
Abstract
Bacteria often reside in sessile communities called biofilms, where they adhere to a variety of surfaces and exist as aggregates in a viscous polymeric matrix. Biofilms are resistant to antimicrobial treatments, and are a major contributor to the persistence and chronicity of many bacterial infections. Herein, we determined that the CpxA-CpxR two-component system influenced the ability of enteropathogenic Yersinia pseudotuberculosis to develop biofilms. Mutant bacteria that accumulated the active CpxR~P isoform failed to form biofilms on plastic or on the surface of the Caenorhabditis elegans nematode. A failure to form biofilms on the worm surface prompted their survival when grown on the lawns of Y. pseudotuberculosis. Exopolysaccharide production by the hms loci is the major driver of biofilms formed by Yersinia. We used a number of molecular genetic approaches to demonstrate that active CpxR~P binds directly to the promoter regulatory elements of the hms loci to activate the repressors of hms expression and to repress the activators of hms expression. Consequently, active Cpx-signalling culminated in a loss of exopolysaccharide production. Hence, the development of Y. pseudotuberculosis biofilms on multiple surfaces is controlled by the Cpx-signalling, and at least in part this occurs through repressive effects on the Hms-dependent exopolysaccharide production.
Similar content being viewed by others
Introduction
Yersinia pseudotuberculosis (Yptb) is a common foodborne pathogen that most often causes mild and self-limiting gastrointestinal illnesses1. This bacteria is the progenitor of the deadly zoonotic pathogen, Yersinia pestis (Ype), the causative agent of the devasting diseases, bubonic and septicemic plague2,3. The two species share a genomic identity of ~97%4, yet are distinguishable by the diverse repertoires of pathophysiological factors that they can encode5.
One of the most crucial pathophysiological features associated with many bacteria is the ability to form biofilms. Biofilms are a viscous polymeric matrix of bacterial communities capable of growth on abiotic and biotic surfaces, and which are inherently resistant to antimicrobial agents that causes significant problems for the clinicians trying to treat the bacterial infections6,7,8. Regardless of the species, Yersinia survival in the environment or during an infection is often dependent upon the ability to develop a biofilm9,10,11.
A core component of Yersinia biofilm is the extracellular matrix material termed exopolysaccharide (EPS). The production and transport of EPS is controlled by the chromosomal hms loci in Yptb9,12 and Ype12,13,14,15,16,17,18,19,20,21,22,23,24. Yet recent ‘omics’ based applications using Yptb25,26 and Ype25,27,28 models has uncovered many additional structural factors, metabolic pathways and integrated regulatory circuits involved in Yersinia biofilm development. It is now known that the quorum sensing, RovA-RovM and RcsA-RcsB regulatory cascades are common processes controlling Yersinia biofilms, albeit they have contrasting affects in Yptb26,29,30,31,32 compared to Ype20,28,31,32,33,34,35. These observations are consistent with Yptb and Ype having evolved in different environments. By extension, it is not surprising that the different species and even serotypes within species have also co-opted specific regulatory circuitry to contribute to biofilm formation. For example, RpoS36, BarA/UvrY37, motility38 and a type VI secretion system25 are associated with Yptb biofilm development. On the other hand, various factors that fine-tune bacterial surface characteristics39,40,41, as well as metabolic adaptation and the function of small regulatory RNAs42,43,44,45,46,47,48 are all associated with Ype biofilm development.
From earlier work on Yptb, the classical CpxAR two-component signalling system is involved in modulating the regulatory cascade outputs of RovA-RovM49,50 and the Rcs phosphorelay system51. These couplings suggests that Cpx-signalling might also be involved in controlling aspects of biofilm development. The CpxAR system has historically been implicated in maintaining the integrity of the bacterial envelope during exposure to extracytoplasmic stresses52. CpxA functions as both a sensor kinase and a phosphatase for the cognate response regulator, CpxR. In response to particular extracyoplasmic stresses, CpxA is first an autokinase and then a phosphoryl donor to CpxR. The active phosphorylated CpxR (CpxR~P) isoform subsequently serves as a transcription activator or repressor for the controlled expression of many genes53,54,55,56. In the absence of stress, CpxA phosphatase activity predominates, and this serves to dephosporylate CpxR~P keeping it inactive. While the mechanisms of Cpx-signalling have been mostly established in a laboratory strain of Escherichia coli, it has emerged recently that this pathway is an important molecular switch controlling virulence gene expression in pathogenic Yersinia and many other clinically and agriculturally relevant Gram-negative bacterial pathogens57.
Establishment of cohesive biofilms require bacteria to sense the surfaces they encounter58. Exactly how this might occur is largely mysterious. There is some suggestion that it is accomplished by certain cell appendages, including curli, pili/fimbriae and flagella, or via two-component and phosphorelay signalling systems59. The fact that the Cpx- and Rcs- signalling systems are both activated upon hydrophobic and hydrophilic surface contact, respectively, is consistent with roles in biofilm development60,61,62. However, it has been suggested recently that Rcs-signalling, but not the Cpx-signalling, is activated upon surface contact and involved in promoting initial biofilm formation63.
Hence, ambiguity in the literature surrounds the role of Cpx-signalling during initial surface sensing and subsequent biofilm development. The goal of this study was therefore to assess the role of Cpx-signalling in the homoeostasis of biofilms formed by Yptb. This is further motivated by earlier observations that Cpx-signalling reduces adhesion of Yptb to eukaryotic cells64. Indeed, we report that mutant Yptb lacking the dual functional CpxA sensor kinase and phosphatase totally aborted biofilm formation on a plastic surface and on the surface of the Caenorhabditis elegans nematode. This substantial reduction in biofilm development was caused in part by an excess of active CpxR~P deregulating expression from the hms loci. Therefore, Cpx-signalling prevents biofilm formation by restricting EPS production, the core component of Yersinia biofilm extracellular matrix material.
Results
Intact Cpx-signalling is crucial for the biofilm formation on diverse surfaces
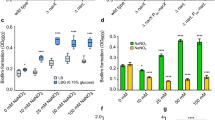
Due to the ambiguity surrounding the role of Cpx-signalling in surface contact and sensing during biofilm formation63,64, we examined the role of Yptb-YPIII Cpx-signalling in biofilm formation on two different surfaces—one was an abiotic surface and the other a biotic surface. To monitor biofilm development on an abiotic surface, 96-well polystyrene microtiter plates were used. Biofilm biomass dynamic was assessed following the incubation of 6-fold serially diluted cultures of the parental Yptb-YPIII serotype O:3, a food-borne clinical isolate, and the Cpx-signalling isogenic mutants, ΔcpxA and ΔcpxR. Strikingly, loss of CpxA (ΔcpxA) prevents biofilm formation (Fig. 1a, left panel). We noted that planktonic growth of the ΔcpxA mutant was slightly compromised as measured by optical density measurements at a wavelength of 600 nm (Fig. 1a, right panel). Since differences in cell wall composition could result in different light scattering characteristics, we also tested the actual number of living cells at similar OD600 for the different strains. Regardless of the strain tested, similar numbers of viable bacteria were recovered, although the colonies formed by mutants lacking cpxA were smaller and exhibited a different pigmentation (Supplementary Fig. 1). Nevertheless, since biofilm is growth dependent, we repeated the assay with a 10-fold increase in initial culture density. Despite an anticipated increase in planktonic growth (Fig. 1b, right panel), this mutant was still unable to form a biofilm (Fig. 1b, left panel). Importantly, the biofilm defect in the ΔcpxA mutant was partially restored via the ectopic expression of cpxA (ΔcpxA/pCpxA), supporting the crucial role of CpxA in biofilm formation (Fig. 1c). These results suggest a vital role for Cpx-signalling in the cellular response to surface contact and sensing by Yptb during biofilm development.

Dynamic biofilm formation was analysed in 96-well round-bottom microtiter plates. A serial dilution-based scheme (6-fold, 9 times) was applied to monitor development of sessile biofilm and growth of planktonic cells from the LB-cultured (24 h incubation at 26 °C, 125 rpm shaking) strains of Yptb-YPIII. Each dot in the graph represents a different dilution of the same culture. Parental, wild-type (WT) or mutants within the Cpx-signalling were seeded either at 0.1 OD600 or both at 0.1 and 1.0 OD600. (a) Loss of CpxA (ΔcpxA null-mutant) prevents biofilm formation. (b) Poor growth of the ΔcpxA null-mutant does not account for this biofilm formation defect. (c) Biofilm formation is restored to the ΔcpxA null-mutant via ectopic expression of wild-type copy of cpxA in ΔcpxA null-mutant of the Cpx-signalling. Error bars on the graphs represent standard error of mean from three biological and three technical replicates of each strain. Statistical differences between sessile biofilm formation of each strain uses a total biofilm biomass counts (represented by the area under each curve) in comparison with a respective reference control (WT for a, WT_01 OD for b and WT/Evct for c). Standard error of mean for each strain was calculated from three independent biological experiments containing three technical replicates. Extent of significance from the parent or negative control was determined using One-way ANOVA with Tukey’s multiple comparisons test, with a single pooled variance. The difference in variance with a p-value of <0.05 was considered significant. The p-values are indicated by <0.001 (***), <0.01 (**) and > 0.05 (ns).
To explore further a role for Cpx-signalling in biofilm formation, we utilised the same set of isogenic Yptb-YPIII Cpx-signalling variants, but now expressing GFP, to examine their ability to form biofilm on the biotic surface of the Caenorhabditis elegans nematode. Larvae at L-4 stage from a standard laboratory strain (N2) of C. elegans were transferred (n = 30) on to normalised lawns of Gfp-expressing Yptb-YPIII Cpx-signalling variants. After 24 h growth at room temperature, C. elegans were examined by fluorescent microscopy. When exposed to a lawn of parental bacteria or bacteria lacking CpxR, a distinct biofilm biomass was formed on the worm surface (Fig. 2a). In contrast, the surface of worms that had been grown on bacteria lacking CpxA remained completely free of the biofilm biomass (Fig. 2a). However, the ΔcpxA mutant strain complemented by ectopic expression of CpxA (ΔcpxA/pCpxA) regained the ability to establish a robust biofilm on the surface of exposed C. elegans (Fig. 2a). These data corroborate the plastic plate assay, confirming that Cpx-signalling is crucial for Yptb-YPIII biofilm development on at least two different surfaces—one being abiotic and the other being biotic. Additionally, we monitored the survival rate of C. elegans on the lawns of these strains over a 5-day period. We observed that by the end of day 3, all worms had died on the parental Yptb-YPIII (WT) lawn (Fig. 2b). Moreover, growth on the lawns of ΔcpxR and ΔcpxA/pCpxA caused the death of all worms after 4 days (Fig. 2b). In contrast, all worms survived on the ΔcpxA lawn throughout the incubation period (Fig. 2b). Hence, the biofilm biomass formed by WT, ΔcpxR, and ΔcpxA/pCpxA strains on the worm surface resulted in worm starvation and death. On the other hand, an inability of the ΔcpxA mutant to form a biofilm permitted the worms to continue feeding and remain alive. Taking these observations all together, intact Cpx-signalling is clearly crucial for the ability of Yptb-YPIII to form biofilms on diverse surfaces.

Infection and survival of N2, a standard laboratory strain of Caenorhabditis elegans nematode on the lawn of Cpx-signalling strains. (a) L4 stage (larval) nematodes were seeded on the equalised OD600 lawn of Gfp-expressing parental Yptb-YPIII (WT), ΔcpxA, ΔcpxR, ΔcpxP and ΔcpxA/pCpxA strains of Cpx-signalling and imaged C. elegans after 24 h of growth on each respective lawn. White arrowheads indicate Gfp-coloured dense mass of biofilm from the corresponding strain. The ∆cpxA null-mutant failed to form biofilm on the surface of C. elegans, which was restored upon ectopic expression of pCpxA. H- head and T- tail of C. elegans. A representative image from three independent biological replicates of each strain is shown. Pictures were acquired by Nikon Stereoscopic Microscope SMZ1500 with Gfp-fluorescence (excitation ƛ 470 nm and emission ƛ 525 nm) at ×50 magnification, equivalent to a 10 µm scale bar. The Pre-installed NIS Elements V4.0 imaging software was used to capture the pictures. (b) Survivability percentage of C. elegans on parental wild-type (WT) and mutant strains of Cpx-signalling. Thirty L4 stage (larval) nematodes were seeded on the equalised OD600 lawn of WT, ΔcpxA, ΔcpxA/pCpxA, ΔcpxR and ΔcpxP strains and scoring of live nematodes was recorded every 24 h, continuous for 5 days.
Accumulation of active phosphorylated CpxR prevents biofilm formation
In several different bacteria it has been demonstrated that a deletion of cpxA leads to an excess of CpxR~P as CpxR is phosphorylated via alternate pathways such as through the low molecular weight phosphodonor Acetyl-phosphate49,65,66,67,68. Hence, we wondered if an excess of active CpxR~P in the cpxA strain of Yptb-YPIII prevents biofilm formation. Protein lysates were prepared from the biofilm biomass derived from the microtitre plate serial dilution assay. When analysed by the Phos-tagTM acrylamide gel system it was evident that this material contained active phosphorylatable CpxR~P accumulated in the cpxA null-mutant (Supplementary Fig. 2). Specificity of this assay was validated by using an isogenic mutant encoding cpxRD8A, D9A, D51A, M53A, K100A that produces a non-phosphorylated CpxR (CpxRPneg). This CpxRPneg variant migrated only as the inactive non-phosphorylated isomer (Supplementary Fig. 2). Thus, to address if this accumulation prevented biofilm formation, we utilised a set of isogenic mutations lacking the ackA and pta genes responsible for the production of the high-energy phospho intermediate, Acetyl-phosphate—one in the CpxA-plus (WT) background and one in the CpxA-minus (ΔcpxA) background. Significantly, the triple mutant lacking cpxA, ackA and pta did not accumulate CpxR~P to high levels as judged by the Phos-tagTM acrylamide gel system (Fig. 3a). Hence, an inability to produce the high-energy phospho intermediate, Acetyl-phosphate, reduces the amount of active phosphorylatable CpxR~P accumulated in the cpxA null-mutant. When these strains were assessed for biofilm formation by microtitre plate serial dilution assay, introduction of the ackA and pta mutations into the cpxA mutant restored the ability to produced biofilms, albeit not to the extent observed for parental (WT) bacteria (Fig. 3b). Thus, this data provides compelling evidence for a role of active phosphorylated CpxR~P in preventing biofilm formation by Yptb.

The Phos-tagTM acrylamide system was used to measure accumulated CpxR~P in bacteria grown in LB broth to late-stationary phase at 26 °C in 96-well round-bottomed microtiter plate (a). Samples were recovered from the planktonic portion of the culture from the mature stage of biofilm (equivalent to 4th dilution of the 6-fold dilution series). Lysed bacteria were electrophoresed on a freshly prepared 12% Phos-tagTM Acrylamide AAL-107 gel, immunoblotted, and detected with anti-CpxR antiserum. The cytoplasmic molecular chaperone DnaJ, served as a loading control and derived from the same samples, but analysed on a conventional 12% acrylamide SDS-PAGE and immunoblotted with anti-DnaJ antiserum as described in Methods section. The representative unprocessed (raw) blot image of each can be seen in the Supplementary file. Strains: parent (WT), YPIII/pIB102; cpxR null-mutant, YPIII08/pIB102; nlpE null-mutant, YPIII34/pIB102; cpxA null-mutant, YPIII07/pIB102; ackA, pta null mutant, YPIII69/pIB102; ackA, pta and cpxA null-mutant, YPIII49/pIB102. The red arrow reflects the active phosphorylated CpxR~P isoform accumulated in the Yptb-YPIII cytoplasm, while the black arrow indicates the accumulated inactive non-phosphorylated CpxR isoform. An unknown non-specific product (NSP) is indicated by a grey arrow. These same strains were monitored for biofilm formation using the serial dilution scheme (6-fold, 9 times) in 96-well round-bottom microtiter plates (b). Error bars on the graphs represent standard error of mean from three independent biological and three technical replicates of each strain. Statistical differences between sessile biofilm formation of each strain uses a total biofilm biomass counts (represented by the area under each curve) in comparison with WT as the reference control. Statistical significance with respect to the parent (WT) was determined using One-way ANOVA with Tukey’s multiple comparisons test, with a single pooled variance. The difference in variance with a p-value of <0.05 was considered significant. The p-values are indicated by <0.001 (***), <0.01 (**) and >0.05 (ns).
The hms loci of Yptb-YPIII are transcriptionally regulated by active Cpx-signalling
As demonstrated above, Cpx-signalling is crucial for biofilm formation, irrespective of the surface. We hypothesised that active CpxR~P acts as a transcription factor regulating one or more aspects of biofilm formation. EPS production controlled by the hms loci is a vital component of Yptb biofilm9,12. We could also verify this in our experimental systems. A Hms-EPS defective strain created by deletion of the hmsS (∆hmsS) that encodes for the biofilm PGA (Poly-β-1, 6-GlcNAc) synthesis protein, PgaD/HmsS was unable to form any measurable biofilm biomass (Fig. 1a), in agreement with previous observations20. The lack of biofilm biomass was equivalent to the ∆cpxA mutant (Fig. 1a). Hence, we investigated whether the ∆cpxA mutant phenotype was caused by active CpxR~P controlling transcription from the hms loci. We inspected the transcriptional profile of four hms loci—hmsHFRS, hmsCDE, hmsT and hmsP (Fig. 4a). In the first approach, qRT-PCR was used to measure endogenous transcriptional expression of the hms loci located in cis in Yptb of the locked-on Cpx-signalling mutant, ΔcpxA and the locked-off CpxR defective mutant, ΔcpxR, in comparison with parent (WT) bacteria. Following late-stationary phase growth, it was observed that active CpxR~P (in ΔcpxA) significantly reduced the expression of the Hms-EPS transporter gene, hmsH (~50% reduction cf WT) (Fig. 4b), and the diguanylate cyclase encoding genes, hmsT (~45% reduction cf WT) (Fig. 4c) and hmsC (~50% reduction cf WT) (Fig. 4d), which synthesise c-di-GMP, a vital molecule for the synthesis and export of Hms-EPS on the cell surface16. This repressive effect was relieved in ΔcpxR null-mutant, confirming the active contribution of CpxR~P as a transcriptional repressor of expression from the hmsHFRS, hmsT and hmsCDE loci (Fig. 4b–d—in cis). This CpxAR-dependent collective loss of expression from these three loci would be expected to reduce EPS synthesis and transport to the bacterial surface. In contrast, we also observed that active CpxR~P increased the relative expression of hmsP (Fig. 4e), which encodes a diguanylate phosphodiesterase that hydrolyses c-di-GMP19,24. Critically, a CpxAR-dependent increase of HmsP will decrease c-di-GMP levels, which will also hinder EPS production.

(a) Operon structure of the Yptb-YPIII hms loci. Genetic organisation of hmsHFRS and hmsCDE operons and unlinked hmsT and hmsP genes. The locus-tag (YPK_XXXX) of each loci mentioned underneath. PCR-amplified gene-specific (for qRT-PCR) and 5' UTR (for EMSA) fragments of corresponding loci are represented by respective vertical blue and horizontal black rectangle box. Numbers in parentheses indicate amino acid sequence identity with isofunctional homologues from Ype strain KIM10+. (b–e) Cpx-signalling mediated differential expression of hms loci. Quantitative RT-PCR was performed on the cDNA template, synthesised from the total RNA of Yptb-YPIII Cpx-signalling in cis strains, parental WT (intact Cpx-signalling), ΔcpxA (locked-on Cpx-signalling producing excessive active phosphorylated CpxR~P) and ΔcpxR (locked-off Cpx-signalling unable to produce CpxR) and from in trans strains, ΔcpxR/pMMB208 (ectopic expression of IPTG-inducible pMMB208 plasmid in ΔcpxR null-mutant, negative control), ΔcpxR/pCpxR-WT (ectopic expression of wild-type CpxR from pMMB208 plasmid in ΔcpxR null-mutant) and ΔcpxR/pCpxRPneg (ectopic expression of phosphorylation defective CpxRPneg mutant from pMMB208 plasmid in ΔcpxR null-mutant). Both in cis and in trans strains were cultured in LB with shaking (150 rpm) at 26 °C. The in cis strains were grown for 24 h (no IPTG) while the in trans strains were grown up to 6 h with IPTG-induction (10 µM) following subculture (1/20 dilutions). Expression of the hms loci within the in cis strains, ΔcpxA and ΔcpxR was calculated relative to WT (b–e; left panel) whereas expression of corresponding hms gene within the in trans strains, ΔcpxR/pCpxR-WT and ΔcpxR/pCpxRPneg was calculated relative to ΔcpxR/pMMB208 (b–e; right panel). Relative expression of hms loci from both in cis and in trans was monitored from three biological and three technical replicates of each strain with two house-keeping internal standards, gyrB and rpoC. Statistical significance was determined using One-way ANOVA with Tukey’s multiple comparisons test, with a single pooled variance. The difference in variance with a p-value of < 0.05 was considered significant. The p-values are indicated by <0.0001 (****), <0.001 (***), <0.01 (**), <0.05 (*) and >0.05 (ns).
To validate these data, we performed qRT-PCR analysis of hms loci within the ΔcpxR mutant ectopically producing from an expression plasmid either an active wild-type (pCpxR+) or phosphorylation defective inactive isoform of CpxR (pCpxRPneg). Mirroring the “in cis” generated data, accumulation of active CpxR~P by overexpression of CpxR~P resulted in repression of hmsH, hmsT and hmsC expression (Fig. 4b–d—in trans), and activation of hmsP expression (Fig. 4e—in trans). On the other hand, accumulation of inactive non-phosphorylated CpxRPneg resulted in activation of hmsH, hmsT and hmsC expression (Fig. 4b–d—in trans). Interestingly, this phosphorylation defective CpxRPneg also lead to robust de-repression of hmsP (Fig. 4e—in trans). This indicates that the CpxR-HmsP intrinsic negative feedback loop might be eliminated by ectopic expression of an IPTG-inducible form of inactive CpxR.
In parallel, we also established translational gene fusions of hmsH, hmsT and hmsP with gfpmut3 in the backgrounds of parent Yptb-YPIII (WT), ΔcpxA mutant and ΔcpxR mutant. Fluorescence intensity output from the PhmsH::Gfp (Supplementary Fig. 3a) and PhmsT::Gfp (Supplementary Fig. 3b) fusions were considerably lower in the ΔcpxA null-mutant compared to the ΔcpxR null-mutant. Moreover, fluorescence intensity output from the PhmsP::Gfp fusion was considerably higher in the ΔcpxA null-mutant compared to the ΔcpxR null-mutant (Supplementary Fig. 3c). These data corroborated observations from qRT-PCR experiments. Taken altogether, we show clearly that active CpxR~P represses transcriptional output from the hmsHFRS, hmsT and hmsCDE loci, but induces output from the hmsP locus. This differential control is explained by the opposing roles these loci have in EPS synthesis in Yptb-YPIII.
CpxR~P-dependent control of hms transcription is direct
Having established that active CpxR~P represses transcriptional output from the hmsHFRS, hmsT and hmsCDE loci, but induces output from the hmsP locus, we wondered if this regulatory control was direct. Hence, we examined the in vitro binding of an active phosphorylated form of purified CpxR (CpxRwt) and an inactive non-phosphorylated form of purified CpxR (CpxRPneg) at the promoter regions of the four hms loci, PhmsH, PhmsT, PhmsC and PhmsP as depicted in Fig. 4a. Two concentrations of purified CpxRHis6 −50 µM and 100 µM, were tested for specific binding. In all cases, the higher concentration of active phosphorylated CpxRwt caused a shift of all four specific hms DNA fragments (Fig. 5). This was specific targeted binding because inactive non-phosphorylated CpxRPneg failed to induce any shift in the four specific hms DNA fragments under identical conditions, and neither CpxRwt nor CpxRPneg induced a shift of the 16 S rRNA encoding DNA fragment that was used as another specificity control (Fig. 5). Hence, phosphorylated CpxR~P is required to bind to the promoters of the four hms loci. Further, this direct binding is likely a major mechanism driving CpxR~P dependent repression of hmsHFRS, hmsT and hmsCDE transcription, and induction of hmsP transcription.

An EMSA with complete 5' intergenic regulatory DNA of indicated hms loci with active CpxR~P was used to measure specific protein-nucleic acid interactions. Red asterisks (*) indicate the target promoter DNA-CpxRwt complex. Unbound promoter DNA is indicated with an arrowhead. The inactive non-phosphorylatable CpxRPneg isoform was unable to bind target DNA at these same conditions. A 16S rDNA fragment (148 bp) used as ‘non-specific’ negative control and its running location is indicated by an arrow (←). Lane-1: target promoter DNA, Lane-2: target promoter plus non-specific 16S rDNA, Lane-3: target promoter, non-specific 16S rDNA and CpxRHis6 (100 µM), Lane-4: target promoter, non-specific 16S rDNA and CpxRHis6 (50 µM) and Lane-5: target promoter, non-specific 16S rDNA and phosphorylation defective Mt7-CpxRHis6 (100 µM). Each reaction contained 200 mM Acetyl-phosphate to phosphorylate CpxRHis6 in the respective lane. Gel were stained with 1× GelRed DNA-staining dye solution. A representative image from three independent experiments for each target is shown. The representative unprocessed (raw) image of each EMSA-gel can be seen in the Supplementary file.
Cpx-signalling dependent transcriptional repression of the hms loci reduces EPS production
Having established that active CpxR~P differentially regulates transcription of the hms loci, we investigated if this correlated with altered Hms-EPS synthesis and export—an essential component of a cohesive Yersinia biofilm16. The Yersinia-Congo Red-binding assay is a traditional way to determine whether Yersinia strains manufacture and export Hms-EPS on the cell surface18,20,37. Colonies of the locked-on CpxA defective strain (∆cpxA) were unable to bind Congo Red (Fig. 6). This mirrored the phenotype of colonies of the Hms-EPS defective strain, ∆hmsS, which were also unable to bind Congo Red (Fig. 6). Crucially, colonies of parental Yptb-YPIII (WT), the ∆cpxR and ∆cpxP null-mutants, and the complemented ΔcpxA/pCpxA+ strain were all found to adsorb the Congo Red dye giving a distinctive red appearance (Fig. 6). Moreover, the cpxA null-mutant that also lacked the ackA and pta genes, regained the ability to bind Congo Red (Fig. 6). This emphasises that it is the accumulation of active phosphorylated CpxR~P that limits the ability of Yptb to bind Congo Red. Taken together, these results signify that Yptb-YPIII Cpx-signalling differentially regulates hms loci transcription, and this combined repression of hmsHFRS, hmsT and hmsCDE transcription, and induction of hmsP transcription prevents the manufacture and export of EPS to the cell surface. Hence, the lack of EPS represents one major reason why the Yptb-YPIII ΔcpxA null-mutant is unable to develop a biofilm.

The Congo Red binding assay was used as an indicator of EPS production by various Yptb-YPIII strains. Indicated Yptb strains were grown (with shaking) at 26 °C for 18 h in LB broth lacking NaCl. A 3.5 µL culture containing an equal number of cells via normalisation of optical density at 600 nm was spotted on NaCl-lacking LA that was supplemented with the dye 0.01% (w/v) Congo Red. Images were taken after growth at 26 °C for 24 h. A representative colony from three independent biological and three technical triplicates of each strain is shown. Used as a negative control was the ∆hmsS mutant that contains an in-frame deletion of codons 11–135 in hmsS, a crucial gene for the Hms-EPS matrix synthesis and export. Indicated scale bar (green line) is 1000 µm. Strains: parent (WT), YPIII/pIB102; cpxA null-mutant, YPIII07/pIB102; complemented cpxA/pCpxA+, YPIII07/pIB102, pJF067; cpxR null-mutant, YPIII08/pIB102; cpxP null-mutant, YPIII41/pIB102; hmsS null- mutant, YPIII_2238/pIB102; ackA, pta null-mutant, YPIII69/pIB102; ackA, pta and cpxA null-mutant, YPIII49/pIB102; nlpE null-mutant, YPIII34/pIB102.
Contribution of auxiliary Cpx-signalling molecules, CpxP and NlpE, on biofilm formation
CpxP and NlpE are auxiliary proteins involved in environmental signal integration in the Cpx-signalling pathway. The periplasmic located CpxP inhibits the histidine kinase ‘sensor’ function of CpxA through a direct dynamic interaction69,70, whereas the outer membrane located NlpE contributes to system activation60,62. To address if CpxP and NlpE of Yptb-YPIII contribute to biofilm formation we examined the phenotype of a full-length in-frame cpxP mutant and a full-length in-frame nlpE mutant in our biofilm assays. The cpxP mutant formed biofilm on abiotic surface as judged by the plastic microtitre plate serial dilution assay, albeit to a reduced extent compared to parental Yptb (Fig. 1a). On the other hand, robust biofilms were also formed on the biotic surface according to the C. elegans grazing assay (Fig. 2a). Critically, the biofilms formed by the cpxP null-mutant on the worm body quickly resulted in worm starvation as was similarly observed for worms exposed to wild-type bacteria (Fig. 2b). Moreover, the cpxP mutant retained the ability to produce EPS as indicated by colonies stained with Congo red (Fig. 6). The ability of the cpxP mutant to form biofilms and maintain EPS production correlates with this bacteria accumulating very little active phosphorylated form of CpxR (Supplementary Fig. 2). Additionally, the nlpE null-mutant formed considerable biofilm on the abiotic surface that could not be distinguished from the biofilms formed by parental bacteria (Fig. 3b). Expectantly, the nlpE null-mutant retained the ability to produce EPS as judged by colonies stained with Congo Red (Fig. 6). Furthermore, the ability of the nlpE null-mutant to form biofilms and maintain EPS production correlates with this bacteria accumulating very little active phosphorylated form of CpxR (Fig. 3a). Hence, the mechanism by which Cpx-signalling regulates biofilms formed by Yptb to a minor extent involves CpxP, but is independent of NlpE.
CpxAR can act through RpoE signalling
In some bacteria, alternative sigma factor, RpoE contributes to biofilm formation71,72,73. It is not known if this is true also for Yersinia spp. because a growth dependence on RpoE makes deficient mutants difficult to study74,75,76. Nevertheless, CpxAR has been shown to act through RpoE in several bacteria66,77,78,79. Hence, here we aimed to determine the impact of cpxA and cpxR deletions on rpoE transcription in Yptb-YPIII, with a view that this might be a mechanism through which Cpx-signalling can act. We performed qRT-PCR to measure differential transcriptional expression of the rpoE gene in the intrinsically active Cpx-signalling mutant, ΔcpxA, and the CpxR defective mutant, ΔcpxR, in comparison with parent (WT) Yptb, following late-stationary phase growth. It was observed that active CpxR~P (in ΔcpxA) significantly reduced the expression of rpoE (~50% reduction cf WT) (Fig. 7a). This repressive effect was relieved in the ΔcpxR null-mutant, indicating an active contribution of CpxR~P as a transcriptional repressor of expression from rpoE (Fig. 7a). Consistent with this, active phosphorylated form of purified CpxR (CpxRwt) bound to the rpoE promoter region in vitro, whereas the inactive non-phosphorylated form of purified CpxR (CpxRPneg) did not (Fig. 7b). Correspondingly, we identified a putative CpxR~P binding motif reminiscent of the reported consensus sequence GTAAA-N(4-8)-GTAAA78,80, which may represent the CpxR binding site within the rpoE promoter (Supplementary Fig. 4). Taken together, this data establishes that CpxAR can act through RpoE signalling, which indicates that this Cpx-RpoE regulatory cascade may influence biofilms formed by Yptb.

Cpx-signalling mediated repression of rpoE transcription (a). Quantitative RT-PCR was performed on the cDNA template, synthesised from the total RNA isolated from the Yptb-YPIII isogenic ‘in cis’ mutant strains: parental WT (intact Cpx-signalling), ΔcpxA (locked-on Cpx-signalling with excessive accumulation of active phosphorylated CpxR~P) and ΔcpxR (locked-off Cpx-signalling unable to produce CpxR), and from ‘in trans’ strains: ΔcpxR/pMMB208 (ectopic expression of IPTG-inducible pMMB208 plasmid in ΔcpxR null mutant—negative control), ΔcpxR/pCpxRWT (ectopic expression of wild-type CpxR from pMMB208 plasmid in ΔcpxR null mutant) and ΔcpxR/pCpxRPneg (ectopic expression of phosphorylation defective CpxRPneg mutant from pMMB208 plasmid in ΔcpxR null mutant). Both in cis and in trans strains were cultured in LB with shaking (150 rpm) at 26 °C. The in cis strains were grown for 24 h (no IPTG) while the in trans strains were grown up to 6 h with IPTG-induction (10 µM) following subculture (1/20 dilutions). Expression of rpoE within the in cis strains, ΔcpxA and ΔcpxR was calculated relative to WT (left panel) whereas expression within the in trans strains, ΔcpxR/pCpxRWT and ΔcpxR/pCpxRPneg was calculated relative to ΔcpxR/pMMB208 (right panel). Relative expression of rpoE from both in cis and in trans was monitored from three independent biological and three technical replicates of each strain with two house-keeping internal standards, gyrB and rpoC. Statistical significance was determined using One-way ANOVA with Tukey’s multiple comparisons test, with a single pooled variance. The difference in variance with a p-value of <0.05 was considered significant. The p-values are indicated by, <0.001 (***), <0.01 (**) and <0.05 (*). The active phosphorylated form of CpxR (CpxR~P) binds at the promoter of rpoE (b). EMSA with complete 5' intergenic regulatory DNA of rpoE with active CpxR~P. A single asterisk (*) indicates the target promoter DNA-CpxRwt complex. Unbound promoter DNA is indicated with an arrowhead. A 16S rDNA fragment (148 bp) used as ‘non-specific’ negative control and its running location is indicated by an arrow (←). Inactive non-phosphorylated CpxR (CpxRPneg) was unable to bind target DNA. Lane-1: target promoter DNA, Lane-2: target promoter plus non-specific 16S rDNA, Lane-3: target promoter, non-specific 16S rDNA and CpxRHis6 (100 µM), Lane-4: target promoter, non-specific 16S rDNA and CpxRwt (50 µM) and Lane-5: target promoter, non-specific 16S rDNA and phosphorylation defective CpxRPneg (100 µM). Each reaction contained 200 mM Acetyl phosphate to phosphorylate CpxRwt in the respective lane. Gel were stained with 1× GelRed DNA-staining dye solution. A representative image from three independent experiments is shown. The representative unprocessed (raw) image of EMSA-gel can be seen in the Supplementary file.
Discussion
In this study, we established that Cpx-signalling is vital for the initiation, maturation and maintenance of biofilms formed by Yptb. This connection remained intact irrespective of using an abiotic or biotic surface for the development of biofilm. Although Yersinia spp. can generate different biofilm matrices, the Hms-EPS—Poly-β-1,6-GlcNAc—is a predominant factor of biofilms formed by Yersinia spp.11,16. Controlled Hms-EPS production and assembly requires four distinct loci; hmsHFRS, hmsT, hmsCDE and hmsP. The hmsHFRS operon encodes for four membrane components that are essential for EPS biosynthesis and export14,16,18,21. The hmsD and hmsT genes encode for two diguanylate cyclase enzymes required for the production of c-di-GMP22,23. The c-di-GMP is a second messenger molecule critical for Yersinia biofilm production22. On the other hand, hmsP encodes a phosphodiesterase that acts as a counter balance, enabling Yersinia spp. to hydrolyse c-di-GMP to restrict biofilm formation19,24. We could demonstrate that active CpxR~P isoform directly targets promoters of the hmsHFRS, hmsT and hmsCDE operons causing down-regulation of gene expression. Active CpxR~P isoform also directly targets the promoter of hmsP causing up-regulation of gene expression. The net effect of this CpxR-mediated differential repression and activation is reduced synthesis and export of Hms-EPS to the cell surface, which severely restricts biofilm development. This suggests that Yptb utilises the CpxA-CpxR two-component signalling pathway for surface contact and sensing, which is consistent with initial evidence of a similar role in E. coli60. These findings add to the many examples that demonstrate the ability of CpxR~P to act as a transcriptional repressor and as a transcriptional activator in diverse bacteria53,54,55,56.
Biofilm development is a multifactorial process involving a large number of structural components, metabolic processes and regulatory circuitry. This enables bacteria to transition through intial bacterial attachment to a surface, microcolony formation, biofilm maturation, and dispersal to permit bacteria to establish new biofilms in favourable conditions elsewhere. These processes require a number of surface associated factors including pili/fimbriae, flagella, other adhesive fibers as well as carbohydrate-binding proteins81,82,83. This is true also for Yersinia biofilm formation, which involves a number of structural factors in addition to Hms-EPS production11,25,27. Hence, future work will need to investigate the possibility that Cpx-signalling influences the expression of biofilm structural components other than Hms-EPS.
Similarly, we expect that regulators and signalling cascades other than Cpx-signalling would also work to mediate the development, maintenance and renewal of Yptb biofilms. As illustrated in our model (Fig. 8), the involvement of other regulatory elements in controlling biofilm development has already been established using Yptb. RovA-RovM is a global regulatory system that likely influences multiple aspects of biofilm development, such as motility and EPS production30. The RcsA-RcsB phosphorelay system moderates EPS production by targeting the hmsT and hmsCDE operons32, and also influences biofilm stability through control of the YadE protein84. Moreover, the BarA/UvrY two-component system also impacts on biofilm production and stability, possibly through cascade regulation of CsrB and RcsB37. In addition, RpoS can influence biofilm formation via effects on motility through control of flhDC gene expression, and by regulating EPS synthesis36. Furthermore, N-Acyl homoserine lactone-mediated quorum sensing influences biofilm formation at least in part via repression of the plasmid encoded Ysc-Yop type III secretion system29. Follow-up work will need to establish whether these pathways work independently or through Cpx-signalling. We have already established existence of a CpxR-RovM-RovA regulatory cascade for the control of Yptb adhesion49,50 and a CpxR-RcsA-RcsB regulatory cascade for the control of Yptb Ysc-Yop type III secretion51. Hence, it could be that these regulatory cascades may extend the influence of Cpx-signalling to fine-tuning biofilm development.

A regulatory cascade involving the second messenger signalling molecule, c-di-GMP, is a vital component for the synthesis of Hms-EPS on the cell surface. The hmsCDE operon and hmsT gene encode diguanylate cyclase (synthesise c-di-GMP), which then helps in the biosynthesis and export of EPS on the cell surface by the hmsHFRS operon. The level of c-di-GMP is controlled by the product of hmsP, which encodes phosphodiesterase and assists in the hydrolysis of c-di-GMP. Active CpxR~P influences expression of the hms loci, activating hmsP (represented by green arrow) and repressing both hmsCDE and hmsHFRS operons along with hmsT gene (indicated by red hammer). For clarity, other regulatory cascades controlling Hms-EPS and subsequent biofilm formation are not shown. As biofilm is complex, incorporating many regulatory cascades, these regulatory cascades controlling biofilm (dotted line) could work through Cpx-signalling. These potential connections were not the focus of this study; they remain to be investigated, and this fact is indicated by the question mark ‘?’ symbol.
In this study, C. elegans nematodes were used as a model biotic surface for biofilm development by Yptb. We noted over the course of several independent experiments that Yptb parental bacteria, the ΔcpxR mutant and the ΔcpxP, which developed robust biofilms on the nematode surface, and resulting in nematode death, appeared to render alterations in nematode surface appearance, including partial disintegration of the worm. These observations were not observed with C. elegans exposed to the ∆cpxA mutant. These symptoms could well be associated with an invasive Yptb infection with pathological consequences. This is not without precendent for there are studies reporting on Yersinia colonisation of the nematode gastrointestinal tract85,86. This is interesting because the symptoms we observed are analogous to an acute systemic infection. It is tempting to speculate that these symptoms could relate to the production of factors that would promote biofilm-independent invasion such as the degradation activity of NghA, a β-N-acetylglucosaminidase17, or the toxigenic activity of the CNFY toxin87. The Yptb genome also enodes a potential chitinase, ChiC (YPK_0693). Hence, our follow-up work intends to examine the existence of biofilm-independent Yptb killing of infected nematodes.
We used the food-borne clinical isolate Yptb-YPIII serotype O:388,89 as a model system for these studies. On the basis that conserved Cpx-signalling has been verified experimentally in other Yptb isolates51,90, and also in two other Yersinia species—Y. pestis91,92 and Y. enterocolitica79,93,94, we assume that Cpx-signalling controls biofilm development in other Yersinia. Follow-up experimental work with other Yersinia isolates will attempt to verify this predicted connection. A comparison of protein sequence derived from Yptb-YPIII and Y. pestis (Ype-KIM10+) revealed a fully intact CpxAR system in Ype, including the periplasmic auxiliary signalling molecules, CpxP and NlpE (Supplementary Fig. 5). The Hms components were also represented in both bacteria (Fig. 4a). Moreover, the intergenic regulatory region between the divergent cpxP and cpxRA operons (Supplementary Fig. 5), as well as upstream of nlpE (Supplementary Fig. 5), hmsHFRS (Supplementary Fig. 6), hmsT (Supplementary Fig. 7), hmsCDE (Supplementary Fig. 8) and hmsP (Supplementary Fig. 9) all display high degrees of sequence conservation between Yptb and Ype. Significantly, these intergenic regions contained identifiable CpxR~P binding motifs reminiscent of the reported consensus sequence GTAAA-N(4-8)-GTAAA78,80. Hence, it seems probable that Ype derived sequence with minimal nucleotide differences would also support the in vitro binding by active CpxR~P. Follow-up work will strive to confirm this with a suite of experiments designed to explore a conserved role of Cpx-signalling in control of Hms-dependent EPS production in both Yptb and Ype.
Materials and methods
Bacterial strains, plasmids and growth conditions
Bacterial strains and plasmids used in this study are listed in Supplementary Table 1. Unless otherwise specified, bacteria were routinely grown in Luria-Bertani agar or Lysogeny broth95 at 26 ˚C with aeration using a shaking incubator. As appropriate, antibiotics—Ampicillin (100 μg/mL), Chloramphenicol (25 μg/mL) or Kanamycin (50 μg/mL) were added to the media/broth.
Mutants construction
The ∆hmsS, ∆cpxP and ∆nlpE null-mutants were constructed by overlap PCR technique96 using the relevant primer combinations listed in Supplementary Table 2. The PCR-amplified fragments were cloned into pJET1.2/Blunt plasmid (Thermo Scientific), and the mutated alleles of hmsS (representing a deletion of codons 11–135), cpxP (15–151), and nlpE (12–213) were confirmed by sequencing (Eurofins MWG Operon, Ebersberg, Germany) with pJET1.2F and pJET1.2R primers (Supplementary Table 2). The confirmed fragments were subcloned into the suicide plasmid, pDM4, following XbaI-XhoI restriction enzyme digestion. The ligated plasmids were transformed and maintained in E. coli SY327λpir. Plasmids with correct insert were sequenced using the gene-specific ‘A’ and ‘D’ primers, respectively (Supplementary Table 2). Confirmed plasmids were transformed into E. coli S17-1λpir, which served as the donor in conjugal matings with parental Yptb-YPIII. Mutated alleles were introduced into the Yptb-YPIII genome by a double cross-over homologous recombination event, and the ∆hmsS, ∆cpxP and ∆nlpE genotypes were recovered by sacB-dependent sucrose sensitivity97. Presence of these mutations in the genome of Yptb was confirmed by PCR using the gene-specific ‘A’ and ‘D’ primer pair, respectively (Supplementary Table 2) combined with subsequent sequence analysis of the amplified fragment with the same primer pair.
A mutated cpxR (YPK_4132) gene containing the site specific mutations—D8A, D9A, D51A, M53A, K100A—was synthetically generated by GenScript Biotech (Piscataway, New Jersey, USA) and contained within the plasmid pDK1011 (Supplementary Table 1). The synthetic DNA fragment contained the full length cpxR sequence as well as flanking DNA that was 215 bp upstream and 106 bp downstream of cpxR. This DNA fragment of 1032 bp was excised from pDK1011 by restriction digestion with XhoI/XbaI, and then cloned into the XhoI/XbaI restricted pDM4, giving rise to the mutagenesis vector pDM-DK1011 plasmid (Supplementary Table 1). The inserted sequence in pDM-DK1011 was confirmed by sequencing using the forward and reverse primers, CatR2 and R6KR, respectively (Supplementary Table 2). Generation of the in cis cpxRD8A, D9A, D51A, M53A, K100A mutant encoding a non-phoshorylated CpxRPneg variant occurred via allelic exchange to reconstitute the ∆cpxR mutant (YPIII08/pIB102) as described for the ∆hmsS, ∆cpxP and ∆nlpE mutants. The reconstituted CpxRPneg-producing mutant—YPIII_4132pneg/pIB102—was confirmed by colony-PCR using DK1011-12A and DK1011-12D primers (Supplementary Table 2) and subsequent sequencing of the amplified fragment with two additional primers, DK1011-12B and DK1011-12C (Supplementary Table 2).
Gfp-translational reporter construction
Promoter fragments, PhmsH (708 bp) PhmsT (315 bp) and PhmsP (378 bp), were PCR-amplified from Yptb-YPIII genomic DNA, using corresponding primer pairs (Supplementary Table 2). Respective PCR-fragments were subcloned into a commercial shuttle vector, pJET1.2/blunt (Thermo Scientific) and sequenced using pJET1.2F and pJET1.2R sequencing primers (Supplementary Table 2). Sequence-confirmed fragments were lifted from the shuttle vector by SacI-SphI restriction enzyme digestion, subsequently cloned into SacI and SphI restricted destination plasmid, pNQ705-1, which was then transformed into the E. coli SY327λpir. Each promoter fusion, in-frame with Gfp of pNQ705-1 was confirmed by sequence analysis using the CatR2 and GfpR2 primers pair (Supplementary Table 2). The sequence-confirmed Gfp-reporter plasmids were transformed into E. coli S17-1λpir (donor) and mobilised by conjugal mating into parental, ∆cpxA and ∆cpxR strains of Yptb. Genome integrated single copies of each Gfp-reporter fusion in recipient Yptb-YPIII strains was confirmed by colony PCR using respective genomic-integration primer pairs (Supplementary Table 2).
Serial dilution-based biofilm dynamic assay
To assay for dynamic of biofilm formation in a plastic microtiter dish, a Crystal violet based serial dilution method was employed with modifications98. Briefly, optical density at 600 nm wavelength (OD600) of the overnight grown cultures was measured using a DU® 730 Life Science UV/Vis spectrophotometer (Beckman Coulter). The number of cells were standardised to an OD600 of 0.1 using LB broth and then serially diluted 6-fold a total of nine times in sterile Eppendorf tubes. A volume of 150 μL from each serially diluted tube along with undiluted first tube was seeded in triplicate in the sterile 96-well round-bottom microtiter plate (NunclonTM ∆ Surface, Denmark). LB broth devoid of any bacteria was seeded in two separate columns as a negative control. The microtiter plates were incubated at 26 °C for 24 h with gentle agitation at 125 rpm. A duplicate plate was prepared and used for measuring the planktonic growth (OD600 nm). To determine if low bacterial concentration was the cause of an absence of biofilm formation, the initial bacterial-inoculum concentration was increased to an OD600 of 1.0. On the following day, seeded microtiter plates for biofilm biomass measurement were washed repeatedly with tap water and passed over an open Bunsen burner flame for 2–3 s to heat-fix the biofilm biomass. Biomass was stained with 200 µL of 0.5% (w/v) Crystal violet stain (Sigma Aldrich), and plates were incubated at room temperature for 15 min. The unbound stain was removed by repeated gentle rinsing with tap water. Crystal violet stained biofilm biomass was solubilised with 200 µL of 33% (v/v) Glacial acetic acid (Sigma Aldrich). Plates were incubated at 26 °C for 20 min with gentle agitation. Solubilised biofilm biomass (150 µL) was transferred into a sterile 96-well flat-bottom microtiter plate (NunclonTM ∆ Surface, Denmark) and absorbance measured at 570 nm using a TECAN spectrophotometer plate reader. In parallel, viability of bacteria from each seeded well was monitored before and after the formation of biofilm by spotting 3.5 µL culture from each on selective LA plates containing desired antibiotic(s) and further incubation at 26 °C for 24 h.
Biofilm analysis on the surface of nematode, Caenorhabditis elegans
The N2 Bristol strain of nematode Caenorhabditis elegans was used throughout. The C. elegans were maintained on lawns of E. coli OP50 at room temperature in Petri dishes (55 mm diameter) containing NGM agar (Per liter: 17 g Difco bacto-agar, 25 g Difco bactopeptone, 0.3% NaCl, 0.5% Cholesterol, 1 M CaCl2, 1 M MgSO4 and 1 M Postssium phosphate buffer- pH 6.0)99. For the biofilm assay, an equal number of Yptb bacteria tagged with Gfp, expressed from pFPV25.1 (a gift from Raphael Valdivia via Addgene plasmid # 20668; http://n2t.net/addgene:20668; RRID:Addgene_20668) in 50 µL volumes (from overnight cultures equalised to the lowest OD600 measurement) were seeded on the NGM agar plates and incubated at 26 °C overnight. Thirty L4-stage (larval) of nematodes were transferred to the NGM plates containing the bacterial lawns and left at room temperature. Development of biofilm was examined by Gfp-expressing Yptb on the surface of the C. elegans every 24 h using fluorescence microscopy. Scoring of at least 30 live worms was carried out continuously for 5 days. This required that worms were transferred on sterile glass coverslips, rinsed gently with Phosphate buffered saline, and fixed in 50% sterile glycerol and viewed with Gfp-fluorescence at ×50 magnification, equivalent to a 10 µm scale bar.
Gfp reporter assay
A bacterial inoculum was prepared from a single isolated colony and then grown in selective LB broth at 26 °C for 18 h with gentle agitation. On the following day, a sterile 96-well flat-bottom µCLEAR® black polystyrene microtiter plate (Greiner Bio-one, Germany) was seeded (in triplicate) with 150 μL of an overnight inoculum standardised to an OD600 of 0.01. The lid of the plate was sealed with sterile Parafilm, and the plate was incubated at 26 °C for 24 h with gentle agitation at 125 rpm. LB broth devoid of any bacteria was seeded in two separate columns as a negative control. Extent of bacterial growth at a wavelength of 600 nm, and and Gfp-fluorescence (excitation ƛ 485 nm and emission ƛ 515 nm) were recorded after 24 h by a TECAN spectrophotometer with following kinetic settings established via the Infinite-200 Tecan i-control software (version 1.12.4.0): shaking (linear) every 5 min for 3 s, number of flashes-25, bandwidth- 9 nm for Absorbance A600 nm and fluorescence-excitation, and for fluorescence-emission- 20 nm, integration time- 20 µs, Gains- 70 and 90. Readings for both growth and Gfp-fluorescence were recorded from the top of the plate. The mean of gained Gfp-fluorescence was normalised with corresponding growth (OD600 nm) and calculated Gfp-fluorescence unit (FU/OD).
Real-time qPCR
Total RNA from cultures standardised to the lowest OD600 was isolated using the Nucleo Spin RNA isolation kit (Macherey-Nagal, Germany) as per the manufacturer’s protocol. Isolated total RNA was treated with Turbo DNase (Thermo Scientific) and inactivated as recommended using DNase inactivation reagent. Turbo DNase treated total RNA template (200 ng) was used to synthesise cDNA using RevertAid H-minus reverse transcriptase (Thermo Scientific) as per the manufacturer’s protocol. A negative reaction (without reverse transcriptase) was also prepared to further check for DNA contamination. Both reverse transcriptase + /− reactions were incubated in PCR machine at 25 °C for 10 min, 42 °C for 60 min, 70 °C for 10 min and 4 °C for 10 min. Synthesised cDNA was quantified by a Nano-drop spectrophotometer (Thermo Scientific) and stored at −20 °C. Real-time qPCR was performed on iQ5 Thermocycler (Bio-Rad) using 50 ng of cDNA template combined with 1× qPCRBIO SyGreen mix with fluorescein and 400 nM of each gene-specific qRT-primer pair (Supplementary Table 2). PCR reactions (20 µL size) were performed as per the manufacturer’s protocol, considering melting temperature of respective primers pair and the in-built melting analysis for optimum annealing of each primer. Relative expression of each gene was calculated as 2Λ−∆∆Ct100. The expression of each gene was examined from three biological replicates having three technical replicates. To enhance the reliability of expression, each targeted gene was normalised with two house-keeping internal standards, gyrB and rpoC.
Purification of wild-type and phosphorylation defective CpxR
An established recombinant E. coli BL21(DE3) pLysS strain expressing pKECO17 plasmid64 that contains cpxR gene from the parental Yptb-YPIII, cloned under the control of IPTG-inducible promoter of pET22b plasmid (Invitrogen) was used to express and purify C-terminally His6-tag fused CpxRwt. The phosphorylation defective CpxRPneg was designed by substituting Asp8, Asp9, Asp51, Met53 and Lys100 with Ala amino acid on Yptb-YPIII cpxR synthetic DNA (obtained from GenScript, USA) and subcloned into pET22b plasmid (Invitrogen). Subsequent expression, purification and storage of phosphorylation defective CpxRPneg was carried out by the Protein Expertise Platform (Umeå University, Sweden). Briefly, an overnight culture was prepared in 15 mL selective LB broth at 37 °C for 16 h. On the following day, 250 mL selective LB broth was sub-cultured (1/20) with overnight inoculum and grown for 2.5 h at 37 °C with aeration. The culture was induced with IPTG at a final concentration of 0.5 mM and the incubation continued for a further 4 h at 30 °C. Bacterial cells were harvested by centrifugation at 8000 × g for 15 min and mixed into 50 mL lysis buffer (20 mM Tris-HCl pH 7.5, 500 mM NaCl, 30 mM imidazole, 0.9% Triton X-100, 1% Glycerol, 8.75 mM β-mercaptoethanol and 5 mM EDTA-free PMSF proteases inhibitor). Cell lysate was prepared by sonication (pulse on/off- 10 sec, Amp- 50%) for 5 min ×4 and clarified by ultracentrifugation at 104,000 × g for 60 min at 4 °C to isolate soluble ‘active’ CpxRwt and ‘inactive’ CpxRPneg from the supernatant fraction. The CpxR variants were purified manually using 1 mL HisTrapTM HP column (GE Healthcare) as per the manufacturers protocol. Five protein fractions (1 mL each) were eluted sequentially with 150, 250 and 350 mM imidazole in the column binding buffer (20 mM Tris-HCl pH 7.5, 500 mM NaCl) and analysed by SDS-PAGE followed by Coomassie blue staining. Based on Coomassie blue staining, fraction 7 contained sufficiently pure CpxRHis6, and was dialysed in a D-tube dialyser (Novagen, MWCO 6-8 kD) against dialysis buffer (10 mM Tris-HCl pH 7.5, 50 mM KCl and 1.0 mM β-mercaptoethanol) overnight at 4 °C with three changes of the dialysis buffer. Dialysed CpxRHis6 was quantified by Nanodrop spectrophotometer (Thermo Scientific) and by classical Bradford assay. Glycerol (2.5%) and EDTA-free PMSF proteases inhibitor (1.0 mM) were added to the quantified CpxR variants and stored in aliquots at −20 °C.
Electrophoretic mobility shift assay
DNA fragments encompassing the entire promoter regions of the target genes, and for control purposes the non-specific 16S rDNA (148 bp from YPK_R0086; rRNA), were PCR-amplified from the genomic DNA of parental Yptb-YPIII using EMSA primers, listed in Supplementary Table 2. The amplified DNA fragments were analysed by agarose gel electrophoresis and purified using the GenJET PCR purification kit (Thermo Scientific). For EMSA, either 50 µM or 100 µM of dialysed CpxRwt or CpxRPneg variant was combined with 40 ng of non-specific 16S rDNA and 40 ng of target promoter fragment in EMSA binding buffer (50 mM Tris-HCl pH 8.0, 30 mM NaCl, 20 mM Acetyl-P and 1% β-mercaptoethanol). EMSA reactions (10 µL) were incubated for 30 min at 30 °C without agitation. Completed reactions were mixed with 2.5 µL of 5× EMSA loading dye (50 mM Tris-HCl pH 6.8, 0.01% Bromophenol blue, 25% glycerol) and 10 µL aliquots were loaded on homemade 1.0 mm thick 6% native Polyacrylamide gel [30% Acrylamide (37.5:1) with 2.6% cross-linker (1. 2 mL), 50% glycerol (300 µL), 1× TBE (300 µL), ddH2O (4.2 mL), 10% APS (60 µL) and TEMED (6.0 µL)]. The gel was electrophoresed in 1× TBE buffer at 100 volts at room temperature until the dye front had migrated to the end of the gel. Gels were subsequently stained with 1× GelRed (Cambridge Bioscience, UK), diluted in 1× TBE, at room temperature for 30 min with gentle agitation. The excessive stain was removed by with three successive 10 min washes with sterile ddH2O. The gel was imaged using GelDoc2000 (Bio-Rad) imager with an automatic exposure of UV-light.
Congo Red-binding colony morphotype on salt-free Luria agar
The Yptb-YPIII strains were routinely grown in 2.5 mL LB broth lacking NaCl with shaking (150 rpm) at 26 °C-18 h. On the following day, an equal number of cells in 3.5 µL volumes (equalised to the lowest OD600 measurement) were spotted on LA (without NaCl) plates, supplemented with 0.01% (w/v) Congo Red (Sigma Aldrich) and appropriate antibiotic(s). Plates were incubated at 26 °C-24 h. Colony morphotype was imaged on an inverted microscope with an epi-light source.
Phos-tagTM of in vivo accumulated CpxR~P in the biofilm-planktonic cells
Equal numbers of cells were harvested from the desired Yptb-YPIII strains, grown in LB broth to late-stationary phase at 26 °C, in 96-well round-bottomed microtiter plate. This was achieved by combining planktonic culture from 8 wells of respective strain from the mature stage of biofilm (equivalent to 4th dilution of the 6-fold dilution series). Harvested cells (by centrifugation at 16,000 × g for 20 min) were mixed in 100 μL of BugBuster® Master Mix (Novagen®, Merck Millipore, Sweden) and cell lysis was performed by incubation at 26 °C for 30 min with gentle agitation. Cell lysis was halted by adding 100 μL 2x SDS-PAGE sample buffer lacking β-Mercapoethanol (100 mMTris-HCl; pH 6.8, 4% SDS, 20% Glycerol and 0.01% Bromophenol blue). Following heat-denaturation at 95 °C for 5 min, 5 μL total cell-lysate was electrophoresed on a freshly prepared 12% Phos-tagTM Acrylamide AAL-107 gel (Wako Nard Institute, Japan) at 80 volts at room temperature, until dye front reaches at the bottom of gel. Preparation of Phos-tagTM gel and subsequent processing for Western immunoblotting were as per the manufacturer’s suggestions. Following wet electrotransfer onto PVDF membrane (53 volts for 2 h at 4 °C), the two CpxR isoforms were bound with rabbit polyclonal anti-CpxR antibody (1: 2000 dilutions in TBST plus 5% skimmed milk) for overnight at 4 °C, followed by anti-rabbit-HRP antibody (1: 6000 in TBST plus 5% skimmed milk) for 1 h at room temprature, and then detected with PierceTM ECL Plus Western blotting system as per the manufacturer’s instructions using ImageQuant™ LAS 4000 imager (GE Healthcare).
Quantification and statistical analysis
The level of active phosphorylated CpxR~P on Phos-tagTM Western blot images was quantified using ImageJ101. The active level of CpxR~P was inferred as percentage as follow: Protein band intensity of CpxR~P divided by Protein band intensity of CpxR~P plus full-length CpxR at ~26.5 kDa, and multiplied by 100. Throughout, standard error of mean ± was calculated for at least three biological replicates. Significance from the parent or negative control was determined using One-way ANOVA with Tukey’s multiple comparisons test, with a single pooled variance. The difference in variance with a p-value of < 0.05 was considered significant. Analysis was performed using GraphPad Prism-7, for MacBook Pro (GraphPad Software, Inc. La Jolla, CA, USA).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The datasets generated and/or analysed during the current study are available from the corresponding authors on reasonable request. The NCBI database codes for retrieving the whole genome sequence of Yersinia pseudotuberculosis YPIII and Yersinia pestis KIM10 + are CP000950.1 and AE009952.1, respectively. The locus tag of the studied genes of Y. pseudotuberculosis YPIII are, YPK_4133 (cpxA), YPK_4132 (cpxR), YPK_4131 (cpxP), YPK_2241 (hmsH),YPK_3638 (hmsT), YPK_0094 (hmsP), YPK_3615 (hmsC), YPK_2238 (hmsS), YPK_1182 (rpoE), YPK_1090 (nlpE), YPK_1551 (ackA), YPK_1550 (pta), YPK_0004 (gyrB), YPK_0341 (rpoC) and YPK_R0086 (16 S rDNA). Software used for image quantification was ImageJ101. Software used for statistical analysis was contained within GraphPad Prism-7, for MacBook Pro (GraphPad Software, Inc. La Jolla, CA, USA).
References
Galindo, C. L., Rosenzweig, J. A., Kirtley, M. L. & Chopra, A. K. Pathogenesis of Y. enterocolitica and Y. pseudotuberculosis in Human Yersiniosis. J. Pathog. 2011, 182051 (2011).
Barbieri, R. et al. Yersinia pestis: the Natural History of Plague. Clin. Microbiol. Rev. 34, 1–44 (2020).
Valles, X. et al. Human plague: An old scourge that needs new answers. PLoS Negl. Trop. Dis. 14, e0008251 (2020).
Califf, K. J., Keim, P. S., Wagner, D. M. & Sahl, J. W. Redefining the differences in gene content between Yersinia pestis and Yersinia pseudotuberculosis using large-scale comparative genomics. Micro. Genom. 1, e000028 (2015).
Atkinson S., Williams P. Yersinia virulence factors—a sophisticated arsenal for combating host defences. F1000Res. 5,1–10 (2016).
Vestby L. K., Gronseth T., Simm R., Nesse L. L. Bacterial Biofilm and its Role in the Pathogenesis of Disease. Antibiotics. 9, 1–29 (2020).
Donne, J. & Dewilde, S. The Challenging World of Biofilm Physiology. Adv. Micro. Physiol. 67, 235–292 (2015).
Del Pozo, J. L. Biofilm-related disease. Expert Rev. Anti Infect. Ther. 16, 51–65 (2018).
Joshua, G. W. P. et al. Caenorhabditis elegans model of Yersinia infection: biofilm formation on a biotic surface. Microbiology 149, 3221–3229 (2003).
Erickson, D. L., Jarrett, C. O., Wren, B. W. & Hinnebusch, B. J. Serotype differences and lack of biofilm formation characterize Yersinia pseudotuberculosis infection of the Xenopsylla cheopis flea vector of Yersinia pestis. J. Bacteriol. 188, 1113–1119 (2006).
Zhou, D. & Yang, R. Formation and regulation of Yersinia biofilms. Protein Cell. 2, 173–179 (2011).
Darby, C., Hsu, J. W., Ghori, N. & Falkow, S. Caenorhabditis elegans: plague bacteria biofilm blocks food intake. Nature 417, 243–244 (2002).
Hinnebusch, B. J., Perry, R. D. & Schwan, T. G. Role of the Yersinia pestis hemin storage (hms) locus in the transmission of plague by fleas. Science 273, 367–370 (1996).
Lillard, J. W. Jr., Fetherston, J. D., Pedersen, L., Pendrak, M. L. & Perry, R. D. Sequence and genetic analysis of the hemin storage (hms) system of Yersinia pestis. Gene 193, 13–21 (1997).
Jarrett, C. O. et al. Transmission of Yersinia pestis from an infectious biofilm in the flea vector. J. Infect. Dis. 190, 783–792 (2004).
Bobrov, A. G., Kirillina, O., Forman, S., Mack, D. & Perry, R. D. Insights into Yersinia pestis biofilm development: topology and co-interaction of Hms inner membrane proteins involved in exopolysaccharide production. Environ. Microbiol. 10, 1419–1432 (2008).
Erickson, D. L., Jarrett, C. O., Callison, J. A., Fischer, E. R. & Hinnebusch, B. J. Loss of a biofilm-inhibiting glycosyl hydrolase during the emergence of Yersinia pestis. J. Bacteriol. 190, 8163–8170 (2008).
Forman, S. et al. Identification of critical amino acid residues in the plague biofilm Hms proteins. Microbiology 152, 3399–3410 (2006).
Kirillina, O., Fetherston, J. D., Bobrov, A. G., Abney, J. & Perry, R. D. HmsP, a putative phosphodiesterase, and HmsT, a putative diguanylate cyclase, control Hms-dependent biofilm formation in Yersinia pestis. Mol. Microbiol. 54, 75–88 (2004).
Sun, Y. C., Hinnebusch, B. J. & Darby, C. Experimental evidence for negative selection in the evolution of a Yersinia pestis pseudogene. Proc. Natl Acad. Sci. USA. 105, 8097–8101 (2008).
Abu Khweek, A., Fetherston, J. D. & Perry, R. D. Analysis of HmsH and its role in plague biofilm formation. Microbiology 156, 1424–1438 (2010).
Bobrov, A. G. et al. Systematic analysis of cyclic di-GMP signalling enzymes and their role in biofilm formation and virulence in Yersinia pestis. Mol. Microbiol. 79, 533–551 (2011).
Sun, Y. C. et al. Differential control of Yersinia pestis biofilm formation in vitro and in the flea vector by two c-di-GMP diguanylate cyclases. PLoS One 6, e19267 (2011).
Bobrov, A. G., Kirillina, O. & Perry, R. D. The phosphodiesterase activity of the HmsP EAL domain is required for negative regulation of biofilm formation in Yersinia pestis. FEMS Microbiol Lett. 247, 123–130 (2005).
Chouikha, I., Sturdevant, D. E., Jarrett, C., Sun, Y. C., Hinnebusch, B. J. Differential Gene Expression Patterns of Yersinia pestis and Yersinia pseudotuberculosis during Infection and Biofilm Formation in the Flea Digestive Tract. mSystems. 4, 1–19 (2019).
Joshua, G. W. et al. Genome-wide evaluation of the interplay between Caenorhabditis elegans and Yersinia pseudotuberculosis during in vivo biofilm formation. Infect. Immun. 83, 17–27 (2015).
Liu, L. & Zheng, S. Transcriptional regulation of Yersinia pestis biofilm formation. Micro. Pathog. 131, 212–217 (2019).
Ritzert, J. T., Minasov, G., Embry, R., Schipma, M. J., Satchell, K. J. F. The Cyclic AMP Receptor Protein Regulates Quorum Sensing and Global Gene Expression in Yersinia pestis during Planktonic Growth and Growth in Biofilms. mBio. 10, 1–18 (2019).
Atkinson, S. et al. Biofilm development on Caenorhabditis elegans by Yersinia is facilitated by quorum sensing-dependent repression of type III secretion. PLoS Pathog. 7, e1001250 (2011).
Zhao, R. et al. A starvation-induced regulator, RovM, acts as a switch for planktonic/biofilm state transition in Yersinia pseudotuberculosis. Sci. Rep. 7, 639 (2017).
Fang, N. et al. RcsAB is a major repressor of Yersinia biofilm development through directly acting on hmsCDE, hmsT, and hmsHFRS. Sci. Rep. 5, 9566 (2015).
Guo, X. P., Ren, G. X., Zhu, H., Mao, X. J. & Sun, Y. C. Differential regulation of the hmsCDE operon in Yersinia pestis and Yersinia pseudotuberculosis by the Rcs phosphorelay system. Sci. Rep. 5, 8412 (2015).
Liu, L., et al. Reciprocal regulation of Yersinia pestis biofilm formation and virulence by RovM and RovA. Open Biol. 6, 1–10 (2016).
Vadyvaloo, V. & Hinz, A. K. A LysR-Type Transcriptional Regulator, RovM, Senses Nutritional Cues Suggesting that It Is Involved in Metabolic Adaptation of Yersinia pestis to the Flea Gut. PLoS One 10, e0137508 (2015).
Sun, Y. C., Guo, X. P., Hinnebusch, B. J. & Darby, C. The Yersinia pestis Rcs phosphorelay inhibits biofilm formation by repressing transcription of the diguanylate cyclase gene hmsT. J. Bacteriol. 194, 2020–2026 (2012).
Guan, J. et al. Roles of RpoS in Yersinia pseudotuberculosis stress survival, motility, biofilm formation and type VI secretion system expression. J. Microbiol. 53, 633–642 (2015).
Schachterle, J. K. et al. Yersinia pseudotuberculosis BarA-UvrY Two-Component Regulatory System Represses Biofilms via CsrB. Front Cell Infect. Microbiol. 8, 323 (2018).
Xu, S. et al. FliS modulates FlgM activity by acting as a non-canonical chaperone to control late flagellar gene expression, motility and biofilm formation in Yersinia pseudotuberculosis. Environ. Microbiol. 16, 1090–1104 (2014).
Bontemps-Gallo, S. et al. Nutrient depletion may trigger the Yersinia pestis OmpR-EnvZ regulatory system to promote flea-borne plague transmission. Mol. Microbiol. 112, 1471–1482 (2019).
Vadyvaloo, V. et al. Role of the PhoP-PhoQ gene regulatory system in adaptation of Yersinia pestis to environmental stress in the flea digestive tract. Microbiology 161, 1198–1210 (2015).
Rebeil, R. et al. Induction of the Yersinia pestis PhoP-PhoQ regulatory system in the flea and its role in producing a transmissible infection. J. Bacteriol. 195, 1920–1930 (2013).
Silva-Rohwer, A. R. et al. CsrA Enhances Cyclic-di-GMP Biosynthesis and Yersinia pestis Biofilm Blockage of the Flea Foregut by Alleviating Hfq-Dependent Repression of the hmsT mRNA. mBio. 12, e0135821 (2021).
Bellows, L. E., Koestler, B. J., Karaba, S. M., Waters, C. M. & Lathem, W. W. Hfq-dependent, co-ordinate control of cyclic diguanylate synthesis and catabolism in the plague pathogen Yersinia pestis. Mol. Microbiol. 86, 661–674 (2012).
Liu, L. et al. CRP Is an Activator of Yersinia pestis Biofilm Formation that Operates via a Mechanism Involving gmhA and waaAE-coaD. Front. Microbiol. 7, 295 (2016).
Willias, S. P., Chauhan, S., Lo, C. C., Chain, P. S. & Motin, V. L. CRP-Mediated Carbon Catabolite Regulation of Yersinia pestis Biofilm Formation Is Enhanced by the Carbon Storage Regulator Protein, CsrA. PLoS One 10, e0135481 (2015).
Sun, F. et al. Fur is a repressor of biofilm formation in Yersinia pestis. PLoS One 7, e52392 (2012).
Liu, Z. et al. Plasmid pPCP1-derived sRNA HmsA promotes biofilm formation of Yersinia pestis. BMC Microbiol. 16, 176 (2016).
Ni, B., Wu, H. S., Xin, Y. Q., Zhang, Q. W. & Zhang, Y. Q. Reciprocal Regulation between Fur and Two RyhB Homologs in Yersinia pestis, and Roles of RyhBs in Biofilm Formation. Biomed. Environ. Sci. 34, 299–308 (2021).
Liu, J., Obi, I. R., Thanikkal, E. J., Kieselbach, T. & Francis, M. S. Phosphorylated CpxR restricts production of the RovA global regulator in Yersinia pseudotuberculosis. PLoS One 6, e23314 (2011).
Thanikkal, E. J. et al. The Yersinia pseudotuberculosis Cpx envelope stress system contributes to transcriptional activation of rovM. Virulence 10, 37–57 (2019).
Fei, K., Chao, H. J., Hu, Y., Francis, M. S., Chen, S. CpxR regulates the Rcs phosphorelay system in controlling the Ysc-Yop type III secretion system in Yersinia pseudotuberculosis. Microbiology. 167, 1–11 (2021).
Raivio, T. L. Everything old is new again: an update on current research on the Cpx envelope stress response. Biochim. Biophys. Acta. 1843, 1529–1541 (2014).
Bury-Mone, S. et al. Global analysis of extracytoplasmic stress signaling in Escherichia coli. PLoS Genet. 5, e1000651 (2009).
Labandeira-Rey, M., Brautigam, C. A. & Hansen, E. J. Characterization of the CpxRA regulon in Haemophilus ducreyi. Infect. Immun. 78, 4779–4791 (2010).
Choudhary, K. S., et al. Elucidation of Regulatory Modes for Five Two-Component Systems in Escherichia coli Reveals Novel Relationships. mSystems. 5, 1–20 (2020).
Dbeibo, L., et al. Evaluation of CpxRA as a Therapeutic Target for Uropathogenic Escherichia coli Infections. Infect. Immun. 86, 1–16 (2018).
Hews, C. L., Cho, T., Rowley, G. & Raivio, T. L. Maintaining Integrity Under Stress: Envelope Stress Response Regulation of Pathogenesis in Gram-Negative Bacteria. Front. Cell Infect. Microbiol. 9, 313 (2019).
Laventie, B. J. & Jenal, U. Surface Sensing and Adaptation in Bacteria. Annu. Rev. Microbiol. 74, 735–760 (2020).
Kimkes, T. E. P. & Heinemann, M. How bacteria recognise and respond to surface contact. FEMS Microbiol. Rev. 44, 106–122 (2020).
Otto, K. & Silhavy, T. J. Surface sensing and adhesion of Escherichia coli controlled by the Cpx-signaling pathway. Proc. Natl Acad. Sci. USA. 99, 2287–2292 (2002).
Ferrieres, L. & Clarke, D. J. The RcsC sensor kinase is required for normal biofilm formation in Escherichia coli K-12 and controls the expression of a regulon in response to growth on a solid surface. Mol. Microbiol. 50, 1665–1682 (2003).
Shimizu, T., Ichimura, K. & Noda, M. The Surface Sensor NlpE of Enterohemorrhagic Escherichia coli Contributes to Regulation of the Type III Secretion System and Flagella by the Cpx Response to Adhesion. Infect. Immun. 84, 537–549 (2016).
Kimkes, T. E. P. & Heinemann, M. Reassessing the role of the Escherichia coli CpxAR system in sensing surface contact. PLoS One 13, e0207181 (2018).
Carlsson, K. E., Liu, J., Edqvist, P. J. & Francis, M. S. Influence of the Cpx extracytoplasmic-stress-responsive pathway on Yersinia sp.-eukaryotic cell contact. Infect. Immun. 75, 4386–4399 (2007).
Bontemps-Gallo, S., Madec, E. & Lacroix, J. M. The two-component system CpxAR is essential for virulence in the phytopathogen bacteria Dickeya dadantii EC3937. Environ. Microbiol. 17, 4415–4428 (2015).
Humphreys, S. et al. Role of the two-component regulator CpxAR in the virulence of Salmonella enterica serotype Typhimurium. Infect. Immun. 72, 4654–4661 (2004).
Dorel, C., Vidal, O., Prigent-Combaret, C., Vallet, I. & Lejeune, P. Involvement of the Cpx signal transduction pathway of E. coli in biofilm formation. FEMS Microbiol. Lett. 178, 169–175 (1999).
Spinola, S. M. et al. Activation of the CpxRA system by deletion of cpxA impairs the ability of Haemophilus ducreyi to infect humans. Infect. Immun. 78, 3898–3904 (2010).
Buelow, D. R. & Raivio, T. L. Cpx signal transduction is influenced by a conserved N-terminal domain in the novel inhibitor CpxP and the periplasmic protease DegP. J. Bacteriol. 187, 6622–6630 (2005).
Tschauner, K., Hornschemeyer, P., Muller, V. S. & Hunke, S. Dynamic interaction between the CpxA sensor kinase and the periplasmic accessory protein CpxP mediates signal recognition in E. coli. PLoS One 9, e107383 (2014).
Huang, L. et al. Phenotypic characterization, virulence, and immunogenicity of Pseudomonas plecoglossicida rpoE knock-down strain. Fish. Shellfish Immunol. 87, 772–777 (2019).
Bosse, J. T. et al. Regulation of pga operon expression and biofilm formation in Actinobacillus pleuropneumoniae by sigmaE and H-NS. J. Bacteriol. 192, 2414–2423 (2010).
Korbsrisate, S. et al. The Burkholderia pseudomallei RpoE (AlgU) operon is involved in environmental stress tolerance and biofilm formation. FEMS Microbiol. Lett. 252, 243–249 (2005).
Heusipp, G., Schmidt, M. A. & Miller, V. L. Identification of rpoE and nadB as host responsive elements of Yersinia enterocolitica. FEMS Microbiol. Lett. 226, 291–298 (2003).
Carlsson, K. E., Liu, J., Edqvist, P. J. & Francis, M. S. Extracytoplasmic-stress-responsive pathways modulate type III secretion in Yersinia pseudotuberculosis. Infect. Immun. 75, 3913–3924 (2007).
Palonen, E., Lindstrom, M., Somervuo, P. & Korkeala, H. Alternative sigma factor sigmaE has an important role in stress tolerance of Yersinia pseudotuberculosis IP32953. Appl. Environ. Microbiol. 79, 5970–5977 (2013).
Li, H. et al. The CpxA/CpxR Two-Component System Affects Biofilm Formation and Virulence in Actinobacillus pleuropneumoniae. Front. Cell Infect. Microbiol. 8, 72 (2018).
De Wulf, P., McGuire, A. M., Liu, X. & Lin, E. C. Genome-wide profiling of promoter recognition by the two-component response regulator CpxR-P in Escherichia coli. J. Biol. Chem. 277, 26652–26661 (2002).
Ronnebaumer, K., Sander, G., Shutinoski, B., Schmidt, M. A. & Heusipp, G. Controlled activation of the Cpx system is essential for growth of Yersinia enterocolitica. FEMS Microbiol. Lett. 296, 274–281 (2009).
Yamamoto, K. & Ishihama, A. Characterization of copper-inducible promoters regulated by CpxA/CpxR in Escherichia coli. Biosci. Biotechnol. Biochem. 70, 1688–1695 (2006).
Karatan, E. & Watnick, P. Signals, regulatory networks, and materials that build and break bacterial biofilms. Microbiol. Mol. Biol. Rev. 73, 310–347 (2009).
Karygianni, L., Ren, Z., Koo, H. & Thurnheer, T. Biofilm Matrixome: Extracellular Components in Structured Microbial Communities. Trends Microbiol. 28, 668–681 (2020).
Ruhal, R. & Kataria, R. Biofilm patterns in gram-positive and gram-negative bacteria. Microbiol. Res. 251, 126829 (2021).
Calder, J. T., Christman, N. D., Hawkins, J. M., Erickson, D. L. A Trimeric Autotransporter Enhances Biofilm Cohesiveness in Yersinia pseudotuberculosis but Not in Yersinia pestis. J. Bacteriol. 202, 1–13 (2020).
Gengler, S., Laudisoit, A., Batoko, H. & Wattiau, P. Long-term persistence of Yersinia pseudotuberculosis in entomopathogenic nematodes. PLoS One 10, e0116818 (2015).
Styer, K. L. et al. Yersinia pestis kills Caenorhabditis elegans by a biofilm-independent process that involves novel virulence factors. EMBO Rep. 6, 992–997 (2005).
Schweer, J. et al. The cytotoxic necrotizing factor of Yersinia pseudotuberculosis (CNFY) enhances inflammation and Yop delivery during infection by activation of Rho GTPases. PLoS Pathog. 9, e1003746 (2013).
Gemski, P., Lazere, J. R., Casey, T. & Wohlhieter, J. A. Presence of a virulence-associated plasmid in Yersinia pseudotuberculosis. Infect. Immun. 28, 1044–1047 (1980).
Bolin, I., Norlander, L. & Wolf-Watz, H. Temperature-inducible outer membrane protein of Yersinia pseudotuberculosis and Yersinia enterocolitica is associated with the virulence plasmid. Infect. Immun. 37, 506–512 (1982).
Flamez, C., Ricard, I., Arafah, S., Simonet, M. & Marceau, M. Phenotypic analysis of Yersinia pseudotuberculosis 32777 response regulator mutants: new insights into two-component system regulon plasticity in bacteria. Int J. Med. Microbiol. 298, 193–207 (2008).
Tidhar, A. et al. Disruption of the NlpD lipoprotein of the plague pathogen Yersinia pestis affects iron acquisition and the activity of the twin-arginine translocation system. PLoS Negl. Trop. Dis. 13, e0007449 (2019).
O’Loughlin, J. L., Spinner, J. L., Minnich, S. A. & Kobayashi, S. D. Yersinia pestis two-component gene regulatory systems promote survival in human neutrophils. Infect. Immun. 78, 773–782 (2010).
Leskinen, K., Varjosalo, M., Li, Z., Li, C. M. & Skurnik, M. Expression of the Yersinia enterocolitica O:3 LPS O-antigen and outer core gene clusters is RfaH-dependent. Microbiology 161, 1282–1294 (2015).
Maxson, M. E. & Darwin, A. J. Identification of inducers of the Yersinia enterocolitica phage shock protein system and comparison to the regulation of the RpoE and Cpx extracytoplasmic stress responses. J. Bacteriol. 186, 4199–4208 (2004).
Bertani, G. Lysogeny at mid-twentieth century: P1, P2, and other experimental systems. J. Bacteriol. 186, 595–600 (2004).
Higuchi, R., Krummel, B. & Saiki, R. K. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 16, 7351–7367 (1988).
Francis, M. S., Amer, A. A., Milton, D. L. & Costa, T. R. Site-Directed Mutagenesis and Its Application in Studying the Interactions of T3S Components. Methods Mol. Biol. 1531, 11–31 (2017).
Lopez-Sanchez, A., Jimenez-Fernandez, A., Calero, P., Gallego, L. D. & Govantes, F. New methods for the isolation and characterization of biofilm-persistent mutants in Pseudomonas putida. Environ. Microbiol. Rep. 5, 679–685 (2013).
Girard, L. R. et al. WormBook: the online review of Caenorhabditis elegans biology. Nucleic Acids Res. 35, D472–D475 (2007). Database issue.
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 (2001).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Acknowledgements
We acknowledge Bachelor degree project students, Rushiil Ravichandran and Lincy Marino for their assistance with optimising the abiotic and biotic surface biofilm development methods, respectively. We are also grateful to Katrin Carlsson and Junfa Liu, both of whom participated in preliminary exploratory work in this area. We greatly appreciate assistance of Dr Mikael Lindberg, Protein Expertise Platform (PEP), Umeå University, Sweden for cloning and purification of phosphorylation defective CpxRPneg. We express our gratitude to the Swedish Research Council (Vetenskapsrådet) (M.F., S.N.W.), Foundation for the Medical Research (M.F.), and Faculty of Science & Technology (M.F.), Umeå University, Sweden and BYU College of Life Sciences (D.E.) for project funding.
Funding
Open access funding provided by Umea University.
Author information
Authors and Affiliations
Contributions
Study conception and design (D.K.G., M.F.). Acquisition of data and preparation of figures (D.K.G.). Writing original manuscript draft (D.K.G.). Critical revision of the manuscript (D.K.G., M.F.). Analysis and interpretation of data (D.K.G., S.N.W., D.E., M.F.). Final approval of the manuscript (D.K.G., S.N.W., D.E., M.F.).
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gahlot, D.K., Wai, S.N., Erickson, D.L. et al. Cpx-signalling facilitates Hms-dependent biofilm formation by Yersinia pseudotuberculosis. npj Biofilms Microbiomes 8, 13 (2022). https://doi.org/10.1038/s41522-022-00281-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41522-022-00281-4
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
