
Abstract
Endocrine therapy (ET) with cyclin-dependent kinase 4/6 inhibitor (CDK4/6i) is currently the standard first-line treatment for most patients with hormone receptor (HR) positive, human epidermal growth factor receptor (HER2) negative advanced breast cancer. However, resistance to ET and CDK4/6i inevitably ensues. The optimal post-progression treatment regimens and their sequencing continue to evolve in the rapidly changing treatment landscape. In this review, we summarize the mechanisms of resistance to ET and CDK4/6i, which can be broadly classified as alterations affecting cell cycle mediators and activation of alternative signaling pathways. Recent clinical trials have been directed at the targets and pathways implicated, including estrogen and androgen receptors, PI3K/AKT/mTOR and MAPK pathways, tyrosine kinase receptors such as FGFR and HER2, homologous recombination repair pathway, other components of the cell cycle and cell death. We describe the findings from these clinical trials using small molecule inhibitors, antibody–drug conjugates and immunotherapy, providing insights into how these novel strategies may circumvent treatment resistance, and discuss how some have not translated into clinical benefit. The challenges posed by tumor heterogeneity, adaptive rewiring of signaling pathways and dose-limiting toxicities underscore the need to elucidate the latest tumor biology in each patient, and develop treatments with improved therapeutic index in the era of precision medicine.
Similar content being viewed by others

Introduction
The hormone receptor-positive (HR + ) and human epidermal growth factor receptor-2 negative (HER2-) subtype comprises ~68% of all breast cancers (BCs)1. Endocrine therapy (ET) with cyclin-dependent kinase 4/6 inhibitor (CDK4/6i) is currently the standard treatment for HR + /HER2− advanced BC (ABC)2,3, with the demonstration of overall survival (OS) benefit in several trials4,5,6,7. However, treatment resistance inevitably ensues, and the optimal management after prior CDK4/6i plus ET continues to be refined, especially with the increasing use of adjuvant CDK4/6i8, as well as adjuvant poly ADP-ribose polymerase inhibitors (PARPi)9, changing the profile of patients with recurrent metastatic disease. Currently, there is a lack of data on the tumor biology and the treatment strategies after relapse in these patients. Tumor and/or liquid biopsy upon relapse or progression to determine the latest HR and HER2 status as well as genomic profile may provide insight into the underlying resistance mechanisms, and allow an individualized approach to treatment. New therapeutic targets may be discovered from profiling the resistant metastatic lesions, which harbor acquired alterations absent in the primary tumor10,11,12.
There is considerable heterogeneity among the HR + /HER2− BCs. With gene expression profiling, distinct intrinsic subtypes (IS) of BCs may be identified, which predict for differing prognosis and response to endocrine therapy. Despite being HR+ by immunohistochemistry (IHC), luminal B tumors have a higher expression of proliferation/cell cycle-related genes, and predict a worse prognosis compared to luminal A tumors13,14. IS classification also predicts for response to treatment, with a shorter time to progression on ET with CDK4/6i in luminal B, HER2 enriched, and basal subtypes compared to luminal A tumors15. Furthermore, upon disease progression, conversion of IS from a luminal type to a nonluminal type can occur, leading to a more aggressive endocrine-resistant biology13.
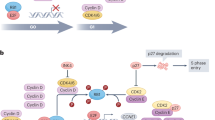
Mechanisms of resistance to CDK4/6 blockade can be broadly categorized as aberrations affecting cell cycle progression and activation of other signaling pathways as described previously12,16 (Fig. 1). The alterations which affect cell cycle mediators include loss-of-function alterations in RB1 and upregulation of CDK6, cyclin E1/E2, and Aurora kinase A. Activation of the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) and the mitogen-activated protein kinase (MAPK) pathways can be effected through activating mutations of oncogenes or loss of function of tumor suppressors, while activating mutations or amplification involving other growth factor receptor genes such as ERBB2 (HER2) and FGFR (fibroblast growth factor receptor)16,17,18 can lead to signaling through alternative oncogenic pathways. Primary or secondary resistance to the ET backbone can occur concurrently18,19. Given the differential properties of the three CDK4/6is which may influence the mode of action, the underlying mechanisms of resistance may potentially vary. Data from such research is awaited.

The potential treatment strategies are shown in pink boxes, while crosstalk is indicated by dotted lines (figure created with BioRender.com). AKT V-akt murine thymoma viral oncogene homolog 1, AR androgen receptor, ARE androgen response element, AURKA aurora kinase A, BCL2 B-cell lymphoma 2, BCLXL B-cell lymphoma-extra large, CDK cyclin-dependent kinase, CHK checkpoint kinase, ER estrogen receptor, ERE estrogen response element, ERK extracellular signal‑regulated kinase, FGFR fibroblast growth factor receptor, GAP GTPase-activating protein, GDP guanosine diphosphate, GTP guanosine triphosphate, HER human epidermal growth factor receptor, INPP4B inositol polyphosphate 4-phosphatase type II, LATS large tumor suppressor kinase, MEK mitogen‑activated protein kinase, MST mammalian STE20-like kinase, mTOR mammalian target of rapamycin, PARP poly(ADP-ribose) polymerase, PD-1 programmed cell death protein 1, PD-L1 programmed death-ligand 1, PI3K phosphatidylinositol-3-kinase, PTEN phosphatase and tensin homolog, RAS rat sarcoma viral oncogene homolog, RAF rapidly accelerated fibrosarcoma kinase, RB retinoblastoma tumor suppressor, TAZ transcriptional co-activator with PDZ-binding motif, Trop-2 trophoblast cell-surface antigen 2, TTK spindle assembly checkpoint kinase, YAP Yes-associated protein.
While mechanisms involving other hallmarks of cancer such as deregulation of cellular metabolism, epigenetic reprogramming, pro-tumorigenic inflammation, and microenvironment18,20, may be implicated, this review will focus on the therapeutic strategies with reported clinical trial activity that have the potential to transform our treatment paradigm over the next decade (Figs. 1 and 2). For the various strategies, we describe the preclinical and clinical rationale, summarize the efficacy demonstrated in clinical trials, highlight the challenges and discuss the implications for patient management with future directions.

Blue boxes indicate current treatment options based on FDA-approved drugs and the latest NCCN162, ESMO, ASCO guidelines2,3. Pink boxes indicate the potential therapeutic strategies in the future, which are currently still investigational. Dotted pink arrows indicate the possibility of switching between treatments, or change in sequence of the treatment options, pending further data. ADC antibody–drug conjugate, AR androgen receptor, DDRi DNA damage repair inhibitor, dMMR deficient mismatch repair, ET endocrine therapy, ESR1 estrogen receptor 1 gene, gBRCA mut germline BRCA1/BRCA2 mutation (now termed variant), HRD homologous recombination deficiency, MAPK mitogen-activated protein kinase, MSI-H microsatellite instability high, NTRK neutotrophic tyrosine receptor kinase, PALB2 mut partner and localizer of BRCA2 mutation, RET rearranged during transfection, T-DXd trastuzumab deruxtecan, TMB-H tumor mutational burden high, TP53 transformation-related protein 53.
Prospective therapeutic strategies post cdk4/6 inhibition
Targeting the estrogen receptor (ER) pathway
Estrogen receptor 1 (ESR1) activating mutations at the ligand binding domain of ER are rare in untreated primary BCs, but are typically enriched after exposure to aromatase inhibitors (AIs) with a prevalence of ~20–40% in pretreated ABC21,22,23,24, promoting estrogen-independent constitutional activation and downstream transcription of ER-controlled genes25. Other effects of ESR1 mutations include the induction of metabolic alterations26, distinct cistromes, and transcriptional changes to stimulate growth and metastases27.
Tamoxifen is a SERM (selective estrogen receptor modulator) which competes with estrogen in binding to ER and partially inhibits mutant ERα transcription. However, more potent ER antagonists may be more efficacious. Preclinical studies showed that lasofoxifene, a potent antagonist of both wild-type and mutant ER28, was effective in reducing tumor growth in an endocrine-resistant ESR1-mutated xenograft model29. However, ELAINE 1, an open-label phase II trial of patients with HR + /HER2− ESR1-mutated ABC with progression after at least 12 months of AI and CDK4/6i, failed to show a statistically significant improvement in progression free survival (PFS) of lasofoxifene over fulvestrant30. Otherwise, in the single-arm phase II ELAINE 2 trial, among 29 women with HR + /HER2− ESR1-mutated metastatic BC (MBC) whose disease progressed after 1–2 prior lines of ET with or without CDK4/6i, lasofoxifene plus abemaciclib was well-tolerated and demonstrated activity with median PFS (mPFS) of 13.9 months and objective response rate (ORR) of 33.3%31.
Fulvestrant, a pure ER antagonist and the only clinically approved SERD (selective estrogen receptor degrader) until recently, not only reduces ER dimerization and transcription, but also induces ER degradation via a proteasome-dependent system25. Fulvestrant is superior to AI in the presence of ESR1 mutations32, but higher drug levels are required for optimal activity, especially in Y537S-mutant cells10,25. Fulvestrant is poorly soluble, requiring administration via intramuscular injections and limiting the volume or dose that can be delivered. The standard high dose of 500 mg fulvestrant failed to completely abolish ER availability in tumors of 38% of patients in a [18F]fluoroestradiol (FES) positron emission tomography/computed tomography (PET/CT) study33. Hence there is great interest in developing oral SERDs for which the dose may be escalated to optimize antitumor activity, as well as complete ER antagonists (CERANs), selective ER covalent antagonists (SERCAs) and proteolysis targeting chimeras (PROTACs). PROTACs facilitate proteosomal degradation of target proteins via ubiquitination34, while SERCAs covalently inactivate both wild-type and mutant ER by inducing conformational changes in the ER, reducing binding of co-activators35. These agents are being tested in ongoing trials, including combination trials with CDK4/6 or PI3K/AKT/mTOR inhibition (Table 1).
ARV-471 is a novel PROTAC evaluated in the phase I/II VERITAC study enrolling patients with HR + /HER2− ABC. ARV-471 monotherapy showed a clinical benefit rate (CBR) of 40% in the phase I dose-escalation study in patients who received prior ET and CDK4/6i. Phase II dose expansion confirmed its CBR of 37.1% with 200 mg QD dose and 38.9% with 500 mg QD dose, with the suggestion of enhanced activity in ESR1-mutated subgroup36. Part C of the study is looking at ARV-471 in combination with palbociclib in pretreated HR + /HER2− ABC and is currently recruiting patients (Clinical Trials.gov identifier NCT04072952), while the ongoing phase 3 VERITAC-2 trial will be comparing ARV-471 against fulvestrant after progression on first-line ET with CDK4/6i (NCT05654623).
Results from randomized trials on several novel SERDs were recently presented. Elacestrant demonstrated a significant progression-free survival (PFS) benefit over physician’s choice of standard-of-care (SOC) ET (fulvestrant or AI) in the open-label randomized phase III EMERALD trial, where patients had progressed after 1–2 lines of ET for HR + /HER2− ABC, including prior CDK4/6i, and maximum of one line of palliative chemotherapy. Primary endpoints of centrally reviewed PFS in all patients (intent-to-treat (ITT) population, n = 477) and in patients with detectable ESR1 mutations from circulating tumor DNA (ctDNA) were met (hazard ratio (HR) 0.70, P = 0.002 and HR 0.55, P = 0.0005, respectively). The Kaplan–Meier survival curves dipped significantly during the first 3 months in both arms, reflecting the limited benefit from endocrine monotherapy in majority of patients. However, the 12-month PFS achieved with elacestrant in the ITT population was 22.3% versus 9.4% with SOC ET, supporting its activity in endocrine-sensitive tumors. In addition, the magnitude of PFS improvement was higher in patients with detectable ESR1 mutation24. Elacestrant was generally well-tolerated. Nausea of any grade was reported in 35.0% of patients receiving elacestrant compared to 18.8% receiving SOC; grade 3/4 nausea was uncommon at 2.5% and 0.9% respectively. Ocular or cardiac toxicities which have been reported with other SERDs, SERMs or SERCAs, such as giredestrant, camizestrant37, tamoxifen38, and H3B-6545, were not observed with elacestrant24,39.
Camizestrant is a next-generation oral SERD and pure ER antagonist40. The phase II SERENA-2 trial reported superiority of camizestrant over fulvestrant in patients with recurrence or progression on 1 line of ET and no more than 1 line of chemotherapy. Investigator-assessed mPFS was 7.2 months with 75 mg camizestrant and 7.7 months with 150 mg dose, compared to 3.7 months with fulvestrant (HRs 0.58, P = 0.0124 and 0.67, P = 0.0161, respectively)41. These positive results contrast with the negative randomized phase II studies of two other oral SERDs, amcenestrant and giredestrant. They did not meet their primary objective of PFS improvement in pretreated HR + /HER2− ABC over ET of physician’s choice42,43, although a numerical trend suggested greater benefit in the presence of ESR1 mutations. A prespecified interim analysis of the phase III AMEERA-5 trial (NCT04478266) comparing first-line amcenestrant and palbociclib with letrozole and palbociclib did not meet the prespecified boundary for continuation in patients with HR + /HER2- ABC, leading to termination of the amcenestrant clinical development program.
While the discordant results may be related to different intrinsic properties influencing the potency of the various compounds39,44, patient selection criteria or imbalance of tumor characteristics in small randomized phase 2 trials may also impact on the efficacy results. The benefit of SERD over SOC ET appears to be more pronounced in patients with endocrine-sensitive tumors harboring ESR1 mutations, without significant activation of other pathways. The Food and Drug Administration (FDA) approved elacestrant for postmenopausal women or adult men with ER + /HER2−, ESR1-mutated ABC after progression on at least one line of ET in January 2023, and the Guardant360 CDx assay as a companion diagnostic. As of May 2023, to aid in treatment selection, routine testing for ESR1 mutations from blood or tumor biopsy at progression, is now recommended based on the updated American Society of Clinical Oncology (ASCO) guidelines45.
Continuing CDK4/6 inhibition beyond progression with switch in ET
In the phase III PADA-1 trial, 172 patients with rising circulating ESR1 mutation before radiological progression on first-line AI and palbociclib were randomized to continuing their initial treatment or to receive palbociclib with fulvestrant. After median follow-up of 26.0 months from randomization, mPFS from assignment improved from 5.7 months in the control arm to 11.9 months with early switch to fulvestrant (stratified HR = 0.61, P = 0.0040)23. Data are awaited on whether this “lead-time” PFS advantage from early switch will translate into any meaningful OS benefit, warranting change in clinical practice. SERENA-6 (NCT04964934) is designed to assess the switching to camizestrant versus continuing an AI for HR + /HER2- ABC patients on first-line AI and CDK4/6i with ESR1 mutation detected on ctDNA without clinical progression.
In BioPER, a single-arm phase II trial of continuing palbociclib with switch to ET of physician’s choice upon progression on prior palbociclib + ET, CBR was 34.4% with mPFS of 2.6 months46. Biomarker analysis showed loss of Rb in 13% of tumors, which did not benefit from continuing palbociclib. More recently, preliminary results from the randomized phase II PACE trial failed to show PFS improvement in continuing palbociclib with fulvestrant (n = 111) over fulvestrant alone (n = 55) after progression on AI and CDK4/6i; >90% of patients had received prior palbociclib47.
In the phase II MAINTAIN trial, 119 patients whose HR + /HER2− ABC progressed on ET plus any CDK4/6i and ≤1 prior line of chemotherapy were randomized to ribociclib versus placebo with fulvestrant or exemestane. In terms of prior CDK4/6i, 87% had received palbociclib. In the ITT population, mPFS was 5.29 months in the ribociclib arm compared to 2.76 months in the placebo arm (HR 0.57, P = 0.006). Results were similar in the fulvestrant subgroup48. Taken together, the data seems to show a possible benefit of switching from one CDK4/6i to another, versus re-exposure to the same CDK4/6i upon progression. While these results suggest benefit from ribociclib, a confirmatory phase III trial is essential before this strategy is adopted as routine clinical practice. Moreover, such an approach will not be effective if the tumors have lost expression of ER, developed RB1 mutation or loss, or are driven by activation of alternate pathways. The tumor biology should be reassessed in each patient with serial biopsies or liquid biopsy if feasible, and provided it may alter clinical management.
Targeting the androgen receptor pathway
Androgen receptor (AR) is expressed in up to 90% of ER+ BCs49. AR expression portends a favorable prognosis with its role as a tumor suppressor by antagonizing ER target genes and repressing expression of cell cycle genes50. In a randomized phase II trial, adding enzalutamide, an AR inhibitor, to exemestane failed to improve PFS compared to exemestane alone in patients with 0-1 prior line of endocrine therapy. Low levels of ESR1 mRNA expression with high AR expression predicted benefit from the addition of enzalutamide in an exploratory analysis51. However, a recent study uncovered the differential effects of AR inhibition depending on the AR to ER ratio. In AR-low BC cells, enzalutamide displaced estrogen from ER binding sites, functioning as ER antagonist. In the AR-high setting where AR repressed ERα signaling, enzalutamide antagonized AR, promoting ERα signaling, while RAD140, a selective AR agonist, activated AR signaling and suppressed AR-high tumor growth by inhibiting ER52.
Enobosarm, a selective androgen receptor modulator (SARM) which activates AR in breast, muscle and bone without the virilizing side effects associated with steroidal androgens, was assessed in the open-label phase II G200802 trial among patients with AR+ (nuclear staining >10%), ER + ABC. The primary endpoint of CBR at 24 weeks was 32% in the 9 mg group (n = 72) and 29% in the 18 mg group (n = 64). ORR was 48% in tumors that were AR ≥ 40% positive versus 0% with AR < 40%53. Enobosarm was granted fast-track designation by the FDA for the treatment of AR + /HR + /HER2− ABC in January 2022; randomized phase III monotherapy and combination therapy trials are ongoing (NCT04869943, NCT05065411).
Targeting the PI3K/AKT/mTOR pathway
The PI3K/AKT/mTOR pathway can be activated by PIK3CA activating mutations and amplification, loss of tumor suppressors such as PTEN, INPP4B, and overexpression and/or mutation of upstream receptor tyrosine kinases (RTKs). PI3K/AKT/mTOR signaling promotes cellular proliferation and resistance to apoptosis; AKT can also activate ER-mediated transcription independently of estrogen, leading to endocrine resistance54,55. In HR + /HER2− ABC, the prevalence of activating PIK3CA mutations is estimated at ~30–40%, and 5–10% for activating AKT1 mutations and inactivating PTEN alterations separately11,56,57,58,59. PIK3CA-mutated HR + /HER2− ABC have also been associated with inferior survival outcomes compared to wild-type tumors60,61.
In the SOLAR-1 trial, addition of alpelisib, an alpha-specific PI3K inhibitor—better tolerated than the older generation of pan-PI3K inhibitors, to fulvestrant in the cohort of patients with PIK3CA-mutated ABC improved the mPFS from 5.7 months to 11 months (HR 0.65, P < 0.001). The 7.9-month numeric improvement in median OS was not statistically significant. No benefit was observed in patients with PIK3CA-wild-type tumors. FDA’s approval of alpelisib and fulvestrant in PIK3CA-mutated HR + /HER2− ABC, along with the updated ASCO guidelines, recognized PIK3CA mutation (tumor or liquid biopsy) as a biomarker to guide systemic therapy in ABC62,63. However, merely 6% of subjects had received prior CDK4/6i in the SOLAR-1 trial58,64.
BYLieve is a phase II, open-label, three-cohort, non-comparative study of alpelisib plus either fulvestrant or letrozole in patients with HR + /HER2− PIK3CA mutant ABC progressing on or after prior treatments, including CDK4/6i (Table 2). All patients in cohorts A and B had received CDK4/6i with AI and fulvestrant, respectively, as the immediate past treatment, whilst 66.7% of those in cohort C received prior CDK4/6i65,66,67. At 6 months, 50.4% of patients in cohort A, 46.1% in cohort B and 48.7% in cohort C were alive and without disease progression. mPFS was slightly lower than that in the predominantly CDK4/6i naive SOLAR-1 study population. The most frequent grade 3/4 adverse events (AEs) included hyperglycemia and rash which can affect dose delivery and efficacy. Frequency of AEs leading to discontinuation of alpelisib was lower compared to in SOLAR-1, possibly due to increasing familiarity and improved management of toxicities with measures such as prophylactic antihistamines for rash. Prophylactic metformin use was able to reduce the severity and incidence of alpelisib-induced hyperglycemia in the METALLICA study68. Other challenges include the development of inactivating PTEN mutations, adaptive rewiring, epigenetic and metabolic reprogramming, rendering the cancer cells resistant to PI3K inhibition69,70. Given the importance of PI3K inhibition, novel PI3K inhibitors which degrade the mutant oncoprotein selectively such as inavolisib (GDC-0077)71, RLY-2608 (NCT05216432), and LOXO-783, an allosteric PIK3CA H1047R-mutant-specific inhibitor72, and may be better tolerated, are being tested in clinical trials currently.
The results from the randomized double-blind phase III CAPItello-291 trial which tested the addition of capivasertib, an AKT inhibitor (starting dose of 400 mg twice daily for 4 days, followed by 3 days off), to fulvestrant in 708 patients after progression on ≤2 lines of prior endocrine therapy and ≤1 line of prior chemotherapy appear promising (Table 2). Unlike the randomized Phase II FAKTION trial where the benefit of capivasertib was predominantly in patients with PI3K/AKT/PTEN pathway alterations59,73, mPFS was doubled with the addition of capivasertib from 3.6 months to 7.2 months (HR 0.60, two-sided P < 0.001) in the overall ITT population, and increased from 3.1 months to 7.3 months (HR 0.50, two-sided P < 0.001) in the AKT-pathway altered population74. While this suggests that the benefit of capivasertib may not be restricted to tumors harboring PIK3CA, AKT1, or PTEN alteration, the study was not powered to detect the benefit in non-altered tumors. Diarrhea, rather than hyperglycemia, was the most common all-grade AE (72.4% versus 16.3%) despite a less stringent criteria of baseline HbA1c < 8.0%. On the other hand, 38.0% of patients in the capivasertib arm experienced any type of rash, compared to only 7.1% in control arm. The positioning of capivasertib with fulvestrant is currently unclear, without direct comparison of its efficacy with alpelisib or everolimus. Moreover, there is no prospective data on the activity of each agent after prior exposure or resistance to another drug targeting the same pathway. However, capivasertib, with its intermittent dosing schedule, may play an increasing role if it is better tolerated than the current PI3K or mTOR inhibitors, or should there be an overall survival benefit.
In contrast, IPATUNITY130 did not show improved efficacy of addition of ipatasertib, another AKT inhibitor, to paclitaxel in patients with HR + /HER2- PIK3CA/AKT1/PTEN-altered ABC. This was postulated to be due to lack of endocrine blockade75. Results from randomized trials testing the efficacy of adding ipatasertib to fulvestrant are awaited (NCT04650581, NCT04060862).
Although the everolimus trials were conducted in the pre-CDK4/6i era, mTOR inhibition remains a valid treatment option for patients regardless of PIK3CA mutation status2,3. While alpelisib with ET is often the preferred option post CDK4/6i for PIK3CA-mutated tumors, mTORC1 activation by feedback loop limits the sensitivity to alpha-specific PI3K inhibitors69. Hence mTOR inhibition may potentially be considered after progression on PI3K inhibitor. The triplet combination of ET with CDK4/6i and PI3K/AKT/mTOR inhibitors was explored in several early-phase trials (Table 3), based on the rationale that PI3K/AKT/mTOR pathway aberrations may confer resistance to CDK4/6i, and preclinical evidence of CDK4/6i re-sensitizing resistant cells to PI3K inhibitors76. TRINITI-1 investigated exemestane, everolimus, and ribociclib in patients who progressed on CDK4/6i; the CBR was 41.1% and mPFS was 5.7 months77. Although the triplet combination of fulvestrant, ribociclib, and alpelisib was not feasible due to toxicities78, the combinations of fulvestrant and palbociclib with ipatasertib, capivasertib or inavolisib continue to be tested in ongoing trials (NCT04060862, NCT04862663, NCT04191499).
Targeting the Ras/Raf/MAPK pathway
Alterations in the Ras/Raf/MAPK pathway such as NF1, KRAS, HRAS, BRAF, ERBB2, and EGFR, are enriched in endocrine-resistant HR + /HER2− tumors. They appear to be mutually exclusive with ESR1 mutations, occurring in 16% of ESR1-wild-type BCs post-ET11. Increased signaling promoted cellular growth and proliferation, and induced an ER-negative phenotype in vitro11,79.
Selumetinib, a MEK1/2 inhibitor, was tested in a randomized phase II trial with fulvestrant against placebo and fulvestrant in HR + /HER2− ABC post 1st line AI. Trial recruitment was halted after the selumetinib combination failed to reach the prespecified disease control rate (DCR) at the interim analysis, faring worse than fulvestrant/placebo with DCR of 23% vs 50%, respectively80. Possible reasons include significant toxicities such as mucocutaneous disorders, fatigue, nausea/vomiting, edema, diarrhea impacting dose delivery, compensatory activation of alternative signaling pathways, lack of efficacy in a biomarker-unselected population, and imbalance of other factors due to chance with the small sample size. As such, the role of targeting the MAPK pathway remains to be explored. Current trials are evaluating newer-generation MEK inhibitors in ABC or solid tumors harboring NF1 or MAPK-activating mutations (NCT05554354, NCT05054374).
Targeting receptor tyrosine kinases
Overexpression or activating mutation of RTKs such as FGFR and HER2 lead to activation of downstream oncogenic signaling pathways, including the PI3K/AKT/mTOR and RAF/RAS/MAPK pathways. Aberrant FGFR signaling has been shown to mediate resistance to ET, CDK4/6, and PI3K inhibitors. PFS was shorter in patients with FGFR1 amplification on ET and CDK4/6i81,82. FGFR axis alterations such as FGFR1, FGFR2, or FGF3 can also be acquired, detected in up to around 40% of resistant specimens12,81,83.
Phase II trials of FGFR inhibitors dovitinib84, lucitanib85, and AZD454786 in unselected patients showed modest activity, and were complicated by toxicities such as hypertension, hypothyroidism and retinal detachments. The benefit appeared to be restricted to tumors with FGFR1 amplification or overexpression, or dependence on FGFR signaling. In the preliminary report of a phase Ib trial testing fulvestrant, palbociclib and erdafitinib, a pan-FGFR inhibitor in FGFR-amplified/ER + /HER2-negative pretreated ABC, toxicities led to treatment discontinuation in several patients. No objective responses were observed, though PFS was longer (6 months) in patients with high levels of FGFR1 amplification or FGFR3 amplification87.
Lenvatinib, a multi-kinase inhibitor with activity against VEGFR1-3 (vascular endothelial growth factor receptor), FGFR1-4, RET (rearranged during transfection), PDGFR (platelet-derived growth factor receptor) and KIT, was investigated with letrozole in a phase Ib/II trial. ORR was 23.3% in 31 patients who had received median 5 lines of prior therapy, with median duration of response lasting 6.9 months88. RET expression levels on immunohistochemistry were not predictive of response; further biomarker analyses are awaited.
The prevalence of activating HER2 mutations varies from 2 to 4% in primary BCs to >5% in lobular cancers and metastatic lesions89,90,91. These mutations may span across the extracellular, transmembrane or juxta-membrane domain90. Neratinib, an irreversible pan-HER inhibitor, inhibited cell growth90 and restored sensitivity to fulvestrant in estrogen-resistant HER2-mutant cancer cells in vitro91.
In the phase II MutHER study, combination of neratinib and fulvestrant achieved CBR of 38% and 30%, respectively, in the fulvestrant-treated and fulvestrant-naive patients with HR + /HER2− HER2-mutated ABC92. Upon progression, addition of trastuzumab was further able to induce a partial response in 3 out of 5 patients, supporting role of dual-HER2 blockade in overcoming resistance to neratinib92. The SUMMIT trial included 33 patients with HR + /HER2− HER2-mutated ABC on fulvestrant, trastuzumab and neratinib; ORR was 42.4% and mPFS was 7.0 months93. HER2-directed antibody–drug conjugates (ADC) may be useful, but the indication in HR + /HER2− ABC is for HER2-low (details below), unlike lung cancer where FDA approval is for HER2-mutated non-small cell lung cancer94.
Targeting cancer epithelial antigens via antibody–drug conjugates (ADCs)
Beyond targeting pathways, novel ways of delivering chemotherapy have afforded new therapeutic options in the endocrine-resistant setting. ADCs allow the delivery of cytotoxic drugs directly to cancer cells via targeting specific cell-surface antigens. Novel ADCs with enhanced properties such as higher payload to antibody ratio, stable tumor-selective cleavable linker and potent, membrane-permeable payload with short systemic half-life and bystander killing effect have achieved breakthroughs in the cancer treatment landscape95.
The positive results of the phase III DESTINY-Breast04 led to FDA approval of trastuzumab deruxtecan (T-DXd) in August 2022 for treatment of HER2-low ABC in patients who have received prior chemotherapy in the metastatic setting or developed disease recurrence during or within six months of completing adjuvant chemotherapy96. T-DXd contains a novel topoisomerase I inhibitor payload, which is seldom used in ABC, with a drug-to-antibody ratio of 8:197. HER2-low BC, defined by immunohistochemical scoring of IHC 1+ or 2+ with negative in situ hybridization (ISH), accounts for ~60% of HER2- ABC98. HER2-low BCs do not respond to traditional HER2-targeted therapy and are classified as HER2 negative. However, novel HER2-directed ADCs can deliver the cytotoxic payload to the surrounding tumor cells through the bystander effect95.
DESTINY-Breast04 randomized 557 patients with HER2-low ABC in a 2:1 ratio to T-DXd versus the physician’s choice of chemotherapy. Subjects with HR + /HER2-low ABC (n = 494) had received a median of 3 lines of prior palliative therapy, including 1–2 lines of palliative chemotherapy, with prior CDK4/6i in approximately 70% of the cohort (Table 4). The primary endpoint of PFS in the HR + /HER2- cohort was significantly superior with T-DXd compared to control: median 10.1 months versus 5.4 months (HR 0.51, P < 0.001), with OS improvement as well (median 23.9 months versus 17.9 months, HR = 0.64, P = 0.003). Drug-related interstitial lung disease or pneumonitis, an AE of special interest, was observed in 12.1% of patients; 0.8% were grade 5 events, underscoring the need for close surveillance and prompt management.
DAISY, an open-label phase II trial, evaluated the efficacy of single-agent T-DXd with extensive biomarker analysis in 3 cohorts of ABC patients. The primary endpoint of ORR was 70.6% in Cohort 1 (HER2 overexpressing), 37.5% in Cohort 2 (HER2-low) and 29.7% in cohort 3 (HER2-null: IHC 0). Although clinically meaningful activity was seen in HER2-null BC, the mPFS was 4.2 months in Cohort 3, compared to 6.7 months in HER2-low and 11.1 months in HER2-overexpressing cohorts. T-DXd antitumor activity was associated with levels of HER2 expression although other mechanisms could be involved99. The activity of T-DXd in HER2-null in this trial and the interobserver reproducibility of HER2 immunohistochemical scoring100,101 underscore the limitations of the current HER2-low definition based on immunohistochemical testing. Quantitative HER2 assays with greater sensitivity for lower range of HER2 expression have been suggested as a means to better predict response to T-DXd102,103.
HER3 is another RTK belonging to the HER family. Whilst HER3 is not oncogenic when expressed alone, it can be activated through the formation of heterodimers with other receptors and members of the EGFR, effecting downstream PI3K/AKT-pathway signaling. HER3 expression is associated with disease progression and increased metastatic rate. In one study, 30% of primary BCs expressed HER3, but the expression increased to 60% of the matched metastatic specimens104,105. Patritumab deruxtecan (HER3-DXd), a novel ADC against HER3, exhibited cytotoxic activity in vitro via HER3-specific binding to cancer cells and release of its topoisomerase payload intracellularly106. In the U31402-A-J101 phase I/II study of HER3-DXd in HER3-expressing ABC, ORR was 30.1% with mPFS of 7.4 months in the HR + /HER2− cohort, despite being heavily pretreated (Table 4)107.
TROP2, a transmembrane calcium signal transducer, is highly expressed in TNBC and HR + /HER2- BC cells, with prevalence exceeding 90%108. Sacituzumab govitecan (SG), an anti-TROP2 (trophoblast cell-surface antigen 2) ADC with a topoisomerase inhibitor payload (drug-to-antibody ratio 8:1), was first tested in triple-negative BC (TNBC)109. The subsequent phase III TROPiCS-02 trial in HR + /HER2- ABC showed PFS and OS improvement with SG over the physician’s choice of chemotherapy (mPFS 5.5 months vs 4.0 months; HR 0.66, P = 0.0003; mOS 14.4 months vs 11.2 months; HR 0.79, P = 0.02). While the benefit of SG may seem less impressive compared to T-DXd in DESTINY-Breast04, the TROPiCS-02 trial population was more heavily pretreated (Table 4)110,111. Although PFS and OS favored SG over TPC across TROP2 expression levels (H-score <100 and ≥100), including those with H-score ≤10, only 17% of the trial population showed very low TROP2 expression112. In February 2023, FDA-approved SG for patients with HR + /HER2− ABC who have received endocrine-based therapy and at least two additional systemic therapies in the metastatic setting. Datopotamab–deruxtecan (Dato-DXd)—an ADC targeting TROP2, with the same cytotoxic payload as T-DXd, showed an ORR of 29% and a DCR of 85% in a heavily pretreated HR + /HER2− ABC population (median 5 prior lines of treatment) in the phase I TROPION-PanTumor01 trial113. The phase III TROPION-Breast01 (NCT05104866) is currently recruiting patients with HR + /HER2− ABC after 1–2 lines of prior chemotherapy.
Table 5 outlines the ongoing trials of ADCs being evaluated for HR + /HER2− or HER2-low ABC. While ADCs appear promising, different ADCs vary considerably in terms of potency and toxicity profiles. Many questions remain to be explored—the optimal predictive biomarker, the sequencing of one ADC after another, the mechanisms of resistance and the cross-resistance between ADCs with similar target antigen or payload, among others. In view of the promising activity, phase 3 trials comparing T-DXd or SG with chemotherapy of the physician’s choice in the first-line chemotherapy setting are ongoing. Apart from improvements in survival outcomes, the impact on cumulative toxicities and quality of life (QOL) will be important, especially for less heavily pretreated patients, who will be receiving the treatment over a longer period of time. Cost-effectiveness, financial toxicities and inequities in access should also be considered.
Targeting the homologous recombination repair pathway
BRCA1/2 tumor suppressor genes are involved in the homologous recombination repair (HRR) pathway. HR+ BCs arising in germline BRCA (gBRCA) mutation carriers frequently exhibit genomic instability with higher histological grade and Oncotype Dx recurrence score than sporadic tumors114. gBRCA pathogenic variants were also less likely to co-occur with PIK3CA somatic mutations in a recent study115, suggesting distinct tumor biology. Real-world studies have reported inferior PFS and OS on ET with CDK4/6i in patients with germline pathogenic variants in DNA repair genes116,117, reflecting an area of unmet treatment need.
PARP inhibition in cells with homologous recombination deficiency leads to synthetic lethality. Olaparib and talazoparib are approved for use in patients with gBRCA mutations and HER2- ABC in the post-chemotherapy setting following the OlympiAD118 and EMBRCA119 trials which demonstrated PFS benefit over physician’s choice of chemotherapy. HR + /HER2− patients accounted for around half of the population in both trials; absolute mPFS improvement was around 3 months with HR 0.54–0.58 over TPC, albeit without OS benefit.
In the phase II TBCRC048 trial, 54 patients with ABC and germline or somatic mutations in HRR pathway genes other than gBRCA mutations (such as PALB2, somatic BRCA1/2, ATM, or CHEK2) received olaparib120. Out of 19 HR + /HER2− patients who had received prior CDK4/6i, 58% achieved a response. Activity was also reported in the smaller Talazoparib Beyond BRCA trial121. An ongoing phase II trial (NCT03990896) is recruiting patients with HR + /HER2− and triple-negative ABC with somatic BRCA1/2 mutations to investigate the effectiveness of talazoparib.
Mechanisms of resistance to PARP inhibitors include target-related effects (e.g., mutations in PARP, upregulation of drug efflux pumps), reversion mutations restoring BRCA-dependent homologous recombination, and loss of DNA end-protection and restoration of replication fork stability122. The latest research efforts are focusing on the next generation of selective PARP1 inhibitors which may be less myelotoxic, other agents targeting the DNA repair pathway, synergistic combinations and identification of predictive biomarkers beyond gBRCA mutations.
Targeting other components of the cell cycle and cell death
Cyclin E1 (CCNE1) drives the progression of cells from G1 into S and M phases of the cell cycle by binding to and activating cyclin-dependent kinase 2 (CDK2). High CCNE1 mRNA expression was associated with relative resistance to palbociclib in the exploratory biomarker analysis of PALOMA 3 with shorter PFS, and poorer anti-proliferative activity in the Preoperative Palbociclib (POP) trial123. Activation of the MYC oncogene has also been identified as a mechanism of resistance to palbociclib via compensatory CDK2 activation; CDK2 inhibition suppressed cellular proliferation in vitro124. BLU222, a selective CDK2 inhibitor, showed sustained antitumor activity in combination with ribociclib in in vivo HR + /HER2− BC models125. Early-phase trials on BLU222 and PF3600 (CDK2/4/6i) in advanced solid tumors including HR + /HER2− ABC are ongoing (NCT05252416, NCT03519178).
Sensitivity of cell lines with overactivation of CDK2 in S phase to checkpoint kinase 1 (CHK1) inhibition has been reported. CHK1 is activated during DNA damage, and facilitates cell cycle arrest to permit DNA damage repair; CHK1 inhibition thus allows sensitive cells to accumulate DNA breaks, leading to cytotoxicity126. To our knowledge, there is currently no clinical data in HR + /HER2− BC.
CDK7 is integral in (i) initiation and regulation of gene transcription via activation of RNA polymerase II, (ii) regulating the activity of transcription factors including AR and ER, and (iii) directing cell cycle progression via phosphorylation of other CDKs127. Samuraciclib (ICEC0942), a CDK7 inhibitor, induced cell cycle arrest and apoptosis in a preclinical study128. Subsequently, samuraciclib with fulvestrant showed a 24-week CBR of 36% in a single-arm study of 31 patients with HR + /HER2− ABC post progression on AI plus CDK4/6i, leading to FDA granting fast-track status for the compound129. Interestingly, tumors with TP53 mutation have a significantly shorter mPFS than tumors with wild-type TP53 (HR 0.17, P = 0.0008), consistent with findings that TP53 activation is essential to induce transcriptional dependency, rendering the cancer cells susceptible to CDK7 inhibition130. Hence there may be limitations with CDK7 inhibitors in aggressive luminal tumors that harbor TP53 alterations.
Aurora-A kinase (AURKA) is a serine/threonine kinase important for the G2/mitosis transition, regulating centrosome function and mitotic spindle assembly131. Amplification or overexpression of AURKA enhances its role in tumorigenesis, and potentially serves as an antitumor target12. AURKA inhibition mediated mitotic arrest and apoptosis, exhibiting synthetic lethality with RB1 loss in vitro and in vivo12,132. In an older phase I/II trial, alisertib, an AURKA inhibitor, demonstrated 23% ORR with mPFS of 7.9 months in 26 patients with HR + /HER2− ABC133. The addition of alisertib to weekly paclitaxel improved mPFS from 7.1 months with paclitaxel alone to 10.2 months (HR 0.56, P = 0.005) in the HR + /HER2− ABC cohort of a randomized phase II trial134. However, grade 3/4 stomatitis or myelosuppression limited the tolerability of the combination134. A recent randomized phase II trial on 91 patients with prior CDK4/6i and ET showed that addition of fulvestrant to alisertib did not increase the ORR (19.6% in monotherapy arm versus 20.0% in combination arm). Nevertheless, the treatment was well-tolerated and the activity of alisertib monotherapy was considered promising in this pretreated population135.
Induction of apoptosis is another potential strategy, given that BCL2, an estrogen-responsive anti-apoptosis protein, is overexpressed in 85% of HR + /HER2− BC136. Preclinical models of BCL2 expressing HR + /HER2− cancer cells shows enhanced apoptosis with the use of a BCL2 inhibitor ABT-199 (venetoclax) in combination with tamoxifen136, as well as triple therapy of venetoclax with fulvestrant and palbociclib137. However, while the activity of venetoclax with tamoxifen appeared promising in a phase 1B trial with an ORR of 54% and CBR 75% for 24 patients with ER + BCL2 + ABC treated at RP2D138; the addition of venetoclax to fulvestrant in the randomized phase II VERONICA trial failed to show CBR or PFS benefit over fulvestrant alone in HR + /HER2− ABC post CDK4/6i139. The increased BCLXL expression observed post CDK4/6i may have resulted in reduced dependency on BCL2 to evade apoptosis139; targeting BCLXL alone or in combination with BCL2 may merit further investigation.
The spindle assembly checkpoint kinase TTK, also known as monopolar spindle 1(MPS1), is a key regulator of chromosomal segregation. CDK4/6i-resistant ER + ABC cells with RB1 loss harbor mitotic defects and are hypersensitive to TTK inhibitor CFI-402257, with induction of premature chromosome segregation, DNA damage, and cell death140. CFI-402257 is being tested in a phase I/II study, with an HR + /HER2− ABC cohort receiving combination with fulvestrant post CDK4/6i (NCT05251714).
Immunotherapy
While immune checkpoint inhibitors (ICIs) have demonstrated efficacy and transformed the treatment paradigms in TNBC, the efficacy of single-agent PD-1 or PD-L1 blockade in HR + /HER2− ABC has been limited141,142. Possible reasons include the lower levels of tumor infiltrating lymphocytes (TILs)143, lower tumor mutational burden (TMB) and lower frequency of programmed death-ligand 1 (PD-L1) expression. Biomarkers predicting for response to immunotherapy include positive PD-L1 status142, high TMB144, APOBEC mutational signatures which have been associated with less favorable responses to ET with CDK4/6i145,146, and mismatch repair deficiency which is observed in only 1% of BCs147.
Combining immunotherapy with other agents such as ET, small molecule inhibitors, chemotherapy or ADCs have the potential to augment tumor response to immunotherapy. Major challenges remain in overcoming resistance to immunotherapy. In preclinical studies of HR + BC, abemaciclib was able to increase tumor immunogenicity with evidence of suppression of regulatory T-cell proliferation, increased expression of antigen presentation genes, and synergism with PD-1 blockade148. However, the high rates of toxicities such as grade ≥3 transaminitis have prevented further development of such combinations149,150,151. Other promising preclinical data using histone deacetylase inhibitors as a priming modulator for immunotherapy152 also did not translate into meaningful clinical efficacy when used in combination with ICI153.
Although chemotherapy has the potential to stimulate cytotoxic T-cell activation and deplete regulatory T cells with induction of cell death and release of tumor-related antigens154, a phase II randomized trial investigating the addition of pembrolizumab to eribulin compared to eribulin alone in HR + /HER2− ABC did not improve PFS, ORR or OS155. A randomized phase III trial is currently investigating the addition of pembrolizumab to investigator’s choice of chemotherapy in patients with PD-L1 CPS-positive, CDK4/6i-resistant HR + /HER2− ABC who have not received prior chemotherapy (NCT04895358), presumably with less immune exhaustion.
Discussion: future directions
In summary, while various systemic therapeutic strategies have emerged in the post-CDK4/6i era, HR + /HER2− ABC remains incurable. There is a pressing need to improve our understanding of resistance mechanisms to not only ET and CDK4/6i, but also other agents discussed in this review. With the loss of commercial interest in developing certain compounds after failure to meet the clinical efficacy endpoint(s), efforts to interrogate the discordance between preclinical and clinical activity have stalled, missing the opportunity to elucidate the biological underpinnings. Treatment resistance is often complicated by dynamic adaptive changes, tumor heterogeneity and toxicities affecting dose delivery in the clinical setting. Loss of ER expression and change in HER2 expression may also occur12,156,157. Given the diverse mechanisms of resistance, a one-size-fits-all approach may not always be appropriate. Tumor profiling upon progression may help to characterize the latest biology, and assist in the development of novel therapies to circumvent the various mechanisms of resistance in the optimal sequence (Fig. 2).
Optimal sequencing of treatment options depends on (1) the presence of specific molecular aberrations at the specific timepoint, such as acquired ESR1 mutations, MAP kinase pathway alterations; (2) the comparative efficacy of selected treatment relative to current gold standard treatment paradigms; (3) the setting for which clinical efficacy of specific treatment is proven in adequately powered clinical trial(s); and (4) the balance between maximizing patient-derived survival benefits versus QOL, financial and other toxicities, when compared to alternative therapy options in the patient’s overall breast cancer treatment journey. While earlier introduction of a highly efficacious drug may prolong PFS and OS to a greater extent, the impact on long-term toxicities, including financial burden, cost-effectiveness and QOL, should be considered.
The SAFIR02-BREAST trial examined the maintenance strategy of matched targeted therapies to genomic alterations versus standard-of-care chemotherapy in patients with HER2- ABC. Although PFS improvement was observed with matched targeted therapies only for genomic alterations classified as level I/II according to the ESMO Scale for Clinical Actionability of Molecular Targets (ESCAT)158, this may change with the rapidly evolving treatment landscape. ESR1 mutation testing at recurrence or progression on ET in HR + /HER2− ABC has now been incorporated into the latest ASCO guidelines45, and evaluation of other tumor genomic mutations with the option of ctDNA testing may be increasingly adopted as biomarkers become prospectively validated in clinical studies62,45. With the increasing discovery of molecular resistance drivers, the development of novel therapeutics will need to be accelerated, ideally with greater inclusion and diversity in clinical trials. While preliminary results from some early-phase studies may seem promising, adequately powered randomized clinical trials will generally still be required to confirm the efficacy over current standard of care to transform our clinical practice. Patients with resistant or refractory disease continue to be under-represented in industry-sponsored registration trials which focus on less heavily pretreated patients with better prognosis for achieving the primary endpoints. Moving forward, concerted efforts to design and conduct academic trials are instrumental for addressing the unmet needs of various challenging patient subpopulations and dissecting complex biology.
Conclusion
Currently, ET with CDK4/6i is the standard of care for most patients with HR + /HER2− MBC in the first-line setting, though the treatment paradigm for patients post relapse on adjuvant CDK4/6i is unclear. Based on our understanding of the diverse mechanisms of resistance post tumor progression on ET and CDK4/6i, a personalized rather than one-size-fits-all approach will be the optimal strategy. Molecular profiling at time of progression helps to elucidate the specific molecular aberrations and activated pathways, allowing for better tailoring of systemic therapy. Ongoing efforts to identify new therapeutic targets with predictive biomarkers and development of novel therapies will continue to shape the treatment paradigm in HR + /HER2− MBC.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
References
Giaquinto, A. N. et al. Breast cancer statistics, 2022. CA Cancer J. Clin. 72, 524–541 (2022).
Gennari, A. et al. ESMO Clinical Practice Guideline for the diagnosis, staging and treatment of patients with metastatic breast cancer. Ann. Oncol. 32, 1475–1495 (2021).
Burstein, H. J. et al. Endocrine treatment and targeted therapy for hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer: ASCO guideline update. J. Clin. Oncol. 39, 3959–3977 (2021).
Lu, Y. S. et al. Updated overall survival of ribociclib plus endocrine therapy versus endocrine therapy alone in pre- and perimenopausal patients with HR+/HER2- advanced breast cancer in MONALEESA-7: a phase III randomized clinical trial. Clin. Cancer Res. 28, 851–859 (2022).
Hortobagyi, G. N. et al. Overall survival with ribociclib plus letrozole in advanced breast cancer. N. Engl. J. Med. 386, 942–950 (2022).
Sledge, G. W. Jr et al. The effect of abemaciclib plus fulvestrant on overall survival in hormone receptor-positive, ERBB2-negative breast cancer that progressed on endocrine therapy—MONARCH 2: a randomized clinical trial. JAMA Oncol. 6, 116–124 (2020).
Slamon, D. J. et al. Ribociclib plus fulvestrant for postmenopausal women with hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer in the phase III randomized MONALEESA-3 trial: updated overall survival. Ann. Oncol. 32, 1015–1024 (2021).
Harbeck, N. et al. Adjuvant abemaciclib combined with endocrine therapy for high-risk early breast cancer: updated efficacy and Ki-67 analysis from the monarchE study. Ann. Oncol. 32, 1571–1581 (2021).
Tutt, A. N. J. et al. Adjuvant olaparib for patients with BRCA1- or BRCA2-mutated breast cancer. N. Engl. J. Med. 384, 2394–2405 (2021).
Toy, W. et al. Activating ESR1 mutations differentially affect the efficacy of ER antagonists. Cancer Discov. 7, 277–287 (2017).
Razavi, P. et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell 34, 427–438.e6 (2018).
Wander, S. A. et al. The genomic landscape of intrinsic and acquired resistance to cyclin-dependent kinase 4/6 inhibitors in patients with hormone receptor-positive metastatic breast cancer. Cancer Discov. 10, 1174–1193 (2020).
Falato, C., Schettini, F., Pascual, T., Brasó-Maristany, F. & Prat, A. Clinical implications of the intrinsic molecular subtypes in hormone receptor-positive and HER2-negative metastatic breast cancer. Cancer Treat. Rev. 112, 102496 (2023).
Prat, A. et al. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast 24, S26–S35 (2015).
Prat, A. et al. Correlative biomarker analysis of intrinsic subtypes and efficacy across the MONALEESA phase III studies. J. Clin. Oncol. 39, 1458–1467 (2021).
Lloyd, M. R., Spring, L. M., Bardia, A. & Wander, S. A. Mechanisms of resistance to CDK4/6 blockade in advanced hormone receptor-positive, HER2-negative breast cancer and emerging therapeutic opportunities. Clin. Cancer Res. 28, 821–830 (2022).
Watt, A. C. & Goel, S. Cellular mechanisms underlying response and resistance to CDK4/6 inhibitors in the treatment of hormone receptor-positive breast cancer. Breast Cancer Res. 24, 17 (2022).
Hanker, A. B., Sudhan, D. R. & Arteaga, C. L. Overcoming endocrine resistance in breast cancer. Cancer Cell 37, 496–513 (2020).
Cardoso, F. et al. 5th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 5). Ann. Oncol. 31, 1623–1649 (2020).
Hanahan, D. Hallmarks of cancer: new dimensions. Cancer Discov. 12, 31–46 (2022).
Jeselsohn, R. et al. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin. Cancer Res. 20, 1757–1767 (2014).
Brett, J. O., Spring, L. M., Bardia, A. & Wander, S. A. ESR1 mutation as an emerging clinical biomarker in metastatic hormone receptor-positive breast cancer. Breast Cancer Res. 23, 85 (2021).
Bidard, F. C. et al. Switch to fulvestrant and palbociclib versus no switch in advanced breast cancer with rising —ESR1—mutation during aromatase inhibitor and palbociclib therapy (PADA-1): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 23, 1367–1377 (2022).
Bidard, F. C. et al. Elacestrant (oral selective estrogen receptor degrader) versus standard endocrine therapy for estrogen receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: results from the randomized phase III EMERALD trial. J. Clin. Oncol. 40, 3246–3256 (2022).
Toy, W. et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 45, 1439–1445 (2013).
Zinger, L. et al. Ligand-binding domain-activating mutations of ESR1 rewire cellular metabolism of breast cancer cells. Clin. Cancer Res. 25, 2900–2914 (2019).
Jeselsohn, R. et al. Allele-specific chromatin recruitment and therapeutic vulnerabilities of ESR1 activating mutations. Cancer Cell 33, 173–186.e5 (2018).
Andreano, K. J. et al. The dysregulated pharmacology of clinically relevant ESR1 mutants is normalized by ligand-activated WT receptor. Mol. Cancer Ther. 19, 1395–1405 (2020).
Lainé, M. et al. Lasofoxifene as a potential treatment for therapy-resistant ER-positive metastatic breast cancer. Breast Cancer Res. 23, 54 (2021).
Goetz, M. P. et al. Open-label, randomized study of lasofoxifene (LAS) vs fulvestrant (Fulv) for women with locally advanced/metastatic ER+/HER2- breast cancer (mBC), an estrogen receptor 1 (ESR1) mutation, and disease progression on aromatase (AI) and cyclin-dependent kinas. Ann. Oncol. 33, S808–S869 (2022).
Damodaran, S., Plourde, P. V., Moore, H. C. F., Anderson, I. C. & Portman, D. J. Open-label, phase 2, multicenter study of lasofoxifene (LAS) combined with abemaciclib (Abema) for treating pre- and postmenopausal women with locally advanced or metastatic ER+/HER2− breast cancer and an ESR1 mutation after progression on prior therapies. J. Clin. Oncol. 40, 1022–1022 (2022).
Fribbens, C. et al. Plasma ESR1 mutations and the treatment of estrogen receptor-positive advanced breast cancer. J. Clin. Oncol. 34, 2961–2968 (2016).
van Kruchten, M. et al. Measuring residual estrogen receptor availability during fulvestrant therapy in patients with metastatic breast cancer. Cancer Discov. 5, 72–81 (2015).
Qi, S. M. et al. PROTAC: an effective targeted protein degradation strategy for cancer therapy. Front. Pharmacol. 12, 692574 (2021).
Puyang, X. et al. Discovery of selective estrogen receptor covalent antagonists for the treatment of ERα(WT) and ERα(MUT) breast cancer. Cancer Discov. 8, 1176–1193 (2018).
Schott, A. F. et al. ARV-471, A PROTAC® Estrogen Receptor (ER) Degrader in Advanced ER-Positive/human Epidermal Growth Factor Receptor 2 (HER2)-Negative Breast Cancer: Phase 2 Expansion (VERITAC) of a Phase 1/2 Study (San Antonio Breast Cancer Symposium, 2022).
Oliveira, M. et al. Serena-1: updated analyses from a phase 1 study (parts C/D) of the next-generation oral SERD camizestrant (AZD9833) in combination with palbociclib, in women with ER-positive, HER2-negative advanced breast cancer. J. Clin. Oncol. 40, 1032–1032 (2022).
Tripathy, D. et al. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): a randomised phase 3 trial. Lancet Oncol. 19, 904–915 (2018).
Ferraro, E., Walsh, E. M., Tao, J. J., Chandarlapaty, S. & Jhaveri, K. Accelerating drug development in breast cancer: new frontiers for ER inhibition. Cancer Treat. Rev. 109, 102432 (2022).
Scott, J. S. et al. Discovery of AZD9833, a potent and orally bioavailable selective estrogen receptor degrader and antagonist. J. Med Chem. 63, 14530–14559 (2020).
Oliveira, M. et al. Camizestrant, A Next-generation Oral SERD vs Fulvestrant in Post-menopausal Women with Advanced ER-Positive HER2-Negative Breast Cancer: Results of the Randomized, Multi-dose Phase 2 SERENA-2 Trial (San Antonio Breast Cancer Symposium, 2022).
Tolaney, S. M. et al. AMEERA-3: Randomized Phase II Study of Amcenestrant (Oral Selective Estrogen Receptor Degrader) Versus Standard Endocrine Monotherapy in Estrogen Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer. J Clin Oncol. 41, 4014–4024 (2023).
Martin, M. et al. Giredestrant (GDC-9545) vs physician choice of endocrine monotherapy (PCET) in patients (pts) with ER+, HER2– locally advanced/metastatic breast cancer (LA/mBC): primary analysis of the phase II, randomised, open-label acelERA BC study. Ann. Oncol. 33, S88–S121 (2022).
McDonnell, D. P., Wardell, S. E., Chang, C. Y. & Norris, J. D. Next-generation endocrine therapies for breast cancer. J. Clin. Oncol. 39, 1383–1388 (2021).
Burstein, H. J. et al. Testing for ESR1 mutations to guide therapy for hormone receptor–positive, human epidermal growth factor receptor 2–negative metastatic breast cancer: ASCO guideline rapid recommendation update. J. Clin. Oncol. 41, 3423–3425 (2023).
Albanell, J. et al. Palbociclib rechallenge for hormone receptor-positive/HER-negative advanced breast cancer: findings from the phase II BioPER trial. Clin. Cancer Res. 29, 67–80 (2023).
Mayer, E. et al. Palbociclib After CDK4/6i and Endocrine Therapy (PACE): A Randomized Phase II Study of Fulvestrant, Palbociclib, and Avelumab for Endocrine Pre-treated ER + /HER2- Metastatic Breast Cancer (San Antonio Breast Cancer Symposium, 2022).
Kalinsky, K. et al. Randomized phase II trial of endocrine therapy with or without ribociclib after progression on cyclin-dependent kinase 4/6 inhibition in hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer: MAINTAIN trial. J. Clin. Oncol. 41, 4004–4013 (2023).
Ricciardelli, C. et al. The magnitude of androgen receptor positivity in breast cancer is critical for reliable prediction of disease outcome. Clin. Cancer Res. 24, 2328–2341 (2018).
Hickey, T. E. et al. The androgen receptor is a tumor suppressor in estrogen receptor-positive breast cancer. Nat. Med. 27, 310–320 (2021).
Krop, I. et al. A randomized placebo controlled phase II trial evaluating exemestane with or without enzalutamide in patients with hormone receptor-positive breast cancer. Clin. Cancer Res. 26, 6149–6157 (2020).
Wei, L. et al. Pharmacological targeting of androgen receptor elicits context-specific effects in estrogen receptor-positive breast cancer. Cancer Res. 83, 456–470 (2023).
Palmieri, C. et al. Efficacy of enobosarm, a selective androgen receptor (AR) targeting agent, correlates with the degree of AR positivity in advanced AR+/estrogen receptor (ER)+ breast cancer in an international phase 2 clinical study. J. Clin. Oncol. 39, 1020–1020 (2021).
Campbell, R. A. et al. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: a new model for anti-estrogen resistance. J. Biol. Chem. 276, 9817–9824 (2001).
Miller, T. W. et al. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J. Clin. Investig. 120, 2406–2413 (2010).
Rugo, H. S. et al. Biology and targetability of the extended spectrum of PIK3CA mutations detected in breast carcinoma. Clin. Cancer Res. 29, 1056–1067 (2023).
Bertucci, F. et al. Genomic characterization of metastatic breast cancers. Nature 569, 560–564 (2019).
André, F. et al. Alpelisib for PIK3CA-mutated, hormone receptor–positive advanced breast cancer. N. Engl. J. Med. 380, 1929–1940 (2019).
Howell, S. J. et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive, HER2-negative breast cancer (FAKTION): overall survival, updated progression-free survival, and expanded biomarker analysis from a randomised, phase 2 trial. Lancet Oncol. 23, 851–864 (2022).
Mosele, F. et al. Outcome and molecular landscape of patients with PIK3CA-mutated metastatic breast cancer. Ann. Oncol. 31, 377–386 (2020).
Tolaney, S. M. et al. Clinical significance of PIK3CA and ESR1 mutations in circulating tumor DNA: analysis from the MONARCH 2 study of abemaciclib plus fulvestrant. Clin. Cancer Res. 28, 1500–1506 (2022).
Henry, N. L. et al. Biomarkers for systemic therapy in metastatic breast cancer: ASCO guideline update. J. Clin. Oncol. 40, 3205–3221 (2022).
Narayan, P. et al. FDA approval summary: alpelisib plus fulvestrant for patients with HR-positive, HER2-negative, PIK3CA-mutated, advanced or metastatic breast cancer. Clin. Cancer Res. 27, 1842–1849 (2021).
André, F. et al. Alpelisib plus fulvestrant for PIK3CA-mutated, hormone receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: final overall survival results from SOLAR-1. Ann. Oncol. 32, 208–217 (2021).
Rugo, H. S. et al. Alpelisib plus fulvestrant in PIK3CA-mutated, hormone receptor-positive advanced breast cancer after a CDK4/6 inhibitor (BYLieve): one cohort of a phase 2, multicentre, open-label, non-comparative study. Lancet Oncol. 22, 489–498 (2021).
Rugo, H. S. et al. Abstract PD2-07: Alpelisib + letrozole in patients with PIK3CA-mutated, hormone-receptor positive (HR+), human epidermal growth factor receptor-2-negative (HER2-) advanced breast cancer (ABC) previously treated with a cyclin-dependent kinase 4/6 inhibitor (CDK4/6i) + fulvestrant: BYLieve study results. Cancer Res. 81, PD2-07 (2021).
Rugo, H. S. et al. Abstract PD13-05: Alpelisib + fulvestrant in patients with PIK3CA-mutated, HR+, HER2- advanced breast cancer (ABC) who received chemotherapy or endocrine therapy (ET) as immediate prior treatment: BYLieve Cohort C primary results and exploratory biomarker analyses. Cancer Res. 82, PD13-05-PD13-05 (2022).
Borrego, M. R., et al. Abstract PD8-02: metformin (MET) for the prevention of Alpelisib (ALP)-related hyperglycemia (HG) in PIK3CA-mutated, hormone receptor-positive (HR[+]) HER2-negative (HER2[-]) advanced breast cancer (ABC): the METALLICA study. Cancer Res. 83, PD8-02-PD8-02 (2023).
Cai, Y. et al. Genomic alterations in PIK3CA-mutated breast cancer result in mTORC1 activation and limit the sensitivity to PI3Kalpha inhibitors. Cancer Res. 81, 2470–2480 (2021).
Vasan, N. & Cantley, L. C. At a crossroads: how to translate the roles of PI3K in oncogenic and metabolic signalling into improvements in cancer therapy. Nat. Rev. Clin. Oncol. 19, 471–485 (2022).
Song, K. W. et al. RTK-dependent inducible degradation of mutant PI3Kα drives GDC-0077 (inavolisib) efficacy. Cancer Discov. 12, 204–219 (2022).
Juric, D. et al. A Phase 1 Trial of LOXO-783, A Potent, Highly Mutant-selective, Brain-penetrant Allosteric PI3Kα H1047R Inhibitor in PIK3CA H1047R-Mutant Advanced Breast Cancer (aBC) and Other Solid Tumors (PIKASSO-01, Trial in Progress) (San Antonio Breast Cancer Symposium, 2022).
Jones, R. H. et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive breast cancer (FAKTION): a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 21, 345–357 (2020).
Turner, N. et al. Capivasertib in hormone receptor-positive advanced breast cancer. N. Engl. J. Med. 388, 2058–2070 (2023).
Turner, N. et al. Ipatasertib plus paclitaxel for PIK3CA/AKT1/PTEN-altered hormone receptor-positive HER2-negative advanced breast cancer: primary results from cohort B of the IPATunity130 randomized phase 3 trial. Breast Cancer Res. Treat. 191, 565–576 (2022).
Vora, S. R. et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell 26, 136–149 (2014).
Bardia, A. et al. Phase I/II trial of triplet therapy (exemestane, ribociclib, and everolimus) after progression on a CDK4/6 inhibitor in HR+/HER2– advanced breast cancer (TRINITI-1). Clin. Cancer Res. 27, 4177–4185 (2021).
Tolaney, S. M. et al. Phase Ib study of ribociclib plus fulvestrant and ribociclib plus fulvestrant plus PI3K inhibitor (alpelisib or buparlisib) for HR(+) advanced breast cancer. Clin. Cancer Res. 27, 418–428 (2021).
Creighton, C. J. et al. Activation of mitogen-activated protein kinase in estrogen receptor alpha-positive breast cancer cells in vitro induces an in vivo molecular phenotype of estrogen receptor alpha-negative human breast tumors. Cancer Res. 66, 3903–3911 (2006).
Zaman, K. et al. Fulvestrant with or without selumetinib, a MEK 1/2 inhibitor, in breast cancer progressing after aromatase inhibitor therapy: a multicentre randomised placebo-controlled double-blind phase II trial, SAKK 21/08. Eur. J. Cancer 51, 1212–1220 (2015).
Formisano, L. et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat. Commun. 10, 1373 (2019).
Drago, J. Z. et al. FGFR1 amplification mediates endocrine resistance but retains TORC sensitivity in metastatic hormone receptor-positive (HR(+)) breast cancer. Clin. Cancer Res. 25, 6443–6451 (2019).
Mao, P. et al. Acquired FGFR and FGF alterations confer resistance to estrogen receptor (ER) targeted therapy in ER(+) metastatic breast cancer. Clin. Cancer Res. 26, 5974–5989 (2020).
Musolino, A. et al. Phase II, randomized, placebo-controlled study of dovitinib in combination with fulvestrant in postmenopausal patients with HR(+), HER2(-) breast cancer that had progressed during or after prior endocrine therapy. Breast Cancer Res. 19, 18 (2017).
Hui, R. et al. Lucitanib for the treatment of HR(+)/HER2(-) metastatic breast cancer: results from the multicohort phase II FINESSE study. Clin. Cancer Res. 26, 354–363 (2020).
Coombes, R. C. et al. Results of the phase IIa RADICAL trial of the FGFR inhibitor AZD4547 in endocrine resistant breast cancer. Nat. Commun. 13, 3246 (2022).
Mayer, I. A. et al. A phase Ib trial of fulvestrant + CDK4/6 inhibitor (CDK4/6i) palbociclib + pan-FGFR tyrosine kinase inhibitor (TKI) erdafitinib in FGFR-amplified/ ER+/ HER2-negative metastatic breast cancer (MBC). Cancer Res. 81, PD1–PD03 (2021).
Lim, J. S. J. et al. Phase Ib/II dose expansion study of lenvatinib combined with letrozole in postmenopausal women with hormone receptor-positive breast cancer. Clin. Cancer Res. 28, 2248–2256 (2022).
Nayar, U. et al. Acquired HER2 mutations in ER(+) metastatic breast cancer confer resistance to estrogen receptor-directed therapies. Nat. Genet. 51, 207–216 (2019).
Bose, R. et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 3, 224–237 (2013).
Croessmann, S. et al. Combined blockade of activating ERBB2 mutations and ER results in synthetic lethality of ER+/HER2 mutant breast cancer. Clin. Cancer Res. 25, 277–289 (2019).
Ma, C. X. et al. The Phase II MutHER study of neratinib alone and in combination with fulvestrant in HER2-mutated, non-amplified metastatic breast cancer. Clin. Cancer Res. 28, 1258–1267 (2022).
Jhaveri, K. et al. Neratinib + Fulvestrant+ Trastuzumab for Hormone-receptor Positive, HER2-Mutant Metastatic Breast Cancer, And Neratinib + Trastuzumab for Metastatic Triple-negative Disease: Latest Updates from the SUMMIT Trial (San Antonio Breast Cancer Symposium, 2021).
Li, B. T. et al. Trastuzumab deruxtecan in HER2-mutant non-small-cell lung cancer. N. Engl. J. Med. 386, 241–251 (2021).
Drago, J. Z., Modi, S. & Chandarlapaty, S. Unlocking the potential of antibody-drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 18, 327–344 (2021).
Modi, S. et al. Trastuzumab deruxtecan in previously treated HER2-low advanced breast cancer. N. Engl. J. Med. 387, 9–20 (2022).
Ogitani, Y. et al. DS-8201a, a novel HER2-targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T-DM1. Clin. Cancer Res. 22, 5097–5108 (2016).
Tarantino, P. et al. HER2-low breast cancer: pathological and clinical landscape. J. Clin. Oncol. 38, 1951–1962 (2020).
Mosele, F. et al. Trastuzumab deruxtecan in metastatic breast cancer with variable HER2 expression: the phase 2 DAISY trial. Nat Med. 29, 2110–2120 (2023).
Schettini, F. et al. Clinical, pathological, and PAM50 gene expression features of HER2-low breast cancer. NPJ Breast Cancer 7, 1 (2021).
Fernandez, A. I. et al. Examination of low ERBB2 protein expression in breast cancer tissue. JAMA Oncol. 8, 607–610 (2022).
Sajjadi, E. et al. Pathological identification of HER2-low breast cancer: tips, tricks, and troubleshooting for the optimal test. Front. Mol. Biosci. 10, 1176309 (2023).
Moutafi, M. et al. Quantitative measurement of HER2 expression to subclassify ERBB2 unamplified breast cancer. Lab. Investig. 102, 1101–1108 (2022).
Haikala, H. J. P. 30 years of HER3: from basic biology to therapeutic interventions. Clin. Cancer Res. 27, 3528–3539 (2021).
Da Silva, L., Simpson, PT., Smart, CE., Cocciardi, S., Waddell, N. & Lane, A. et al. HER3 and downstream pathways are involved in colonization of brain metastases from breast cancer. Breast Cancer Res. 12, R46 (2010).
Hashimoto, Y. et al. A novel HER3-targeting antibody-drug conjugate, U3-1402, exhibits potent therapeutic efficacy through the delivery of cytotoxic payload by efficient internalization. Clin. Cancer Res. 25, 7151–7161 (2019).
Ian, K. et al. Results from the phase 1/2 study of patritumab deruxtecan, a HER3-directed antibody-drug conjugate (ADC), in patients with HER3-expressing metastatic breast cancer (MBC). J. Clin. Oncol. 40, 1002–1002 (2022).
Zaman, S., Jadid, H., Denson, A. C. & Gray, J. E. Targeting Trop-2 in solid tumors: future prospects. Onco Targets Ther. 12, 1781–1790 (2019).
Bardia, A. et al. Sacituzumab govitecan in metastatic triple-negative breast cancer. N. Engl. J. Med. 384, 1529–1541 (2021).
Rugo, H. S. et al. Sacituzumab govitecan in hormone receptor-positive/human epidermal growth factor receptor 2-negative metastatic breast cancer. J. Clin. Oncol. 40, 3365–3376 (2022).
Rugo, H. S. et al. Overall survival (OS) results from the phase III TROPiCS-02 study of sacituzumab govitecan (SG) vs treatment of physician’s choice (TPC) in patients (pts) with HR+/HER2- metastatic breast cancer (mBC). Ann. Oncol. 33, S808–S869 (2022).
Rugo, H. S. et al. Sacituzumab Govitecan (SG) vs Treatment of Physician’s Choice (TPC): Efficacy by Trop-2 Expression in the TROPiCS-02 Study of Patients (Pts) With HR + /HER2– Metastatic Breast Cancer (mBC) (San Antonio Breast Cancer Symposium, 2022).
Meric-Bernstam, F. et al. Abstract PD13-08: PD13-08 phase 1 TROPION-PanTumor01 study evaluating datopotamab deruxtecan (Dato-DXd) in unresectable or metastatic hormone receptor-positive/HER2–negative breast cancer (BC). Cancer Res. 83, PD13-08-PD13-08 (2023).
Shah, P. D. et al. Twenty-one-gene recurrence score assay in BRCA-associated versus sporadic breast cancers: differences based on germline mutation status. Cancer 122, 1178–1184 (2016).
Park, S. et al. Clinical characteristics and exploratory genomic analyses of germline BRCA1 or BRCA2 mutations in breast cancer. Mol. Cancer Res. 18, 1315–1325 (2020).
Park, S. Y. et al. Prognostic role of tumor subtype and germline BRCA mutation in advanced breast cancer patients treated with palbociclib plus endocrine therapy. Breast Cancer Res. Treat. 196, 121–128 (2022).
Bruno, L. et al. Cyclin-dependent kinase 4/6 inhibitor outcomes in patients with advanced breast cancer carrying germline pathogenic variants in DNA repair-related genes. JCO Precis. Oncol. 6, e2100140 (2022).
Robson, M. et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N. Engl. J. Med. 377, 523–533 (2017).
Litton, J. K. et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N. Engl. J. Med. 379, 753–763 (2018).
Tung, N. M. et al. TBCRC 048: phase II study of olaparib for metastatic breast cancer and mutations in homologous recombination-related genes. J. Clin. Oncol. 38, 4274–4282 (2020).
Gruber, J. J. et al. A phase II study of talazoparib monotherapy in patients with wild-type BRCA1 and BRCA2 with a mutation in other homologous recombination genes. Nat. Cancer 3, 1181–1191 (2022).
Dias, M. P., Moser, S. C., Ganesan, S. & Jonkers, J. Understanding and overcoming resistance to PARP inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 18, 773–791 (2021).
Turner, N. C. et al. Cyclin E1 expression and palbociclib efficacy in previously treated hormone receptor-positive metastatic breast cancer. J. Clin. Oncol. 37, 1169–1178 (2019).
Freeman-Cook, K. et al. Expanding control of the tumor cell cycle with a CDK2/4/6 inhibitor. Cancer Cell 39, 1404–1421.e11 (2021).
Brown, V. et al. CDK2 Inhibition With BLU-222 in Combination With Ribociclib Demonstrates Robust Antitumour Activity in Pre-clinical Models of CDK4/6 Inhibitor-naive and Resistant HR + /HER2- Breast Cancer (San Antonio Breast Cancer Symposium, 2022).
Sakurikar, N., Thompson, R., Montano, R. & Eastman, A. A subset of cancer cell lines is acutely sensitive to the Chk1 inhibitor MK-8776 as monotherapy due to CDK2 activation in S phase. Oncotarget 7, 1380–1394 (2016).
Chou, J., Quigley, D. A., Robinson, T. M., Feng, F. Y. & Ashworth, A. Transcription-associated cyclin-dependent kinases as targets and biomarkers for cancer therapy. Cancer Discov. 10, 351–370 (2020).
Patel, H. et al. ICEC0942, an orally bioavailable selective inhibitor of CDK7 for cancer treatment. Mol. Cancer Ther. 17, 1156–1166 (2018).
Coombes, R. C. et al. Dose escalation and expansion cohorts in patients with advanced breast cancer in a Phase I study of the CDK7-inhibitor samuraciclib. Nat Commun. 14, 4444 (2023).
Kalan, S. et al. Activation of the p53 transcriptional program sensitizes cancer cells to Cdk7 inhibitors. Cell Rep. 21, 467–481 (2017).
Tang, A. et al. Aurora kinases: novel therapy targets in cancers. Oncotarget 8, 23937–23954 (2017).
Thrane, S. et al. A kinase inhibitor screen identifies Mcl-1 and Aurora kinase A as novel treatment targets in antiestrogen-resistant breast cancer cells. Oncogene 34, 4199–4210 (2015).
Melichar, B. et al. Safety and activity of alisertib, an investigational aurora kinase A inhibitor, in patients with breast cancer, small-cell lung cancer, non-small-cell lung cancer, head and neck squamous-cell carcinoma, and gastro-oesophageal adenocarcinoma: a five-arm phase 2 study. Lancet Oncol. 16, 395–405 (2015).
O’Shaughnessy, J. et al. Efficacy and safety of weekly paclitaxel with or without oral alisertib in patients with metastatic breast cancer: a randomized clinical trial. JAMA Netw. Open 4, e214103 (2021).
Haddad, T. C. et al. Evaluation of alisertib alone or combined with fulvestrant in patients with endocrine-resistant advanced breast cancer: the phase 2 TBCRC041 randomized clinical trial. JAMA Oncol. 9, 815–824 (2023).
Vaillant, F. et al. Targeting BCL-2 with the BH3 mimetic ABT-199 in estrogen receptor-positive breast cancer. Cancer Cell 24, 120–129 (2013).
Whittle, J. R. et al. Dual targeting of CDK4/6 and BCL2 pathways augments tumor response in estrogen receptor-positive breast cancer. Clin. Cancer Res. 26, 4120–4134 (2020).
Lok, S. W. et al. A phase Ib dose-escalation and expansion study of the BCL2 inhibitor venetoclax combined with tamoxifen in ER and BCL2-positive metastatic breast cancer. Cancer Discov. 9, 354–369 (2019).
Lindeman, G. J. et al. VERONICA: randomized phase II study of fulvestrant and venetoclax in ER-positive metastatic breast cancer post-CDK4/6 inhibitors—efficacy, safety, and biomarker results. Clin. Cancer Res. 28, 3256–3267 (2022).
Soria-Bretones, I. et al. The spindle assembly checkpoint is a therapeutic vulnerability of CDK4/6 inhibitor-resistant ER(+) breast cancer with mitotic aberrations. Sci. Adv. 8, eabq4293 (2022).
Dirix, L. Y. et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: a phase 1b JAVELIN Solid Tumor study. Breast Cancer Res. Treat. 167, 671–686 (2018).
Rugo, H. S. et al. Safety and antitumor activity of pembrolizumab in patients with estrogen receptor-positive/human epidermal growth factor receptor 2-negative advanced breast cancer. Clin. Cancer Res. 24, 2804–2811 (2018).
El Bairi, K. et al. The tale of TILs in breast cancer: a report from The International Immuno-Oncology Biomarker Working Group. NPJ Breast Cancer 7, 150 (2021).
Alva, A. S. et al. Pembrolizumab in Patients With Metastatic Breast Cancer With High Tumor Mutational Burden: Results From The Targeted Agent And Profiling Utilization Registry (TAPUR) study. J. Clin. Oncol. 39, 2443–2451 (2021).
DiMarco, A. V. et al. APOBEC mutagenesis inhibits breast cancer growth through induction of T cell-mediated antitumor immune responses. Cancer Immunol. Res. 10, 70–86 (2022).
Sammons, S. et al. APOBEC mutational signatures in hormone receptor-positive human epidermal growth factor receptor 2-negative breast cancers are associated with poor outcomes on CDK4/6 inhibitors and endocrine therapy. JCO Precis. Oncol. 6, e2200149 (2022).
Davies, H. et al. Whole-genome sequencing reveals breast cancers with mismatch repair deficiency. Cancer Res. 77, 4755–4762 (2017).
Goel, S. et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 548, 471–475 (2017).
Rugo, H. S. et al. Abemaciclib in combination with pembrolizumab for HR+, HER2− metastatic breast cancer: Phase 1b study. NPJ Breast Cancer 8, 118 (2022).
Jerusalem, G. et al. 92MO Neoadjuvant nivolumab (NIVO) + palbociclib (PALBO) + anastrozole (ANA) for estrogen receptor-positive (ER+)/human epidermal growth factor receptor 2-negative (HER2−) primary breast cancer (BC): CheckMate 7A8. Ann. Oncol. 33, S165–S166 (2022).
Herold, C. et al. A Phase 1b Study of the CDK4/6 Inhibitor Ribociclib in Combination with the PD-1 Inhibitor Spartalizumab in Patients with Hormone Receptor-positive Metastatic Breast Cancer (HR + MBC) and Metastatic Ovarian Cancer (MOC) (San Antonio Breast Cancer Symposium, 2019).
Terranova-Barberio, M. et al. HDAC inhibition potentiates immunotherapy in triple negative breast cancer. Oncotarget 8, 114156–114172 (2017).
Terranova-Barberio, M. et al. Exhausted T cell signature predicts immunotherapy response in ER-positive breast cancer. Nat. Commun. 11, 3584 (2020).
Murciano-Goroff, Y. R., Warner, A. B. & Wolchok, J. D. The future of cancer immunotherapy: microenvironment-targeting combinations. Cell Res. 30, 507–519 (2020).
Tolaney, S. M. et al. Effect of eribulin with or without pembrolizumab on progression-free survival for patients with hormone receptor-positive, ERBB2-negative metastatic breast cancer: a randomized clinical trial. JAMA Oncol. 6, 1598–1605 (2020).
Bergeron, A. et al. Anticipating changes in the HER2 status of breast tumours with disease progression-towards better treatment decisions in the new era of HER2-low breast cancers. Br J Cancer. 129, 122–134 (2023).
Miglietta, F. et al. Evolution of HER2-low expression from primary to recurrent breast cancer. NPJ Breast Cancer 7, 137 (2021).
Andre, F. et al. Genomics to select treatment for patients with metastatic breast cancer. Nature 610, 343–348 (2022).
Oliveria, M. et al. Ipatasertib in Combination with Palbociclib and Fulvestrant in Patients with Hormone Receptor-Positive HER2-Negative Advanced Breast Cancer (San Antonio Breast Cancer Symposium, 2021).
Bedard, P. L. et al. A Phase I/Ib Study Evaluating GDC-0077 + Palbociclib (palbo) + Fulvestrant in Patients (pts) with PIK3CA-Mutant (mut), Hormone Receptor-Positive/HER2-Negative Metastatic Breast Cancer (HR + /HER2- mBC) (San Antonio Breast Cancer Symposium, 2020).
Layman, R. et al. Phase Ib Expansion Study of Gedatolisib in Combination with Palbociclib and Endocrine Therapy in Women with ER+ Advanced Breast Cancer (San Antonio Breast Cancer Symposium, 2021).
National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology: Breast Cancer. 23 March 2023. https://www.nccn.org/professionals/physician_gls/pdf/breast.pdf (2023).
Acknowledgements
This work was not supported by any specific grant from any funding agency in the public, commercial or not-for-profit sectors. Y.-S.Y. is supported by Singapore National Medical Research Council (NMRC) Clinician Scientist Award (MOH-CSAINV18nov-0009).
Author information
Authors and Affiliations
Contributions
Conception and design: Y.-S.Y. Collection and assembly of the data: all authors (J.M., J.J.C., C.H.T., and Y.-S.Y.). Data analysis and interpretation: all authors (J.M., J.J.C., C.H.T., and Y.-S.Y.). Manuscript writing: all authors (J.M., J.J.C., C.H.T., and Y.-S.Y.). All authors reviewed the results and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
J.M.—travel, accommodations, expenses: AstraZeneca. J.J.C.—honoraria: AstraZeneca, GlaxoSmithKline, Merck Sharp & Dohme, Novartis, Eisai, DKSH Singapore; Consulting or advisory role: Pfizer, Novartis, AstraZeneca, DKSH Singapore, Merck Sharp & Dohme, GlaxoSmithKline; Research funding: OncoQuest (institution), Bristol-Myers Squibb (institution); travel, accommodations, expenses: Novartis, Merck KGaA, DKSH Singapore, Roche, Pfizer. C.H.T.—none. Y.-S.Y.—honoraria: Novartis, Pfizer, Lilly/DKSH, AstraZeneca, Eisai, MSD, Specialized Therapeutics, Roche; research funding: Merck Sharp & Dohme; Travel, accommodations, expenses: DKSH, AstraZeneca.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ma, J., Chan, J.J., Toh, C.H. et al. Emerging systemic therapy options beyond CDK4/6 inhibitors for hormone receptor-positive HER2-negative advanced breast cancer. npj Breast Cancer 9, 74 (2023). https://doi.org/10.1038/s41523-023-00578-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41523-023-00578-3
This article is cited by
-
A network meta-analysis of efficacy and safety for first-line and second/further-line therapies in postmenopausal women with hormone receptor-positive, HER2-negative, advanced breast cancer
BMC Medicine (2024)
-
The two sides of chromosomal instability: drivers and brakes in cancer
Signal Transduction and Targeted Therapy (2024)
-
CDK4/6i-treated HR+/HER2- breast cancer tumors show higher ESR1 mutation prevalence and more altered genomic landscape
npj Breast Cancer (2024)
