
Abstract
Layered lithium intercalating transition metal oxides are promising cathode materials for Li-ion batteries. Here, we scrutinize the recently developed strongly constrained and appropriately normed (SCAN) density functional method to study structural, magnetic, and electrochemical properties of prototype cathode materials LiNiO2, LiCoO2, and LiMnO2 at different Li-intercalation limits. We show that SCAN outperforms earlier popular functional combinations, providing results in considerably better agreement with experiment without the use of Hubbard parameters, and dispersion corrections are found to have a small effect. In particular, SCAN fares better than Perdew–Burke–Ernzerhof (PBE) functional for the prediction of band-gaps and absolute voltages, better than PBE+U for the electronic density of states and voltage profiles, and better than both PBE and PBE+U for electron densities and in operando lattice parameters. This overall better performance of SCAN may be ascribed to improved treatment of localized states and a better description of short-range dispersion interactions.
Similar content being viewed by others
Introduction
Renewable energy has emerged as one of the most important areas of research in recent years, due to growing concerns related to pollution, sustainability, and geopolitics. Leading green energy candidates today include solar cells, fuel cells, supercapacitors, and rechargeable batteries.1,2,3,4,5,6,7,8,9,10 These technologies are in increasing demand, due to a global surge in energy consumption, widening dependence on electronic gadgets, and the desire to replace combustion engines in vehicles with more benign environmental alternatives. In these regards, Li-ion batteries (LIBs) have earned their place as one of the most promising technologies in the renewable energy field, and are currently employed in a variety of applications, ranging from cell phones and laptops to electrical vehicles.1,2,3,4,5,6,7,8,9,10
LIBs are multi-component devices and each component comes with scientific and technological challenges.6 The performance in LIBs, such as energy density and capacity retention, may be greatly enhanced by improving the cathode.3,6,11 Various types of cathode materials exist, including layered, spinel, olivine, and tavorite structures.3,9,12,13,14 In particular, intercalating layered metal oxides have surfaced as a very promising family of materials.15,16 These two-dimensional Li-intercalating materials are superior to other available cathode materials (e.g., spinel, olivine, and tavorite), as they cycle at elevated voltages, provide high capacity, and have good volumetric density.10,15,17,18,19,20 LiCoO2 (LCO) was first introduced in 1980, and was the first layered transition metal oxide to be successfully incorporated into commercial rechargeable LIBs.21 Today, LCO and other layered transition metal oxide cathodes are still widely used in portable electronic devices as positive electrodes.15 It has long been realized that mixing different transition metals (TM) in layered materials can be beneficial.22,23 For instance, in Ni–Co–Mn layered materials, Ni provides favorable capacity, Co kinetics, and Mn stability.15,16 To improve the specific capacity of cathodes, which is important for electrical vehicle applications, increasing the Ni-amount is a promising strategy.24 The current state-of-the-art for these materials stands at capacities close to 300 mAh g−1 for so-called Ni-rich materials.15,16,25 However, Ni-rich layered cathode materials suffer from serious capacity fading during electrochemical cycling due to Ni-ion migrations and oxygen release.25,26,27,28
An important tool in studying cathode materials is density functional theory (DFT).9,11,29,30,31 Using DFT, one can predict the structure, energetics, magnetism, electrochemical properties, and degradation mechanisms.30 Early work in this area employed the local density approximation (LDA),32,33,34,35 although the generalized gradient approximation (GGA)36,37 was quickly adopted as the gold-standard in this area.38 In particular, the Perdew–Burke–Ernzerhof (PBE) functional39 has become the functional of choice.38 In both LDA and GGA approaches, it is customary to include the so-called U Hubbard parameter for strongly correlated systems, which corrects for some of the electron self-interaction in DFT, by localizing the electrons.40,41 Although this is a widely used approach, it does have numerous disadvantages. A trivial disadvantage is that the correct parameter must be derived, typically on a per element basis. A subtler disadvantage is that this parameter will not be identical for different oxidation states of a given element.42 In the case of electrochemical application, this can be a problem as the oxidation states of TM change during Li-ion intercalation.14,25,43 Additionally, use of a U parameter can significantly perturb the electronic structure of materials.44 Hence, a U-free density functional approach, that better accounts for self-interaction, would be of great use. Hence, a more appropriate functional for electrochemical applications is sought.45
Recently, a new functional, dubbed strongly constrained and appropriately normed (SCAN),46,47,48,49 was suggested. This functional, belongs to the non-empirical meta-GGA family of functionals, which includes the gradient of the kinetic energy density. In general, non-empirical functionals should satisfy a number of exact constraints. SCAN is the first semilocal meta-GGA exchange-correlation functional that satisfies 17 known possible exact constraints.46 An important constraint is the exchange enhancement factor (Fx), which is defined as Fx = Ex/\(E_x^{\mathrm {LDA}}\), where Ex and \(E_x^{\mathrm {LDA}}\) are the exchange energy part for SCAN and LDA, respectively. The requirement is that Fx should be less than 1.174, and this constraint is satisfied by SCAN.50 Furthermore, this functional accurately captures intermediate range weak interactions in non-bonded systems and rare-gas atoms due to appropriate norming.46 See Section S1 in the Supporting Information for a more detailed discussion of the different functionals employed here.
Peng and Perdew recently reported excellent binding energies and structural parameters for layered chalcogenides using the SCAN functional and an additional nonlocal correlation functional.51 It has been also shown that the SCAN functional is a better choice for energetics and structural parameters of binary oxides than other functionals, such as PBE, LDA+U, and PBEsol.52 Further, the SCAN functional has been applied to conventional perovskite ferroelectrics, like BaTiO3, CdTiO3, and PbTiO3, showing improved prediction of structural, electric, and energetic properties for these materials.53,54 It was shown in a recent study that random phase approximation (RPA) and SCAN give good agreement for materials like hybrid perovskite.55 The use of SCAN also provides a good description of the instability of the demagnesiated battery material Cr2O4.56 There are also studies that suggest good performance of meta-GGA functionals like SCAN for energetics and electrochemical properties of Ni, Mn, and Ti oxides.57,58,59
Isaacs and Wolverton recently performed a benchmark study for nearly 1000 inorganic solid systems.50 These authors showed that SCAN reproduces formation energies for strongly bound systems significantly better than PBE, but performs slightly worse than PBE for intermetallic, weakly bound, compounds. The slightly worse results of SCAN for weakly bound systems were ascribed to mild over-binding due to exchange interactions. These results suggest that the performance of SCAN should be carefully benchmarked for different groups of materials and types of properties.
In the current work, we apply the SCAN functional to lithium intercalating layered transition metal oxide cathode materials to investigate the electronic, thermodynamic, and electrochemical properties of three basic and widely used LIB cathode materials, namely, LiNiO2 (LNO), LCO, and LiMnO2 (LMO). We demonstrate that the SCAN functional largely outperforms both the GGA and GGA+U methods, offering better agreement with experimental data for most studied properties. Importantly, we show that the SCAN functional obviates the need for the notorious U parameter in these basic LIB cathode materials, while dispersion corrections are not necessary.
Results and discussion
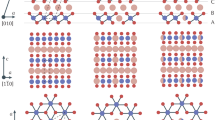
In our study, we considered an R-3m rhombohedral unit cell for LNO and LCO, and a Pmmn orthorhombic unit cell for LMO (see Fig. 1a, b, c, respectively).

Structure of a LNO and b LCO in the hexagonal R-3m space group and c LMO in the Pmmn space group
Structural parameters
The electrochemical properties of electrode materials depend significantly on the changes in structural parameters during cycling. We analyzed the changes in structural parameters of LNO, LCO, and LMO at different delithiation levels (x = 1.0, 0.5, and 0.0). From Fig. 2, we observe that the in-Li-plane a parameter changes only slightly during delithiation, whereas the orthogonal vector, c, changes considerably, in line with previous studies on layered cathode materials.14,25,28,43 Specifically, the a parameter decreases slightly on delithiation for LNO and LCO, while it increases for LMO. This effect may be attributed to differences in the structure of LMO (orthorhombic), and LNO and LCO (rhombohedral). For all studied systems, the DFT methods give similar changes for the a lattice vector on delithiation. The a parameter represents the in-layer distance between two TMs in LNO and LCO, and decreases with delithiation since the ionic radius of the TMs decreases with increasing oxidation state of the TMs. We further observe a systematic monotonous increase in the c lattice vector for LNO and LCO using the PBE and PBE+U methods. Interestingly, on including dispersion correction to PBE and PBE+U, the c vector shows the expected and characteristic dip in the fully delithiated limit.14,60,61 The c parameter represents the interlayer distance between two TM layers in LNO and LCO, and it initially increases with delithiation due to electrostatic repulsion between adjacent O-layers, while close to the fully delithiated limit there is a decrease in interlayer slab distance. Interestingly, using the SCAN functional, this correct behavior is observed both with and without dispersion correction, as this nonlocal functional captures short-range weak interactions.46,47 The described variations of a and c lattice parameters agree with recent reports.14,62,63 The changes in volume with delithiation is a combined effect of a and c parameters, but the c parameter provides the dominating effect. Therefore, the stability of the structure, and hence the capacity of a LIB cell depends significantly on the c parameter.63 A comparison of calculated (using different DFT functionals) and experimental lattice parameters for fully lithiated systems is presented in Table 1. All approaches studied here perform well against the experimental data, with SCAN and PBE-D3+U giving the best agreement for the three systems discussed here. We note that earlier reports also showed that SCAN predicts volumes of layered MgI2 and ZrCl2 materials better than PBE, and this may be ascribed to improved treatment of intermediate van der Waals interactions in SCAN.50 Finally, we note that the calculations are performed at a temperature of 0 K and do not include any thermal breathing of the lattice.

Variation in unit cell lattice parameters, a, c and volume, at different intercalation levels (x) for LixNiO2 (a-c), LixCoO2 (d-f), and LixMnO2 (g-i) using different functional combinations
Band gap
To assess the electronic conductivity, we calculated the band gap for pristine LNO, LCO, and LMO. LDA and GGA functionals, like PBE, are based on semilocal approximations to Exc, and are known to underestimate the bandgap.64 In contrast, SCAN contains some non-locality via the non-local kinetic energy density, which has been shown to slightly improve band-gaps for some materials.50,65 It is therefore of interest to investigate whether the same holds true for the current layered materials.
LNO is predicted to be a half-metal for all functional combinations employed (Table 2), and this is in agreement with earlier computational data40 and in reasonable agreement with experimental observations, which predict a very small band-gap.66 For LCO, we obtain a band-gap of 1.08 eV using PBE, while adding a U parameter increases the band gap to 2.2 eV. The reported experimental band gap for LCO ranges from 2.1 to 2.7 eV. The SCAN functional gives a band-gap of 1.74 eV, showing better agreement with the experimental data than PBE, but not quite as good as PBE+U. The computed band gaps for LMO are 0.92, 1.42, and 1.19 eV using PBE, PBE+U, and SCAN, respectively, which may be compared with the experimental band-gap of 1.64 eV.67 Overall, we find that SCAN improves the band gap relative to PBE, while PBE+U reproduces the experimental values better. As we will see in the following section, the addition of the U parameters comes at a cost of significant perturbation to the electronic structure.
Magnetic and electronic properties
To examine the electronic and magnetic properties of LNO, we considered both parallel and antiparallel configurations for the Ni ions. Our total energy calculations show that the ferromagnetic (parallel), configuration is energetically favorable by 16 meV per formula unit (using PBE) and this result is in agreement with earlier computational studies.42,66 In fully lithiated LNO, all the Ni ions are in a 3+ oxidation state and the low-spin (LS) electronic configuration for Ni3+ is t2g6(|↑↓|↑↓|↑↓|) eg1(|↑ | |). Hence, the calculated local magnetic moment of Ni in LNO is expected to be ~1 µB, and all methods give ca. 1 µB as shown in Table 3, in agreement with earlier reports.40,66 The SCAN functional gives a magnetic moment slightly closer to unity than PBE, and we ascribe this to the more accurate treatment of localized states by SCAN as mentioned by Perdew et al.48 The local magnetic moments of Ni sites in partially and fully delithiated states are different because of the presence of Ni4+, which has a t2g6(|↑↓|↑↓|↑↓|) eg0(| | |) configuration.
We further analyzed the electronic structure of pristine LNO using the different functionals (Fig. 3a–c). We observe hybridization between Ni-d and O-p states and there is a finite density of states (DOS) at the Fermi level for all methods. Inspection of the DOS in Fig. 3b reveals that both up and down t2g spin channels are completely occupied, while the up channel of eg is partially occupied, indicating that Ni3+ is in LS, as discussed above. Interestingly, using PBE and PBE+U, the t2g and eg states are similarly occupied, whereas with SCAN t2g is largely occupied and eg is unoccupied.

Density of states for LNO a–c, LCO d–f, and LMO g–i obtained with the PBE, PBE+U, and SCAN functionals. The systems were rotated to align the MO6 octahedra with the global z-axis of the unit-cell to assign the t2g and eg states
In the experimental valence electron XPS for LNO it was observed that the band near the Fermi level (at ~−1.4 eV) is predominantly composed of Ni-3d, while O-2p states were found at ~−3.8 eV.68 Here, we find that both PBE and SCAN predict dominant contribution of Ni-3d states near the Fermi level (−1 to −2 eV) for LNO, in agreement with experimental data,68 while PBE+U predicts dominant contribution of O-2p states in this range. The deleterious effect of U on the relative d- and p-band positions in pristine layered oxide materials has been noted in our previous study on Ni-rich mixed transition metal oxides.43 We underscore that the dominant contribution of Ni-3d states near the Fermi level, that is correctly predicted by SCAN, explains classical redox behavior of LiNiO2. We further note that the electronic structure predicted by SCAN also suggests a slightly higher degree of hybridization than that of the PBE method.
The calculated local magnetic moment on Co sites in the fully lithiated LCO is 0.0, because the Co3+ ions are in a low-spin state (LS) (t2g6(|↑↓|↑↓|↑↓|) eg0(| | |)), in agreement with earlier studies on LCO.66 The electronic structure (DOS) is displayed in Fig. 3d, e, f for the PBE, PBE+U, and SCAN functionals, respectively. From Fig. 3d–f, we observe strong hybridization between Co-3d and O-2p. Further, we note that with the SCAN functional, the t2g band of Co is completely occupied in both spin channels, while eg is unoccupied, reflecting the LS state of Co, as explained above. The DOS is in agreement with XPS studies of LCO, which showed similar hybridization between Co-3d and O-p states.69 Ensling et al. performed valence XPS of LCO and noted a dominant contribution of Co-3d states in the upper valence band region (~−1 to 3 eV). They also suggested that a broad band near −2.5 to 7.5 eV has prevalent O-2p character, but with some Co-3d admixtures.70 We note that the electronic structure obtained using both PBE and SCAN suggest dominant contribution of Co-3d states in the upper valence region, in agreement with experiments,70 and suggest a classical redox behavior for Co in the fully intercalated limit. However, PBE+U suggest an almost equal contribution of Co-3d and O-2p states near the Fermi level. We note that on increasing the U value, the contribution of the O-2p states near the Fermi increases. We further note that the occupied and unoccupied bands are more separated in PBE+U compared to PBE and it effectively increases the band gap of the system. However, using PBE+U the valence band was found to be pinned to the Fermi level in contrast to the results obtained with PBE, SCAN, and the experimental valence XPS.70
For LMO, the antiferromagnetic (AFM) spin configuration is energetically favorable for all functional combinations investigated. Using PBE, the difference in total energy between AFM and ferromagnetic configurations per formula unit is 0.125 meV, while using SCAN it is 0.085 meV. The local magnetic moment of Mn (~3.6 µB) implies that Mn is in a high spin state t2g3(|↑ |↑ |↑ |) eg0(|↑ | |). In Fig. 3g–i, we display the DOS for LMO in the AFM configuration, and we observe strong hybridization between Mn-d and O-p near the Fermi level using SCAN, in agreement with the experimental XPS data.71 However, PBE+U suggests a significantly greater contribution of O-2p states in the upper valence band region.
Overall, both PBE and SCAN improve the electronic DOS for these layered materials over PBE+U, while SCAN slightly improves the magnetic structure over PBE.46,48
We also assessed the quality of the electron density of the different methods (Fig. 4). To this end we compared the electron density of PBE, PBE+U, and SCAN to the electron density from the hybrid density functional PBE0,72 which has recently been shown to give excellent electron density relative to high-level ab-initio methods.73,74 Visual inspection of the density difference figures clearly shows that SCAN gives better agreement with PBE0 than the other functionals. The quantitative difference in density (DD) was estimated according to:

Density difference isosurfaces (blue/yellow colors) between the three target DFT methods PBE, PBE+U, and SCAN, and the density of the hybrid functional PBE0. The isodensity value was set to 0.003352 a.u. The quantitative difference is shown in orange boxes (a.u.)
Here, ρi is the density at grid point i, XC is one of the functionals PBE, PBE+U, or SCAN, while N is the number of grid points. The quantitative analysis (orange boxes in Fig. 4) confirms the impression from the visual inspection, that the electron density from SCAN is the most accurate among the functionals tested.
Formation energy
To understand the formation of solid solutions of lithiated and delithiated LNO, LCO, and LMO, we calculated the formation energy per formula unit:75
Here, x is the fractional amount of Li present in the system, E(LixMO2) is the energy of the partially delithiated material, while E(LiMO2) and E(MO2) represent the energies of the pristine and fully delithiated structures, respectively. The calculated formation energy for different intercalation limits using different functionals is shown in Fig. 5a, b, c for LNO, LCO, and LMO respectively. We observe that the formation energy of partially lithiated states is negative for all materials, and hence a solid solution is predicted. This preference is minute using SCAN, while more significant using PBE. In an earlier study using PBE, the FE for LCO is calculated to be ca. 0.22 eV/f.u.75 Here, we observe that the FE for partially lithiated (x = 0.5) LCO using PBE and SCAN are calculated to be 0.21 and 0.16 eV/f.u., respectively. Similarly, our calculated value of 103 meV using PBE for partially lithiated (x = 0.5) LNO is in good agreement with the value reported by Ceder and co-workers using the same functional (~130 meV/f.u.).29 Using SCAN this value is somewhat lower. We note that all the applied functionals predict a solid solution behavior in agreement with experiments.60,76,77

The formation energy for a LixNiO2, b LixCoO2, c LixMnO2 at different intercalation limits (x) using different functional combinations
Quantitative comparison with experiment is not possible for the formation energies (Eq. (2)) of LixMO2, as such values are not available. To better quantify the error in each method, we also compute the formation energy for the individual pristine and delithiated states, which may be compared with available experiments (Table S1). Based on the computed formation energies we find that overall PBE and SCAN perform similarly, while PBE+U does slightly better. In all cases, SCAN overbinds and we may ascribe this to the exchange energy, as suggested before.50
Intercalation voltage
We calculated the intercalation voltage using the following formula:14,33
where E(Lix+dxMO2) and E(LixMO2) represent the total energy per formula unit of the system before and after lithium deintercalation. E(Libcc) is the energy per formula unit of bulk Li. The calculated intercalation potentials are displayed in Fig. 6. For LNO, the experimental voltage profile for the fully lithiated to fully delithiated states varies from 3 to ~4.3 V in LNO.29,78 The calculated intercalation profile for LNO ranges from 2.8 to 3.2 V and 3.6 to 3.8 V using PBE and SCAN, respectively, while using PBE+U results in a significant overestimation. The observed intercalation profile for LCO ranges from 3.0 to 3.8 V and 4.1 to 4.7 V using PBE and SCAN, respectively, whereas the experimental profile varies from 3.6 to 4.8 V.79 The observed intercalation profile for LMO ranges from 2.4 to 3.2 V and 2.9 to 3.4 V using PBE and SCAN, respectively, while PBE+U is within the experimental range. Experimentally, the voltage ranges from 2.0 to 4.6 V for LMO.16,80 In conclusion, the intercalation voltage is underestimated in case of PBE, while using the SCAN functional the results are in better agreement with experiments. The improved performance of SCAN may be ascribed to the improved ground state geometries and electronic structure using this functional over the PBE and PBE+U, due to a better description of localized states and dispersion interactions.56,81

Intercalation potentials (V) for a LixNiO2, b LixCoO2, and c LixMnO2 at different intercalation limits (x)
We studied the structural details, band-gap, magnetic and electronic structure, formation energy, and intercalation profiles for the prototype layered cathode materials LNO, LCO, and LMO using standard GGA density functional approaches and the recently developed meta-GGA SCAN density functional. The computed data were compared with available experimental data, and we find that the SCAN functional performs similarly or better than PBE and PBE+U for most properties studied. We ascribe the overall superior performance of the semilocal SCAN density functional to its design principle, whereby the functional, which belongs to the meta-GGA family, satisfies all 17 possible exact constraints. Practically, this results in two key improvements over PBE and PBE+U that are crucial in layered cathode materials. First, SCAN’s improved treatment of localized states obviates the need for the notorious U parameter, which should not be used when comparing layered materials in different lithiation states. Second, SCAN provides a better description of short-range van der Waals interactions, which are key to describing the intercalation process in layered materials, hence obviating the need for dispersion corrections. Specifically, SCAN performs better than PBE for the prediction of band-gaps and absolute voltages, better than PBE+U for electronic DOS and voltage profiles, and better than both PBE and PBE+U for electron densities and in operando (i.e., during electrochemical cycling) lattice parameters. In addition, the inclusion of dispersion corrections to SCAN does not improve performance significantly, suggesting that the functional includes some dispersion interactions. In conclusion, SCAN is a versatile functional that provides good all-round performance for all relevant electrochemical properties benchmarked in this study for prototype layered cathode materials.
Methodology
All DFT electronic structure calculations were performed using the plane wave based Vienna ab-initio Simulation Package (VASP).82,83 All calculations employed projector augmented wave (PAW)84 potentials for all elements. An energy cut-off of 520 eV was imposed for the plane wave basis. In LNO, LCO, and LMO, the supercells were constructed from the primitive cell by doubling the system along the a direction (see Fig. 1). A Gamma-centered 4 × 8 × 2 k-mesh grid was used for the LCO and LNO supercells and a 6 × 4 × 3 k-mesh grid was used for LMO. Geometry and cell parameter optimizations were performed using the conjugate-gradient method and the convergence criteria were set to 0.01 eV/Å. We employed the PBE functional39 with and without onsite Coulomb interaction, U. The effective U parameters were 5.96, 3.0, and 5.10 eV for Ni, Co, and Mn, respectively,14 and Dudarev’s method85 was used in GGA+U. Further, we employed the recently developed SCAN46,47 exchange-correlation functional. Dispersion corrections were included using Grimme’s DFT-D3 method.86 Overall, we considered the following functional combinations: PBE, PBE+U, PBE+U+D3, SCAN, and SCAN+D3. We note that the computational cost of adding U or dispersion correction to DFT calculations is negligible, while the cost of SCAN can in practice be slightly greater than for PBE, but significantly lower than hybrid functionals (Table S2).
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Tarascon, J. M. & Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 414, 359–367 (2001).
Winter, M. & Brodd, R. J. What are batteries, fuel cells, and supercapacitors? Chem. Rev. 104, 4245–4269 (2004).
Whittingham, M. S. Lithium batteries and cathode materials. Chem. Rev. 104, 4271–4302 (2004).
Bruce, P. G. Energy storage beyond the horizon: rechargeable lithium batteries. Solid State Ion. 179, 752–760 (2008).
Goodenough, J. B. & Kim, Y. Challenges for rechargeable Li batteries. Chem. Mater. 22, 587–603 (2010).
Etacheri, V., Marom, R., Elazari, R., Salitra, G. & Aurbach, D. Challenges in the development of advanced Li-ion batteries: a review. Energy Environ. Sci. 4, 3243–3262 (2011).
Thackeray, M. M., Wolverton, C. & Isaacs, E. D. Electrical energy storage for transportation-approaching the limits of, and going beyond, lithium-ion batteries. Energy Environ. Sci. 5, 7854–7863 (2012).
Meng, Y. S. & Arroyo-de Dompablo, M. E. Recent advances in first principles computational research of cathode materials for lithium-ion batteries. Acc. Chem. Res. 46, 1171–1180 (2013).
Islam, M. S. & Fisher, C. A. Lithium and sodium battery cathode materials: computational insights into voltage, diffusion and nanostructural properties. Chem. Soc. Rev. 43, 185–204 (2014).
Nitta, N., Wu, F., Lee, J. T. & Yushin, G. Li-ion battery materials: present and future. Mater. Today 18, 252–264 (2015).
Ceder, G. Opportunities and challenges for first-principles materials design and applications to Li battery materials. MRS Bull. 35, 693–701 (2010).
Goodenough, J. B. & Park, K.-S. The Li-ion rechargeable battery: a perspective. J. Am. Chem. Soc. 135, 1167–1176 (2013).
Singh, V. et al. Magnetism in olivine-type LiCo(1−x)Fe(x)PO4 cathode materials: bridging theory and experiment. Phys. Chem. Chem. Phys. 17, 31202–31215 (2015).
Dixit, M. et al. Thermodynamic and kinetic studies of LiNi0.5Co0.2Mn0.3O2 as a positive electrode material for Li-ion batteries using first principles. Phys. Chem. Chem. Phys. 18, 6799–6812 (2016).
Liu, W. et al. Nickel-rich layered lithium transition-metal oxide for high-energy lithium-ion batteries. Angew. Chem. 54, 4440–4457 (2015).
Schipper, F. et al. Review—recent advances and remaining challenges for lithium ion battery cathodes I. Nickel-rich, LiNixCoyMnzO2. J. Electrochem. Soc. 164, A6220–A6228 (2017).
Delmas, C. et al. An overview of the Li(Ni,M)O2 systems: syntheses, structures and properties. Electrochim. Acta 45, 243–253 (1999).
Ohzuku, T. & Brodd, R. J. An overview of positive-electrode materials for advanced lithium-ion batteries. J. Power Sources 174, 449–456 (2007).
He, P., Yu, H., Li, D. & Zhou, H. Layered lithium transition metal oxide cathodes towards high energy lithium-ion batteries. J. Mater. Chem. 22, 3680–3695 (2012).
Myung, S.-T. et al. Nickel-rich layered cathode materials for automotive lithium-ion batteries: achievements and perspectives. ACS Energy Lett. 2, 196–223 (2017).
Mizushima, K., Jones, P. C., Wiseman, P. J. & Goodenough, J. B. LixCoO2 (0 < x ≤ 1): a new cathode material for batteries of high energy density. Mater. Res. Bull. 15, 783–789 (1980).
Ohzuku, T. & Makimura, Y. Layered lithium insertion material of LiCo1/3Ni1/3Mn1/3O2 for lithium-ion batteries. Chem. Lett. 30, 642–643 (2001).
Belharouak, I., Sun, Y. K., Liu, J. & Amine, K. Li(Ni1/3Co1/3Mn1/3)O2 as a suitable cathode for high power applications. J. Power Sources 123, 247–252 (2003).
Noh, H.-J., Youn, S., Yoon, C. S. & Sun, Y.-K. Comparison of the structural and electrochemical properties of layered Li[NixCoyMnz]O2 (x = 1/3, 0.5, 0.6, 0.7, 0.8 and 0.85) cathode material for lithium-ion batteries. J. Power Sources 233, 121–130 (2013).
Dixit, M., Markovsky, B., Schipper, F., Aurbach, D. & Major, D. T. Origin of structural degradation during cycling and low thermal stability of Ni-rich layered transition metal-based electrode materials. J. Phys. Chem. C 121, 22628–22636 (2017).
Hwang, S. et al. Investigating local degradation and thermal stability of charged nickel-based cathode materials through real-time electron microscopy. ACS Appl. Mater. Interfaces 6, 15140–15147 (2014).
Yang, J. & Xia, Y. Suppressing the phase transition of the layered Ni-rich oxide cathode during high-voltage cycling by introducing low-content Li2MnO3. ACS Appl. Mater. Interfaces 8, 1297–1308 (2016).
Schipper, F. et al. Stabilizing nickel-rich layered cathode materials by a high-charge cation doping strategy: zirconium-doped LiNi0.6Co0.2Mn0.2O2. J. Mater. Chem. A 4, 16073–16084 (2016).
Arroyo y de Dompablo, M. E., Van der Ven, A. & Ceder, G. First-principles calculations of lithium ordering and phase stability on LixNiO2. Phys. Rev. B 66, 064112 (2002).
Seo, D.-H. et al. The structural and chemical origin of the oxygen redox activity in layered and cation-disordered Li-excess cathode materials. Nat. Chem. 8, 692–697 (2016).
Ceder, G., Hautier, G., Jain, A. & Ong, S. P. Recharging lithium battery research with first-principles methods. MRS Bull. 36, 185–191 (2011).
Kohn, W. & Sham, L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 140(4A), A1133–A1138 (1965).
Aydinol, M. K., Kohan, A. F., Ceder, G., Cho, K. & Joannopoulos, J. Ab initio study of lithium intercalation in metal oxides and metal dichalcogenides. Phys. Rev. B 56, 1354–1365 (1997).
Aydinol, M. K., Kohan, A. F. & Ceder, G. Ab initio calculation of the intercalation voltage of lithium-transition-metal oxide electrodes for rechargeable batteries. J. Power Sources 68, 664–668 (1997).
Aydinol, M. K. & Ceder, G. First-principles prediction of insertion potentials in Li–Mn oxides for secondary Li batteries. J. Electrochem. Soc. 144, 3832–3835 (1997).
Langreth, D. C. & Mehl, M. J. Beyond the local-density approximation in calculations of ground-state electronic properties. Phys. Rev. B 28, 1809–1834 (1983).
Deiss, E., Wokaun, A., Barras, J. L., Daul, C. & Dufek, P. Average voltage, energy density, and specific energy of lithium-ion batteries. J. Electrochem. Soc. 144, 3877–3881 (1997).
Meng, Y. S. & Arroyo-de Dompablo, M. E. First principles computational materials design for energy storage materials in lithium ion batteries. Energy Environ. Sci. 2, 589–609 (2009).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Anisimov, V. I., Zaanen, J. & Andersen, O. K. Band theory and Mott insulators: Hubbard U instead of Stoner I. Phys. Rev. B 44, 943–954 (1991).
Vladimir, I. A., Aryasetiawan, F. & Lichtenstein, A. I. First-principles calculations of the electronic structure and spectra of strongly correlated systems: the LDA+U method. J. Phys. Condens. Matter 9, 767–808 (1997).
Zhou, F., Cococcioni, M., Marianetti, C. A., Morgan, D. & Ceder, G. First-principles prediction of redox potentials in transition-metal compounds with LDA+U. Phys. Rev. B 70, 235121 (2004).
Dixit, M., Markovsky, B., Aurbach, D. & Major, D. T. Unraveling the effects of Al doping on the electrochemical properties of LiNi0.5Co0.2Mn0.3O2 using first principles. J. Electrochem. Soc. 164, A6359–A6365 (2017).
Singh, V., Kosa, M., Majhi, K. & Major, D. T. Putting DFT to the test: a first principles study of electronic, magnetic and optical properties of Co3O4. J. Chem. Theory Comput. 11, 64–72 (2015).
Mattsson, A. E. In pursuit of the “Divine” functional. Science 298, 759–760 (2002).
Sun, J. W., Ruzsinszky, A. & Perdew, J. P. Strongly constrained and appropriately normed semilocal density functional. Phys. Rev. Lett. 115, 036402 (2015).
Sun, J. et al. Accurate first-principles structures and energies of diversely bonded systems from an efficient density functional. Nat. Chem. 8, 831–836 (2016).
Perdew, J. P., Sun, J., Martin, R. M. & Delley, B. Semilocal density functionals and constraint satisfaction. Int. J. Quantum Chem. 116, 847–851 (2016).
Zhang, Y. et al. Efficient first-principles prediction of solid stability: towards chemical accuracy. npj Comput. Mater. 4, 9 (2018).
Isaacs, E. B. & Wolverton, C. Performance of the strongly constrained and appropriately normed density functional for solid-state materials. Phys. Rev. Mater. 2, 063801 (2018).
Peng, H. & Perdew, J. P. Rehabilitation of the Perdew–Burke–Ernzerhof generalized gradient approximation for layered materials. Phys. Rev. B 95, 081105 (2017).
Hinuma, Y., Hayashi, H., Kumagai, Y., Tanaka, I. & Oba, F. Comparison of approximations in density functional theory calculations: energetics and structure of binary oxides. Phys. Rev. B 96, 094102 (2017).
Zhang, Y., Sun, J., Perdew, J. P. & Wu, X. Comparative first-principles studies of prototypical ferroelectric materials by LDA, GGA, and SCAN meta-GGA. Phys. Rev. B 96, 035143 (2017).
Kennedy, B. J. et al. Low-temperature structure and the ferroelectric phase transitions in the CdTiO3 perovskite. Phys. Rev. B 96, 214105 (2017).
Bokdam, M., Lahnsteiner, J., Ramberger, B., Schafer, T. & Kresse, G. Assessing density functionals using many body theory for hybrid perovskites. Phys. Rev. Lett. 119, 145501 (2017).
Chen, T., Sai Gautam, G., Huang, W. & Ceder, G. First-principles study of the voltage profile and mobility of Mg intercalation in a chromium oxide spinel. Chem. Mater. 30, 153–162 (2018).
Huang, L. F. & Rondinelli, J. M. Electrochemical phase diagrams for Ti oxides from density functional calculations. Phys. Rev. B 92, 245126 (2015).
Kitchaev, D. A. et al. Energetics of MnO2 polymorphs in density functional theory. Phys. Rev. B 93, 045132 (2016).
Huang, L. F. & Rondinelli, J. M. Electrochemical phase diagrams of Ni from ab initio simulations: role of exchange interactions on accuracy. J. Phys. Condens. Matter 29, 475501 (2017).
Amatucci, G. G., Tarascon, J. M. & Klein, L. C. CoO2, the end member of the LixCoO2 solid solution. J. Electrochem. Soc. 143, 1114–1123 (1996).
Koyama, Y., Tanaka, I., Adachi, H., Makimura, Y. & Ohzuku, T. Crystal and electronic structures of superstructural Li1−x[Co1/3Ni1/3Mn1/3]O2 (0 ≤ x ≤ 1). J. Power Sources 119-121, 644–648 (2003).
Aykol, M., Kim, S. & Wolverton, C. van der Waals interactions in layered lithium cobalt oxides. J. Phys. Chem. C 119, 19053–19058 (2015).
de Biasi, L. et al. Between Scylla and Charybdis: balancing among structural stability and energy density of layered NCM cathode materials for advanced lithium-ion batteries. J. Phys. Chem. C 121, 26163–26171 (2017).
Perdew, J. P. Density functional theory and the band gap problem. Int. J. Quantum Chem. 28(S19), 497–523 (1985).
Yang, Z.-h, Peng, H., Sun, J. & Perdew, J. P. More realistic band gaps from meta-generalized gradient approximations: only in a generalized Kohn–Sham scheme. Phys. Rev. B 93, 205205 (2016).
Laubach, S. et al. Changes in the crystal and electronic structure of LiCoO2 and LiNiO2 upon Li intercalation and de-intercalation. Phys. Chem. Chem. Phys. 11, 3278–3289 (2009).
Kong, F. et al. Ab initio study of doping effects on LiMnO2 and Li2MnO3 cathode materials for Li-ion batteries. J. Mater. Chem. A 3, 8489–8500 (2015).
Galakhov, V. R. et al. Electronic structure of LiNiO2, LiFeO2 and LiCrO2: X-ray photoelectron and X-ray emission study. Solid State Commun. 95, 347–351 (1995).
van Elp, J. et al. Electronic structure of CoO, Li-doped CoO, and LiCoO2. Phys. Rev. B 44, 6090–6103 (1991).
Ensling, D., Thissen, A., Laubach, S., Schmidt, P. C. & Jaegermann, W. Electronic structure of LiCoO2 thin films: a combined photoemission spectroscopy and density functional theory study. Phys. Rev. B 82, 195431 (2010).
Galakhov, V. R. et al. Electronic structure of LiMnO: X-ray emission and photoelectron spectra and band structure calculations. Eur. Phys. J. B 14, 281–286 (2000).
Adamo, C. & Barone, V. Toward reliable density functional methods without adjustable parameters. J. Chem. Phys. 110, 6158–6170 (1999).
Medvedev, M. G., Bushmarinov, I. S., Sun, J., Perdew, J. P. & Lyssenko, K. A. Density functional theory is straying from the path toward the exact functional. Science 355, 49–52 (2017).
Brorsen, K. R., Yang, Y., Pak, M. V. & Hammes-Schiffer, S. Is the accuracy of density functional theory for atomization energies and densities in bonding regions correlated? J. Phys. Chem. Lett. 8, 2076–2081 (2017).
Van der Ven, A., Aydinol, M. K., Ceder, G., Kresse, G. & Hafner, J. First-principles investigation of phase stability in LixCoO2. Phys. Rev. B 58, 2975–2987 (1998).
Mizutani, A., Sugiura, K., Ohta, H. & Koumoto, K. Epitaxial film growth of LixCoO2 (0.6 ≤ x ≤ 0.9) via topotactic ion exchange of Na0.8CoO2. Cryst. Growth Des. 8, 755–758 (2008).
Moshtev, R. V., Zlatilova, P., Manev, V. & Sato, A. The LiNiO2 solid solution as a cathode material for rechargeable lithium batteries. J. Power Sources 54, 329–333 (1995).
Ohzuku, T., Ueda, A. & Nagayama, M. Electrochemistry and structural chemistry of LiNiO2 (R3̅m) for 4 Volt secondary lithium cells. J. Electrochem. Soc. 140, 1862–1870 (1993).
Julien, C., Mauger, A., Zaghib, K. & Groult, H. Comparative Issues of cathode materials for Li-ion batteries. Inorganics 2, 132–154 (2014).
Bruce, P. G., Armstrong, A. R. & Gitzendanner, R. L. New intercalation compounds for lithium batteries: layered LiMnO2. J. Mater. Chem. 9, 193–198 (1999).
Shi, Y. et al. Understanding the electrochemical properties of naphthalene diimide: implication for stable and high-rate lithium-ion battery electrodes. Chem. Mater. 30, 3508–3517 (2018).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+U study. Phys. Rev. B 57, 1505 (1998).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Croguennec, L., Deniard, P., Brec, R. & Lecerf, A. Nature of the stacking faults in orthorhombic LiMnO2. J. Mater. Chem. 7, 511–516 (1997).
Acknowledgements
This work was partially supported by the Israel Science Foundation (ISF) in the framework of the INREP project.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chakraborty, A., Dixit, M., Aurbach, D. et al. Predicting accurate cathode properties of layered oxide materials using the SCAN meta-GGA density functional. npj Comput Mater 4, 60 (2018). https://doi.org/10.1038/s41524-018-0117-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-018-0117-4
This article is cited by
-
Combined study of phase transitions in the P2-type NaXNi1/3Mn2/3O2 cathode material: experimental, ab-initio and multiphase-field results
npj Computational Materials (2024)
-
Realizing unipolar and bipolar intrinsic skyrmions in MXenes from high-fidelity first-principles calculations
npj Computational Materials (2023)
-
Calculating electron-phonon coupling matrix: Theory introduction, code development and preliminary application
Science China Technological Sciences (2023)
-
Effect of exchange-correlation functionals on the estimation of migration barriers in battery materials
npj Computational Materials (2022)
-
Stability and electronic properties of layered NaMnO2 using the SCAN(+U)
Journal of the Korean Physical Society (2022)
