
Abstract
Following the first characterization of circulating tumor DNA (ctDNA) in the 1990s, recent advances led to its introduction in the clinics. At present, the European Society Of Medical Oncology (ESMO) recommendations endorse ctDNA testing in routine clinical practice for tumor genotyping to direct molecularly targeted therapies in patients with metastatic cancer. In studies on metastatic breast cancer, ctDNA has been utilized for treatment tailoring, tracking mechanisms of drug resistance, and for predicting disease response before imaging. We review the available evidence regarding ctDNA applications in metastatic breast cancer.
Similar content being viewed by others

Introduction
Tumor characterization has historically been based on tissue analysis, most often limited to a single biopsy of either the primary tumor or a metastasis. Recently, the discovery of nucleic acids of tumor origin in the bloodstream provided a different means of describing tumors’ molecular landscape. The term “liquid biopsy” refers to the analysis of neoplastic material isolated in blood or other fluids, shed by tumor cells residing in different body niches: the possible analytes include nucleic acids, circulating tumor cells, extracellular vesicles, tumor-educated platelets and proteins and metabolites1,2.
The presence of cell-free DNA (cfDNA) in plasma from healthy individuals, mainly shed from senescent haematopoietic cells, was first reported in 19483. Only in the 1990s circulating tumor DNA (ctDNA) was characterized as an admixture of DNA released from cancer cells, representing the cfDNA fraction of tumor origin. At present, ctDNA testing is exploited in multiple research and clinical settings in oncology: as a tool for treatment tailoring and prognostication in the metastatic setting, detection of minimal residual disease after curative-intent therapy, and for cancer screening and diagnosis1. However, no single ctDNA assay appears suitable for all these purposes.
According to recent recommendations by the European Society of Medical Oncology (ESMO), there is sufficient evidence to support the routine use of ctDNA assays in clinical practice for tumor genotyping to direct molecularly targeted therapies in patients with metastatic cancer4. In fact, blood-based tumor genotyping may be more informative than tissue-based approaches in identifying molecular targets for precision medicine strategies, by reflecting variants from different disease sites, and might turn especially useful when rapid results are needed, by circumventing the delays and risks of realizing a tissue biopsy. Also, liquid biopsy can be easily performed serially, allowing to appraise temporal tumor heterogeneity and the emergence of resistance mechanisms. Nevertheless, tissue-based testing remains the gold-standard, also considering the limits of ctDNA assays in detecting genomic abnormalities other than single nucleotide variations (SNVs), such as gene fusions and copy number alterations. Moreover, a low fraction of cfDNA of tumor origin may lead to false-negative results, i.e. a variant present in the tumor cannot be detected in peripheral blood, while false-positive results may arise due to plasma DNA from apoptotic haematopoietic cells bearing stochastic somatic alterations, a phenomenon known as clonal haematopoiesis of indeterminate potential (CHIP)4,5. Thus, while the evidence supporting ctDNA assays to direct targeted treatments is now strong, their limitations should be taken into account, and a non-informative ctDNA test result should prompt reflex tissue-based testing4.
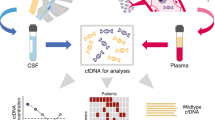
In this article, we will review available evidence supporting the use of ctDNA assays to direct clinical decisions in metastatic breast cancer (mBC) (Fig. 1).

The figure summarizes different clinical situations in which ctDNA is currently being exploited or might be exploited in the near future for the treatment of metastatic breast cancer: a treatment selection; b treatment switch; c detection of emerging resistance mechanisms; d prediction of complex phenotypes; e integration of ctDNA dynamics with imaging response criteria.
Tumor genotyping for treatment selection
Molecular screening programs in mBC
Diverse studies across cancers explored the concept of sequencing tumor genomes to identify potential therapeutic targets6. The plasmaMATCH trial was a large prospective study evaluating the clinical validity and utility of ctDNA assessment in directing molecularly targeted treatments for mBC patients7. In this phase 2 trial, patients with mutations of ESR1, ERBB2, AKT1 or PTEN identified on ctDNA were offered treatment with increased-dose fulvestrant, neratinib +/− fulvestrant, capivasertib + fulvestrant or capivasertib monotherapy, respectively, plus a fifth cohort enrolling patients with triple-negative mBC treated with olaparib + the ATR inhibitor AZD67387. The primary endpoint was the overall response rate (ORR) in each cohort. The study not only showed that ctDNA testing is highly accurate and sensitive compared to tissue-based mutation testing, but also confirmed a relevant clinical activity of treatment strategies targeting rare driver mutations in mBC7.
Consistent results came from the SAFIR02-BREAST trial, suggesting that a precision medicine approach matching drugs to genomic alterations classified as level I or II according to the ESMO Scale of Actionability of Molecular Targets (ESCAT) improves outcomes for mBC patients. However, most of the enrolled patients were treated based on tissue genotyping, and ctDNA was an option in case of not feasible tissue biopsy8.
Recently, in a large trial investigating the clinical utility of ctDNA profiling for directing targeted treatments in advanced cancers, mBC was one of the five most represented neoplasms, with 171 mBC patients out of 1772 enrolled overall9. The authors demonstrated the feasibility of treatment matching based on ctDNA genotyping, with about 33% of screened patients receiving a matched targeted agent based on molecular tumor board’s discussions and ESCAT guidance9.
ctDNA validated targets for treatment selection
Large trials of targeted therapies for mBC have demonstrated the clinical utility of ctDNA for detecting specific tumor aberrations and selecting treatments, leading to integration of certain ctDNA assays into standard clinical practice.
Mutations of PIK3CA represent the second most common single molecular alteration isolated in mBC following TP53 SNVs, and are identified in 28%-46% of HR+/HER2- mBC cases10,11. The SOLAR-1 trial demonstrated that the combination of the α-selective PI3K inhibitor alpelisib with fulvestrant significantly improves progression-free survival (PFS) over placebo + fulvestrant in HR+/HER2- mBC progressed to first-line endocrine therapy (ET) bearing PIK3CA mutations. No PFS benefit was observed in the PIK3CA-wild type group, as well as no statistically significant impact on overall survival (OS) in either group at a median follow-up of 42.4 months11,12. Patients were enrolled in the PIK3CA-mutated cohort based on the identification on tumor tissue of one of 11 PIK3CA mutations by the Therascreen test11. However, a subsequent subgroup analysis demonstrated a relevant OS improvement for patients with PIK3CA-mutant ctDNA, irrespective of their PIK3CA testing result on tissue at study screening11. In a large study of comprehensive genome profiling (CGP) on mBC tumor tissue and liquid biopsy samples from the Flatiron Health-Foundation Medicine clinico-genomic database, the agreement between FoundationOne CDx on tissue and FoundationOne Liquid CDx on liquid biopsy for PIK3CA mutation detection was 77%, increasing to 95% for samples with a cfDNA tumor fraction ≥2%13. Moreover, the study showed that 20% of patients with a PIK3CA mutation identified by the two CGP panels, testing the whole PIK3CA exome either in tumor tissue or ctDNA, had different mutations from the 11 identified by Therascreen. This confirms the previous report by Martìnez-Sàez et al., who suggested that Therascreen misses up to 30% of PIK3CA-mutated patients compared to whole-exome sequencing assays14. Also, in a phase 2 study of alpelisib monotherapy for HER2- mBC, the addition of ctDNA testing to tissue genotyping increased the number of different PIK3CA aberrations detected from 29 to 45 variants13,15, highlighting the utility of ctDNA evaluation in improving PIK3CA alterations detection, expanding access to alpelisib. Accordingly, the 2022 update to the American Society of Clinical Oncology (ASCO) guidelines on biomarkers in mBC recommends that patients potentially candidate to alpelisib + ET are tested for PIK3CA mutations via ctDNA sequencing, with reflex testing on tumor tissue in case of negative ctDNA results16.
The ESR1 mutation has long been known as an acquired resistance mechanism to endocrine treatment with aromatase inhibitors (AIs), by inducing constitutive activation of the estrogen receptor (ER). Virtually absent in untreated mBC, its incidence averages 20-40% in mBCs previously exposed to AIs17. Exploratory analyses from the SoFEA and EFECT studies, evaluating the effectiveness of exemestane versus fulvestrant following progression to nonsteroidal AIs in HR+/HER2- mBC, suggested the clinical utility of ESR1 mutation detection in ctDNA to select patients more likely to benefit from fulvestrant18,19,20,21. The phase 3 EMERALD trial demonstrated a PFS benefit from treatment with the oral selective estrogen receptor degrader (SERD) elacestrant compared to fulvestrant for patients with ESR1 mutant mBC progressed to at least one line of ET including a CDK4/6 inhibitor22. Based on these results, in January 2023 the US Food and Drugs Administration (FDA) approved elacestrant, together with the Guardant360 CDx companion diagnostic for ESR1 mutation detection in ctDNA. More recently, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) issued a positive opinion for elacestrant in the same indication. Since June 2023, ASCO recommends ESR1 mutation testing in blood or tumor tissue obtained at the time of disease progression to ET23.
Another potential use of ctDNA is the detection of BRCA1 and 2 somatic mutations4. The PARP inhibitors olaparib and talazoparib have been approved for the treatment of HER2-negative mBC in patients bearing a germline BRCA1/2 mutation, based on results of OlympiAD and EMBRACA, respectively24,25. Even though liquid biopsy has currently no role in selecting mBC patients who could benefit from this treatment, the detection of somatic BRCA1/2 mutations in ctDNA might have clinical relevance26. In fact, there are reports of PARP inhibitors effectiveness in mBC with somatic BRCA1/2 mutations identified in ctDNA27, and phase 2 studies are currently evaluating their possible role as predictive biomarker in this setting (NCT03344965 and NCT03990896). In addition, ctDNA monitoring during treatment with PARP inhibitors can reveal the emergence of resistance via BRCA2 reversion28,29. Moreover, given that routine germline BRCA1/2 testing in all mBC patients is not yet implemented in several real-world settings, the detection of a somatic BRCA1/2 mutation in ctDNA could prompt clinicians to search for germline variants30.
HER2 amplification can also be estimated in ctDNA, even though with suboptimal detection by available assays, but needs to be confirmed with a validated assay such as fluorescent in situ hybridisation in tissue4,31.
Finally, ctDNA has clinical utility in selecting patients for tumor-type agnostic drugs. Based on data from KEYNOTE-158, pembrolizumab obtained FDA and EMA approval for the treatment of solid neoplasms characterized by microsatellite instability-high (MSI-H) status or high tumor mutational burden (TMB-H) defined as ≥10 mutations/megabase, including mBC32,33. Moreover, the TAPUR study demonstrated clinically relevant activity of pembrolizumab in heavily pre-treated mBC patients with TMB-H, evaluated either in tumor tissue or ctDNA34. While the detection of MSI status in ctDNA has been adequately validated and this can be used for treatment selection, there is currently limited evidence that blood TMB (bTMB) alone can be used for treatment with pembrolizumab4,35. Finally, mBC patients might receive additional tumor-agnostic treatments, such as entrectinib and larotrectinib in the presence of NTRK gene fusions, or selpercatinib and pralsetinib in case of RET fusions; however, these agents target structural variants, with less studied diagnostic yields in ctDNA4,36,37,38,39,40.
Predicting patient benefit from CDK4/6 inhibition
The association of CDK4/6 inhibitors and ET is the preferred first-line treatment for HR+/HER2- mBC, where this approach has demonstrated to improve both PFS and OS41,42,43,44. Translational analyses from registration studies of CDK4/6 inhibitors evaluated several biomarkers, including ctDNA, for their ability to predict treatment response; however, no validated biomarker has yet been identified to improve patients selection, beyond ER expression in the tumor45,46.
An exploratory analysis from PALOMA-3, evaluating fulvestrant +/− palbociclib for HR+/HER2- mBC, demonstrated a worse PFS and OS for patients with either a baseline ctDNA fraction >10%, a TP53 mutation, or a FGFR1 amplification, in both treatment arms47,48. In contrast, patients with a low circulating tumor fraction (≤10%) seemed to have a greater PFS and OS gain from palbociclib, irrespective of the mutational status of ESR1, PIK3CA or TP53, even though patients harboring these mutations had a worse outcome48. Similarly, the presence of ctDNA PIK3CA mutation or ESR1 mutation in baseline plasma samples was not related to the benefit of adding abemaciclib to fulvestrant in patients progressing on previous ET in the MONARCH-2 study49. Also in the MONARCH-3 study, evaluating the efficacy of adding abemaciclib to AIs in post-menopausal HR+/HER2- mBC patients, a shorter median PFS was observed for patients harboring one or more of 70 cancer-related gene alterations in baseline ctDNA, regardless of the treatment arm50.
Conversely, a recent pooled analysis of MONALEESA-2, −3 and −7, evaluating ET +/− ribociclib in different HR+/HER2- mBC populations, suggested a greater PFS benefit from ribociclib in patients harboring alterations in ERBB2, FAT3, FRS2, MDM2, SFRP1 and ZNF217, while alterations in ANO1, CDKN2A/2B/2C and RB1 seemed to predict decreased sensitivity to ribociclib51. Of note, the above results are hypothesis generating, as for none of the above genes a statistical significant interaction with treatment was observed after correction of multiple testing52.
In addition, the mutational status of 21 genes in ctDNA was evaluated in exploratory analyses of the PEARL study, which couldn’t demonstrate any superiority of palbociclib + ET compared to capecitabine in the treatment of AI-resistant HR+/HER2- mBC53. Comparing plasma samples at baseline and after two weeks of treatment, capecitabine induced a greater extent of ctDNA suppression, and lack of ctDNA suppression was associated with worse outcome in both treatment arms. Also, TP53 mutations had a poor prognostic value in both patient groups, suggesting an aggressive tumor behavior unrelated to endocrine resistance, while PIK3CA mutations apparently conferred a worse OS only to patients treated with palbociclib + fulvestrant53,54.
Taken together, these results suggest that ctDNA detection is an unfavorable prognostic biomarker in mBC, likely reflecting a higher tumor burden, while no single molecular aberration can be used in the clinic to predict benefit/resistance from ET+CDK4/6 inhibitors. Further trials are warranted, to validate the findings from the pooled MONALEESA trials.
Selecting treatment post CDK4/6 inhibitors
At progression to CDK4/6 inhibitors + ET, international guidelines recommend testing for somatic PIK3CA and ESR1 mutations and germline BRCA1/2 mutations, plus optional PALB2 testing16,23,43. The optimal treatment in this setting should consider the agents previously administered, the duration of response obtained, disease burden, ctDNA genotyping, treatment availability and patient preferences.
In addition, resistance mechanisms might be identified among molecular aberrations isolated in ctDNA. Retinoblastoma (RB1) is a tumor suppressor gene, acting downstream in the cyclin D1-CDK4/6-retinoblastoma pathway targeted by CDK4/6 inhibitors. Loss of function of the Rb protein mediates resistance to these agents, as demonstrated by preclinical models and clinical reports55. Rb loss is well characterized as a resistance mechanism to CDK4/6 inhibitors, and retrospective biomarker analyses from registration trials confirmed the emergence of RB1 mutations in 2–9% of exposed patients55,56,57. Recently, the phase 2 MAINTAIN trial provided evidence of a PFS benefit from ribobicilib with a switch in endocrine backbone following disease progression to 1st line ET + CDK4/6 inhibitors58. Since most patients were previously treated with palbociclib, this phase 2 trial provided preliminary evidence of the value of switching endocrine therapy and adding ribociclib in patients previously treated with palbociclib. Also, the BioPER phase 2 trial evaluating palbociclib post disease progression (PD) to palbociclib failed to demonstrate a meaningful clinical benefit59. Other ongoing phase 3 trials evaluate the role of the new SERD imlunestrant with or without abemaciclib (Ember-3: NCT04975308) or of abemaciclib plus fulvestrant (PostMonarch: NCT05169567) post CDK4/6 inhibitors. The identification of RB1 alterations in ctDNA should urge clinicians against a rechallenge with CDK4/6 inhibitors.
On the other hand, ctDNA alterations could provide hints on intracellular pathways activity. For instance, besides defining patients eligibility for treatment with elacestrant, the presence of ESR1 mutations in ctDNA suggests that the disease retains endocrine sensitivity, once constitutive activation of the ER is circumvented by treatment17. This concept is also supported by the evidence of a greater PFS benefit from elacestrant in patients with longer response to CDK4/6 inhibition, suggesting a greater endocrine sensitivity of the disease60. Also translational analyses from the BOLERO-2 study – demonstrating a shorter PFS and OS for patients with a Y537S or D538G ESR1 mutation isolated in plasma via droplet-digital PCR – could suggest a reduced efficacy of adding everolimus to exemestane in cases where the disease was driven by a strong constitutive ER activation61. Therefore, the detection of ESR1-mutant ctDNA after progression to CDK4/6 inhibitors might suggest possible benefit from an endocrine-based second-line treatment.
Moreover, different ESR1 mutations seem to have variable impact on disease behavior. Both in BOLERO-2 and in plasmaMATCH, different clinical outcomes were observed for patients harboring different ESR1 variants in ctDNA61,62. Translational analyses from plasmaMATCH demonstrated the coexistence in ctDNA of polyclonal alterations of the same gene as well as of molecular alterations known to be mutually exclusive in mBC, with variable clonal dominance63. The evidence of divergent tumor evolutionary routes in single patients highlights the relevance of combination approaches to HR+/HER2- mBC treatment, and the potential for ctDNA in directing them.
Directing an early switch in endocrine backbone
As previously discussed, ESR1 alterations are acquired in HR+/HER2- BC under the selective pressure of AIs, and predict poor sensitivity to further aromatase inhibition17. Given the dynamic and subclonal nature of the alteration, liquid biopsy represents the appropriate tool for ESR1 mutations detection during ET. Since ESR1-mutated tumors might retain sensitivity to SERDs, the PADA-1 study evaluated the efficacy of an early change in ET in case of a rising ESR1 mutation in the blood of patients treated with palbociclib + AIs for HR+/HER2- mBC64. Those patients who presented a new ESR1 mutation in ctDNA, or an increased level of a known mutation, with no radiologic disease progression, were randomized to continue AI vs switching to fulvestrant, while continuing palbociclib. The median PFS from random assignment was 11.9 months for fulvestrant + palbociclib and 5.7 months for AI + palbociclib64. Besides showing the clinical benefit of an early switch in endocrine backbone for these patients, the trial proved the feasibility of serial ctDNA monitoring to track resistance mechanisms and guide treatment in mBC. Applying the same principle, the ongoing SERENA-6 study (NCT04964934) is currently exploring the benefit of a switch from AIs to the oral SERD camizestrant as endocrine backbone to palbociclib upon detection of ESR1 mutations in ctDNA65.
Integrating ctDNA and imaging response
Plasma ctDNA levels largely depend on tumor burden and tumor cells turnover66. Given the short half-life of ctDNA in plasma – about 2 hours – it has the potential to reflect tumor dynamics almost in real time2. Currently available data support the assumptions that a fall in ctDNA corresponds to a positive effect of treatment on cancer, and that such measurement can provide an adequate surrogate of the overall aggregated response of a patient to treatment67.
In 2013, Dawson et al. demonstrated, in a group of mBC patients treated with chemotherapy (CT) or ET, that ctDNA had a greater correlation with radiologic changes in tumor burden compared to both CA15-3 and circulating tumor cells, also providing a more timely measure of disease response68. Another study on 420 patients with metastatic colorectal, ovarian or non-small cell lung cancer treated with CT, evaluated the relationship of ORR and ctDNA response – defined as the fraction of patients converting from measurable ctDNA at baseline to unmeasurable levels at first radiologic evaluation – with OS69. ctDNA response outperformed ORR as a predictor of OS, with higher sensitivity and reproducibility. Furthermore, Stover et al. developed an algorithm, ichorCNA, to quantify the cfDNA tumor fraction (TFx) based on 0.1x coverage whole-genome sequencing. Applying this method to 506 plasma samples from 164 patients with advanced TNBC, they observed a significantly worse survival among patients with TFx ≥10%70.
Similar evidence came from studies of targeted treatments + ET. Davis et al. collected serial blood samples at baseline and during treatment with CDK4/6 inhibitors + ET from 54 patients, to determine the bTMB and copy-number burden (CNB). They observed a decrease in blood CNB after 15–30 days of treatment, while in over 66% of patients an increase from previous nadir levels preceded radiologic detection of PD by ≥3 months, and in 4 cases by 9 months71. Similarly, in a phase 2 study of alpelisib monotherapy for HER2- mBC, levels of mutant PIK3CA in ctDNA fell in patients achieving a radiologic response, while rising prior to detection of PD, with a median lead time of 54 days from first rise to overt radiologic progression (range, 1-247 days)15. Another retrospective proof-of-concept study on 82 patients evaluated ctDNA fluctuations as a potential biomarker of early PD in mBC72. The total variant allele fraction (VAF) was calculated by summing up the VAFs of all mutations detected via Guardant360 in ctDNA at different patient time points – i.e. at the onset of a new line of treatment and during treatment – and dividing these by the number of mutations at each time point, to account for polyclonality. No association was detected between baseline ctDNA levels and radiologic progression, but patients showing a rise in ctDNA during treatment had twice the risk of a PD in their subsequent CT scan and a shorter radiologic PFS. Molecular progression could predict radiologic PD in advance of an average 5.8 weeks (range, 4–12 weeks)72. Moreover, in a Belgian study, 45 mBC patients underwent 18F-FGD PET/CT and ctDNA assessment after 14 days of treatment with everolimus + exemestane for mBC: either the absence of a 18F-FDG PET/CT response or the detection of ctDNA were associated with a shorter PFS. The group of patients presenting absence of a 18F-FDG PET/CT response and detection of ctDNA at day 14 had the worst outcome73.
Additionally, several studies of CDK4/6 inhibitors demonstrated that ctDNA dynamics over the first month of treatment might inform later outcomes. An exploratory analysis from PALOMA-3 demonstrated that the relative change in PIK3CA-mutant ctDNA levels after 15 days on treatment was a strong predictor of PFS, and a fall in ctDNA levels was observed after 15 days of treatment with palbociclib but not with ET + placebo74. In a study on 50 patients treated with ET + CDK4/6 inhibitors for HR+/HER2- mBC, targeted NGS for 74 cancer genes was performed on plasma samples taken at baseline and after 4 weeks of treatment. For genetic mutations isolated both at baseline and after one cycle of treatment, the authors calculated the ratio of the VAFs at the two timepoints, obtaining a ratio <1 where the VAF for a specific mutation was lower at C2D1 compared to baseline, and vice versa. Next, they calculated the mean of these ratios for each patient (mVAFR), demonstrating a significant association between individual mVAFR and PFS75. Similar results were obtained by Darrigues and colleagues, who applied a tumor-informed approach to evaluate ctDNA mutations at baseline and after 15 and 30 days of treatment with fulvestrant + palbociclib in HR+/HER2- mBC76. They observed no correlation between baseline ctDNA levels and PFS, but there was a strong association of ctDNA clearance at day 30 with disease response after 3 months of treatment and PFS76.
These studies suggest that early variations in the mutational burden of ctDNA have the potential to predict clinical outcomes already few weeks after the onset of CDK4/6 inhibition. Further evidence is needed to fully understand the predictive value of early ctDNA dynamics.
Similar data are now accumulating for other solid neoplasms77, leading to initiatives for the structural integration of ctDNA dynamics into imaging-based Response Evaluation Criteria in Solid Tumors (RECIST) – such as the recently published LB-RECIST criteria78 – in order to optimize the detection of disease progression79. In the future, ctDNA dynamics could inform clinical decisions in situations where the disease is non-evaluable per RECIST criteria, as in bone-only metastatic disease. While RECIST evaluation criteria are tumor agnostic and to some extent treatment agnostic80 (with the exception of immunotherapy81), it is likely that ctDNA dynamics could be tumor and treatment dependent. Currently, there are several questions for the role of ctDNA dynamics to predict clinical outcome such as: 1. what is the optimal time to assess ctDNA response? 2. What is the level of ctDNA decrease that is associated with outcome? 3. Will adding ctDNA response to RECIST response provide clinically meaningful information to RECIST that can be cost-effective and scalable to most hospitals? The value of ctDNA response using well-validated assays in addition to standard imaging needs to be evaluated in well-conducted prospective clinical trials.
Conclusions and future perspectives
The data we presented point to an increasing role of ctDNA evaluation in informing clinical decision-making, with some applications already integrated into clinical care. Thanks to technological advances, the amount of information extracted from a single ctDNA test will increase, providing information beyond SNVs, while the same fidelity of genotyping will be achieved from less genomic material. Recent works used low-coverage whole genome sequencing of cfDNA and applied computational approaches to ctDNA processing in mBC, predicting complex tumor phenotypes such as proliferation status, ER expression, histology82,83.
On the other hand, the integration of ctDNA assays into real-world settings is still lagging behind1. The implementation of ctDNA testing in clinical practice requires setting up dedicated operating procedures for test prescription, sample collection, handling, and analysis, reporting and interpretation of results. More specifically, the standardized reporting of the results needs to include pre-analytical parameters and evidence-based annotations, to facilitate the translation into evidence-based treatment recommendations1,4. Databases ranking molecular aberrations based on their actionability by targeted treatments will also facilitate this process, such as ESCAT, ranking aberrations into class I to IV with decreasing level of evidence, and the OncoKB database developed by the Memorial Sloan Kettering Cancer Center, sorting aberrations into 4 levels of evidence84,85,86. The increased integration of ctDNA evaluation in drug development87 will further support its increased use in the clinics. Finally, incorporating ctDNA assays into clinical workflows requires considerable investment, administrative vision and ad hoc training for the involved staff1. The increased integration of ctDNA assays in clinical practice will help accelerate the delivery of precision medicine.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
References
Ignatiadis, M., Sledge, G. W. & Jeffrey, S. S. Liquid biopsy enters the clinic — implementation issues and future challenges. Nat. Rev. Clin. Oncol. 18, 297–312 (2021).
Tivey, A., Church, M., Rothwell, D., Dive, C. & Cook, N. Circulating tumour DNA — looking beyond the blood. Nat. Rev. Clin. Oncol. 19, 600–612 (2022).
Mandel, P. & Metais, P. [Nuclear acids in human blood plasma]. C. R. Seances Soc. Biol. Fil. 142, 241–243 (1948).
Pascual, J. et al. ESMO recommendations on the use of circulating tumour DNA assays for patients with cancer: a report from the ESMO Precision Medicine Working Group. Ann. Oncol. 33, 750–768 (2022).
Razavi, P. et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat. Med. 25, 1928–1937 (2019).
Arnedos, M. et al. Precision medicine for metastatic breast cancer—limitations and solutions. Nat. Rev. Clin. Oncol. 12, 693–704 (2015).
Turner, N. C. et al. Circulating tumour DNA analysis to direct therapy in advanced breast cancer (plasmaMATCH): a multicentre, multicohort, phase 2a, platform trial. Lancet Oncol. 21, 1296–1308 (2020).
Andre, F. et al. Genomics to select treatment for patients with metastatic breast cancer. Nature 610, 343–348 (2022).
Bayle, A. et al. Clinical utility of circulating tumor DNA sequencing with a large panel: a National Center for Precision Medicine (PRISM) study. Ann. Oncol. 34, 389–396 (2023).
Aftimos, P. et al. Genomic and transcriptomic analyses of breast cancer primaries and matched metastases in AURORA, the Breast International Group (BIG) Molecular Screening Initiative. Cancer Discov. 11, 2796–2811 (2021).
André, F. et al. Alpelisib plus fulvestrant for PIK3CA-mutated, hormone receptor-positive, human epidermal growth factor receptor-2–negative advanced breast cancer: final overall survival results from SOLAR-1. Ann. Oncol. 32, 208–217 (2021).
André, F. et al. Alpelisib for PIK3CA -mutated, hormone receptor–positive advanced breast cancer. N. Engl. J. Med. 380, 1929–1940 (2019).
Rugo, H. S. et al. Biology and targetability of the extended spectrum of PIK3CA mutations detected in breast carcinoma. Clin. Cancer Res. 29, 1056–1067 (2023).
Martínez-Sáez, O. et al. Frequency and spectrum of PIK3CA somatic mutations in breast cancer. Breast Cancer Res. 22, 45–45 (2020).
Savas, P. et al. Alpelisib monotherapy for PI3K-altered, pretreated advanced breast cancer: A Phase II study. Cancer Discov. 12, 2058–2073 (2022).
Henry, N. L. et al. Biomarkers for systemic therapy in metastatic breast cancer: ASCO Guideline Update. J. Clin. Oncol. 40, 3205–3221 (2022).
Will, M., Liang, J., Metcalfe, C. & Chandarlapaty, S. Therapeutic resistance to anti-oestrogen therapy in breast cancer. Nat. Rev. Cancer 23, 673–685 (2023).
Fribbens, C. et al. Plasma ESR1 mutations and the treatment of estrogen receptor–positive advanced breast cancer. J. Clin. Oncol. 34, 2961–2968 (2016).
Turner, N. C. et al. ESR1 mutations and overall survival on fulvestrant versus exemestane in advanced hormone receptor-positive breast cancer: A combined analysis of the Phase III SoFEA and EFECT Trials. Clin. Cancer Res. 26, 5172–5177 (2020).
Fribbens, C. et al. Tracking evolution of aromatase inhibitor resistance with circulating tumour DNA analysis in metastatic breast cancer. Ann. Oncol. 29, 145–153 (2018).
Schiavon, G. et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci. Transl. Med. 7, 313ra182 (2015).
Bidard, F.-C. et al. Elacestrant (oral selective estrogen receptor degrader) Versus Standard Endocrine Therapy for Estrogen Receptor–Positive, Human Epidermal Growth Factor Receptor 2–Negative Advanced Breast Cancer: Results From the Randomized Phase III EMERALD Trial. J. Clin. Oncol. 40, 3246–3256 (2022).
Burstein, H. J., DeMichele, A., Somerfield, M. R. & Henry, N. L., for the Biomarker Testing and Endocrine and Targeted Therapy in Metastatic Breast Cancer Expert Panels. Testing for ESR1 Mutations to Guide Therapy for Hormone Receptor–Positive, Human Epidermal Growth Factor Receptor 2–Negative Metastatic Breast Cancer: ASCO Guideline Rapid Recommendation Update. J. Clin. Oncol. 41, 3423–3425 (2023).
Litton, J. K. et al. Talazoparib versus chemotherapy in patients with germline BRCA1/2-mutated HER2-negative advanced breast cancer: final overall survival results from the EMBRACA trial. Ann. Oncol. 31, 1526–1535 (2020).
Robson, M. E. et al. OlympiAD final overall survival and tolerability results: Olaparib versus chemotherapy treatment of physician’s choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann. Oncol. 30, 558–566 (2019).
Vidula, N. et al. Identification of somatically acquired BRCA1/2 mutations by cfDNA analysis in patients with metastatic breast cancer. Clin. Cancer Res. 26, 4852–4862 (2020).
Yoon, J.-K. et al. Efficacy of Olaparib in treatment-refractory, metastatic breast cancer with uncommon somatic BRCA mutations detected in circulating tumor DNA. Cancer Res. Treat. 55, 1048–1052 (2023).
Weigelt, B. et al. Diverse BRCA1 and BRCA2 reversion mutations in circulating cell-free DNA of therapy-resistant breast or ovarian cancer. Clin. Cancer Res. 23, 6708–6720 (2017).
Quigley, D. et al. Analysis of circulating cell-free DNA identifies multiclonal heterogeneity of BRCA2 Reversion Mutations Associated with Resistance to PARP Inhibitors. Cancer Discov. 7, 999–1005 (2017).
Mahtani, R. et al. Real-World Study of Regional Differences in Patient Demographics, Clinical Characteristics, and BRCA1/2 Mutation Testing in Patients with Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer in the United States, Europe, and Israel. Adv. Ther. 40, 331–348 (2023).
Verschoor, N. et al. Validity and utility of HER2/ERBB2 copy number variation assessed in liquid biopsies from breast cancer patients: A systematic review. Cancer Treat. Rev. 106, 102384 (2022).
Marabelle, A. et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 21, 1353–1365 (2020).
Marabelle, A. et al. Efficacy of Pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair–deficient cancer: Results from the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 38, 1–10 (2020).
Alva, A. S. et al. Pembrolizumab in patients with metastatic breast cancer with high tumor mutational burden: results from the Targeted Agent and Profiling Utilization Registry (TAPUR) Study. J. Clin. Oncol. 39, 2443–2451 (2021).
Willis, J. et al. Validation of microsatellite instability detection using a comprehensive plasma-based genotyping panel. Clin. Cancer Res. 25, 7035–7045 (2019).
Rolfo, C. et al. NTRK1 Fusions identified by non-invasive plasma next-generation sequencing (NGS) across 9 cancer types. Br. J. Cancer 126, 514–520 (2022).
Subbiah, V. et al. Pan-cancer efficacy of pralsetinib in patients with RET fusion–positive solid tumors from the phase 1/2 ARROW trial. Nat. Med. 28, 1640–1645 (2022).
Subbiah, V. et al. Tumour-agnostic efficacy and safety of selpercatinib in patients with RET fusion-positive solid tumours other than lung or thyroid tumours (LIBRETTO-001): a phase 1/2, open-label, basket trial. Lancet Oncol. 23, 1261–1273 (2022).
Doebele, R. C. et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1-2 trials. Lancet Oncol. 21, 271–282 (2020).
Hong, D. S. et al. Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 21, 531–540 (2020).
Grinshpun, A., Tolaney, S. M., Burstein, H. J., Jeselsohn, R. & Mayer, E. L. The dilemma of selecting a first line CDK4/6 inhibitor for hormone receptor-positive/HER2-negative metastatic breast cancer. Npj Breast Cancer 9, 15 (2023).
Spring, L. M. et al. Cyclin-dependent kinase 4 and 6 inhibitors for hormone receptor-positive breast cancer: past, present, and future. Lancet 395, 817–827 (2020).
Gennari, A. et al. ESMO Clinical Practice Guideline for the diagnosis, staging and treatment of patients with metastatic breast cancer. Ann. Oncol. 32, 1475–1495 (2021).
Burstein, H. J. et al. Endocrine treatment and targeted therapy for hormone receptor–positive, human epidermal growth factor receptor 2–negative metastatic breast cancer: ASCO Guideline Update. J. Clin. Oncol. 39, 3959–3977 (2021).
Finn, R. S. et al. Biomarker analyses of response to cyclin-dependent Kinase 4/6 inhibition and endocrine therapy in women with treatment-naïve metastatic breast cancer. Clin. Cancer Res. 26, 110–121 (2020).
Finn, R. S. et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 16, 25–35 (2015).
O’Leary, B. et al. Circulating tumor DNA markers for early progression on fulvestrant with or without Palbociclib in ER+ advanced breast cancer. J. Natl Cancer Inst. 113, 309–317 (2021).
Cristofanilli, M. et al. Overall survival with Palbociclib and Fulvestrant in women with HR+/HER2− ABC: Updated exploratory analyses of PALOMA-3, a double-blind, Phase III randomized study. Clin. Cancer Res 28, 3433–3442 (2022).
Tolaney, S. M. et al. Clinical Significance of PIK3CA and ESR1 mutations in circulating tumor DNA: Analysis from the MONARCH 2 study of Abemaciclib plus Fulvestrant. Clin. Cancer Res. 28, 1500–1506 (2022).
Goetz, M. P. et al. Landscape of baseline and acquired genomic alterations in circulating tumor DNA with abemaciclib alone or with endocrine therapy in advanced breast cancer. Clin. Cancer Res., OF1–OF12 (2023).
André, F. et al. Pooled ctDNA analysis of MONALEESA phase III advanced breast cancer trials. Ann. Oncol. 34, 1003–1014 (2023).
Agostinetto, E. & Ignatiadis, M. ctDNA as a predictive biomarker in advanced breast cancer: Lessons from the MONALEESA studies. Ann. Oncol. 34, 955–959 (2023).
Pascual, J. et al. Baseline and longitudinal ctDNA biomarkers in GEICAM/2013-02 (PEARL) trial cohort 2 comparing palbociclib and fulvestrant (PAL + FUL) versus capecitabine (CAPE). J. Clin. Oncol. 40, 1019–1019 (2022).
Pascual, J. et al. Baseline mutations and ctDNA dynamics as prognostic and predictive factors in ER-positive/HER2-negative metastatic breast cancer patients. Clin. Cancer Res. 29, 4166–4177 (2023).
Asghar, U. S., Kanani, R., Roylance, R. & Mittnacht, S. Systematic review of molecular biomarkers predictive of resistance to CDK4/6 inhibition in metastatic breast cancer. JCO Precis. Oncol. 6, e2100002 (2022).
Condorelli, R. et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann. Oncol. 29, 640–645 (2018).
O’Leary, B. et al. The genetic landscape and clonal evolution of breast cancer resistance to Palbociclib plus Fulvestrant in the PALOMA-3 Trial. Cancer Discov. 8, 1390–1403 (2018).
Kalinsky, K. et al. Randomized Phase II trial of endocrine therapy with or without Ribociclib after progression on cyclin-dependent Kinase 4/6 inhibition in hormone receptor–positive, human epidermal growth factor Receptor 2–negative metastatic breast cancer: MAINTAIN trial. J. Clin. Oncol. 41, 4004–4013 (2023).
Albanell, J. et al. Palbociclib rechallenge for hormone receptor–positive/HER-negative advanced breast cancer: Findings from the Phase II BioPER trial. Clin. Cancer Res. 29, 67–80 (2023).
Kaklamani, V. G. et al. Oral elacestrant vs standard-of-care in estrogen receptor-positive, HER2-negative (ER+/HER2-) advanced or metastatic breast cancer (mBC) without detectable ESR1 mutation (EMERALD): Subgroup analysis by prior duration of CDK4/6i plus endocrine therapy (ET). J. Clin. Oncol. 41, 1070–1070 (2023).
Chandarlapaty, S. et al. Prevalence of ESR1 mutations in Cell-free DNA and outcomes in metastatic breast cancer: A secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol. 2, 1310–1315 (2016).
Kingston, B. et al. ESR1 F404 mutations and acquired resistance to fulvestrant in ESR1 mutant breast cancer. Cancer Discov. 14, 274–289 (2024).
Kingston, B. et al. Genomic profile of advanced breast cancer in circulating tumour DNA. Nat. Commun. 12, 2423 (2021).
Bidard, F.-C. et al. Switch to fulvestrant and palbociclib versus no switch in advanced breast cancer with rising ESR1 mutation during aromatase inhibitor and palbociclib therapy (PADA-1): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 23, 1367–1377 (2022).
Turner, N. et al. Design of SERENA-6, a phase III switching trial of camizestrant in ESR1-mutant breast cancer during first-line treatment. Future Oncol. 19, 559–573 (2023).
Diehl, F. et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 14, 985–990 (2008).
Jacob, S. et al. The use of serial circulating tumor DNA to detect resistance alterations in progressive metastatic breast cancer. Clin. Cancer Res. 27, 1361–1370 (2021).
Dawson, S.-J. et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 368, 1199–1209 (2013).
Jakobsen, A. et al. Early ctDNA response to chemotherapy. A potential surrogate marker for overall survival. Eur. J. Cancer 149, 128–133 (2021).
Stover, D. G. et al. Association of cell-free DNA tumor fraction and somatic copy number alterations with survival in metastatic triple-negative breast. Cancer J. Clin. Oncol. 36, 543–553 (2018).
Davis, A. A. et al. Genomic complexity predicts resistance to endocrine therapy and CDK4/6 inhibition in Hormone Receptor-Positive (HR+)/HER2-negative metastatic breast cancer. Clin. Cancer Res 29, 1719–1729 (2023).
Velimirovic, M. et al. Rising circulating tumor DNA as a molecular biomarker of early disease progression in metastatic breast cancer. JCO Precis. Oncol. 4, 1246–1262 (2020).
Gombos, A. et al. FDG positron emission tomography imaging and ctDNA detection as an early dynamic biomarker of everolimus efficacy in advanced luminal breast cancer. Npj Breast Cancer 7, 125 (2021).
O’Leary, B. et al. Early circulating tumor DNA dynamics and clonal selection with palbociclib and fulvestrant for breast cancer. Nat. Commun. 9, 896 (2018).
Martínez-Sáez, O. et al. Circulating tumor DNA dynamics in advanced breast cancer treated with CDK4/6 inhibition and endocrine therapy. Npj Breast Cancer 7, 8 (2021).
Darrigues, L. et al. Circulating tumor DNA as a dynamic biomarker of response to palbociclib and fulvestrant in metastatic breast cancer patients. Breast Cancer Res 23, 31 (2021).
Reichert, Z. R. et al. Prognostic value of plasma circulating tumor DNA fraction across four common cancer types: a real-world outcomes study. Ann. Oncol. 34, 111–120 (2023).
Gouda, M. A. et al. Liquid Biopsy Response Evaluation Criteria in Solid Tumors (LB-RECIST). Ann. Oncol. S0923-7534, 05114–1 (2023).
Jakobsen, A. K. M. & Spindler, K.-L. G. ctDNA-Response evaluation criteria in solid tumors – a new measure in medical oncology. Eur. J. Cancer 180, 180–183 (2023).
Litière, S. et al. RECIST 1.1 for response evaluation apply not only to chemotherapy-treated patients but also to targeted cancer agents: a pooled database analysis. J. Clin. Oncol. 37, 1102–1110 (2019).
Seymour, L. et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 18, e143–e152 (2017).
Prat, A. et al. Circulating tumor DNA reveals complex biological features with clinical relevance in metastatic breast cancer. Nat. Commun. 14, 1157 (2023).
Doebley, A.-L. et al. A framework for clinical cancer subtyping from nucleosome profiling of cell-free DNA. Nat. Commun. 13, 7475 (2022).
Mateo, J. et al. A framework to rank genomic alterations as targets for cancer precision medicine: the ESMO Scale for Clinical Actionability of molecular Targets (ESCAT). Ann. Oncol. 29, 1895–1902 (2018).
Chakravarty, D. et al. OncoKB: A precision oncology knowledge base. JCO Precis. Oncol. 1, 1–16 (2017).
Condorelli, R. et al. Genomic alterations in breast cancer: level of evidence for actionability according to ESMO Scale for Clinical Actionability of molecular Targets (ESCAT). Ann. Oncol. 30, 365–373 (2019).
Araujo, D. et al. Oncology phase I trial design and conduct: time for a change - MDICT Guidelines 2022. Ann. Oncol. 34, 48–60 (2023).
Acknowledgements
M.I. was supported by a grant from “L’Association Jules Bordet”.
Author information
Authors and Affiliations
Contributions
O.A. and N.G. retrieved and critically reviewed the literature and drafted the manuscript. O.A. designed the figure. M.I. critically reviewed, edited and supervised the work. All authors contributed to conceiving the work, read and approved the final version of the manuscript, and are accountable for all its aspects.
Corresponding author
Ethics declarations
Competing interests
O.A. and N.G. declare no competing financial or non-financial interests. M.I. declares the following competing financial interests: Consultant for: Seattle Genetics, Rejuveron senescence therapeutics, Gilead Sciences, Menarini/Stemline, Daichi; Speaker’s Bureau for: Novartis; Grant/Research support from: Roche (my institution), Inivata Inc (my institution), Natera Inc (my institution), Pfizer (my institution). M.I. is Chair of the EORTC Breast Cancer Group.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Amato, O., Giannopoulou, N. & Ignatiadis, M. Circulating tumor DNA validity and potential uses in metastatic breast cancer. npj Breast Cancer 10, 21 (2024). https://doi.org/10.1038/s41523-024-00626-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41523-024-00626-6
