
Abstract
The correct calculation of formation enthalpy is one of the enablers of ab-initio computational materials design. For several classes of systems (e.g. oxides) standard density functional theory produces incorrect values. Here we propose the “coordination corrected enthalpies” method (CCE), based on the number of nearest neighbor cation–anion bonds, and also capable of correcting relative stability of polymorphs. CCE uses calculations employing the Perdew, Burke and Ernzerhof (PBE), local density approximation (LDA) and strongly constrained and appropriately normed (SCAN) exchange correlation functionals, in conjunction with a quasiharmonic Debye model to treat zero-point vibrational and thermal effects. The benchmark, performed on binary and ternary oxides (halides), shows very accurate room temperature results for all functionals, with the smallest mean absolute error of 27(24) meV/atom obtained with SCAN. The zero-point vibrational and thermal contributions to the formation enthalpies are small and with different signs—largely canceling each other.
Similar content being viewed by others

Introduction
The accurate prediction of the thermodynamic stability of a compound—crucial in computational materials design1—mostly relies on the calculation of the formation enthalpy: the enthalpy change with respect to elemental reference phases. Using density functional theory (DFT), the formation energy, neglecting pressure–volume contributions, is routinely computed ab initio. For systems where elements and compounds are metallic, i.e. chemically similar, accurate results are usually obtained by using standard (semi)local approximations to DFT.2,3 They include the local density approximation (LDA)4,5 or the generalized gradient approximation (GGA), for instance PBE.6 In this way, formation energies for millions of metal alloys have already been calculated in materials databases such as AFLOW,7,8,9,10 the Materials Project,11,12 and OQMD.13,14
When the compound and the elements have a different chemical character, as for example in the case of oxides, nitrides, or sulfides, the situation is less favorable. For oxides, the compound is typically an ionic insulator while the elements are metals or semiconductors and a diatomic gas. When comparing to experimental enthalpies,15,16,17,18 standard approximations of DFT lead to mean absolute errors (MAEs) of the order of several hundred meV/atom. For reaction energies between binary and ternary oxides, within a similar chemical realm, a smaller average error of about 24–35 meV/atom has been observed.19
Different attempts have been made to calculate more accurate formation energies ab initio. A modified version of PBE was proposed by Sarmiento-Pérez et al.:20 three functional parameters were optimized, improving results by about a factor of two. The hybrid functional HSE06 yields only a slight improvement for transition metal oxides.21 The recently developed strongly constrained and appropriately normed (SCAN) meta-generalized-gradient approximation22 has an accuracy limited to about 100 meV/atom.23,24
Non-self-consistent exact exchange plus random phase approximation (EXX + RPA) calculations can lead to more accurate formation energies by about a factor of two–three compared to PBE.25,26 The renormalized adiabatic PBE method improves the results based on RPA for 19 main group and two transition metal oxides by about a factor of two.27 A Bayesian error estimation functional (mBEEF) systematically improves PBE results reaching an MAE of about 120 meV/atom for a test set of 24 compounds.28 Applying a correction method on top of the functional could reduce the MAE to 90 meV/atom, which is 20–60 meV/atom less than if the correction is applied on top of other functionals. Unfortunately, such computationally expensive approaches are not suitable for screening large materials sets and do not, in general, reach the necessary chemical accuracy of 1 kcal/mol (≈40 meV/atom).
Several empirical correction schemes have been established for formation energies calculated with DFT by comparing to experimentally measured formation enthalpies. Wang et al.29 suggested an oxygen correction of 1.36 eV per O2 to be subtracted from formation energies calculated with PBE. The approach was extended to H2, N2, F2, and Cl2 for different functionals.30 For sulfides, a different correction is found depending on whether the anion is S2− or \({\mathrm{S}}_2^{2 - }\).31 Jain et al. suggested an empirical scheme for mixing GGA and GGA + U calculations to compute formation enthalpies for compounds containing transition metal elements.32 An MAE of 45 meV/atom was achieved for a test set of 49 ternary oxides with respect to experimental values.32 A local environment dependent GGA + U method based on the GGA/GGA + U mixing scheme was also developed.33 It introduced significantly more parameters and achieved an MAE of 19 meV/atom for a test set of 52 transition metal oxides. In the fitted elemental-phase reference energies (FERE) method,34,35 element-specific corrections were used to optimize the error cancellation when calculating total energy differences between chemically dissimilar materials. Corrected formation energies calculated for a test set of 55 ternary compounds gave an MAE of 48 meV/atom.35 In conclusion, existing correction schemes and advanced theoretical approaches do not, in general, reach an accuracy of the order of the thermal energy at room temperature (~25 meV) for formation enthalpies.
Here, we propose a physically motivated correction scheme — coordination corrected enthalpies (CCE), based on the number of bonds between each cation and surrounding anions. Compared to previous approaches, it leads to systematically more accurate results. The smallest MAE of 27 (24) meV/atom for a test set of ternary oxides (halides) is reached when starting from SCAN calculations. Contrary to earlier approaches, the ansatz also allows correction of the relative stability of polymorphs with different number of cation–anion bonds.
The article mainly focuses on oxides because of: (i) high technological relevance, (ii) abundance of experimental thermochemical data, especially for ternary oxides, and (iii) generally low error bars of the experimental values allowing accurate corrections and predictions. Calculated room temperature formation enthalpies for a set of 79 binary and 71 ternary oxides are presented employing the three main approximations to the DFT exchange-correlation functional: LDA, PBE, and SCAN.
In other schemes, temperature effects have been completely neglected,32,34,35 or room temperature experimental values were interpolated to 0 K using a Debye model parameterized with the measured room temperature heat capacities and entropies.19,33 Here, the thermal contributions to the formation enthalpy are calculated via a quasiharmonic Debye model.36,37,38,39,40 Our approach includes the contribution due to zero-point vibrational energies.
First, the methodology of calculating coordination corrected room temperature formation enthalpies is presented. Then, the DFT derived and CCE results are discussed. Additional comparisons and tables with structure data, values of the corrections, of calculated, corrected, and experimental formation enthalpies/energies, as well as the vibrational contributions are listed in the supplementary information.
Results
Room temperature formation enthalpies
The formation enthalpy includes contributions due to the pressure–volume term (e.g. for O2). The formation energy takes into account only internal energy contributions. The formalism, introduced for oxides, works equivalently for other polar systems.
From DFT, an approximate formation energy \({\mathrm{\Delta }}_{\mathrm{f}}E^{0,{\mathrm{DFT}}}\) of an oxide \(A_{x_1}B_{x_2} \ldots {\mathrm{O}}_{x_n}\) at zero T and p, without zero-point vibrational energies, can be calculated:
where \(U_{\begin{array}{c}{A_{x_1} \ldots {\mathrm{O}}_{x_n}}\end{array}}^{0,{\mathrm{DFT}}}\), \(U_i^{0,{\mathrm{DFT}}}\), and \(U_{{\mathrm{O}}_2}^{0,{\mathrm{DFT}}}\) are the total energies of the compound per formula unit, the i-element reference phase per atom, and O2, respectively, and x1, …, xn are stoichiometries.
The tabulated experimentally measured standard formation enthalpy at the reference temperature Tr = 298.15 K, \(\Delta _{\mathrm{f}}H^{ \circ ,T_{\mathrm{r}},{\mathrm{exp}}}\), corresponds to:
where \(H_{\begin{array}{c}{A_{x_1} \ldots {\mathrm{O}}_{x_n}}\end{array}}^{ \circ ,T_{\mathrm{r}}}\), \(H_i^{ \circ ,T_{\mathrm{r}}}\), and \(H_{{\mathrm{O}}_2}^{ \circ ,T_{\mathrm{r}}}\) are the standard enthalpies of the compound per formula unit, the i-element reference phase per atom and O2, respectively, all at Tr.
Using H = U + pV and neglecting the pV terms for the compound and the elements (less O2), the formation enthalpy becomes:
Generally, neglecting pV is a very good approximation: pressures are small and the molar volumes of condensed systems are typically three orders of magnitude smaller than gases—the contribution to the formation enthalpy is expected to be well below 1 meV/atom.
Writing the total energies and the standard enthalpy of O2 at Tr as the value at 0 K plus the difference between Tr and 0 K, and separating the zero-point vibrational energy for each system, gives:
where \(U_{\begin{array}{c}{A_{x_1} \ldots {\mathrm{O}}_{x_n}}\end{array}}^{\begin{array}{c}{\mathrm{ZP}}\end{array}}\), \(U_i^{\begin{array}{c}{\mathrm{ZP}}\end{array}}\), and \(U_{{\mathrm{O}}_2}^{\begin{array}{c}{\mathrm{ZP}}\end{array}}\) are the zero-point vibrational energies of the compound, the i-element reference and O2, respectively. \({\mathrm{\Delta }}_{\mathrm{f}}H_{\begin{array}{c}{A_{x_1} \ldots {\mathrm{O}}_{x_n}}\end{array}}^{ \circ ,T_{\mathrm{r}},{\mathrm{cal}}}\) stands for the calculated standard formation enthalpy at Tr. The terms are:
is the internal energy contribution excluding vibrational effects;
collects all zero-point (ZP) contributions;
is the overall thermal contribution (TC).
The internal energy contribution to \({\mathrm{\Delta }}_{\mathrm{f}}H_{\begin{array}{c}{A_{x_1} \ldots {\mathrm{O}}_{x_n}}\end{array}}^{ \circ ,T_{\mathrm{r}},{\mathrm{exp}}}\) can be identified with \({\mathrm{\Delta }}_{\mathrm{f}}E_{\begin{array}{c}{A_{x_1} \ldots {\mathrm{O}}_{x_n}}\end{array}}^{0,{\mathrm{DFT}}}\) calculated with DFT according to Eq. (1). The pressure dependence is negligible at the standard value of 1 bar.
For the thermal contribution, the internal energy differences between 0 K and Tr are almost entirely due to vibrations. The quantity is estimated by using the AFLOW Automatic GIBBS Library (AGL) via a quasiharmonic Debye model36,37,38,39,40 with default parameters (28 strained structures, 1% lattice strain increments39). The approach is tested by comparing the calculated internal energy difference between 0 K and Tr with experimental enthalpy differences (see Section 1 in the supplementary information), indicating good agreement for both compounds and references.
The AGL calculations also provide a zero-point vibrational energy, which is used to treat the zero-point contribution. Notably, e.g. for BeO the energy is calculated to be 0.11 eV/atom for all three functionals, which agrees exactly with the value reported in ref. 41 obtained from more expensive phonon calculations. In the rest of the article, the sum of the zero-point and thermal contributions is denoted as the vibrational contribution.
For O2, the enthalpy difference between 0 K and Tr can be estimated from a perfect diatomic gas with five degrees of freedom where the bond-stretching vibrational mode is not excited at Tr, leading to 90 meV/O2.42 The value agrees exactly with the tabulated enthalpy difference from the NIST-JANAF thermochemical tables.16 For F2, Cl2, BF3, and SiF4, the enthalpy differences from NIST-JANAF corresponding to 91, 95, 121, and 159 meV are taken, respectively. The zero-point vibrational energy of O2 is calculated, using the experimental oxygen vibrational frequency of 1580.1932 cm−1,16 to be 98 meV/O2. For F2, Cl2, BF3, and SiF4, the calculated zero-point energies are 55, 35, 339, and 346 meV. Similarly, for Hg the total energy at 0 K is calculated for the low-temperature rhombohedral structure, with the zero-point vibrational energy obtained from AGL. The experimental enthalpy difference from 0 K to Tr of 97 meV/Hg atom from the NIST-JANAF tables,16 including fusion at 234.29 K, is used to account for thermal effects.
Coordination corrected enthalpies scheme
The remaining deviation between calculated and measured room temperature formation enthalpies is almost entirely due to the internal energy contribution \({\mathrm{\Delta }}_{\mathrm{f}}E_{\begin{array}{c}{A_{x_1} \ldots {\mathrm{O}}_{x_n}}\end{array}}^{0,{\mathrm{DFT}}}\) obtained with DFT. Compounds with strong polar bonds are chemically different from elements—mostly metallic plus a diatomic gas. As already noted by Lany34 and Stevanović et al.,35 this leads to an incomplete error cancellation when calculating total energy differences—standard semilocal functionals do not allow calculation of accurate total energies.
Since a reliable description of the bonding in a material is central for capturing its properties, it seems reasonable to assume in first approximation that DFT makes errors per bond. As such, the CCE scheme considers the number of nearest-neighbor bonds (coordination number) formed between the cation and oxygen. The approach enables accounting for coordination changes, as illustrated in Fig. 1 for the case of CaO, rutile TiO2, and perovskite CaTiO3. For the binary oxides, Ca is six-fold (octahedrally) coordinated by O in the rocksalt structure of CaO, while Ti is six-fold in rutile TiO2. For Ti, the coordination number remains the same in CaTiO3, but the number of nearest neighbor Ca–O bonds changes to eight. The phenomenon is quite common for several elements when going from binary to ternary oxides, and can be captured within CCE.

Coordination change. Crystal structures of a CaO, b rutile TiO2, and c CaTiO3 (perovskite). The coordination polyhedra of Ca and Ti are shown in green and blue, respectively. Note: Ca is six-fold (octahedrally) coordinated with oxygen in CaO and eight-fold coordinated in CaTiO3, requiring coordination corrections. Colors: Ca black, Ti light gray, and O red66
The corrections per bond \(\delta H_{A - {\mathrm{O}}}^{A^{ + \alpha }}\) are extracted from the deviation between the calculated and experimental formation enthalpies of binary oxides \(A_{x_1}{\mathrm{O}}_{x_2}\) for each functional:
where NA–O is the number of nearest neighbor A–O bonds of element A in oxidation state +α. CCE is constructed to be dependent on +α: the energetic position of the bonding states and hence also the correction are expected to be oxidation state specific. In AFLOW, oxidation numbers can be determined by a Bader analysis,43,44 while ensuring that the sum over all atoms equals zero. When counting bonds for distorted or low-symmetry environments, a length variation up to 0.5 Å is allowed. After trying different tolerances, this value is found to lead to the best results. In the case of CaTiO3 (see Fig. 1c) the nearest-neighbor Ca–O bond length varies between 2.36 and 2.69 Å for the relaxed PBE structure.
As mentioned before, DFT errors do not only originate from the inaccurate treatment of the bonding in the compound, but also from the lack of error cancellation with the different reference phases. CCE corrections per bond implicitly include those of the elemental references—for a given bonded pair of atoms, reference phases are constant and the lack of error cancellation is then “absorbed” into corrections per bond. It especially applies to the molecular O2 reference, for which the atomization energy is known to be poorly described in DFT.29,45
The energy corrections extracted from binary oxides are then applied to the test-set of ternary oxides \(A_{x_1}B_{x_2}{\mathrm{O}}_{x_3}\) to calculate the corrected formation enthalpies:
where Ni–O is the number of nearest-neighbor bonds between the cation i-species and oxygen.
Compared to other approaches,29,32,33,34,35 it is important to note that at fixed composition, CCE is capable of correcting the relative stability of polymorphs with different coordination numbers.
Comparisons are performed with a quasi-FERE approach following the ideas of refs. 34,35 A least-squares problem for all binary oxides in the fitting set is solved for the element-specific corrections \(\delta H_i^{{\mathrm{qFERE}}}\):
The corrections are then added to the calculated reference enthalpies used to calculate the corrected formation enthalpies. Contrary to the original FERE,34,35 here (i) no Hubbard-U term is used, (ii) only oxides are considered in the fitting set, (iii) the corrections are determined and applied with respect to the calculated room temperature formation enthalpies rather than DFT formation energies, and (iv) in part different experimental data are used.
Principal thermodynamic considerations
There is also another caveat. Corrections depending linearly on the concentration (like the previously proposed renormalization of the chemical potential of one or more species) are equivalent to tilting the whole Gibbs landscape, and might—in some cases—lead to thermodynamic paradoxes. For example, consider the case of non-ideal activity vs. concentration, differing from the Raoult’s law with a negative(positive) deviation at low(high) concentration.46 Any linear interpolation tends to balance the deviations and erroneusly correct the chemical potential by decreasing its non-ideal behavior. This is a rare scenario. Yet, phase diagrams having a very-high monotectoid and very-low eutectoid do exist, and the accuracy of calculated critical temperatures would be reduced with inappropriately corrected enthalpies. The problem can be solved only by including more information in the DFT correction, introducing nonlinearity and/or considering topology and oxidation states like in the case of CCE.
Selection of structural inputs
All room temperature structures are obtained from the AFLOW-ICSD online library.7,9,10,47 The selection is based on the structure information in the Kubaschewski et al. tables.15 If it is insufficient, it is taken from the Springer Materials database.48 The ICSD numbers, space groups and Pearson symbols are listed in Tables 2 and 3 in the supplementary information. Space-groups and Pearson symbols are calculated with AFLOW-SYM.49 For SiO2, both the α-quartz (space group P3121 #152; Pearson symbol hP9; AFLOW prototype A2B_hP9_152_c_a [http://aflow.org/CrystalDatabase/A2B_hP9_152_c_a.html]50,51) and α-cristobalite (P41212 #92; tP12; A2B_tP12_92_b_a [http://aflow.org/CrystalDatabase/A2B_tP12_92_b_a.html]50,51) prototypes are considered. TiO2 is calculated in the rutile (P42/mnm #136; tP6; A2B_tP6_136_f_a [http://aflow.org/CrystalDatabase/A2B_tP6_136_f_a.html]50,51), and anatase (I41/amd #141; tI12; A2B_tI12_141_e_a [http://aflow.org/CrystalDatabase/A2B_tI12_141_e_a.html]50,51) structures. Al2SiO5 is represented in the kyanite (P1̄ #2; aP32) and andalusite (Pnnm #58; oP32) structures. CaSiO3 is treated as wollastonite (P1̄ #2; aP30) and pseudowollastonite (C2/c #15; mS60). For O2, F2, Cl2, BF3, and SiF4, a 10 × 10 × 10 Å3 cubic box is used, the intermolecular bond length is relaxed until the forces are smaller than 10 meV/Å, and the Brillouin zone is sampled only at the Γ-point.
Selection of computational inputs
The accuracy of experimental data used is crucial. For oxides and halides, several reliable thermochemical libraries do exist, and here, we rely on the collections of Kubaschewski et al.,15 NIST-JANAF,16 Barin,17 and NBS.18
For the validation of the experimental room temperature enthalpies, a procedure similar to Hautier et al.19 is applied. Each \({\mathrm{\Delta }}_{\mathrm{f}}H^{ \circ ,T_{\mathrm{r}},{\mathrm{exp}}}\) of Kubaschewski et al.15 is first compared to the values from the NIST-JANAF database,16 which is believed to be the most accurate.19 If the deviation exceeds 5 meV/atom, the value from ref. 16 is used. For the oxides with no corresponding entry in NIST-JANAF, the formation enthalpies are compared with the Barin ones. If the values differ by more than 10 meV/atom, Barin’s \({\mathrm{\Delta }}_{\mathrm{f}}H^{ \circ ,T_{\mathrm{r}},{\mathrm{exp}}}\) is used. NaCrO2 is an exception: the Kubaschewski formation enthalpy is taken, since the Barin value deviates by 0.15 eV/atom from the Kubaschewski and NBS data. Both Hautier et al.19 and Aykol & Wolverton33 used the \({\mathrm{\Delta }}_{\mathrm{f}}H^{ \circ ,T_{\mathrm{r}},{\mathrm{exp}}}\) from Kubaschewski and obtained good agreement with the calculated reaction energies and formation enthalpies—this would not have been possible with the Barin value. In general, the NBS collection might not be considered as a suitable source for comparisons: When compared to all others, it exhibits several examples with significant deviations (see Section 2 in the supplementary information). This might be at least partially due to the special consistency requirements within NBS.18 Oxides from Kubaschewski with no corresponding formation enthalpy in Barin are therefore excluded. For halides, the procedure is relaxed for NaBF4 and Na2SiF6 due to the scarcity of experimental data for polar ternaries other than oxides. In these two cases, the Kubaschewski formation enthalpy is taken, which could only be verified by NBS.
Room temperature DFT + AGL results
The difference between calculated DFT + AGL and experimental room temperature formation enthalpies for 79 binary and 71 ternary oxides for the three functionals employed are illustrated in Fig. 2. The vibrational (zero-point + thermal) contribution is shown in the lower panels of panels a and b. MAEs are included in Table 1. The calculated formation enthalpies for each functional, together with the experimental values, are included in Tables 4 and 6 in the supplementary information. The vibrational, zero-point and thermal contributions are listed in Tables 9 and 11 in the supplementary information.

Uncorrected enthalpies. Differences between calculated (Eq. (4)) and experimental room temperature formation enthalpies of binary oxides (a upper panel) and vibrational (zero-point + thermal) contribution to the calculated formation enthalpy (a lower panel). Vertical blue lines separate the different l-blocks with respect to the position of the non-O element of the compound in the periodic table. Differences between calculated and experimental room temperature formation enthalpies of ternary oxides (b upper panel) and vibrational contribution to the calculated formation enthalpy (b lower panel)
Vibrational contribution
In general the vibrational term is very small (lower panels of Fig. 2a, b), and decreases with increasing atomic number of the non-O elements. The maximum value of 23(23) meV/atom is reached for Al2O3(kyanite—Al2SiO5) with SCAN. The minimum of −22(−4) meV/atom occurs for HgO(PbWO4) with LDA(PBE and SCAN). For HgO, this is due to the heat of fusion of Hg being about 24 meV/atom at 234.29 K.16 On average, the absolute vibrational value for binaries(ternaries) is very small: 5, 7, and 6(7, 9, and 8) meV/atom for PBE, LDA, and SCAN, respectively, due to partial cancellations of the zero-point and thermal contributions (Section 3 in the supplementary information provides additional insights). Unless stated otherwise, our PBE, LDA, and SCAN formation enthalpies include vibrational contributions, which, despite the often negligible values, consistently improve the MAEs of LDA and SCAN for binaries and ternaries by 2–5 meV/atom (Table 1). For PBE, the MAE increases when including the vibrational value—most likely an artifact for the functional having the largest errors.
Comparison of calculated and experimental results
In Fig. 2a, the compounds are grouped according to the l-block of the non-O element in the periodic table. Materials are ordered with respect to increasing atomic number of the non-O element. PBE tends to underestimate the formation enthalpy leading to the largest deviations from the experimental values (MAE 235 meV/atom). Both LDA and SCAN show an increasingly better performance with total MAEs of 176 and 105 meV/atom, respectively. The findings are in agreement with previous reports23,35 including similar compounds. LDA was found to systematically yield better formation energies than PBE34 for a much smaller set of 13(9 binary, 4 ternary) oxides.
Results indicate a pronounced dependence on the l-character of the non-O element. For s-oxides, SCAN gives very accurate formation enthalpies with an MAE of 27 meV/atom, with LDA and PBE showing increasing deviations. For p-oxides, all functionals display a decreasing trend in \({\mathrm{\Delta }}_{\mathrm{f}}H^{ \circ ,T_{\mathrm{r}},{\mathrm{cal}}}\) with respect to \({\mathrm{\Delta }}_{\mathrm{f}}H^{ \circ ,T_{\mathrm{r}},{\mathrm{exp}}}\) with increasing atomic number of the non-O species, the trend being weakest for SCAN. Spin–orbit coupling could be the culprit, although often the effect largely cancels out when calculating formation energies.35,52 Instead, the trend might be caused by an increasing degree of covalency. MAEs for the combined set of all s- and p- (main group) oxides of 223, 113, and 46 meV/atom are obtained for PBE, LDA, and SCAN, respectively. The values are in good agreement with ref. 23, where a largely similar set of main group oxides was investigated. For transition metal, i.e. d-oxides, all functionals show large errors of several hundred meV/atom, with SCAN having the smallest MAE of 163 meV/atom. For the ternary oxides, deviations similar to the binaries are shown in Fig. 2b: MAEs are 279, 107, and 105 meV/atom for PBE, LDA, and SCAN.
Further improvements on a semilocal DFT level might be difficult considering that SCAN already fulfills all known constraints required for the exact functional.22 A promising direction might be provided by the recently developed size-extensive self-interaction correction scheme53,54,55,56 potentially leading to more accurate formation enthalpies.
Coordination corrected enthalpies
This section compares the two correction schemes described above: CCE and the quasi-FERE approach. The oxygen correction introduced by Wang et al.29 is not considered as it shows a strong dependency on the fitting set when p-oxides are included (see Section 4 in the supplementary information).
CCE uses the deviation between calculated and experimental room temperature formation enthalpies of single-valence binary oxides to obtain corrections per cation–O bond for each functional. They are then applied to the calculated formation enthalpies of ternary and mixed-valence binary oxides. The quasi-FERE method uses the binary data to obtain element-specific corrections, optimizing the systematic error cancellation between the total energies/enthalpies of the references with respect to the compound.34,35
Corrected binary results
For the binary fit set, CCE gives almost exact solutions, as indicated by the small MAEs of 5, 4, and 3 meV/atom for PBE, LDA, and SCAN (Table 1). The corrections per bond are included in Table 2 and in Table 4 in the supplementary information. The quality is not surprising: the scheme is constructed to reproduce the experimental formation enthalpies of the single-valence binary oxides. The few other cases include mixed-valence compounds, multiple polymorphs at the same composition and per- as well as superoxides, leading to non-zero MAE for the binary set (Table 1) and allowing assessment of CCE reliability. For SbO2, the corrections obtained from Sb2O3 and Sb2O5 are used. Pb3O4 is refined based on PbO and PbO2, and for Ti3O5 the corrections from Ti2O3 and TiO2 (rutile) are taken. For SiO2 (α-cristobalite) and TiO2 (anatase), the \(\delta H_{A - {\mathrm{O}}}^{A^{ + \alpha }}\) determined from SiO2 (α-quartz) and TiO2 (rutile) are applied, respectively. The results are included in Table 4 in the supplementary information. For all cases, the corrected values agree well with the experimental data—typically within 20 meV/atom—and a maximum systematic deviation of about 50 meV/atom is observed for Pb3O4.
The per- and superoxides cannot be corrected exactly, since their structure incorporates bonds between the cation and O, as well as an internal O–O bond. The values are corrected based on the assumption that for the cation–O bond the correction of the normal (O2−) oxide can be taken, and the O–O bond correction is transferable between (su)peroxides: O–O correction for peroxides is obtained from Li2O2; the O–O correction for superoxides is derived from KO2. The two values are listed in Table 4 in the supplementary information. All other per- and superoxides are corrected based on these values. In general, the procedure leads to good agreement with experiment, and the largest absolute deviation occurs for the corrected PBE value of NaO2: 124 meV/atom (the absolute deviations for LDA and SCAN are 17 and 45 meV/atom, respectively).
For quasi-FERE, MAEs of 53, 44, and 48 meV/atom for PBE, LDA, and SCAN are obtained for the binary fit set. They agree well with the MAE of 54 meV/atom reported for the original FERE in ref. 35 using PBE for the fit set of binary compounds. Our calculations indicate that even better agreement is obtainable when using LDA or SCAN in the DFT calculations.
FERE tends to yield large deviations if multivalent p-oxides, such as SnO, SnO2, Sb2O3, SbO2, Sb2O5, Tl2O, Tl2O3, PbO, PbO2, and Pb3O4 are considered.35 Indeed, for these systems, errors for the quasi-FERE corrected values partly exceeding 100 meV/atom are observed, in agreement with ref. 35. CCE circumvents the problem through its explicit dependence on the oxidation state of the cation according Eq. (8).
Corrected ternary results
The differences between CCE and quasi-FERE corrected and experimental room temperature formation enthalpies are displayed in Fig. 3 for the test set of 71 ternary oxides calculated with PBE (panel a), LDA (panel b) and SCAN (panel c). MAEs are included in Table 1 and the formation enthalpies are listed in Tables 6 and 8 in the supplementary information. The importance of using ab-initio data as input for CCE is discussed in Section 5 in the supplementary information. CCE predicts accurate results for almost all ternary compounds: MAE is 38, 29, and 27 meV/atom with PBE, LDA, and SCAN, respectively. Compared to plain DFT + AGL, the errors are decreased by about a factor of 4–7. The mean deviations are significantly smaller than 45 and 48 meV/atom predicted by the GGA/GGA + U mixing and FERE corrections of refs. 32,35. For the quasi-FERE method on the same set of compounds, MAEs of 43, 35, and 44 meV/atom are obtained for the corrected values of PBE, LDA, and SCAN (The MAE of the quasi-FERE method on the test set is smaller than the one on the binary fit set for all functionals in agreement with previous results.35 The fitting set contains a larger variety of elements in oxidation states potentially problematic for DFT. In the test set of ternary oxides there are fewer such cases leading to smaller MAEs.). CCE consistently yields more accurate results than quasi-FERE for all three functionals. The MAEs of CCE are slightly larger than the 19 meV/atom of the local environment-dependent GGA + U method.33 The latter scheme, however, uses about a factor two more parameters and is constructed for transition metal compounds. On the contrary CCE is applicable to all systems. CCE is simpler and more intuitive.

Corrected enthalpies. Differences between corrected and experimental room temperature formation enthalpies of the test set of 71 ternary oxides for the CCE and quasi-FERE correction schemes on top of PBE a, LDA b, and SCAN c. Note the different energy scale compared to Fig. 2. The red lines at ±50 meV/atom indicate the typical MAE of previous correction schemes32,35
The largest single absolute deviation over the whole set is also higher for the quasi-FERE method—182, 152 and 166 meV/atom for PBE (Na2CrO4), LDA (MnTiO3), and SCAN (Na2CrO4)—compared to CCE—118, 135, and 118 meV/atom for PBE, LDA, and SCAN (always FeAl2O4).
When CCE predicts a similar value for all three functionals with a large deviation with respect to the experimental data, the measured \({\mathrm{\Delta }}_{\mathrm{f}}H^{ \circ ,T_{\mathrm{r}},{\mathrm{exp}}}\) might be inaccurate. The conclusion is further confirmed if the quasi-FERE-corrected values predict a similar trend. Based on the analysis, the experimental data of FeAl2O4 and NiAl2O4 might be too low (i.e. too negative) by about 120–140 and 80–110 meV/atom, respectively. SrHfO3 might be too high by about 60–90 meV/atom.
The scarcity of reliable experimental data for polar ternary systems other than oxides restricts the number of compounds available to demonstrate the generality of CCE. In Fig. 4, uncorrected and corrected results are presented for a set of seven ternary halides. The formation enthalpies are listed in Tables 5 and 7 in the supplementary information with the vibrational, zero-point, and thermal contributions in Tables 10 and 12 in the supplementary information. Corrections are given in Table 3 and in Tables 5 and 14 in the supplementary information. It is difficult to ascribe a statistical significance to a set of only seven entries: MAEs amount to 264, 78, and 135, as well as 49, 74, and 24 meV/atom for the uncorrected and corrected results of PBE, LDA, and SCAN, respectively. CCE guarantees a significant improvement in all cases. For Na3AlF6, and potentially also NaAlCl4, the experimental value might be too low by about 70–90 and 20–80 meV/atom. The accuracy of the corrected results for KBF4, NaBF4, and Na2SiF6 is interesting—in these cases part of the corrections are obtained from the gaseous molecular BF3 and SiF4 phases and applied to solid ternaries. For PBE and LDA, the corrected results show rather large errors revealing that for these functionals the corrections per bond are not well transferable from molecules to solids. This biases the MAE particularly for the corrected LDA values. For SCAN, however, the corrected results are accurate, further showcasing the better suitability of this functional for CCE. The functionals‘ different behaviors agree with previous reports.57,58

Uncorrected vs. corrected enthalpies for halides. Differences between calculated a as well as corrected b and experimental room temperature formation enthalpies for seven ternary halides. For the compounds marked with “*”, the experimental formation enthalpy from Kubaschewski et al.15 can only be verified by NBS.18 The red lines at ±50 meV/atom indicate the typical MAE of previous correction schemes32,35
The vibrational (zero-point + thermal) contribution to the formation enthalpy can be largely included in the corrections without explicit calculation, being mostly element specific. For example, for binary oxides the vibrational term is highest for Al2O3, BeO, and SiO2 (α-quartz), ranging from 16 to 23 meV/atom (depending on the functional). For ternaries, the largest value is found for kyanite Al2SiO5 with 19–23 meV/atom. MAEs of the corrected formation energies obtained without vibrational contribution in both the binary-fit and ternary-test sets (as in refs. 32,34,35) are included in brackets in Table 1. They deviate no more than 1 meV/atom from the MAEs of the corrected DFT + AGL results. Thus, \({\mathrm{\Delta }}_{\mathrm{f}}E^{0,{\mathrm{DFT}}}\) corrections can be reliably based on only \({\mathrm{\Delta }}_{\mathrm{f}}H^{ \circ ,T_{\mathrm{r}},{\mathrm{exp}}}\). In addition, the vibrational term usually does not lead to significant differences between two structures at the same composition. This has already been seen with machine learning analysis.59 Therefore, the following discussion is based on results directly obtained with DFT.
Relative stability
CCE can also correct the relative stability of same stoichiometry structures with different number of nearest-neighbor cation–O bonds. Al2SiO5 is an example: kyanite is the experimental ground state and andalusite is higher in energy. PBE falsely predicts kyanite to be 19 meV/atom above andalusite (−2.937 vs. −2.956 eV/atom). CCE correctly gives kyanite to be lower by 4 meV/atom (−3.343 vs. −3.339 eV/atom), in good agreement with the experimental values (−3.361 vs. −3.358 eV/atom).
The situation is more evident with polymorphs having large energy differences. Experimentally, MnO and CoO have rocksalt ground states. In ref. 23, it was reported that PBE and SCAN predict other ground states for both systems with only four cation–O bonds, in disagreement with the experimental finding: 6. CCE solves the issue. We take the DFT ground states provided in ref. 23, relax and primitivize them. PBE/SCAN for MnO and SCAN for CoO find zincblende (space group F4̄3m #216; Pearson symbol cF8; AFLOW prototype AB_cF8_216_c_a [http://aflow.org/CrystalDatabase/AB_cF8_216_c_a.html]50,51). With PBE the final CoO structure is body-centered tetragonal (I4̄m2 #119; tI4; AB_tI4_119_c_a [http://aflow.org/CrystalDatabase/AB_tI4_119_c_a.html]50,51). For CoO, PBE, and SCAN erroneously give the energies of the relaxed geometries to be 164 and 103 meV/atom below rocksalt. CCE solves the dilemma. When corrected, they become 76 and 20 meV/atom above the experimental ground state. For MnO the PBE structure is corrected from being 5 meV/atom more-stable to 246 meV/atom less-stable than the experimental report. The MnO structure given by SCAN is already 44 meV/atom higher than rocksalt. CCE reduces its difference to 11 meV/atom without changing the correct experimental order. CCE succeeds in all examples. Any scheme dealing only with stoichiometry (such as FERE) would not be able to disentangle the relative stability.
Application to Ti–O systems
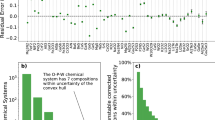
To test whether CCE will also yield quantitatively reliable results for defect energies, the method is applied to Ti–O. The corrections are obtained from Ti2O3 and rutile TiO2, and are then applied to predict the enthalpies of other oxides, including crystallographic shear compounds (Magnéli phases) TinO2n−1. In Fig. 5, the section of the convex hull phase diagram60 between Ti2O3 and TiO2 is presented for both uncorrected and CCE-corrected results. Plain DFT captures well the position of all structures with respect to the individual convex hull for each functional, but yields quantitative errors of the order of several 100 meV/atom in all three cases. When corrected by CCE, all three functionals produce formation enthalpies within 10–20 meV/atom of experiments. Note that for all functionals (corrected and uncorrected) and from the experimental data, Ti3O5 is found to be above the stability hull by up to about 30 meV/atom.

Uncorrected vs. corrected convex hull. Section of the convex hull between Ti2O3 and TiO2 (rutile) for the Ti–O system from plain DFT a and corrected by CCE b
Discussion
We have introduced a coordination corrected enthalpies (CCE) scheme based on the number of nearest-neighbor cation–anion bonds. 71(7) ternary oxides (halides) are used as a test set. CCE gives very accurate corrected formation enthalpies with MAEs of 38(49), 29(74) and 27(24) meV/atom for PBE, LDA, and SCAN, respectively. Zero-point and finite temperature vibrational contributions are treated within a quasiharmonic Debye model and are found to largely cancel out. Errors are significantly smaller than previous approaches.32,34,35 Because CCE considers bonding connectivity and topology, it can also correct the relative stability of different structures at a given composition.
Correction schemes for formation enthalpies are the steps in a ladder of approximations:
-
i.
The oxygen correction of ref. 29 applies a constant energy shift per O2; it can be seen as a 0th order step: one parameter for all oxides. The approach typically leads to MAEs of 100 meV/atom or larger, and can be combined with the GGA/GGA + U mixing scheme for improved accuracy.32
-
ii.
The FERE method34,35 corrects the elemental reference energy of each species of the compound; it is a first-order approximation: one parameter per element. FERE's accuracy is typically limited to about 40–50 meV/atom. Improvements require considering the characteristics of the compounds.
-
iii.
CCE leverages the topology of nearest-neighbor shells. CCE yields accurate formation enthalpies with an average absolute error as small as 20–30 meV/atom. The method is simple and easy to extend to other materials classes, e.g. nitrides, phosphides, or sulfides. It can be used to predict a wide variety of properties relying on accurate formation enthalpies, such as battery voltages, defect energies, and the formation of high-entropy materials.61
Methods
Calculations are performed using the AFLOW framework7,8,9,10,44,47,50,51,62 leveraging the Vienna Ab-initio Simulation Package (VASP)63,64 with projector-augmented-wave pseudopotentials65 of version 5.4. The exchange-correlation functionals LDA,4,5 PBE,6 and SCAN22 are employed. The parameters of the structural relaxation and static calculations largely follow the AFLOW Standard for entries from the ICSD library47 with the internal VASP precision set to ACCURATE. No Hubbard-U term is used, and for the elements Li, Be, Na, and W, pseudopotentials with the labels Li, Be, Na_pv, and W_sv are taken, respectively. For calculating total energy differences between a compound and its references, the kinetic energy cutoff is set to be 40% larger than the highest value recommended among all pseudopotentials for the compound but to at least 560 eV (oxygen cutoff). For magnetic systems, spin-polarized calculations are performed with all possible ferromagnetic, ferrimagnetic, and antiferromagnetic configurations initialized for five different sizes of the induced magnetic moments in the primitive unit cell. For computational efficiency, for Ti4O7, Ti5O9, and Ti6O11, only four different ferromagnetic configurations were initialized. The final magnetic state with the lowest total energy is considered for the formation enthalpy.
References
Curtarolo, S. et al. The high-throughput highway to computational materials design. Nat. Mater. 12, 191–201 (2013).
Wolverton, C. & Ozolinš, V. First-principles aluminum database: energetics of binary Al alloys and compounds. Phys. Rev. B 73, 144104 (2006).
Curtarolo, S., Morgan, D. & Ceder, G. Accuracy of ab initio methods in predicting the crystal structures of metals: a review of 80 binary alloys. Calphad 29, 163–211 (2005).
Kohn, W. & Sham, L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133 (1965).
von Barth, U. & Hedin, L. A local exchange-correlation potential for the spin polarized case: I. J. Phys. C 5, 1629 (1972).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Oses, C., Toher, C. & Curtarolo, S. Data-driven design of inorganic materials with the automatic flow framework for materials discovery. MRS Bull. 43, 670–675 (2018).
Setyawan, W. & Curtarolo, S. High-throughput electronic band structure calculations: challenges and tools. Comput. Mater. Sci. 49, 299–312 (2010).
Taylor, R. H. et al. A RESTful API for exchanging materials data in the AFLOWLIB.org consortium. Comput. Mater. Sci. 93, 178–192 (2014).
Rose, F. et al. AFLUX: The LUX materials search API for the AFLOW data repositories. Comput. Mater. Sci. 137, 362–370 (2017).
Jain, A. et al. A high-throughput infrastructure for density functional theory calculations. Comput. Mater. Sci. 50, 2295–2310 (2011).
Jain, A. et al. Commentary: the materials project: a materials genome approach to accelerating materials innovation. APL Mater. 1, 011002 (2013).
Saal, J. E. et al. Materials design and discovery with high-throughput density functional theory: the open quantum materials database (OQMD). JOM 65, 1501–1509 (2013).
Kirklin, S. et al. The open quantum materials database (OQMD): assessing the accuracy of DFT formation energies. NPJ Comput. Mater. 1, 15010 (2015).
Kubaschewski, O., Alcock, C. B. & Spencer, P. J. Materials Thermochemistry. 6th edn (Pergamon Press, Oxford, UK, 1993).
Chase, M. W. NIST-JANAF Thermochemical Tables. 4th edn (American Chemical Society and American Institute of Physics for the National Institute of Standards and Technology, Woodbury, NY, 1998).
Barin, I. Thermochemical Data of Pure Substances. 3rd edn (VCH, Weinheim, 1995).
Wagman, D. D. The NBS tables of chemical thermodynamic properties. J. Phys. Chem. Ref. Data 11 (Suppl. 2), 1–392 (1982).
Hautier, G. et al. Accuracy of density functional theory in predicting formation energies of ternary oxides from binary oxides and its implication on phase stability. Phys. Rev. B 85, 155208 (2012).
Sarmiento-Pérez, R., Botti, S. & Marques, M. A. L. Optimized exchange and correlation semilocal functional for the calculation of energies of formation. J. Chem. Theory Comput. 11, 3844–3850 (2015).
Chevrier, V. L. et al. Hybrid density functional calculations of redox potentials and formation energies of transition metal compounds. Phys. Rev. B 82, 075122 (2010).
Sun, J., Ruzsinszky, A. & Perdew, J. P. Strongly constrained and appropriately normed semilocal density functional. Phys. Rev. Lett. 115, 036402 (2015).
Zhang, Y. et al. Efficient first-principles prediction of solid stability: towards chemical accuracy. NPJ Comput. Mater. 4, 9 (2018).
Isaacs, E. B. & Wolverton, C. Performance of the strongly constrained and appropriately normed density functional for solid-state materials. Phys. Rev. Mater. 2, 063801 (2018).
Yan, J., Hummelshøj, J. S. & Nørskov, J. K. Formation energies of group I and II metal oxides using random phase approximation. Phys. Rev. B 87, 075207 (2013).
Yan, J. & Nørskov, J. K. Calculated formation and reaction energies of 3d transition metal oxides using a hierarchy of exchange-correlation functionals. Phys. Rev. B 88, 245204 (2013).
Jauho, T. S., Olsen, T., Bligaard, T. & Thygesen, K. S. Improved description of metal oxide stability: beyond the random phase approximation with renormalized kernels. Phys. Rev. B 92, 115140 (2015).
Pandey, M. & Jacobsen, K. W. Heats of formation of solids with error estimation: the mBEEF functional with and without fitted reference energies. Phys. Rev. B 91, 235201 (2015).
Wang, L., Maxisch, T. & Ceder, G. Oxidation energies of transition metal oxides within the GGA+U framework. Phys. Rev. B 73, 195107 (2006).
Grindy, S. et al. Approaching chemical accuracy with density functional calculations: diatomic energy corrections. Phys. Rev. B 87, 075150 (2013).
Yu, Y., Aykol, M. & Wolverton, C. Reaction thermochemistry of metal sulfides with GGA and GGA + U calculations. Phys. Rev. B 92, 195118 (2015).
Jain, A. et al. Formation enthalpies by mixing GGA and GGA + U calculations. Phys. Rev. B 84, 045115 (2011).
Aykol, M. & Wolverton, C. Local environment dependent GGA + U method for accurate thermochemistry of transition metal compounds. Phys. Rev. B 90, 115105 (2014).
Lany, S. Semiconductor thermochemistry in density functional calculations. Phys. Rev. B 78, 245207 (2008).
Stevanović, V., Lany, S., Zhang, X. & Zunger, A. Correcting density functional theory for accurate predictions of compound enthalpies of formation: fitted elemental-phase reference energies. Phys. Rev. B 85, 115104 (2012).
Blanco, M. A. et al. Thermodynamical properties of solids from microscopic theory: applications to MgF2 and Al2O3. J. Mol. Struct.: Theochem. 368, 245–255 (1996).
Blanco, M. A., Francisco, E. & Luaña, V. GIBBS: isothermal–isobaric thermodynamics of solids from energy curves using a quasi-harmonic Debye model. Comput. Phys. Commun. 158, 57–72 (2004).
Toher, C. et al. High-throughput computational screening of thermal conductivity, Debye temperature, and Grüneisen parameter using a quasiharmonic Debye model. Phys. Rev. B 90, 174107 (2014).
Toher, C. et al. Combining the AFLOW GIBBS and elastic libraries to efficiently and robustly screen thermomechanical properties of solids. Phys. Rev. Mater. 1, 015401 (2017).
Poirier, J.-P. Introduction to the Physics of the Earth’s Interior, 2nd edn (Cambridge University Press, Cambridge, UK, 2000).
Hinuma, Y. et al. Comparison of approximations in density functional theory calculations: energetics and structure of binary oxides. Phys. Rev. B 96, 094102 (2017).
Atkins, P. W. & de Paula, J. Atkins’ Physical Chemistry. 8th edn (W.H. Freeman: New York, 2006).
Henkelman, G., Arnaldsson, A. & Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 36, 354–360 (2006).
Toher, C. et al. The AFLOW fleet for materials discovery. In Handbook of Materials Modeling (eds. Andreoni, W. & Yip, S.) 1–28 (Springer International Publishing, Cham, Switzerland, 2018).
Hammer, B., Hansen, L. B. & Nørskov, J. K. Improved adsorption energetics within density-functional theory using revised Perdew–Burke–Ernzerhof functionals. Phys. Rev. B 59, 7413–7421 (1999).
Lupis, C. H. P. Chemical Thermodynamics of Materials. (North-Holland, New York, 1983).
Calderon, C. E. et al. The AFLOW standard for high-throughput materials science calculations. Comput. Mater. Sci. 108, 233–238 (2015).
Springer Materials. https://materials.springer.com. Accessed 20 Nov 2018.
Hicks, D. et al. AFLOW-SYM: platform for the complete, automatic and self-consistent symmetry analysis of crystals. Acta Crystallogr. Sect. A 74, 184–203 (2018).
Mehl, M. J. et al. The AFLOW library of crystallographic prototypes: Part 1. Comput. Mater. Sci. 136, S1–S828 (2017).
Hicks, D. et al. The AFLOW Library of Crystallographic Prototypes: Part 2. Comput. Mater. Sci. https://doi.org/10.1016/j.commatsci.2018.10.043 (2019). (in press)
Levy, O., Hart, G. L. W. & Curtarolo, S. Hafnium binary alloys from experiments and first principles. Acta Mater. 58, 2887–2897 (2010).
Pederson, M. R., Ruzsinszky, A. & Perdew, J. P. Communication: self-interaction correction with unitary invariance in density functional theory. J. Chem. Phys. 140, 121103 (2014).
Yang, Z.-h, Pederson, M. R. & Perdew, J. P. Full self-consistency in the Fermi-orbital self-interaction correction. Phys. Rev. A 95, 052505 (2017).
Kao, D.-y et al. Self-consistent self-interaction corrected density functional theory calculations for atoms using Fermi–Löwdin orbitals: optimized Fermi-orbital descriptors for Li−Kr. J. Chem. Phys. 147, 164107 (2017).
Schwalbe, S. et al. Fermi–Löwdin orbital self-interaction corrected density functional theory: ionization potentials and enthalpies of formation. J. Comput. Chem. 39, 2463–2471 (2018).
Perdew, J. P. et al. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 100, 136406 (2008).
Yang, Z.-h, Peng, H., Sun, J. & Perdew, J. P. More realistic band gaps from meta-generalized gradient approximations: only in a generalized Kohn–Sham scheme. Phys. Rev. B 93, 205205 (2016).
Legrain, F. et al. How chemical composition alone can predict vibrational free energies and entropies of solids. Chem. Mater. 29, 6220–6227 (2017).
Oses, C. et al. AFLOW-CHULL: cloud-oriented platform for autonomous phase stability analysis. J. Chem. Inf. Model. 58, 2477–2490 (2018).
Sarker, P., Harrington, T. et al. High-entropy high-hardness metal carbides discovered by entropy descriptors. Nat. Commun. 9, 4980 (2018).
Supka, A. R. et al. AFLOWπ: a minimalist approach to high-throughput ab initio calculations including the generation of tight-binding hamiltonians. Comput. Mater. Sci. 136, 76–84 (2017).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Momma, K. & Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272–1276 (2011).
Acknowledgements
We thank Ohad Levy, Frisco Rose, Eric Gossett, David Hicks, and Denise Ford for fruitful discussions. Research supported by DOD-ONR (N00014-15-1-2863, N00014-15-1-2266, N00014-17-1-2090, N00014-16-1-2326, N00014-17-1-2876). R.F. acknowledges support from the Alexander von Humboldt foundation under the Feodor Lynen research fellowship. C.O. acknowledges support from the National Science Foundation Graduate Research Fellowship under Grant no. DGF-1106401. S.C. acknowledges financial support from the Alexander von Humboldt foundation.
Author information
Authors and Affiliations
Contributions
R.F. and S.C. proposed the formation enthalpy correction. All authors—R.F., D.U., C.O., A.S., M.F., M.B.N., C.T., S.C.—discussed the results and contributed to the writing of the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Friedrich, R., Usanmaz, D., Oses, C. et al. Coordination corrected ab initio formation enthalpies. npj Comput Mater 5, 59 (2019). https://doi.org/10.1038/s41524-019-0192-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-019-0192-1
This article is cited by
-
Disordered enthalpy–entropy descriptor for high-entropy ceramics discovery
Nature (2024)
-
AFLOW for Alloys
Journal of Phase Equilibria and Diffusion (2024)
-
A flexible and scalable scheme for mixing computed formation energies from different levels of theory
npj Computational Materials (2022)
-
Theoretical prediction of high melting temperature for a Mo–Ru–Ta–W HCP multiprincipal element alloy
npj Computational Materials (2021)
-
A framework for quantifying uncertainty in DFT energy corrections
Scientific Reports (2021)
