 eV for transition-metal compounds. The error on the band gap can even be further decreased if one uses for example the partially self-consistent GW0 (typical errors of 3–5%) or the quasi-particle self-consistent QSGW (~0.1–0.25 eV errors) techniques.68,90 However, hybrid and GW calculations both require a much larger computational time than DFT ones. This is why existing HT studies usually rely on the DFT band gap for the first screening, knowing that it is underestimated. Alternative techniques offering band gap estimation at lower computational cost have been suggested (e.g., the Δ-sol approach from Chan et al.91 or the Tran–Blaha functional92), but have not been used on a large scale yet.
The computation of the band gap (both fundamental and optical) gives an indication of the absorption onset. However, it does not take into account forbidden transitions or the shape of the band structure of the material. In some materials, the absorption can be important for larger energies than the optical band gap only. The absorption coefficient can be estimated within the independent particle approximation (IPA) or including local-field effects in the random phase approximation (RPA). IPA and RPA computations on a given band structure provide the dielectric tensor as a function of the frequency from which the absorption coefficient and reflectivity can be computed. They take into account the matrix elements 〈ψnk|r|ψmk〉 which weight the optical transitions in function of their probability (oscillator strengths). In some cases, transitions have low or zero probability and these so-called forbidden transitions are important to explain the transparency of certain TCOs such as In2O3.93 An estimate of the plasma frequency is usually also given by the software implementing the IPA/RPA. These approaches do not take into account any change of the band structure due to the excitation of electrons nor the interactions between the created holes and excited electrons, the so-called excitons. Taking into account these effects in order to obtain an absorption coefficient in better agreement with experiment requires to solve the Bethe–Salpeter equation.94,95 Due to its large computational load, this task is usually performed only in specific cases,96 where the IPA or RPA alone are unable to explain experimental results. Finally, instead of relying on the Drude model, it is possible to compute the phonon-assisted absorption coefficient from first principles.86,97 Such computations are, however, still quite heavy and not used in HT yet.
HT approach
As outlined in the previous sections, many of the properties of interest for TCMs are reachable using ab initio techniques with different levels of computational cost and accuracy. This naturally opens an avenue for the HT search of high-performance TCMs. Since 2013, several studies have followed this path and we will report on all of them, focusing on the differences in their approach, on the insight they provide and on the following experimental validation. Table 1 summarizes these studies and the techniques used to compute the descriptors. The TCM candidates outlined by these HT screenings are collected in Table 2. A general HT screening funnel for TCMs is depicted in Figure 6. The exact order will vary between different studies but they all somehow address all these properties. The quality of the descriptors has been rigorously assessed by Woods-Robinson et al.58

A generic HT computational screening funnel for transparent conductors. The first four filters can be derived from DFT calculations and are explored in this review. Refinements indicate any other screening parameter that might be utilized, such as additional calculations of optical properties or band alignment calculations. Reprinted with permission from Woods-Robinson et al.58 (Copyright (2018) American Chemical Society)
One of the first computational screenings for p-type TCOs was presented by Peng et al.35 The number of screened materials was very limited as it focused on a series of manganese spinels only. Taking into account a series of metrics (the thermodynamic stability, band gap, hole effective mass, absence of hole small polarons, p-type dopability) obtained from techniques ranging from GGA + U to GW, the study identified Cr2MnO4 which was synthesized later confirming its transparency as well as its dopability.35,98 The conductivity was however found to be relatively low (~3.5 × 10−2 S cm−1) because of the low mobility (<1 cm2 V−1 s−1) of the still too heavy holes in this material (9.8me).98
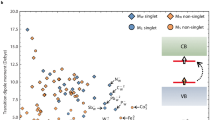
A few months later in 2013, Hautier et al.24 were the first to apply a HT approach to the p-type TCO problem on a large scale and across a very large chemical space. They screened 3052 existing binary and ternary oxides. The screening was based on the GGA hole effective mass, the G0W0 band gap and defect computations combining G0W0 and GGA. Figure 7a shows their results for the band gap and the effective mass, where good candidates lie in the lower right corner. The study clearly indicates the difficulty to find high-mobility p-type oxides due to the inherent O 2p character of the VB leading to flat band and high hole effective mass. The most promising compounds identified by the study are B6O, PbMO3 (M = Ti or Zr), ZrOS and K2Sn2O3. Two design principles for low hole effective mass p-type TCOs could be extracted from this work: using reduced main group elements (Sn2+, Bi3+, Sb3+, or Pb2+) or oxyanions. Both approaches mitigate the flat O 2p character of the VB by either mixing cation s states with oxygen states or replacing oxygen atoms by anions with more delocalized orbitals. This work motivated a series of follow-up studies, e.g., on the defect chemistry of B6O and on the rationale of the exceptional hole effective mass in K2Sn2O3.42,99 Further computational exploration of the Sn2+ and Pb2+ chemistries was also performed by Singh et al. focusing on alkali-earth and phosphates.100,101,102

a Effective mass versus band gap for the p-type TCO candidates. The authors superposed on the band gap axis a color spectrum corresponding to the wavelength associated with a photon energy. The TCO candidates are marked with red dots. A few known p-type (blue diamonds) and n-type (green square) TCOs can be compared to the new candidates. The best TCOs should lie in the lower right corner. For clarity, the authors kept only one representative when polymorphs existed for a given stoechiometry (for example, PbTiO3 and K2Sn2O3) and did not plot Rb2Sn2O3, which is superposed on K2Sn2O3. Reprinted from Hautier et al.24 b Hole effective masses as a function of the HSE band gap for all compounds that are thermodynamically stable. Reprinted from Shi et al.107
Bhatia et al.103 extended this HT screening to quaternary oxides. The screening identified Ba2BiTaO6 as a p-type TCO candidate. The good theoretical transport performances of this material were explained by the efficient mixing of the O 2p states with the Bi 6s states forming the VB. Synthesis of the material both in powder and thin film confirmed the high transparency of the material, with a measured band gap larger than 4.5 eV.103 Extrinsic doping was necessary to obtain p-type conductivity and measure the hole mobility, which was found to be relatively large (>30 cm2 V−1 s−1) especially in comparison with typical hole mobilities in oxides (at most around 5 cm2 V−1 s−1). The carrier concentration remained, however, small (only ~1014 cm−3) despite a large amount of extrinsic dopants introduced. The study demonstrated that Bi3+, which had been considered inappropriate for p-type TCOs in previous studies due to the too low Bi 6s orbitals,104 could lead in an adequate structure to very low hole effective mass. Another HT study leading to an experimental confirmation was performed by Yan et al. focusing on half-Heusler materials. The authors identified TaIrGe as an extremely high mobility material but with also low hole concentration.105
A natural extension using the data generated by the previous work was to study n-type TCOs.28 Focusing mainly on the electron effective mass, Hautier et al. showed that it is unlikely to find a TCO with a significantly lower effective mass than the n-type TCOs already used in applications (In2O3, ZnO, and SnO2). Neverthless, the work reported a long list of low effective mass materials (between 0.15 and 0.3me) which could be alternative high-mobility n-type oxides. The researchers also mined their data set and rationalized the importance of main group elements and late transition metals for n-type TCOs: Zn2+, Ga3+, Ge4+, Cd2+, In3+, Sn4+, and Sb5+.
Other HT screenings of oxides for TCO applications have been performed by Sarmadian et al.30,56 In their first work, the authors focused on the bixbyite structure of In2O3. They generated 1541 bixbyites starting from the known binary structures (A2O3 with A3+ cations) and combining them to create ternary oxides (AxB1−xO3 with A3+ and B3+ cations). They used the HSE functional to compute the band gap of the binary oxides and an interpolation approach for the band gap of ternary oxides. For the materials with the highest band gaps, dopability was assessed using the BPE. The authors found that Ga2O3 could be a good n-type TCO and suggested V2O3 to be a p-type candidate. In their subsequent work, Sarmadian et al.30 screened all the 12,211 binary, ternary, and quaternary oxides using the AFLOWLIB database and the provided semi-empirical band gaps and effective masses (along symmetry lines).17 Using additionally the BPE for a first doping screening followed by HSE band gap, they isolated 4 candidates for p-type TCO applications: X2SeO2 with X = La, Pr, Nd, or Gd. They concluded their study by computationally showing that the doping of La2SeO2 with Na atoms is possible. In view of the similarity of the goals from both studies (Hautier et al. and Samarian et al.), it is interesting to compare the results at the very least after the screening on the effective mass and band gap. While many candidates are identified by both studies (e.g., B6O, K2Sn2O3, NaNbO2, and Ca4As2O), others are only present in one of the studies. For instance, Ba2BiTaO6 appeared to be absent from the AFLOWLIB database or the X2SeO2 was not found by Hautier et al. The first explanation for this discrepancy is the different databases used: AFLOWLIB and the Materials Project, while covering many of the ICSD phases, have small differences. Another more fundamental difference is the definition of the hole effective mass: Samarian et al. use an effective mass fitted along symmetry lines while Hautier et al. consider the complete tensorial data. Finally, the screening thresholds used in the two works are slightly different.
Cerqueira et al.106 combined new structure prediction and HT screening to identify 45 novel Cu-, Ag-, and Au-based TCOs, and characterized them by computing their hole conductivity effective mass and GGA band gaps. In the same spirit, Shi et al.107 searched for novel ternary chalcogenides and found 79 new stable systems not present in the Materials Project. Their purpose was to find new nonoxide based chalcogenides. They computed the HSE band gap and conductivity effective masses of the new compounds and pointed out four materials as potential p-type TCM candidates: MYS2 (M = Ag, K) and MLaO2 (M = Cr, Br).
The HT studies presented so far have assessed the dopability by two approaches: using the BPE or through full defect computations but only for the most promising candidates in view of the cost of defect computations. Yim et al. suggested to rely on the FEH to characterize the p-type dopability of the materials they studied. Focusing on Cu1+ and Sn2+ containing compounds, the authors used the GGA conductivity effective mass and band gaps from HSE followed by their FEH evaluation. In total, they screened 40 oxides and found back a few known materials such as CuAlO2 or SnO. Interestingly, they retrieved K2Sn2O3 as a very highly dopable, low-hole effective mass candidate. They outlined two new p-type TCO candidates: CuLiO with properties comparable to CuAlO2 and La2TeO2 which appears chemically close to the La2SeO2 outlined by Samarian et al.30
Oxides were initially considered by HT studies because they are the most common chemistry for current TCMs. However, as already mentioned and clearly observed in HT studies, the oxygen nature of the VB in most oxides limits the mobility of holes. Therefore, Varley et al. extended the HT search to nonoxides considering more than 30,000 materials, including sulfides, nitrides, and phosphides.108 Figure 8 compares the different chemistries in terms of the hole conductivity effective mass and corrected band gap distribution. It shows that the average hole effective mass is much higher for oxides than for sulfides, nitrides, and phosphides. The lowest hole effective masses are present in phosphides which are therefore attractive for their transport properties. However, Fig. 8 also shows that nonoxide materials have smaller band gaps. The distribution of phosphides is centered on a band gap value of 2 eV, making them on average much less transparent than oxides. A strategy to bypass this fundamental issue with nonoxides was suggested by taking into account the direct or indirect character of the gap. A material with a high direct band gap but a low indirect one would only absorb weakly if used as a thin film and be transparent enough for TCM applications (see Section “Transparency to visible light”). Applying this approach to phosphides and combining effective mass screening with DFT followed by G0W0 band gap computations, they showed that BP exhibits a very low hole effective mass combined with a high direct band gap and p-type dopability. Measured hole mobilities in BP range from 8 to 350 cm2 V−1 s−1, and a conductivity as high as 2800 S cm−1 has been reported in the literature for BP confirming the computational insight and stressing the importance of experimentally considering BP as a p-type TCM.108 Very recently, Ha et al. screened all nonoxide compounds from the ICSD and identified CaTe and Li3Sb as potential p-type TCM candidates.109 They also performed a defect study and computed the phonon-limited mobility for these candidates to further assess their potential. Nonoxides were also screened by Raghupathy et al. with a focus on nontoxic binary nonoxide chalcogenide-based semiconductors from the ICSD and Materials Project (more than 500 materials).29 They screened materials based on their HSE band gap, their stability and their hole effective mass. Finally, they assessed the dopability of the remaining candidates through defect computations, and outlined nine potential p-type TCM candidates. Four of them (ZnS, MgS, MgTe, and Al2Se3) have a band gap (both measured and computed) above 3 eV.

a Violin plot of the hole effective mass for four chemistries: oxides (O), sulfides (S), nitrides (N), and phosphides (P). The dashed white lines indicate median and quartiles. The hole effective mass of the typical p-type TCO CuAlO2 is also indicated. b Hole effective mass versus empirically corrected DFT band gap for the four different chemistries. All masses are scaled relative to the electron mass (me). The number of investigated compounds is indicated at the top for each chemistry. Reprented with permission from Varley et al.108 (Copyright (2017) American Chemical Society)
Sharing of HT data sets to enable other researchers to mine or screen on the data sets is definitely an exciting avenue for HT computing. The data set of conductivity effective mass developed within the Materials Project and used in several screenings has been recently released by Ricci et al.23 A very good demonstration of how data sharing can rapidly be used is provided by the work from Raghupathy et al. who very quickly used this data set to perform a HT search for p-type ternary chalcogenides.110 The authors screened for stability, band gap and hole conductivity effective mass from the shared data set. After investigating the dopability, they finally listed a series of potential p-type TCMs highlighting IrSbS. The use of purely DFT-based defect computations however could lead to an overestimation of the p-type dopability of this material.
Outlook
TCOs are widely used materials. Anyone with a smartphone actually carries a thin layer of TCO as these materials are used in touchscreens. These fascinating materials are emblematic of the typical challenges faced by materials scientists as they combine properties that are antagonistic: transparency and conductivity. Ab initio techniques have reached such a level of maturity that many of the optical, electronic, and defect properties required to understand and predict TCOs properties are nowadays computable with a reasonable accuracy and cost. This led to their use to perform materials prediction especially using HT approaches and mainly tackling the search for p-type TCMs. While there is still much work to be done, exciting first successes have clearly been achieved with a few materials discovered in silico and confirmed experimentally (e.g., Ba2BiTaO6 and TaIrGe). These previous successes make us confident that more confirmation of previous computational predictions from HT studies will emerge in the future. The HT studies have also provided a clearer view of the opportunities for future materials exploration. Moving towards specific oxides (reduced main group elements) and nonoxides chemistries appears to be very promising avenues. As we outlined in this review, the methodological toolbox for computing TCM related materials properties is already very large. The future will see the automation of some of the properties that are still “manually” performed (e.g., defects or scattering time) and the improvement of the understanding of the accuracy of different methods in predicting certain properties. In parallel, new concepts for TCOs design (e.g., the use of correlated metal oxides)77 will percolate to HT computing and offer new screening studies. Last but not least, the vast data generated by these HT efforts will provide an unprecedented resource for machine learning and data mining, improving even further the understanding of TCMs.
Change history
13 August 2019
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Morales-Masis, M., De Wolf, S., Woods-Robinson, R., Ager, J. W. & Ballif, C. Transparent electrodes for efficient optoelectronics. Adv. Electron. Mater. 3, 1600529 (2016).
Ellmer, K. Past achievements and future challenges in the development of optically transparent electrodes. Nat. Photonics 6, 808–816 (2012).
Liu, H., Avrutin, V., Izyumskaya, N., Özgür, Ü. & Morkoç, H. Transparent conducting oxides for electrode applications in light emitting and absorbing devices. Superlattices Microstruct. 48, 458–484 (2010).
Beyer, W., Hüpkes, J. & Stiebig, H. Transparent conducting oxide films for thin film silicon photovoltaics. Thin Solid Films 516, 147–154 (2007).
Gordon, R. G. Criteria for choosing transparent conductors. MRS Bull. 25, 52–57 (2000).
Ohira, S. et al. Characterization of transparent and conducting Sn-doped β-Ga2O3 single crystal after annealing. Thin Solid Films 516, 5763–5767 (2008).
Suzuki, N. et al. Fabrication and characterization of transparent conductive Sn-doped β-Ga2O3 single crystal. Phys. Status Solidi C 4, 2310–2313 (2007).
Prakash, A. et al. Wide bandgap BaSnO3 films with room temperature conductivity exceeding 104 S cm−1. Nat. Commun. 8, 15167 (2017).
Paik, H. et al. Adsorption-controlled growth of La-doped BaSnO3 by molecular-beam epitaxy. APL. APL Mater. 5, 116107 (2017).
Zhang, K. H. L., Xi, K., Blamire, M. G. & Egdell, R. G. P-type transparent conducting oxides. J. Phys. 28, 383002 (2016).
Wang, Z., Nayak, P. K., Caraveo-Frescas, J. A. & Alshareef, H. N. Recent developments in p-type oxide semiconductor materials and devices. Adv. Mater. 28, 3831–3892 (2016).
Fortunato, E., Barquinha, P. & Martins, R. Oxide semiconductor thin-film transistors: a review of recent advances. Adv. Mater. 24, 2945–2986 (2012).
Chen, L. et al. p-Type transparent conducting oxide/n-type semiconductor heterojunctions for efficient and stable solar water oxidation. J. Am. Chem. Soc. 137, 9595–9603 (2015).
Bandara, J. & Yasomanee, J. P-type oxide semiconductors as hole collectors in dye-sensitized solid-state solar cells. Semicond. Sci. Technol. 22, 20 (2006).
Chou, C.-S., Hsiung, C.-M., Wang, C.-P., Yang, R.-Y. & Guo, M.-G. Preparation of a counter electrode with P-type NiO and its applications in dye-sensitized solar cell. Int. J. Photoenergy. 2010, 9 (2010).
Curtarolo, S. et al. The high-throughput highway to computational materials design. Nat. Mater. 12, 191–201 (2013).
Curtarolo, S. et al. AFLOWLIB.ORG: a distributed materials properties repository from high-throughput ab initio calculations. Comput. Mater. Sci. 58, 227–235 (2012).
Jain, A. et al. Commentary: the materials project: a materials genome approach to accelerating materials innovation. APL Mater. 1, 011002 (2013).
Saal, J. E., Kirklin, S., Aykol, M., Meredig, B. & Wolverton, C. Materials design and discovery with high-throughput density functional theory: the open quantum materials database (OQMD). JOM 65, 1501–1509 (2013).
Madsen, G. K. H., Carrete, J. & Verstraete, M. J. BoltzTraP2, a program for interpolating band structures and calculating semi-classical transport coefficients. Comput. Phys. Commun. 231, 140–145 (2018).
Madsen, G. K. & Singh, D. J. BoltzTraP. A code for calculating band-structure dependent quantities. Comput. Phys. Commun. 175, 67–71 (2006).
Ashcroft, N. W. & Mermin, N. D. Solid State Physics (Saunders College, Philadelphia, 1976), 1 edn.
Ricci, F. et al. An ab initio electronic transport database for inorganic materials. Sci. Data 4, 170085 (2017).
Hautier, G., Miglio, A., Ceder, G., Rignanese, G. M. & Gonze, X. Identification and design principles of low hole effective mass p-type transparent conducting oxides. Nat. Commun. 4, 2292 (2013).
Calnan, S. & Tiwari, A. N. High mobility transparent conducting oxides for thin film solar cells. Thin Solid Films 518, 1839–1849 (2010).
Chen, W. et al. Understanding thermoelectric properties from high-throughput calculations: trends, insights, and comparisons with experiment. J. Mater. Chem. C 4, 4414–4426 (2016).
Gibbs, Z. M. et al. Effective mass and Fermi surface complexity factor from ab initio band structure calculations. npj Comput. Mater. 3, 8 (2017).
Hautier, G., Miglio, A., Waroquiers, D., Rignanese, G. M. & Gonze, X. How does chemistry influence electron effective mass in oxides? A high-throughput computational analysis. Chem. Mater. 26, 5447–5458 (2014).
Kormath Madam Raghupathy, R., Kühne, T. D., Felser, C. & Mirhosseini, H. Rational design of transparent p-type conducting non-oxide materials from high-throughput calculations. J. Mater. Chem. C 6, 541–549 (2018).
Sarmadian, N., Saniz, R., Partoens, B. & Lamoen, D. Easily doped p-type, low hole effective mass, transparent oxides. Sci. Rep. 6, 20446 (2016).
Setyawan, W., Gaume, R. M., Feigelson, R. S. & Curtarolo, S. Comparative study of nonproportionality and electronic band structures features in scintillator materials. IEEE Trans. Nucl. Sci. 56, 2989–2996 (2009).
Castelli, I. E. et al. New cubic perovskites for one-and two-photon water splitting using the computational materials repository. Energy Environ. Sci. 5, 9034–9043 (2012).
Kuhar, K. et al. Sulfide perovskites for solar energy conversion applications: computational screening and synthesis of the selected compound LaYS3. Energy Environ. Sci. 10, 2579–2593 (2017).
Kuhar, K., Pandey, M., Thygesen, K. S. & Jacobsen, K. W. High-throughput computational assessment of previously synthesized semiconductors for photovoltaic and photoelectrochemical devices. ACS Energy Lett. 3, 436–446 (2018).
Peng, H. et al. Li-doped Cr2MnO4: a new p-type transparent conducting oxide by computational materials design. Adv. Funct. Mater. 23, 5267–5276 (2013).
Tang, Y. et al. Convergence of multi-valley bands as the electronic origin of high thermoelectric performance in CoSb3 skutterudites. Nat. Mater. 14, 1223 (2015).
Yim, K. et al. Computational discovery of p-type transparent oxide semiconductors using hydrogen descriptor. npj Comput. Mater. 4, 17 (2018).
Peng, H. & Lany, S. Semiconducting transition-metal oxides based on d 5 cations: Theory for MnO and Fe2O3. Phys. Rev. B 85, 201202 (2012).
Zhang, K. H. L. et al. Perovskite Sr-doped LaCrO3 as a new p-type transparent conducting oxide. Adv. Mater. 27, 5191–5195 (2015).
Himmetoglu, B., Janotti, A., Bjaalie, L. & Van de Walle, C. G. Interband and polaronic excitations in YTiO3 from first principles. Phys. Rev. B 90, 161102 (2014).
Bjaalie, L. et al. Small hole polarons in rare-earth titanates. Appl. Phys. Lett. 106, 232103 (2015).
Varley, J. B., Lordi, V., Miglio, A. & Hautier, G. Electronic structure and defect properties of B6O from hybrid functional and many-body perturbation theory calculations: a possible ambipolar transparent conductor. Phys. Rev. B 90, 045205 (2014).
Waroquiers, D. et al. Statistical analysis of coordination environments in oxides. Chem. Mater. 29, 8346–8360 (2017).
Zimmermann, N. E., Horton, M. K., Jain, A. & Haranczyk, M. Assessing local structure motifs using order parameters for motif recognition, interstitial identification, and diffusion path characterization. Front. Mater. 4, 34 (2017).
Freysoldt, C. et al. First-principles calculations for point defects in solids. Rev. Mod. Phys. 86, 253–305 (2014).
Ágoston, P., Albe, K., Nieminen, R. M. & Puska, M. J. Intrinsic n-type behavior in transparent conducting oxides: a comparative hybrid-functional study of In2O3, SnO2, and ZnO. Phys. Rev. Lett. 103, 245501 (2009).
Scanlon, D. O. & Watson, G. W. On the possibility of p-type SnO2. J. Mater. Chem. 22, 25236–25245 (2012).
Robertson, J. & Clark, S. J. Limits to doping in oxides. Phys. Rev. B 83, 075205 (2011).
Varley, J. B., Schleife, A., Janotti, A. & Van De Walle, C. G. Ambipolar doping in SnO. Appl. Phys. Lett. 103, 082118 (2013).
Quackenbush, N. F. et al. Origin of the bipolar doping behavior of SnO from X-ray spectroscopy and density functional theory. Chem. Mater. 25, 3114–3123 (2013).
Zunger, A. Practical doping principles. Appl. Phys. Lett. 83, 57 (2003).
Walsh, A. et al. Limits to doping of wide band gap semiconductors. Chem. Mater. 25, 2924–2926 (2013).
Walsh, A. & Zunger, A. Instilling defect tolerance in new compounds. Nat. Mater. 16, 964 (2017).
Tersoff, J. Theory of semiconductor heterojunctions: the role of quantum dipoles. Phys. Rev. B 30, 4874 (1984).
Schleife, A., Fuchs, F., Rödl, C., Furthmüller, J. & Bechstedt, F. Branch-point energies and band discontinuities of III-nitrides and III-/II-oxides from quasiparticle band-structure calculations. Appl. Phys. Lett. 94, 012104 (2009).
Sarmadian, N. et al. High-throughput first-principles calculations of bixbyite oxides for TCO applications. Phys. Chem. Chem. Phys. 16, 17724–17733 (2014).
Shapera, E. P. & Schleife, A. Database-driven materials selection for semiconductor heterojunction design. Adv. Theory Simul. 1, 1800075 (2018).
Woods-Robinson, R. et al. Assessing high-throughput descriptors for prediction of transparent conductors. Chem. Mater. 30, 8375–8389 (2018).
Drabold, D. A. & Estreicher, S. K. Theory of Defects in Semiconductors (Springer, Berlin, 2007).
Alkauskas, A., Deák, P., Neugebauer, J., Pasquarello, A. & Van de Walle, C. G. Advanced Calculations for Defects in Materials: Electronic Structure Methods (John Wiley & Sons, Weinheim, 2011).
Estreicher, S. K. Hydrogen-related defects in crystalline semiconductors: a theorist’s perspective. Matererials Sci. Eng. 14, 319 (1995).
Peng, H. et al. Convergence of density and hybrid functional defect calculations for compound semiconductors. Phys. Rev. B 88, 115201 (2013).
Heyd, J., Scuseria, G. E. & Ernzerhof, M. Hybrid functionals based on a screened coulomb potential. J. Chem. Phys. 118, 8207–8215 (2003).
Brothers, E. N., Izmaylov, A. F., Normand, J. O., Barone, V. & Scuseria, G. E. Accurate solid-state band gaps via screened hybrid electronic structure calculations. J. Chem. Phys. 129, 011102 (2008).
Batista, E. R. et al. Comparison of screened hybrid density functional theory to diffusion Monte Carlo in calculations of total energies of silicon phases and defects. Phys. Rev. B 74, 121102 (2006).
Hedin, L. New method for calculating the one-particle Green’s function with application to the electron-gas problem. Phys. Rev. 139, A796–A823 (1965).
Aryasetiawan, F. & Gunnarsson, O. The GW method. Rep. Prog. Phys. 61, 237 (1998).
van Schilfgaarde, M., Kotani, T. & Faleev, S. Quasiparticle self-consistent GW theory. Phys. Rev. Lett. 96, 226402 (2006).
Van Setten, M. J., Giantomassi, M., Gonze, X., Rignanese, G. M. & Hautier, G. Automation methodologies and large-scale validation for GW: towards high-throughput GW calculations. Phys. Rev. B 96, 155207 (2017).
Makov, G. & Payne, M. C. Periodic boundary conditions in ab initio calculations. Phys. Rev. B 51, 4014 (1995).
Freysoldt, C., Neugebauer, J. & Van de Walle, C. G. Fully ab initio finite-size corrections for charged-defect supercell calculations. Phys. Rev. Lett. 102, 016402 (2009).
Freysoldt, C., Neugebauer, J. & Van de Walle, C. G. Electrostatic interactions between charged defects in supercells. Phys. Status Solidi B 248, 1067 (2011).
Kumagai, Y. & Oba, F. Electrostatics-based finite-size corrections for first-principles point defect calculations. Phys. Rev. B 89, 195205 (2014).
Broberg, D. et al. PyCDT: a Python toolkit for modeling point defects in semiconductors and insulators. Comput. Phys. Commun. 226, 165–179 (2018).
Péan, E., Vidal, J., Jobic, S. & Latouche, C. Presentation of the PyDEF post-treatment Python software to compute publishable charts for defect energy formation. Chem. Phys. Lett. 671, 124–130 (2017).
Goyal, A., Gorai, P., Peng, H., Lany, S. & Stevanović, V. A computational framework for automation of point defect calculations. Comput. Mater. Sci. 130, 1–9 (2017).
Zhang, L. et al. Correlated metals as transparent conductors. Nat. Mater. 15, 204–210 (2016).
Ogo, Y. et al. p-channel thin-film transistor using p-type oxide semiconductor, SnO. Appl. Phys. Lett. 93, 032113 (2008).
Lee, H.-N., Kim, H.-J. & Kim, C.-K. p-channel tin monoxide thin film transistor fabricated by vacuum thermal evaporation. Jpn. J. Appl. Phys. 49, 020202 (2010).
Lyons, J. L., Janotti, A. & Van de Walle, C. G. Why nitrogen cannot lead to p-type conductivity in ZnO. Appl. Phys. Lett. 95, 252105 (2009).
Ha, V. A., Waroquiers, D., Rignanese, G. M. & Hautier, G. Influence of the “second gap” on the transparency of transparent conducting oxides: an ab initio study. Appl. Phys. Lett. 108, 201902 (2016).
Moss, T. The interpretation of the properties of indium antimonide. Proc. Phys. Soc. Sect. B 67, 775 (1954).
Burstein, E. Anomalous optical absorption limit in InSb. Phys. Rev. 93, 632 (1954).
Kul, M., Zor, M., Aybek, A. S., Irmak, S. & Turan, E. Electrical and optical properties of fluorine-doped CdO films deposited by ultrasonic spray pyrolysis. Sol. Energy Mater. Sol. Cells 91, 882–887 (2007).
Kumaravel, R., Ramamurthi, K. & Krishnakumar, V. Effect of indium doping in CdO thin films prepared by spray pyrolysis technique. J. Phys. Chem. Solids 71, 1545–1549 (2010).
Peelaers, H., Kioupakis, E. & Van De Walle, C. G. Fundamental limits on optical transparency of transparent conducting oxides: free-carrier absorption in SnO2. Appl. Phys. Lett. 100, 011914 (2012).
Edwards, P. P., Porch, A., Jones, M. O., Morgan, D. V. & Perks, R. M. Basic materials physics of transparent conducting oxides. Dalton Trans. 19, 2995–3002 (2004).
Bellingham, J. R., Phillips, W. A. & Adkins, C. J. Intrinsic performance limits in transparent conducting oxides. J. Mater. Sci. Lett. 11, 263–265 (1992).
Fleischer, K., Norton, E., Mullarkey, D., Caffrey, D. & Shvets, I. V. Quantifying the performance of p-type transparent conducting oxides by experimental methods. Materials 10, 1019 (2017).
Shishkin, M. & Kresse, G. Self-consistent GW calculations for semiconductors and insulators. Phys. Rev. B 75, 235102 (2007).
Chan, M. & Ceder, G. Efficient band gap prediction for solids. Phys. Rev. Lett. 105, 196403 (2010).
Tran, F. & Blaha, P. Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential. Phys. Rev. Lett. 102, 226401 (2009).
Walsh, A. et al. Nature of the band gap of In2O3 revealed by first-principles calculations and X-ray spectroscopy. Phys. Rev. Lett. 100, 167402 (2008).
Albrecht, S., Reining, L., Del Sole, R. & Onida, G. Ab initio calculation of excitonic effects in the optical spectra of semiconductors. Phys. Rev. Lett. 80, 4510 (1998).
Onida, G., Reining, L. & Rubio, A. Electronic excitations: density-functional versus many-body Greens-function approaches. Rev. Mod. Phys. 74, 601 (2002).
Gillet, Y., Giantomassi, M. & Gonze, X. Efficient on-the-fly interpolation technique for Bethe-Salpeter calculations of optical spectra. Comput. Phys. Commun. 203, 83–93 (2016).
Peelaers, H., Kioupakis, E. & Van De Walle, C. G. Free-carrier absorption in transparent conducting oxides: phonon and impurity scattering in SnO2. Phys. Rev. B 92, 235201 (2015).
Nagaraja, A. R. et al. Experimental characterization of a theoretically designed candidate p-type transparent conducting oxide: Li-doped Cr2MnO4. Chem. Mater. 26, 4598–4604 (2014).
Ha, V. A., Ricci, F., Rignanese, G. M. & Hautier, G. Structural design principles for low hole effective mass s-orbital-based p-type oxides. J. Mater. Chem. C 5, 5772–5779 (2017).
Li, Y. et al. Design of ternary alkaline-earth metal Sn(II) oxides with potential good p-type conductivity. J. Mater. Chem. C 4, 4592–4599 (2016).
Li, Y., Zhang, L. & Singh, D. J. New stable ternary alkaline-earth metal Pb(II) oxides: Ca/Sr/BaPb2O3 and BaPbO2. Phys. Rev. Mater. 1, 055001 (2017).
Xu, Q. et al. Sn(II)-containing phosphates as optoelectronic materials. Chem. Mater. 29, 2459–2465 (2016).
Bhatia, A. et al. High-mobility bismuth-based transparent p-type oxide from high-throughput material screening. Chem. Mater. 28, 30–34 (2016).
Hiramatsu, H., Yanagi, H., Kamiya, T. & Ueda, K. Crystal structures, optoelectronic properties, and electronic structures of layered oxychalcogenides MCuOCh (M = Bi, La; Ch = S, Se, Te): effects of electronic configurations of M3+ ions. Chem. Mater. 20, 326–334 (2008).
Yan, F. et al. Design and discovery of a novel half-Heusler transparent hole conductor made of all-metallic heavy elements. Nat. Commun. 6, 7308 (2015).
Cerqueira, T. F. et al. Identification of novel Cu, Ag, and Au ternary oxides from global structural prediction. Chem. Mater. 27, 4562–4573 (2015).
Shi, J. et al. High-throughput search of ternary chalcogenides for p-type transparent electrodes. Sci. Rep. 7, 43179 (2017).
Varley, J. B. et al. High-throughput design of non-oxide p-type transparent conducting materials: data mining, search strategy, and identification of boron phosphide. Chem. Mater. 29, 2568–2573 (2017).
Ha, V.-A. et al. Computationally driven high-throughput identification of CaTe and Li3Sb as promising candidates for high-mobility p-type transparent conducting materials. Phys. Rev. Mater. 3, 034601 (2019).
Kormath Madam Raghupathy, R., Wiebeler, H., Kuehne, T. D., Felser, C. & Mirhosseini, H. Database screening of ternary chalcogenides for p-type transparent conductors. Chem. Mater. 30, 6794–6800 (2018).
Acknowledgements
G.B., G.-M.R., and G.H. acknowledge the F.R.S.-FNRS for financial support. G.H, G.-M.R., and F.R. acknowledge the F.R.S.-FNRS project HTBaSE (Contract no. PDR-T.1071.15) for financial support. V.-A.H. was funded through a grant from the FRIA.
Author information
Authors and Affiliations
Contributions
G.B., F.R., and V.-A.H. collected the literature and all the authors contributed to the writing of this review.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Brunin, G., Ricci, F., Ha, VA. et al. Transparent conducting materials discovery using high-throughput computing. npj Comput Mater 5, 63 (2019). https://doi.org/10.1038/s41524-019-0200-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-019-0200-5
This article is cited by
-
A robust, simple, and efficient convergence workflow for GW calculations
npj Computational Materials (2024)
-
Leveraging language representation for materials exploration and discovery
npj Computational Materials (2024)
-
High-throughput calculations of charged point defect properties with semi-local density functional theory—performance benchmarks for materials screening applications
npj Computational Materials (2023)
-
AI powered, automated discovery of polymer membranes for carbon capture
npj Computational Materials (2023)
-
Transparent conductive SnO2 thin films via resonant Ta doping
Science China Materials (2023)
