
Abstract
Vaginal microbial composition is associated with differential risk of urogenital infection. Although Lactobacillus spp. are thought to confer protection against infection, the lack of in vivo models resembling the human vaginal microbiota remains a prominent barrier to mechanistic discovery. Using 16S rRNA amplicon sequencing of C57BL/6J female mice, we found that vaginal microbial composition varies within and between colonies across three vivaria. Noting vaginal microbial plasticity in conventional mice, we assessed the vaginal microbiome of humanized microbiota mice (HMbmice). Like the community structure in conventional mice, HMbmice vaginal microbiota clustered into community state types but, uniquely, HMbmice communities were frequently dominated by Lactobacillus or Enterobacteriaceae. Compared to conventional mice, HMbmice were less susceptible to uterine ascension by urogenital pathobionts group B Streptococcus (GBS) and Prevotella bivia. Although Escherichia and Lactobacillus both correlated with the absence of uterine GBS, vaginal pre-inoculation with exogenous HMbmouse-derived E. coli, but not Ligilactobacillus murinus, reduced vaginal GBS burden. Overall, HMbmice serve as a useful model to elucidate the role of endogenous microbes in conferring protection against urogenital pathogens.
Similar content being viewed by others
Introduction
The vaginal microbiota is inextricably tied to women’s urogenital health. Vaginal microbial perturbations are implicated in risk of adverse outcomes including preterm birth, pelvic inflammatory disease, urinary tract infections, and sexually transmitted infections1,2,3,4,5,6,7,8,9. Across geographic, ethnic, and social demographics, the vaginal microbiota in reproductive-age women is largely comprised of Lactobacillus spp. Benefits from Lactobacillus spp. include lactic acid-mediated vaginal acidification, hydrogen peroxide and bacteriocin production, competitive adherence at the vaginal epithelium, and immunomodulatory activity10,11,12,13,14,15,16. While Lactobacillus dominance is canonically considered a hallmark of vaginal health, approximately 1 in 5 women have a more diverse vaginal microbiota with a greater proportion of facultative or strictly anaerobic taxa including Gardnerella vaginalis, Candidatus Lachnocurva vaginae (BVAB1), and Prevotella17,18. Frequently, women with non-Lactobacillus dominant communities are asymptomatic; however, some women experience symptoms such as abnormal discharge and odor and are diagnosed with bacterial vaginosis (BV), while others experiencing inflammation, elevated pH, and overgrowth of aerobic bacteria are diagnosed with aerobic vaginitis (AV)19,20. These more diverse communities have gained a reputation as non-optimal due to their association with vaginal symptoms and heightened risk of obstetric and gynecologic complications18,21,22,23,24.
Although disease associations with human vaginal microbial compositions and insights into specific protective mechanisms have recently expanded, many questions remain regarding microbe-microbe interactions and their interplay with vaginal physiology and host immunity. A prominent barrier to mechanistic discovery is the lack of an in vivo model system resembling the human vaginal microbiota. While mouse models have served a seminal role in delineating host-microbe interactions in reproductive diseases, the murine vaginal microbiota has only recently been defined. Although community structures (low richness and evenness) are similar to humans, the murine phylogenetic composition is quite distinct; Staphylococcus succinus and Enterococcus spp. are the most frequent taxa in C57BL/6J mice25,26, Enterobacteriaceae and Proteus spp. are dominant in CD-1 mice27, and Streptococcus spp. and Proteus spp. are observed in FVB mice28. Conventional mice are poorly colonized by human vaginal Lactobacillus spp. and require multiple, high-inoculum doses to observe in vivo effects29,30,31. Several studies have evaluated the ability of human vaginal communities to colonize mice with low or no endogenous microbiota, but stable colonization by these communities was not achieved32,33. Not only is there a need to understand vaginal microbial dynamics in conventional mice for disease modeling, but there is also dire need for an animal model that better recapitulates the human vaginal microbiota to provide translational relevance34,35.
Here, we evaluated the impact of environment on the vaginal microbiome by comparing conventional C57BL/6J mice raised at three distinct vivaria. To circumvent challenges with transient colonization of human vaginal microbes, we assessed whether a mouse model stably colonized with human fecal microbes, humanized microbiota mice (HMbmice)36, would exhibit a more human-like vaginal composition. We profiled vaginal communities over multiple cohorts, assessed the impact of estrous on HMbmice vaginal microbiome composition, and determined susceptibility to vaginal colonization by three urogenital pathobionts. We found that the murine vaginal microbiota is malleable in composition and that the distinct HMbmouse vaginal microbiota confers protection against group B Streptococcus and Prevotella bivia compared to mice with conventional vaginal microbiota.
Results
The vaginal microbiota differs between vivaria and demonstrates high intra-colony variability
Human and mouse studies have shown that, despite host genetic selection for certain microbiome features37, environmental factors play a dominant role in gut microbial composition38,39,40,41,42,43,44,45. It remains unknown whether the vaginal microbiota is likewise affected. To test this, non-pregnant conventional C57BL/6J mouse vaginal swabs were collected from three different sources [Jackson Lab and in-house colonies at Baylor College of Medicine (BCM) and the University of California San Diego (UCSD)] and subjected to 16S v4 rRNA amplicon sequencing. BCM and UCSD samples were sequenced at their respective institutions, with Jackson samples sequenced at both sites. Sequencing depth and reference-mapping was comparable across sites (Supplementary Fig. 1a, b). Unrarefied, BCM-sequenced Jackson communities were more rich than other groups, contributing to increased alpha diversity (Supplementary Fig. 1c, d) and Bray-Curtis dissimilarity compared to UCSD-sequenced Jackson mice (Supplementary Fig. 1e). However, neither the centroids nor dispersion were significantly different between Jackson mice sequenced at either site as determined by PERMANOVA and PERMDISP analyses performed on weighted normalized UniFrac distances (Supplementary Fig. 1f). Upon repeated rarefaction46,47, BCM-sequenced and UCSD-sequenced Jackson mice remained clustered by normalized weighted UniFrac distances indicating similar phylogeny and feature abundance (Supplementary Fig. 1g). Jackson mice were therefore grouped in the following analyses. To retain samples and capture the full diversity of the vaginal communities, sequences were filtered of contaminants without use of rarefaction (Supplementary Fig. 2).
BCM, Jackson, and UCSD mice had similar community structure: 67%, 59% and 44% of mice respectively had vaginal communities dominated (>50% relative abundance) by a single taxon (Fig. 1a). Colonies also contained shared and unique taxa. Staphylococcus-dominant communities were present in all three colonies but at differing proportions: 9/21 (43%), 30/71 (42%) and 2/36 (5.6%) of BCM, Jackson, and UCSD samples, respectively (Fig. 1a). Weighted normalized UniFrac distances calculated after repeated rarefaction demonstrated overlap in community composition across colonies, but with several unique clusters of Jackson and UCSD mice (Fig. 1b). Mice at all three sites had similar vaginal OTUs and Shannon diversity (Fig. 1c, d). Intra-colony Bray-Curtis dissimilarity scores indicated that the Jackson colony had the lowest commonality among mice, although all three groups had diverse compositions within their respective colonies (Fig. 1e). Phylogenetically, however, UCSD samples showed more compositional diversity while Jackson samples clustered more closely to one another (Fig. 1f, Supplementary Fig. 1g).

Vaginal swabs were collected from mouse colonies raised at BCM (n = 21), Jackson Lab (n = 71), and UCSD (n = 36, 12 mice repeatedly swabbed). a Vaginal microbial compositions of mice at BCM (blue bar), Jackson Lab (yellow bar), and UCSD (pink bar). b PCoA of vaginal communities clustered by weighted normalized UniFrac distances between mice at different vivaria. Centroids were determined by jackknifed rarefaction (100 reads). c Observed OTUs and (d) Shannon entropy of vaginal swab samples. e Bray-Curtis and (f) weighted normalized UniFrac distances between murine vaginal communities of mice within colonies (intra-site) at BCM, Jackson Lab, or UCSD. Colony instability and inter-site variation (weighted normalized UniFrac distances) in comparison to (g) BCM, (h) Jackson Labs, and (i) UCSD. j ANCOM results identifying unique taxa according to colony origin colored by BCM (blue), Jackson Lab (yellow), and UCSD (pink), or BCM and Jackson Lab (black and white). Each column (a) or symbol (b) represents a unique vaginal swab community. Tukey’s boxplots are displayed (c−i). Data were analyzed by Kruskal-Wallis with a Dunn’s multiple comparison test (c−f) or PERMANOVA followed by PERMDISP (g−i). PERMANOVA P values (P < 0.1) are colored in red if PERMDISP was also statistically significant (P < 0.05). All statistically significant P values are reported.
Inter-colony comparisons demonstrated dissimilar compositions between colonies through multiple metrics including Bray-Curtis dissimilarity (Supplementary Fig. 1e, Supplementary Fig. 3) and weighted normalized UniFrac distances (Fig. 1g−i, Supplementary Fig. 1f) with Jackson and UCSD samples demonstrating the greatest inter-colony dissimilarity [weighted normalized UniFrac pseudo-F = 20.3953, P = 0.003). Taxonomic drivers of colony distinctions were identified by ANCOM and included Muribacter, Streptococcus danieliae and Mesocricetibcacter intestinalis in UCSD samples, Mammaliicoccus fleurettii in BCM samples, and Lactobacillus gasseri and Escherichia in Jackson samples (Fig. 1j). Staphylococcus and Acinetobacter distinguished BCM and Jackson samples from UCSD. Together, these data support that vaginal microbial composition in the C57BL/6J genetic background retains a core community structure, but demonstrates unique taxonomic features that are subject to environmental influence.
HMbmice have distinct vaginal communities compared to conventional mice and are enriched in Lactobacillus-dominant communities in a cohort-specific manner
To determine whether stable colonization of human-derived microbes in mice would alter the vaginal microbiota, we defined the vaginal microbiome of HMbmice. HMbmice, founded from germ-free WT C57BL/6J mice colonized with human fecal microbiota, display more human-like gastrointestinal communities compared to conventionally-raised mice and demonstrate generational stability among offspring36. Vaginal swabs collected from multiple generations of HMbmice were subjected to 16S rRNA v4 amplicon sequencing. Half of the first cohort of mice, twelve generations removed from founder mice, demonstrated Lactobacillus dominance (Fig. 2a). Subsequent cohorts had at least one mouse with Lactobacillus colonization (0.0025−99% relative abundance), though the incidence of Lactobacillus-dominant mice was cohort-specific (Fig. 2a). Importantly, vaginal Lactobacilli in HMbmice represent multiple species according to 16S v4 sequences. The most frequent Lactobacillus sequences identified by Greengenes248 mapped to L. gasseri, L. crispatus, or L. jensenii (Fig. 2a). Ligilactobacillus sequences mapped to L. animalis and L. murinus (Supplementary Table 1), which have been isolated from the murine gastrointestinal and vaginal tracts, the latter of which has also been detected in vaginal tract of women who work in rural settings49,50,51,52. Paired vaginal and fecal microbiota of HMbmice (Cohort 3) shared few taxa (e.g., L. reuteri, L. gasseri, and Ligilactobacillus), but remained compositionally distinct, clustering by sample type on a rarefied PCoA generated from weighted normalized UniFrac distances (Supplementary Fig. 4a, b). Vaginal samples had decreased richness and lower Shannon entropy compared to fecal pellets in unpaired (Supplementary Fig. 4c, d) and paired analyses (Supplementary Fig. 4e). There was no correlation in Shannon entropy between paired fecal and vaginal communities (Supplementary Fig. 4f). Additionally, community variability among site-specific samples was higher in vaginal compared to fecal communities (Supplementary Fig. 4g), and vaginal communities were more phylogenetically diverse than fecal compositions (Supplementary Fig. 4h). Together, these data demonstrate site-specific colonization of human fecal microbes in a murine model.

Vaginal swabs were collected from separate cohorts of HMbmice over the course of two years. a Vaginal microbial compositions of distinct cohorts with the duration since previous sampling noted below. Samples from Cohort 1 – Cohort 3 (n = 10−27) represent baseline vaginal swabs from unique mice. Cohort 4 and 5 (n = 12−23) were swabbed repeatedly at baseline over the course of a week; all samples are displayed. b PCoA of vaginal communities clustered by weighted normalized UniFrac distances between mice in different cohorts. Centroids were determined by jackknifed rarefaction (100 reads). Dissimilarity between consecutive cohorts using (c) Bray-Curtis and (d) weighted normalized UniFrac distances. e Top 20 important features over the course of repeated sampling of Cohort 4 and 5 and (f) the net average change in feature relative abundance. g Changes in vaginal community OTU with increased age. Each column (a) or symbol (b, g) represents a unique vaginal swab community. Tukey’s boxplots are displayed (c−d). Values for each feature are displayed (e−f) and colored as in (a). Data were statistically analyzed by PERMANOVA followed by PERMDISP (c−d), Random Forest Regression on 50 estimators (e), and Spearman Correlation (g). PERMANOVA P values (P < 0.1) are colored in red if PERMDISP was also statistically significant (P < 0.05). All statistically significant P values are reported.
Vaginal microbial alpha diversity was similar in HMbmice and conventional mice (Supplementary Fig. 5a, b), and Bray-Curtis dissimilarity comparisons showed high HMbmouse intra- and inter-colony dissimilarities (Supplementary Fig. 5c). Phylogenetically, HMbmice were most distinct from UCSD mice and Jackson mice (Supplementary Fig. 5d). Overlaid PCoA visualization of HMbmice and conventional mice showed overlap of HMbmice and Jackson samples that was partially driven by Lactobacillus spp. and supports that differences in centroids, not dispersion, primarily drove colony variation (Supplementary Fig. 5d, e). In contrast, differences between HMbmice and BCM mice were influenced by dispersion (Supplementary Fig. 5c). Low vaginal pH is a hallmark of a Lactobacillus-rich environment in humans53; vaginal lavage pH was slightly, but significantly lower in HMbmice (median 6.33) compared to conventional mice (median 6.53)(Supplementary Fig. 5f). To determine whether HMbmice were more permissive to human vaginal species, L. crispatus StrepR was intravaginally administered to β-estradiol synchronized mice. Recovered levels of L. crispatus were not significantly different between conventional BCM and HMbmice across five days post-inoculation (Supplementary Fig. 5g).
Within the HMbmice colony, ANCOM analysis identified Acinetobacter, L. gasseri, E. faecalis, L. crispatus, L. jensenii, E. kobei, L. rhamnosus, Escherichia, A. christensenii, B. xylanisolvens, L, fermentum, S. anginosis, S. danieliae, and B. longum as the greatest drivers of cohort variation. Indeed, L. gasseri and E. kobei were most prevalent in Cohort 4 and drove clustering by weighted normalized UniFrac distances (Fig. 2b). Interestingly, the extent of dissimilarity between cohorts did not correspond with time elapsed between sampling periods. For example, phylogenetic diversity between vaginal samples from Cohort 2 and 3 taken four months apart (pseudo-F = 10.3992) was greater than that between Cohort 3 and 4 taken eight months apart (pseudo-F = 7.3179)(Fig. 2c, d). Significant differences in dispersion were detected between Cohort 2 and 3, and Cohort 4 and 5, but not Cohort 3 and 4 (denoted by the red coloring of the PERMANOVA P-value)(Fig. 2d).
We also examined vaginal composition dynamics over time in a subset of mice (Cohorts 4 and 5) recently transferred between rooms. Random Forest Regression analysis of samples collected daily or every three days for a week revealed that L. gasseri was the most important taxa (feature) (Fig. 2e). Despite L. gasseri having the greatest average increase in frequency, the net average change in features ranged from a slight decrease in L. gasseri, to larger relative changes in E. kobei and S. danieliae (Fig. 2f). To evaluate changes to the vaginal community with age, we compared alpha diversity across baseline samples and found that observed OTUs decreased as age of mice increased (Fig. 2g).
Reproductive parameters vary across HMbmice, conventional, and germ-free colonies
Vaginal microbial composition is implicated in birth outcomes in humans54,55 and fecundity in mice; reproductive success improves when germ-free mice become colonized with bacteria56. To resolve whether HMbmice have altered reproductive capacity, HMbmice colony reproductive data were compared against conventional C57BL/6J mice at Jackson Lab57 and genetically-matched conventional and germ-free mice at BCM. Dams in the HMbmice colony averaged 14.4 weeks when their first litter was successfully weaned, which was significantly older than Jackson and BCM mice, but younger than germ-free BCM mice (P < 0.0001 for all comparisons)(Table 1). HMbdams had an average of 3.5 total litters, falling below germ-free mice, conventional mice, and Jackson mice; however, both starting and terminating breeding ages are in part determined by colony management staff. The average projected number of per-dam litters over six months, on the other hand, indicated that HMbmice (5.1 litters) resembled conventional BCM mice (4.9 litters) and could theoretically produce more litters than germ-free and Jackson mice. The gestational interval in HMbmice averaged at 6.8 weeks, aligning more to the timeline of germ-free mice. The average litter size of HMbmice (5.8 pups) was similar to Jackson and conventional BCM mice (5.9 and 5.6 respectively), and significantly higher than germ-free BCM mice (5.0 pups). Together, HMbmice breeding frequency falls within the range observed for conventional and germ-free mice while litter size most resembles Jackson mice, implying that a humanized microbiota minimally affects reproductive performance.
The vaginal microbiota in HMbmice is minimally influenced by estrous stage
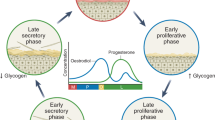
The human vaginal microbiota moderately fluctuates over the course of the menstrual cycle as alpha diversity increases and Lactobacillus relative abundance decreases during menses58,59,60,61,62,63. To determine if vaginal composition in HMbmice is influenced by estrous stage, five HMbmice were swabbed daily for one week. Over the one-week time course, each mouse displayed at least two different dominant (>50% relative abundance) taxa (Fig. 3a). Estrous stages were then assigned by visualizing wet smears of vaginal samples as described previously26(Fig. 3b) and matched to sequencing data for the five mice in Fig. 3a and an additional 30 mice that were sampled periodically. While richness did not differ between stages (Fig. 3c), Shannon entropy was lower in estrus than in both proestrus and diestrus (Fig. 3d). Bray-Curtis dissimilarity approached 1 (highly dissimilar) for all stages, dipping slightly, but significantly, between mice during the diestrus phase compared to proestrus and metestrus (Supplementary Fig. 6a). Parallel results were obtained when comparing weighted normalized UniFrac distances (Fig. 3e). When consecutive transitions from one stage to the next were paired by individual mice, communities transitioning to and from diestrus appeared more similar than in other transition stages, but did not reach statistical significance (Fig. 3f, Supplementary Fig. 6b). Though variability in the vaginal microbiota occurred within each stage, vaginal compositions did not fluctuate drastically between consecutive stages (Fig. 3g, Supplementary Fig. 6c). Together, these results suggest gradual community convergence and increased community stability within mice during diestrus.

Vaginal swabs were collected at baseline and assigned the host’s estrous stage at time of collection. Estrous stage assignment is denoted as P = proestrus, E = estrus, M = metestrus, D = diestrus. a Vaginal microbial compositions of five individual HMbmice from Fig. 2 swabbed daily over the course of a week. Only Mouse 1 and Mouse 2 were co-housed. b Representative microscopic images of vaginal wet smears collected at each stage of the estrous cycle using a 10X objective lens on a brightfield microscope. Scale bar shows 150 μm. c Observed OTUs and (d) Shannon diversity of vaginal swab samples (n = 35). e Weighted normalized UniFrac distances of microbial compositions among samples categorized in the same estrous stage. f Weighted normalized UniFrac distances of microbial compositions between paired, consecutive samples collected from individual mice. g Weighted normalized UniFrac distances of microbial compositions between unpaired samples from sequential estrous stages. Tukey’s boxplots (c−e, g) or individual comparisons marked as symbols (f) are displayed with median and interquartile ranges. Data were statistically analyzed by Kruskal-Wallis with Dunn’s multiple comparisons test (c−f) or PERMANOVA followed by PERMDISP (g). PERMANOVA P values (P < 0.1) are colored in red if PERMDISP was also statistically significant (P < 0.05). All statistically significant P values are reported.
The vaginal microbiota in HMbmice clusters into unique community state types
Vaginal samples were hierarchically clustered into murine community state types (CST) using Ward’s linkage of Euclidean distances as previously described25,26. Because the dominant taxa differed from conventional mice25,26, we designated HMbmice profiles as “humanized murine CST” (hmCST). Two communities resembled mCSTs of conventional mice; hmCST IV (heterogenous taxa with an even composition) and hmCST V (S. succinus-dominant). HMbmice had four unique CSTs: hmCST I and II were Lactobacillus-dominant and Ligilactobacillus-dominant respectively, hmCST III grouped into two Enterobacteriaceae-dominant subsets, and hmCST VI was Streptococcus-dominant (Fig. 4a). Proportions of hmCSTs varied significantly between estrous stages (Fig. 4b). Estrus (estrogen-high and progesterone-low) and diestrus (progesterone high) most starkly contrasted: hmCST I was more prevalent in diestrus than estrus (chi square test, P = 0.0445) and hmCST III-a was more prevalent in estrus compared to diestrus (P < 0.0001), proestrus (P = 0.0009), and metestrus (P = 0.02). Metestrus displayed increased frequency of hmCST III-a compared to diestrus (P = 0.04) and hmCST VI compared to proestrus (P = 0.04). In human longitudinal studies, CSTs self-transition (same CST in consecutive time points) between 35–85% of the time64, and conventional mice typically maintain their mCST between about 20−80% of the time25. Similarly, in HMbmice, self-transitioning frequently occurred in hmCST I and V, whereas hmCST III-b transitioned into hmCST I, and all other communities were less consistent (Fig. 4c). When plotted by weighted normalized UniFrac distances, samples did not separate into discrete clusters based on estrous stage (Figs. 3g, 4d, Supplementary Fig. 6a), but did cluster by hmCST, which is derived from taxonomic classification (Fig. 4e, f, Supplementary Fig. 6d).

Vaginal swabs were collected and categorized by both estrous stage and composition. a Community state type categorization for HMbmice (n = 183 samples) hierarchically clustered into humanized murine CST (upper bar) and its associated estrous stage (lower bar). Estrous staged (n = 35) HMbmice from Figs. 2 and 3 and unstaged (n = 64) HMbmice from Fig. 2 are depicted. b Prevalence of hmCSTs in each estrous stage with total samples per stage noted above. c hmCST transition rates from starting community (left) to community three days later (top) with total numbers per starting hmCST noted on the right. PCoA of vaginal communities clustered by weighted normalized UniFrac distances between mice assigned different (d) estrous stage and (e) hmCST. Centroids were determined by jackknifed rarefaction (100 reads). f Weighted normalized UniFrac distances of microbial compositions between hmCSTs. hmCSTs labeled below are compared to the hmCST of the corresponding background color. Each column (a) or symbol (d−e) represents a unique vaginal swab community. Individual comparisons marked as symbols (f) are displayed with median and interquartile ranges. Data were analyzed by Chi Square analysis of fractions (b) or PERMANOVA followed by PERMDISP (f). PERMANOVA P values (P < 0.1) are colored in red if PERMDISP was also statistically significant (P < 0.05). Statistically significant comparisons are noted in the main text (b); +P < 0.03; ++P < 0.003.
HMbmice exhibit decreased uterine ascension of group B Streptococcus compared to conventional mice
To determine whether the distinct HMbmice vaginal microbiota impacted colonization by pathobionts, we used an established group B Streptococcus (GBS) colonization model65. GBS can asymptomatically colonize the vaginal tract or be associated with AV, a condition characterized by vulvovaginal inflammation and a Lactobacillus-depleted vaginal microbiome19. Additionally, perinatal exposure to GBS during pregnancy or labor and delivery can cause severe disease including stillbirth or neonatal sepsis66. Conventional C57BL/6J mice and HMbmice were vaginally inoculated with GBS (107 CFU) and swabbed daily over seven days (Fig. 5a). At early time points, HMbmice had similar or higher GBS colonization compared to conventional mice. At later time points, HMbmice had significantly lower vaginal GBS burdens than conventional mice with 4/18 HMbmice below the limit of detection for GBS by one-week post-infection (Fig. 5b). To assess GBS ascension, reproductive tract tissues were harvested at day 3 and day 7. Compared to conventional mice, vaginal and cervical GBS burdens were not different between groups, but HMbmice uterine GBS burdens were significantly lower at day 3 and undetectable at day 7 for 6/18 of mice (Fig. 5c, d).

a HMbmice (HMb) and conventional (Conv) mice were vaginally inoculated with 107 CFU of GBS (n = 10−27). b GBS CFU recovered from daily vaginal swabs. Vaginal, cervical, and uterine GBS tissue burdens were collected at (c) day 3 and (d) day 7 post-inoculation. MIP-2 cytokine levels quantified in (e) day 2 vaginal swabs and (f) day 7 uterine tissue homogenates, and (g) IL-17 quantified in day 7 Uterine homogenates. GBS CFU counts from (h) day 2 swabs, (i) day 7 swabs, and (j) combined day 3 and 7 uterine tissues delineated by hmCST assignment of respective mice on day 0 prior to GBS inoculation. Relative abundances of vaginal (k) Escherichia, (l) Lactobacillus gasseri, (m) Acinetobacter, and (n) Pseudomonas across all vaginal swabs in mice grouped into detectable uterine GBS (GBS + ) or no detectable uterine GBS (GBS-) at the time of tissue collection. Symbols represent unique swab or tissue samples with median and interquartile ranges. Data were statistically analyzed by Mann−Whitney test (e−g, k−n) with corrections for multiple comparisons using the two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli and a false discovery rate (<0.05) for (b−b), or Kruskal-Wallis with Dunn’s multiple comparison test (h−j). Statistically significant P values are reported. Adjusted P values < 0.1 are reported for b−d. Schematic (a) was created with BioRender.com.
GBS induces vaginal epithelial production of neutrophil-recruiting chemokines including IL-8, or the mouse ortholog MIP-2, during acute exposure67,68, and vaginal IL-17 is associated with effective GBS clearance in persistent models of colonization68. To determine if these cytokines contributed to the differences between GBS burdens, we measured MIP-2 and IL-17 in day 2 vaginal swab and day 7 uterine tissues by ELISA. In day 2 vaginal swab samples, no differences in vaginal MIP-2 levels were observed between HMbmice and conventional mice (Fig. 5e), and IL-17 levels were below the limit of detection in all samples. In day 7 uterine tissues, no differences were observed in either MIP-2 or IL-17 levels between groups (Fig. 5f, g).
To determine if vaginal microbial composition influenced GBS colonization or ascension, GBS burdens were replotted according to the hmCST assigned at day 0, immediately prior to GBS inoculation. GBS vaginal burdens were not significantly different across hmCST groups at day 2 nor day 7 (Fig. 5h, i). Furthermore, no significant differences in GBS uterine burdens from combined day 3 and 7 samples were detected between hmCSTs (Fig. 5j). To determine whether specific vaginal taxa were associated with GBS uterine ascension, HMbmice were binned into two categories across both time points: those with no detectable GBS uterine CFU (GBS-) or those with detectable GBS uterine CFU (GBS + ). Corresponding vaginal swab 16S sequences from all timepoints were probed for differentially abundant taxa by ANCOM. Mice with no detectable uterine GBS exhibited an enrichment of Escherichia, Lactobacillus, Acinetobacter, and Pseudomonas (Fig. 5k−n).
Ligilactobacillus murinus and E. coli display discordant phenotypes towards GBS in competition assays in vitro and GBS vaginal colonization in vivo
To gain insight into function of differentially abundant taxa between groups, bacterial isolates were collected from HMbmice. Two isolates representing hmCST II and hmCST III-a communities, identified as L. murinus and E. coli by full-length 16S sequencing, were each cultured in MRS broth in competition with GBS at two timepoints across five different starting ratios. Minimal differences in competitive index were observed between L. murinus and GBS, with GBS displaying a competitive advantage at the 1:2 and 1:10 ratios at 3 h (P = 0.01 and 0.0002 respectively) which was retained at 18 h in the 1:10 condition (P = 0.017)(Fig. 6a). To determine how coculture impacted growth of each organism, viable CFU of each organism in coculture was compared to CFU recovered from monoculture. L. murinus was minimally impacted (Fig. 6b); however, GBS growth at 18 h was impaired in the presence at L. murinus at all but the highest GBS starting inoculum (1:10 L. murinus to GBS)(Fig. 6c). Conversely, GBS demonstrated a strong competitive advantage in coculture with E. coli, which was significant in the 1:2 and 1:10 conditions at 3 h (P = 0.001 and <0.0001 respectively) and in all five ratios at the 18 h timepoint (P ≤ 0.029)(Fig. 6d). No differences were observed between E. coli growth in co- or monoculture at 3 h (Fig. 6e). At 18 h, E. coli growth was significantly impaired in the presence of GBS in all conditions (Fig. 6f). Raw viable CFU values for each organism are provided in Supplementary Fig. 7.

In vitro competition assays were performed between GBS and (a−c) L. murinus or (d−f) E. coli. a, d Competitive index (CI) was calculated as the CFU ratio of the treatment microbe over GBS, normalized to the initial ratio of the inoculum. The rate of bacterial growth (viable CFU) in coculture compared to monoculture controls was calculated at timepoints (b, e) 3 h and (c, f) 18 h. Symbols represent the mean of four independent experiments shown with the standard error of the mean (s.e.m.) (a−d) or independent experimental replicates (b−c, e−f) with mean and s.e.m. Data were statistically analyzed by one-sample t-test with a theoretical mean of 1.0 (a, d) and two-way ANOVA with Šídák’s multiple comparisons test for coculture deviation from growth in monoculture (b−c, e−f); *P < 0.05; **P < 0.005; ***P < 0.0005. Raw viable CFU values are reported in Fig. S7.
To determine whether endogenous strains could be re-introduced to confer protection against GBS, L. murinus or E. coli were separately vaginally inoculated into HMbmice twice prior to GBS challenge (Fig. 7a). Despite decreased growth of GBS in the presence of L. murinus in vitro, GBS vaginal colonization and dissemination into the upper reproductive tract was unaffected in vivo (Fig. 7b, c). Prophylactic inoculation with E. coli reduced GBS vaginal burden one day post-inoculation (Fig. 7d) but did not influence swab or tissue burdens by day 7 (Fig. 7e).

a HMbmice were vaginally inoculated with 107 CFU of L. murinus (n = 11/group), 107 CFU of E. coli (n = 8−12/group), or vehicle control twice prior to challenge with 107 CFU of GBS. Recovered GBS CFU from (b) vaginal swabs and (c) day 7 reproductive tract tissues of mice pre-inoculated with L. murinus. Recovered GBS CFU from (d) vaginal swabs and (e) day 7 reproductive tract tissues of mice pre-inoculated with E. coli. Symbols represent individual mice with median and interquartile ranges. Data were statistically analyzed by Mann−Whitney with corrections for multiple comparisons using the two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli and a false discovery rate (<0.05). Adjusted P values < 0.1 are reported. Schematic (a) was created with BioRender.com.
HMbmice exhibit decreased tissue dissemination of Prevotella bivia, but not uropathogenic E. coli, compared to conventional mice
To determine whether HMbmice were protected against multiple pathogens or selectively resistant to GBS, we challenged HMbmice with two additional pathobionts associated with vaginal dysbiosis: Prevotella bivia, which is increased in women diagnosed with BV69, and uropathogenic E. coli (UPEC), a causative agent for urinary tract infection that can establish vaginal reservoirs70 or cause AV20. Unlike GBS, there was no difference in P. bivia vaginal swab burdens between conventional or HMbmice at any timepoint (Fig. 8a). However, at day 7, P. bivia burdens in vaginal, cervical, and uterine tissues were significantly lower in HMbmice compared to conventional mice (Fig. 8b). There were no significant differences in vaginal or uterine MIP-2 (Fig. 8c, d), but IL-17 levels were reduced in HMbmice compared to conventional mice (Fig. 8e). In comparison, no differences in UPEC burdens were observed between HMbmice and conventional mice in vaginal swabs (Fig. 8f) or tissues (Fig. 8g), and cytokine levels were comparable between groups (Fig. 8h−j).

HMbmice (HMb) and conventional (Conv) mice were inoculated with 106 CFU of P. bivia (n = 9−16) or 107 CFU of UPEC (n = 8−16). Recovered P. bivia CFU from (a) vaginal swabs collected on days 1, 2, 3, and 7 post-inoculation and (b) reproductive tract tissues harvested on day 7 (n = 5−12). MIP-2 quantified in (c) day 2 vaginal swabs and (d) day 7 uterine homogenates along with (e) IL-17 in day 7 uterine homogenates of P. bivia-inoculated mice. Recovered UPEC CFU from (f) vaginal swabs collected on days 1, 3, and 7 post-inoculation and (g) reproductive tract tissues harvested on day 7. MIP-2 quantified in (h) day 2 vaginal swabs and (i) day 7 uterine homogenates along with (j) IL-17 in day 7 uterine homogenates of UPEC-inoculated mice. Symbols represent individual mice with median and interquartile ranges. Data were statistically analyzed by Mann−Whitney test (c−e, h−j) with corrections for multiple comparisons using the two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli and a false discovery rate (<0.05)(a−b, f−g). Statistically significant P values are reported. Adjusted P values < 0.1 are reported for (a−b), (f−g). P < 0.1 is shown (d) to be transparent of lack of significance.
Discussion
Despite strong clinical correlations between the vaginal microbiota and women’s health outcomes, ascribing function of the microbiota in vaginal health and disease susceptibility is impaired by the lack of an animal model that recapitulates the human vaginal microbiota. Lactobacillus spp. dominate ~73% of human vaginal communities18 but are rare (<1%) in vaginal communities in other mammals53. Attempts to colonize animal models such as non-human primates71 or laboratory mice29,30,31,32,33 with human vaginal bacteria have failed to achieve long-term colonization. Key goals of this study were to examine the impact of host environment, microbial seeding, and estrous cycle on the vaginal microbiota in mice with the same genetic background and to establish the influence of vaginal microbiota in pathogen control by using conventional mice and mice exposed to human microbes.
The comparison of the vaginal microbiota across mice of the same genetic background within and between vivaria provides insight into both retained features and variability in mouse models. Samples were collected at different times and sequenced through different pipelines; however, comparable sequencing and intra-colony similarities in Jackson mice at both sites demonstrated that inter-colony differences were not artifacts of this confounding variable. An additional limiting factor is the low sample number of UCSD mice; even so, effect sizes were large enough to determine statistical significance.
Similar to murine fecal communities45,72, we observed unique signatures in the otherwise conserved vaginal microbial composition of C57BL/6J mice that gave rise to phylogenetic diversity within and across three distinct facilities. The vaginal microbiota of HMbmice recapitulated the community structure seen in conventional mice characterized by low alpha diversity and dominance by a single taxon in most mice. This finding suggests that host selective pressures drive the vaginal community towards a skewed dominance of a single organism independent of microbial exposure25,26,27. We observed a slight, but significant decrease in HMbmice vaginal pH compared to conventional mice; however, the biological relevance of this difference and how it relates to pathogen colonization warrants further investigation. Clinically, the threshold of pH 4.5 does not affect colonization rates of GBS73; yet, GBS enhances virulence via transcriptional regulation in pH 7.5 compared to pH as high as 5.574. Moreover, we discovered high intra-colony variability in cohorts sampled across multiple years. Counter to prior observations44,75,76, microbial fluctuation was not explained by seasonal changes or convergence of microbial compositions over the two-year sampling period, but rather was likely due to an accumulation of differential colony maintenance, diet, and environmental stimuli45,72,77. Congruent with our study, distinct changes to the vaginal microbiota have also been reported in wild field mice upon captivity78. Age has also been associated with changes in vaginal microbiota, with an increase in alpha diversity in post-menopausal women62,79. HMbmice exhibited decreased richness in vaginal microbial composition with increased age from weaning to 6 months of age; however, composition beyond breeding range were not evaluated in this study.
Reproductive parameter differences between HMbmice, conventional BCM, and Jackson mice likely resulted from colony management rather than biologic divergences as litter size and number of litters per 6 months were similar. An important limitation of this study is that we did not evaluate microbial composition dynamics in pregnancy, of which little is known in mice. Recent human studies have shown an increase in Shannon diversity towards the end of pregnancy51 and similar dynamics were recently described in a longitudinal pregnancy study in conventional mice80. The human vaginal microbiota fluctuates over the menstrual cycle following hormone-mediated changes in glycogen availability, a key nutrient for Lactobacilli and other microbes53,81,82. Observations in other mammals are mixed; reproductive cycle-associated fluctuations have been reported in some non-human primates, cows, and rats83,84,85,86,87 but not other non-human primates, horses, or mini-pigs88,89,90,91. Unlike conventional mice26, we observed correlations between estrous stage and vaginal communities, particularly Escherichia-dominant hmCST III-a, in HMbmice. Similar to human studies58,59,60,61,62,63, we observed modest, but significant, fluctuations in alpha (Shannon) and beta diversity (Bray-Curtis) with the lowest alpha diversity occurring during estrus and a high beta-diversity during metestrus. An important caveat is that we qualitatively determined estrous stage based on vaginal cytology and not hormone levels. Additionally, this study may be underpowered to delineate distinct microbial signatures that could be better resolved by daily tracking of a larger number of individual mice.
Despite intra-colony variability, we observed consistent hmCSTs in HMbmice suggesting continuity of dominant microbes over time. Compared to conventional mice, HMbmice were enriched in colonization by Lactobacillus spp.; Lactobacillus dominance occurred in 2.8% of samples from Jackson mice but 25.3% of samples from HMbmice. However, there remain several limitations to this model. There is likely some “conventionalization” of HMbmice since we detected a Staphylococcus-dominant community (hmCST V) which is the most common community in conventional C57BL/6J Jackson mice25,26. Additionally, HMbmice frequently colonized by Ligilactobacillus-dominant communities (hmCST II) had OTUs mapping to L. murinus. While only a rarely reported human-associated species49, L. murinus is a homofermentative lactic acid bacterium92 isolated from the vaginal tract of wild mice78 and gut of conventional C57BL/6J mice93, having the genetic capacity for glycogen metabolism94 and pathogen inhibition95. It is also important to note that, despite identification of HMbmice sequences mapping to L. crispatus, L. gasseri, and L. jensenii, it is not possible to ascribe the same functionality between the taxa identified in this study and human gut (the source of human microbes for HMbmice) or vaginal species without further characterization. Lastly, another frequently observed community was dominated by Enterobacteriaceae including Escherichia (hmCST III-a). Although E. coli are reported in human vaginal samples, they are typically at low relative abundance96,97,98,99. Importantly, even with overlapping microbial phylogeny, human and HMbmice reproductive tracts remain physiologically dissimilar100.
Because the vaginal microbiota is thought to protect against pathogens and microbial-based disorders, we tested the impact of the HMbmice vaginal microbiota of on representative agents of AV and neonatal disease (GBS and UPEC) and BV (P. bivia)66,101. GBS colonization is correlated with specific taxa including Staphylococcus spp., P. bivia, and E. coli in non-pregnant women73,98,102, and a non-Lactobacillus dominant microbiota in pregnant women103. GBS uterine ascension, a mechanism for pregnancy complications104,105,106, is correlated with Staphylococcus-dominant vaginal microbiota in conventional mice26. Although there were minimal differences in GBS vaginal burdens between conventional and HMbmice, HMbmice demonstrated lower or undetectable uterine burdens and revealed four vaginal taxa (three with very low abundance) that were inversely correlated with detection of uterine GBS. L. murinus, but not E. coli, reduced GBS growth in coculture experiments, while exogenous treatment of E. coli, but not L. murinus, reduced GBS colonization in HMbmice. The discordance between in vitro and in vivo findings could be explained by attenuation of L. murinus anti-GBS activity in vivo due to poor colonization, altered competitive ratios, or insufficient production of anti-GBS factors. Alternatively, the in vivo success of exogenous E. coli could be explained by E. coli outcompeting GBS for key nutrients or attachment to host surfaces or the elicitation of an altered immune response. These studies highlight the complexity of host and microbial factors dictating GBS colonization success and may explain why some probiotics with potent anti-GBS activity in vitro have failed to reduce GBS colonization in clinical trials107,108,109,110.
HMbmice were not consistently resistant to pathobionts compared to conventional mice; HMbmice displayed reduced tissue burdens of P. bivia but not UPEC. P. bivia uterine burdens in conventional mice were comparable to previous studies111 suggesting that HMbmice may actively suppress P. bivia uterine ascension or persistence. Interestingly, only P. bivia-challenged HMbmice exhibited reduced cytokine levels in comparison to conventional mice. Whether uterine IL-17 was decreased because of reduced P. bivia burden or other modulations by the endogenous flora was not determined. No differences in UPEC colonization were seen between HMbmice and conventional mice; tissue burdens were consistent with previous findings70,112,113. Counter to other murine gut colonization models114, exogenous UPEC did not appear to be negatively impacted by the frequent endogenous vaginal E. coli found in HMbmice; however, we did not assess changes to the vaginal microbiome following UPEC inoculation, so it is unknown whether UPEC had any impact on endogenous Enterobacteriaceae including E. coli. An important limitation of our study is that assessment of host immune responses to these pathobionts was limited to two cytokines. Previous studies demonstrate that GBS and UPEC, but not P. bivia, induce vaginal immune responses in conventional mice68,112,115, thus further characterization of the HMbmice model, paired with in vitro models such as a human vagina-on-a-chip116, is needed to delineate the role of the vaginal microbiota in shaping immune responses to vaginal pathobionts.
Our results reveal the plasticity of the mouse vaginal microbiota in response to environmental exposures, perhaps a more potent driver of variability than host genetics. Additionally, we found microbial drift between cohorts, age, and within individual mice over the course of a week. Even so, questions remain regarding the biologic factors driving rapid changes to the vaginal microbiota in mice. Although not an exact representation of the human vaginal microbiota, the HMbmouse model described here is enriched in Lactobacillus-dominant communities and demonstrates the importance of the vaginal microbiota in eliciting immune responses and shaping outcomes of reproductive tract infections. Continued improvement of humanized mouse models will provide a pathway to establish the functional role of the vaginal microbiota in health and disease and serve as an improved preclinical model for microbe-based therapies.
Methods
Bacterial strains
GBS COH1 (ATCC BAA-1176), a neonatal meningitis isolate, was grown overnight in Todd-Hewitt Broth (THB) at 37 °C, diluted 1:10 in fresh THB, and grown to mid-log phase (OD600nm = 0.4). Spontaneous streptomycin-resistant mutants of human oral isolate L. crispatus (ATCC 33820)117 and cystitis isolate UPEC UTI89118 were generated by plating an overnight culture on De Man, Rogosa and Sharpe (MRS) agar or Luria Broth (LB) agar, respectively, containing 1000 μg/mL Streptomycin. L. crispatus StrepR was grown anaerobically in a Coy anaerobic chamber at 37 °C in MRS with 1000 μg/mL Streptomycin, washed twice with PBS and saved in 5% glycerol working aliquots. UPEC StrepR was grown overnight in LB with 1000 μg/mL Streptomycin and washed twice with PBS prior to use. Prevotella bivia ATCC29303 StrepR, an endometrial isolate115, was grown anaerobically in a Coy anaerobic chamber at 37 °C in Tryptic Soy Broth (TSB) with 5% laked, defibrinated sheep blood for three days. E. coli and L. murinus were isolated from Hmbmice vaginal swabs plated on MRS agar and grown anaerobically in MRS over one or two days, respectively.
Animals
Animal experiments were approved by the BCM and University of California San Diego Institutional Animal Care and Use Committees and conducted under accepted veterinary standards and in compliance with all relevant ethical regulations. Mice were given food and water ad libitum. Humanized Microbiota mice (HMbmice) were maintained as described previously36. WT C57BL/6J female mice were purchased from Jackson Labs (#000664) or from C57BL/6J stocks bred at BCM and UCSD. Prior to bacterial infections, mice were acclimated for one week in the biohazard room. Mice were distributed so that each treatment group and timepoint contained a similar age range in mice. Mice ranged in age from 2 to 6 months. At predetermined experimental endpoints day 3 or day 7 post infection, mice were euthanized by C02 according to the Guide for the Care and Use of Laboratory Animals119 and then cervically dislocated as a secondary measure.
Vaginal lavage collection and pH assignment
Vaginal lavages were collected using 10 µL of molecular grade water. Water was pipetted into the vaginal tract five times and spotted onto both a strip of Fisher Brand pH paper measuring pH 3.0−5.5 at 0.5 intervals (Lot: 214220) and another strip measuring pH 6−8 at 0.2 intervals (Lot: 211718 A). Values were assigned, unblinded, by three independent researchers. For measurements that sat between two intervals, the median pH was recorded. Final scores were averaged. Only one independent experiment was performed (n = 9−10).
Vaginal swab collection and estrous stage assignment
Vaginal swabs were collected as previously described65, resuspended in 100 μL of PBS, and stored at -20 °C. Samples from UCSD conventional mice and Jackson mice were collected and sequenced in the same time frame at UCSD. A second set of Jackson mice were swabbed and sequenced at BCM along with BCM conventional and HMbmice. Wet mounts of vaginal samples were observed under brightfield 100X magnification on an Echo Revolve microscope. Estrous stages were delineated, unblinded, by three independent researchers according to parameters described previously120,121 and assigned with a consensus of at least two researchers. Mice were sampled at a single time point (n = 2), every three days (n = 28), or daily (n = 5) over the span of seven days.
DNA extraction and 16S rRNA V4 amplicon sequencing
DNA from thawed vaginal swabs was extracted using the Quick-DNA Fungal/Bacterial Microprep Kit protocol (Zymo Research) and following manufacturer’s instructions with two deviations: samples were homogenized for 15 min during lysis and DNA was eluted in 20 µL of water. Amplification and sequencing of the V4 region of the 16S rRNA gene were carried out by BCM Center for Metagenomics and Microbiome Research or UCSD Institute for Genomic Medicine using the Illumina 16Sv4 and Illumina MiSeq v2 2x250bp protocols as described25,26. Sequences were joined, trimmed to 150 bp reads, and denoised using Deblur through QIIME2 version 2022.8122. Operational Taxonomic Units (OTUs) were assigned using Greengenes2 reference tree48.
Vaginal sequences of Jackson Labs mice from previous work were downloaded from EBI accession numbers PRJEB2573326 and PRJEB4930425, and NCBI Sequence Read Archive BioProject accession number PRJNA988548 as supplement to our present study deposited under EBI accession number PRJEB58804 for Fig. 1 and Supplementary Figs. 1,3,5. Since many samples were low biomass, DNA contaminants from sequencing reagents and kits had a substantial impact on the dataset and necessitated filtering of Feature IDs as presented in Supplementary Fig. 2. First, feature IDs that appeared in less than five samples were removed. Second, negative controls from BCM (n = 15) and UCSD (n = 18) that went through the entire pipeline, from DNA extraction to sequencing, were run through the R package Decontam123(R version 4.3.1 (2023-06-16) – “Beagle Scouts”), which identified 27 Feature IDs that were subsequently removed from the feature table. Lastly, the feature table was re-imported into QIIME2 where other abundant contaminants were filtered out.
Alpha diversity (OTUs and Shannon), beta diversity (Bray-Curtis and weighted UniFrac distances analyzed by PERMANOVA and PERMDISP), and differential abundance (ANCOM) tests were carried out in QIIME2124. PCoA plots, the only data represented from rarefied tables, were produced by UPGMS clustering using the “diversity beta-rarefaction” command at a rarefaction depth of 100 for 100 iterations. To assign hmCSTs and create heatmaps, hierarchical clustering was performed on the filtered feature table with Ward’s linkage of Euclidean distances using the R package stats25. Output files were exported and analyzed in R Studio v1.2.5001 using factoextra125, and Phyloseq126. Data visualization was performed with GraphPad Prism v9.4.0 (GraphPad Software Inc.).
Reproductive parameters and data
Jackson Labs data was acquired from the Handbook on Genetically Standardized JAX Mice57. BCM C57BL/6 J and HMbmice colony data were sourced from colony managers. Ranges were not provided for all measures. Parameters for Jackson, BCM, and germ-free mice were set as theoretical means against which HMbmice were compared.
Murine pathogen colonization models and sample analyses
Vaginal colonization studies were conducted as described previously65,111. Mice were synchronized with 0.5 mg β-estradiol administered intraperitoneally 24 h prior to inoculation with GBS, UPEC StrepR or L. crispatus StrepR (Fig. 5a) and at both 48 h and 24 h prior to inoculation with P. bivia StrepR. Mice were vaginally inoculated with 10 μL of GBS COH1 (107 CFU), UPEC StrepR (107 CFU), P. bivia StrepR (106 CFU), or L. crispatus StrepR. Vaginal swabs were collected at indicated timepoints and, for pathogen challenge experiments, tissues were harvested on day 3 and/or 7 as previously described25. CHROMagar StrepB Select (DRG International Inc.) was used to quantify recovered GBS (identified as pink/mauve colonies). CHROMagar Orientation plates were used to quantify recovered UPEC StrepR (identified as pink colonies). P. bivia StrepR was quantified on blood agar containing 1000 mg/mL Streptomycin. L. crispatus StrepR was quantified on MRS containing 1000 mg/mL Streptomycin. Only one independent experiment (n = 9−10) was performed for L. crispatus colonization. For pre-treatment experiments, 0.5 mg β-estradiol was given on day −5 and −2 and either L. murinus (106 CFU), E. coli (106 CFU), or vehicle were administered on day −4 and −1 before GBS challenge (Fig. 7a). Cardiac puncture was performed to collect serum and to plate for the detection of bacteremia. Blood, swabs and tissues were collected as stated above and plated for GBS CFU on CHROMagar StrepB Select. ELISAs were performed on vaginal swab fluid and tissue homogenates (diluted 1:5 and 1:10 respectively) for mouse MIP-2 and (both diluted at 1:10) IL-17α (R&D Systems) per manufacturers’ instructions.
In vitro competition assays
GBS, L. murinus, and E. coli were grown anaerobically in MRS media as cocultures or monocultures at the indicated concentrations. Samples collected at 3 h and 18 h were plated on THB (L. murinus competition) or CHROMagar Orientation (E. coli competition) and cultured anaerobically at 37 °C for two days.
Statistics
Data were collected from at least two independent experiments unless otherwise stated. Mean values from independent experiment replicates, or biological replicates, are represented by medians with interquartile ranges or 95% confidence intervals, or box-and-whisker plots with Tukey’s whiskers as indicated in figure legends. Independent vaginal samples, some taken at multiple timepoints from the same mouse, are represented by symbols on PCoA plots. Intra-colony alpha and beta-diversity metrics, and GBS burdens by hmCST were analyzed by Kruskal-Wallis with Dunn’s multiple comparisons test or as paired samples by two-tailed Wilcoxon Rank Sum test. Association between age and OTUs was determined by one-tailed Spearman Correlation. Pathogen burdens and cytokine levels between conventional and HMbmice, pre-treatment conditions, or vaginal and fecal communities, were assessed by two-tailed Mann-Whitney test with corrections for multiple comparisons using the two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli and a false discovery rate (<0.05) where necessary. Inter-colony dissimilarity was analyzed using pairwise PERMANOVA and PERMDISP performed with 999 permutations. Reproductive parameters and competitive indices and were analyzed by a two-tailed one-sample t-test with a theoretical mean of corresponding values from conventional mouse colonies for reproductive data or of 1.0 for in vitro data. hmCST frequencies across estrous stages were compared by two-tailed Chi square test. Coculture and monoculture comparisons were assumed to have parametric distribution and were analyzed by two-way ANOVA with Šídák’s multiple comparisons test. Statistical analyses were performed using GraphPad Prism, v9.4.0. For Mann−Whitney tests with corrections for false discovery and PERMANOVA analyses, adjusted P values < 0.1 are shown. Unless otherwise stated, P values < 0.05 were considered statistically significant.
Code availbility
Script and metadata files are accessible at GitHub under project “MouseVaginalMicrobiota-HMb_filtering_CST” at https://github.com/Marlydem/MouseVaginalMicrobiota-HMb_filtering_CST.
Data availability
Sequencing Data generated by this study is available in EBI under accession number PRJEB58804. Other sequencing data was sourced from EBI accession numbers PRJEB25733 and PRJEB49304, and NCBI Sequence Read Archive under BioProject accession number PRJNA988548.
References
France, M., Alizadeh, M., Brown, S., Ma, B. & Ravel, J. Towards a deeper understanding of the vaginal microbiota. Nat. Microbiol. 7, 367–378 (2022).
Feehily, C. et al. Shotgun sequencing of the vaginal microbiome reveals both a species and functional potential signature of preterm birth. NPJ Biofilms Microbiomes 6, 50 (2020).
Nelson, D. B. et al. Early pregnancy changes in bacterial vaginosis-associated bacteria and preterm delivery. Paediatr. Perinat. Epidemiol. 28, 88–96 (2014).
Elovitz, M. A. et al. Cervicovaginal microbiota and local immune response modulate the risk of spontaneous preterm delivery. Nat. Commun. 10, 1305 (2019).
Gosmann, C. et al. Lactobacillus-deficient cervicovaginal bacterial communities are associated with increased hiv acquisition in young South African Women. Immunity 46, 29–37 (2017).
Martin, H. L. et al. Vaginal lactobacilli, microbial flora, and risk of human immunodeficiency virus type 1 and sexually transmitted disease acquisition. J. Infect. Dis. 180, 1863–1868 (1999).
Haggerty, C. L. et al. Identification of novel microbes associated with pelvic inflammatory disease and infertility. Sex. Transm. Infect. 92, 441–446 (2016).
Ness, R. B. et al. A cluster analysis of bacterial vaginosis-associated microflora and pelvic inflammatory disease. Am. J. Epidemiol. 162, 585–590 (2005).
Brotman, R. M. et al. Bacterial vaginosis assessed by gram stain and diminished colonization resistance to incident gonococcal, chlamydial, and trichomonal genital infection. J. Infect. Dis. 202, 1907–1915 (2010).
Kovachev, S. Defence factors of vaginal lactobacilli. Crit. Rev. Microbiol. 44, 31–39 (2018).
Boris, S. & Barbes, C. Role played by lactobacilli in controlling the population of vaginal pathogens. Microbes Infect. 2, 543–546 (2000).
Kobatake, E. & Kabuki, T. S-layer protein of lactobacillus helveticus SBT2171 promotes human beta-defensin 2 expression via TLR2-JNK signaling. Front. Microbiol. 10, 2414 (2019).
Niu, X. X., Li, T., Zhang, X., Wang, S. X. & Liu, Z. H. Lactobacillus crispatus modulates vaginal epithelial cell innate response to Candida albicans. Chin. Med. J. 130, 273–279 (2017).
De Gregorio, P. R., Juarez Tomas, M. S. & Nader-Macias, M. E. Immunomodulation of lactobacillus reuteri CRL1324 on Group B streptococcus vaginal colonization in a murine experimental model. Am. J. Reprod. Immunol. 75, 23–35 (2016).
Doerflinger, S. Y., Throop, A. L. & Herbst-Kralovetz, M. M. Bacteria in the vaginal microbiome alter the innate immune response and barrier properties of the human vaginal epithelia in a species-specific manner. J. Infect. Dis. 209, 1989–1999 (2014).
Anahtar, M. N. et al. Cervicovaginal bacteria are a major modulator of host inflammatory responses in the female genital tract. Immunity 42, 965–976 (2015).
Mu, X. et al. Group B streptococcus colonization induces prevotella and megasphaera abundance-featured vaginal microbiome compositional change in non-pregnant women. PeerJ 7, e7474 (2019).
Ravel, J. et al. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. USA 108, 4680–4687 (2011).
Kaambo, E., Africa, C., Chambuso, R. & Passmore, J. S. Vaginal microbiomes associated with aerobic vaginitis and bacterial vaginosis. Front. Public Health 6, 78 (2018).
Tansarli, G. S., Kostaras, E. K., Athanasiou, S. & Falagas, M. E. Prevalence and treatment of aerobic vaginitis among non-pregnant women: evaluation of the evidence for an underestimated clinical entity. Eur. J. Clin. Microbiol. Infect. Dis. 32, 977–984 (2013).
Mancabelli, L. et al. Vaginotypes of the human vaginal microbiome. Environ. Microbiol. 23, 1780–1792 (2021).
Peebles, K., Velloza, J., Balkus, J. E., McClelland, R. S. & Barnabas, R. V. High global burden and costs of bacterial vaginosis: a systematic review and meta-analysis. Sex. Transm. Dis. 46, 304–311 (2019).
Muzny, C. A. & Schwebke, J. R. Asymptomatic Bacterial Vaginosis: To Treat or Not to Treat? Curr. Infect. Dis. Rep. 22, https://doi.org/10.1007/s11908-020-00740-z (2020).
Reid, G. Is bacterial vaginosis a disease? Appl. Microbiol. Biotechnol. 102, 553–558 (2018).
Mejia, M. E. et al. Human milk oligosaccharides reduce murine Group B streptococcus vaginal colonization with minimal impact on the vaginal microbiota. mSphere 7, e0088521 (2022).
Vrbanac, A. et al. The murine vaginal microbiota and its perturbation by the human pathogen group B Streptococcus. BMC Microbiol. 18, 197 (2018).
Burcham, L. R., Burcham, Z. M., Akbari, M. S., Metcalf, J. L. & Doran, K. S. Interrelated effects of zinc deficiency and the microbiome on Group B streptococcal vaginal colonization. mSphere 7, e0026422 (2022).
Hernandez-Quiroz, F. et al. The vaginal and fecal microbiota of a murine cervical carcinoma model under synergistic effect of 17beta-Estradiol and E7 oncogene expression. Micro. Pathog. 152, 104763 (2021).
De Gregorio, P. R., Juarez Tomas, M. S., Santos, V. & Nader-Macias, M. E. Beneficial lactobacilli: effects on the vaginal tract in a murine experimental model. Antonie Van. Leeuwenhoek 102, 569–580 (2012).
De Gregorio, P. R., Silva, J. A., Marchesi, A. & Nader-Macias, M. E. F. Anti-Candida activity of beneficial vaginal lactobacilli in in vitro assays and in a murine experimental model. FEMS Yeast Res. 19, https://doi.org/10.1093/femsyr/foz008 (2019).
De Gregorio, P. R., Juarez Tomas, M. S., Leccese Terraf, M. C. & Nader-Macias, M. E. Preventive effect of Lactobacillus reuteri CRL1324 on Group B Streptococcus vaginal colonization in an experimental mouse model. J. Appl. Microbiol. 118, 1034–1047 (2015).
Jasarevic, E. et al. The composition of human vaginal microbiota transferred at birth affects offspring health in a mouse model. Nat. Commun. 12, 6289 (2021).
Wolfarth, A. A. et al. A human microbiota-associated murine model for assessing the impact of the vaginal microbiota on pregnancy outcomes. Front. Cell Infect. Microbiol. 10, 570025 (2020).
Ericsson, A. C. & Franklin, C. L. The gut microbiome of laboratory mice: considerations and best practices for translational research. Mamm. Genome 32, 239–250 (2021).
Rosshart, S. P. et al. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science 365, https://doi.org/10.1126/science.aaw4361 (2019).
Collins, J., Auchtung, J. M., Schaefer, L., Eaton, K. A. & Britton, R. A. Humanized microbiota mice as a model of recurrent Clostridium difficile disease. Microbiome 3, 35 (2015).
Goodrich, J. K. et al. Genetic determinants of the gut microbiome in UK Twins. Cell Host Microbe. 19, 731–743 (2016).
Rothschild, D. et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 555, 210–215 (2018).
Abeles, S. R. et al. Microbial diversity in individuals and their household contacts following typical antibiotic courses. Microbiome 4, 39 (2016).
Song, S. J. et al. Cohabiting family members share microbiota with one another and with their dogs. Elife 2, e00458 (2013).
Hildebrand, F. et al. Inflammation-associated enterotypes, host genotype, cage and inter-individual effects drive gut microbiota variation in common laboratory mice. Genome Biol. 14, R4 (2013).
McCafferty, J. et al. Stochastic changes over time and not founder effects drive cage effects in microbial community assembly in a mouse model. ISME J. 7, 2116–2125 (2013).
Singh, G., Brass, A., Cruickshank, S. M. & Knight, C. G. Cage and maternal effects on the bacterial communities of the murine gut. Sci. Rep. 11, 9841 (2021).
Caruso, R., Ono, M., Bunker, M. E., Nunez, G. & Inohara, N. Dynamic and asymmetric changes of the microbial communities after cohousing in laboratory mice. Cell Rep. 27, 3401–3412 e3403 (2019).
Long, L. L. et al. Shared and distinctive features of the gut microbiome of C57BL/6 mice from different vendors and production sites, and in response to a new vivarium. Lab Anim. 50, 185–195 (2021).
Lozupone, C., Lladser, M. E., Knights, D., Stombaugh, J. & Knight, R. UniFrac: an effective distance metric for microbial community comparison. ISME J. 5, 169–172 (2011).
Cameron, E. S., Schmidt, P. J., Tremblay, B. J., Emelko, M. B. & Muller, K. M. Enhancing diversity analysis by repeatedly rarefying next generation sequencing data describing microbial communities. Sci. Rep. 11, 22302 (2021).
McDonald, D. et al. Greengenes2 unifies microbial data in a single reference tree. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-01845-1 (2023).
Fosch, S. E. et al. Contraception: influence on vaginal microbiota and identification of vaginal lactobacilli using MALDI-TOF MS and 16S rDNA sequencing. Open Microbiol. J. 12, 218–229 (2018).
Vasquez, A., Jakobsson, T., Ahrne, S., Forsum, U. & Molin, G. Vaginal lactobacillus flora of healthy Swedish women. J. Clin. Microbiol. 40, 2746–2749 (2002).
Goltsman, D. S. A. et al. Metagenomic analysis with strain-level resolution reveals fine-scale variation in the human pregnancy microbiome. Genome Res. 28, 1467–1480 (2018).
Topisirovic, L. et al. Potential of lactic acid bacteria isolated from specific natural niches in food production and preservation. Int J. Food Microbiol. 112, 230–235 (2006).
Miller, E. A., Beasley, D. E., Dunn, R. R. & Archie, E. A. Lactobacilli dominance and vaginal pH: why is the human vaginal microbiome unique? Front. Microbiol. 7, 1936 (2016).
Donders, G. G. et al. Predictive value for preterm birth of abnormal vaginal flora, bacterial vaginosis and aerobic vaginitis during the first trimester of pregnancy. BJOG 116, 1315–1324 (2009).
van Oostrum, N., De Sutter, P., Meys, J. & Verstraelen, H. Risks associated with bacterial vaginosis in infertility patients: a systematic review and meta-analysis. Hum. Reprod. 28, 1809–1815 (2013).
Shimizu, K. et al. Normalization of reproductive function in germfree mice following bacterial contamination. Exp. Anim. 47, 151–158 (1998).
Flurkey, K. & Currer, J. The Jackson Laboratory Handbook on Genetically Standardized Mice. 6 edn, (The Jackson Laboratory, 2009).
Gajer, P. et al. Temporal dynamics of the human vaginal microbiota. Sci. Transl. Med. 4, 132ra152 (2012).
Chaban, B. et al. Characterization of the vaginal microbiota of healthy Canadian women through the menstrual cycle. Microbiome 2, 23 (2014).
Song, S. D. et al. Daily Vaginal Microbiota Fluctuations Associated with Natural Hormonal Cycle, Contraceptives, Diet, and Exercise. mSphere 5, https://doi.org/10.1128/mSphere.00593-20 (2020).
Santiago, G. L. et al. Longitudinal qPCR study of the dynamics of L. crispatus, L. iners, A. vaginae, (sialidase positive) G. vaginalis, and P. bivia in the vagina. PLoS One 7, e45281 (2012).
Kaur, H., Merchant, M., Haque, M. M. & Mande, S. S. Crosstalk between female gonadal hormones and vaginal microbiota across various phases of women’s gynecological lifecycle. Front. Microbiol. 11, 551 (2020).
Krog, M. C. et al. The healthy female microbiome across body sites: effect of hormonal contraceptives and the menstrual cycle. Hum. Reprod. 37, 1525–1543 (2022).
Brooks, J. P. et al. Changes in vaginal community state types reflect major shifts in the microbiome. Micro. Ecol. Health Dis. 28, 1303265 (2017).
Patras, K. A. & Doran, K. S. A Murine Model of Group B Streptococcus Vaginal Colonization. J. Vis. Exp. https://doi.org/10.3791/54708 (2016).
Patras, K. A. & Nizet, V. Group B Streptococcal maternal colonization and neonatal disease: molecular mechanisms and preventative approaches. Front. Pediatr. 6, 27 (2018).
Patras, K. A. et al. Group B Streptococcus CovR regulation modulates host immune signalling pathways to promote vaginal colonization. Cell Microbiol. 15, 1154–1167 (2013).
Patras, K. A., Rosler, B., Thoman, M. L. & Doran, K. S. Characterization of host immunity during persistent vaginal colonization by Group B Streptococcus. Mucosal Immunol. 8, 1339–1348 (2015).
Zozaya-Hinchliffe, M., Lillis, R., Martin, D. H. & Ferris, M. J. Quantitative PCR assessments of bacterial species in women with and without bacterial vaginosis. J. Clin. Microbiol. 48, 1812–1819 (2010).
Brannon, J. R. et al. Invasion of vaginal epithelial cells by uropathogenic Escherichia coli. Nat. Commun. 11, 2803 (2020).
Langner, C. A. et al. The vaginal microbiome of nonhuman primates can be only transiently altered to become Lactobacillus dominant without reducing inflammation. Microbiol. Spectr. 9, e0107421 (2021).
Rausch, P. et al. Analysis of factors contributing to variation in the C57BL/6J fecal microbiota across German animal facilities. Int J. Med. Microbiol. 306, 343–355 (2016).
Rosen, G. H. et al. Group B Streptococcus and the vaginal microbiota. J. Infect. Dis. 216, 744–751 (2017).
Santi, I. et al. CsrRS regulates group B Streptococcus virulence gene expression in response to environmental pH: a new perspective on vaccine development. J. Bacteriol. 191, 5387–5397 (2009).
Baniel, A. et al. Seasonal shifts in the gut microbiome indicate plastic responses to diet in wild geladas. Microbiome 9, 26 (2021).
Marsh, K. J. et al. Synchronous seasonality in the gut microbiota of wild mouse populations. Front. Microbiol. 13, 809735 (2022).
Kim, Y. M. et al. Light-stress influences the composition of the murine gut microbiome, memory function, and plasma metabolome. Front. Mol. Biosci. 6, 108 (2019).
Matejkova, T. et al. Oral and vaginal microbiota in selected field mice of the genus Apodemus: a wild population study. Sci. Rep. 10, 13246 (2020).
Kim, S. et al. Changes in the microbiome of vaginal fluid after menopause in Korean women. J. Microbiol. Biotechnol. 31, 1490–1500 (2021).
Mercado-Evans, V. et al. Gestational diabetes augments group B Streptococcus perinatal infection through disruptions in maternal immunity and the vaginal microbiota. bioRxiv, https://doi.org/10.1101/2023.06.23.546252 (2023).
Amabebe, E. & Anumba, D. O. C. The vaginal microenvironment: the physiologic role of lactobacilli. Front. Med. 5, 181 (2018).
Mirmonsef, P. et al. Glycogen levels in undiluted genital fluid and their relationship to vaginal pH, estrogen, and progesterone. PLoS One 11, e0153553 (2016).
Adapen, C. et al. Local innate markers and vaginal microbiota composition are influenced by hormonal cycle phases. Front. Immunol. 13, 841723 (2022).
Hallmaier-Wacker, L. K., Luert, S., Roos, C. & Knauf, S. Lactation and menstruation shift the vaginal microbiota in captive rhesus monkeys to be more similar to the male urethral microbiota. Sci. Rep. 9, 17399 (2019).
Quereda, J. J. et al. Vaginal microbiota changes during estrous cycle in dairy heifers. Front. Vet. Sci. 7, 371 (2020).
Gohil, P. et al. Metagenomic analysis of bacterial communities during estrous cycle in Bos indicus. Trop. Anim. Health Prod. 54, 118 (2022).
Levy, M. et al. The rodent vaginal microbiome across the estrous cycle and the effect of genital nerve electrical stimulation. PLoS One 15, e0230170 (2020).
Uchihashi, M. et al. Influence of age, reproductive cycling status, and menstruation on the vaginal microbiome in baboons (Papio anubis). Am. J. Primatol. 77, 563–578 (2015).
Barba, M. et al. Vaginal Microbiota Is Stable throughout the Estrous Cycle in Arabian Maress. Animals (Basel) 10, https://doi.org/10.3390/ani10112020 (2020).
Lorenzen, E. et al. The vaginal microbiome is stable in prepubertal and sexually mature Ellegaard Gottingen Minipigs throughout an estrous cycle. Vet. Res. 46, 125 (2015).
Golinska, E. et al. The vaginal microflora changes in various stages of the estrous cycle of healthy female dogs and the ones with genital tract infections. BMC Vet. Res. 17, 8 (2021).
Brownlie, E. J. E., Chaharlangi, D., Wong, E. O., Kim, D. & Navarre, W. W. Acids produced by lactobacilli inhibit the growth of commensal Lachnospiraceae and S24-7 bacteria. Gut Microbes 14, 2046452 (2022).
Pan, F. et al. Predominant gut Lactobacillus murinus strain mediates anti-inflammaging effects in calorie-restricted mice. Microbiome 6, 54 (2018).
Goh, Y. J. & Klaenhammer, T. R. Insights into glycogen metabolism in Lactobacillus acidophilus: impact on carbohydrate metabolism, stress tolerance and gut retention. Micro. Cell Fact. 13, 94 (2014).
Sandoval-Mosqueda, I. L. et al. Ligilactobacillus murinus Strains Isolated from Mice Intestinal Tract: Molecular Characterization and Antagonistic Activity against Food-Borne Pathogens. Microorganisms 11, https://doi.org/10.3390/microorganisms11040942 (2023).
Safarpoor Dehkordi, F. et al. Uropathogenic Escherichia coli in the high vaginal swab samples of fertile and infertile women: virulence factors, O-serogroups, and phenotyping and genotyping characterization of antibiotic resistance. N. Microbes N. Infect. 38, 100824 (2020).
Meyn, L. A., Krohn, M. A. & Hillier, S. L. Rectal colonization by group B Streptococcus as a predictor of vaginal colonization. Am. J. Obstet. Gynecol. 201, 76.e71–77 (2009).
Cools, P. et al. A multi-country cross-sectional study of vaginal carriage of Group B Streptococci (GBS) and Escherichia coli in resource-poor settings: prevalences and risk factors. PLoS One 11, e0148052 (2016).
Guiral, E., Bosch, J., Vila, J. & Soto, S. M. Prevalence of Escherichia coli among samples collected from the genital tract in pregnant and nonpregnant women: relationship with virulence. FEMS Microbiol. Lett. 314, 170–173 (2011).
Cunha, G. R. et al. Reproductive tract biology: of mice and men. Differentiation 110, 49–63 (2019).
Younes, J. A. et al. Women and their microbes: the unexpected friendship. Trends Microbiol. 26, 16–32 (2018).
Mejia, M. E., Robertson, C. M. & Patras, K. A. Interspecies interactions within the host: the social network of group B Streptococcus. Infect. Immun. 91, e0044022 (2023).
McCoy, J. A. et al. Cervicovaginal Microbial-Immune State and Group B Streptococcus Colonization in Pregnancy. Am. J. Perinatol, https://doi.org/10.1055/s-0043-1772226 (2023).
Sgayer, I. et al. Routine uterine culture swab during cesarean section and its clinical correlations: a retrospective comparative study. Eur. J. Obstet. Gynecol. Reprod. Biol. 249, 42–46 (2020).
Romero, R. et al. Evidence that intra-amniotic infections are often the result of an ascending invasion - a molecular microbiological study. J. Perinat. Med. 47, 915–931 (2019).
Desa, D. J. & Trevenen, C. L. B beta-haemolytic streptococci. Br. J. Obstet. Gynaecol. 91, 237–239 (1984).
Olsen, P., Williamson, M., Traynor, V. & Georgiou, C. The impact of oral probiotics on vaginal Group B Streptococcal colonisation rates in pregnant women: a pilot randomised control study. Women Birth 31, 31–37 (2018).
Sharpe, M. et al. Effectiveness of oral intake of Lactobacillus rhamnosus GR-1 and Lactobacillus reuteri RC-14 on Group B Streptococcus colonization during pregnancy: a midwifery-led double-blind randomized controlled pilot trial. J. Matern. Fetal. Neonatal. Med. 34, 1814–1821 (2021).
Hanson, L., Vandevusse, L., Duster, M., Warrack, S. & Safdar, N. Feasibility of oral prenatal probiotics against maternal group B Streptococcus vaginal and rectal colonization. J. Obstet. Gynecol. Neonatal. Nurs. 43, 294–304 (2014).
Hanson, L. et al. A randomized controlled trial of an oral probiotic to reduce antepartum group B Streptococcus colonization and gastrointestinal symptoms. Am. J. Obstet. Gynecol. MFM 5, 100748 (2023).
Morrill, S. R. et al. Models of murine vaginal colonization by anaerobically grown bacteria. J. Vis. Exp. https://doi.org/10.3791/64032 (2022).
O’Brien, V. P. et al. Low-dose inoculation of Escherichia coli achieves robust vaginal colonization and results in ascending infection accompanied by severe uterine inflammation in mice. PLoS One 14, e0219941 (2019).
Robinson, C. K., Saenkham-Huntsinger, P., Hanson, B. S., Adams, L. G. & Subashchandrabose, S. Vaginal inoculation of uropathogenic Escherichia coli during Estrus leads to genital and renal colonization. Infect. Immun. 90, e0053221 (2022).
Leatham, M. P. et al. Precolonized human commensal Escherichia coli strains serve as a barrier to E. coli O157:H7 growth in the streptomycin-treated mouse intestine. Infect. Immun. 77, 2876–2886 (2009).
Gilbert, N. M. et al. Gardnerella vaginalis and prevotella bivia trigger distinct and overlapping phenotypes in a mouse model of bacterial vaginosis. J. Infect. Dis. 220, 1099–1108 (2019).
Mahajan, G. et al. Vaginal microbiome-host interactions modeled in a human vagina-on-a-chip. Microbiome 10, 201 (2022).
Teodori, L. et al. Complete genome sequence of lactobacillus crispatus type strain ATCC 33820. Microbiol. Resour. Announc 10, e0063421 (2021).
Mulvey, M. A., Schilling, J. D. & Hultgren, S. J. Establishment of a persistent Escherichia coli reservoir during the acute phase of a bladder infection. Infect. Immun. 69, 4572–4579 (2001).
in Guide for the Care and Use of Laboratory Animals The National Academies Collection: Reports funded by National Institutes of Health (2011).
Ajayi, A. F. & Akhigbe, R. E. Staging of the estrous cycle and induction of estrus in experimental rodents: an update. Fertil. Res. Pract. 6, 5 (2020).
Caligioni, C. S. Assessing reproductive status/stages in mice. Curr Protoc Neurosci Appendix 4, Appendix 4I, https://doi.org/10.1002/0471142301.nsa04is48 (2009).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microb/iome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Davis, N. M., Proctor, D. M., Holmes, S. P., Relman, D. A. & Callahan, B. J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6, 226 (2018).
Mandal, S. et al. Analysis of composition of microbiomes: a novel method for studying microbial composition. Micro. Ecol. Health Dis. 26, 27663 (2015).
Kassambara, A. & Mundt, F. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses, https://CRAN.R-project.org/package=factoextra (2020).
McMurdie, P. J. & Holmes, S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217 (2013).
Acknowledgements
We would like to acknowledge Colleen Katherine Ardis of the Britton lab for colony management of the HMbmice, the Baylor College of Medicine Center for Metagenomics and Microbiology Research for 16S rRNA v4 sequencing, Andrew Kyle Brand Ardis and Firas Midani for assistance in troubleshooting the 16S rRNA sequencing analysis and repositing 16S rRNA v4 data, and Amanda Lewis and Nicole Gilbert for the strain of streptomycin resistant Prevotella bivia. The gnotobiotic studies were performed by the GEMS Gnotobiotic Core at Baylor College of Medicine, which is supported in part through the TMC DDC by NIH PHS grant P30DK056338 and the assistance of Margaret E. Conner. M.E.M. and J.J.Z. were supported by an NIH T32 award (T32GM136554) and M.E.M., V.M.E. and S.O. were supported by NIH F31 awards (AI167538, AI167547, and HD111236) respectively. Studies were supported by the Burroughs Wellcome Fund Next Gen Pregnancy Initiative (NGP10103), and NIH R01 (DK128053) and U19 (AI157981) awards to K.A.P. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Author information
Authors and Affiliations
Contributions
K.A.P. and M.E.M. conceived and designed experiments. M.E.M., V.M.E., J.J.Z., S.O., K.R., M.B.B., S.W.F. and K.A.P. performed experiments and collected data. K.A.P. and M.E.M. analyzed and interpreted results. M.E.M., V.M.E. and K.A.P. drafted the manuscript. K.A.P. and R.A.B. secured funding. All authors contributed to the discussion and manuscript edits.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mejia, M.E., Mercado-Evans, V., Zulk, J.J. et al. Vaginal microbial dynamics and pathogen colonization in a humanized microbiota mouse model. npj Biofilms Microbiomes 9, 87 (2023). https://doi.org/10.1038/s41522-023-00454-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41522-023-00454-9
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
