
Abstract
Chronic infections often contain complex mixtures of pathogenic and commensal microorganisms ranging from aerobic and anaerobic bacteria to fungi and viruses. The microbial communities present in infected tissues are not passively co-existing but rather actively interacting with each other via a spectrum of competitive and/or cooperative mechanisms. Competition versus cooperation in these microbial interactions can be driven by both the composition of the microbial community as well as the presence of host defense strategies. These interactions are typically mediated via the production of secreted molecules. In this review, we will explore the possibility that microorganisms competing for nutrients at the host–pathogen interface can evolve seemingly cooperative mechanisms by controlling the production of subsets of secreted virulence factors. We will also address interspecies versus intraspecies utilization of community resources and discuss the impact that this phenomenon might have on co-evolution at the host–pathogen interface.
Similar content being viewed by others

Introduction to life at the host–pathogen interface
Pathogens must overcome host-imposed starvation
The host–pathogen interface is a battleground where reactive molecules and harsh conditions are produced by both the host and the pathogen. One of the harsh conditions faced by invading pathogens is the limitation of essential nutrients which are actively sequestered by the host immune response. These nutrients include metals such as zinc, manganese and iron, which serve as co-factors for several essential enzymes in pathogens and host cells.1,2 Sequestration of these metals and other nutrients by the host immune system during an infection is known as nutritional immunity.2,3 Nutritional immunity leads to starvation in pathogens and affects numerous cellular processes within them. Thus, pathogens are constantly at war with the host as well as other microbial species present at sites of infection to scavenge the resources required to establish infection and persist in a challenging host environment.
Many pathogens have been known to employ different metal acquisition systems to battle against host-imposed starvation and to outcompete nearby microbial species. For example, to combat the iron limitation mediated by host proteins such as lactoferrin, Staphylococcus aureus uses a combination of hemolysins and a high-affinity heme acquisition system to liberate heme from red blood cells for use as an iron source.4 A variety of mechanisms for circumventing this host-mediated starvation extends to many other microorganisms. Organisms such as the fungal pathogen Cryptococcus neoformans possesses enzymes such as ferric reductase that can acquire iron from the surrounding environment.5 Additionally, numerous organisms produce iron-scavenging siderophores6,7 (Fig. 1). Siderophores are small secreted molecules capable of binding iron (and sometimes other ions) with high affinity.8 While this struggle for nutrients intuitively creates competitive interactions, there is also evidence to support the existence of cooperative interactions designed to overcome such starvation. A potential example of this type of cooperation can be observed in the enhancement of the β-hemolytic activity of S. aureus in presence of other Staphylococcus species such as S. epidermidis as well as in the presence of certain Corynebacterium species.9 The increased hemolytic activity would presumably result in increased nutrient availability for each species at the host–pathogen interface, reducing the need for competition. S. aureus and S. epidermidis are both common components of the human microbiome so such in vitro findings support the idea that the evolution of microbial synergism intended to overcome host defenses could potentially occur during an infection.

Selective pressures at the host–pathogen interface select for microbial adaptation and/or evolution. This figure depicts some of the possible stresses encountered by microbial pathogens while invading host tissues. For example, the innate immune system deploys phagocytes and neutrophils to target microbial pathogens at the site of infections. These cells can kill microbial cells by engulfing them and/or releasing antimicrobial molecules such as metal-binding proteins to sequester essential metals from pathogens, antimicrobial peptides such as LL-37, and reactive oxygen species. Pathogens like Staphylococcus aureus and Pseudomonas aeruginosa elicit physiological responses and/or evolve adaptations over the course of acute to chronic infection that allow them to circumvent these stresses and survive in spite of the presence of host offensive molecules. In this example, pathogen A is modeled after P. aeruginosa and pathogen B is modeled after S. aureus, but the disease progression may be applicable to other examples. Depicted here, initial infection of epithelial cells by S. aureus paves the way for opportunistic Gram-negatives such as P. aeruginosa. Microbial interactions of these species lead to formation of aminoglycosidic antibiotic resistant small colony variants (artistically depicted as smaller cells in the diagram) in S. aureus and might provide protection to P. aeruginosa from host immune insults. As the infection advances, the host–pathogen interface becomes populated by mutants that are selected for better survival such as mutants capable of dealing with host reactive oxygen species (such as pyomelanin producers depicted as releasing brown pigment) and mutants with increased biofilm formation to protect from phagocytosis and/or antimicrobial peptides such as LL-37
Mutagenic stressors are encountered at the host–pathogen interface
The stressors that microorganisms encounter at the host–pathogen interface expand beyond nutritional scarcity and include exposure to toxic molecules designed to kill microbial cells. These toxic molecules can be produced by both the host immune response and by other microorganisms (both commensals and pathogens) present within the host. Microorganisms occupying overlapping niches often attempt to outcompete each other to reduce competition in terms of limited nutrients and space. For example, Pseudomonas aeruginosa is known to outcompete co-infecting microorganisms like Burkholderia cepacia through the secretion of molecules such as pyocyanin that can generate reactive oxygen species (ROS).10 A study of P. aeruginosa and Acinetobacter baumannii showed pyocyanin leads to production of catalase and superoxide dismutase via quorum sensing in A. baumannii11 indicating that pyocyanin is capable of inducing oxidative stress in a range of microorganisms. Interestingly, A. baumannii’s response to this onslaught of ROS results in an increase in persister cell formation and antibiotic resistance, indicating that seemingly competitive interactions do not impact all microbial species equivalently.
In the same way that some microorganisms can utilize ROS to eliminate certain competitors, the host immune system possesses mechanisms to target invading pathogens through the release of these toxic molecules. Superoxides and other ROS produced by the host immune system can cause lethal damage to microbial macromolecules such as lipids, proteins and DNA. Hence, these are part of the first line of offensive molecules presented by host cells.12,13 Though many pathogens contain enzymes such as catalase and superoxide dismutase to counteract such oxidative stresses,14,15 often these enzymes require iron or manganese as key cofactors.16 Therefore, the nutritional immunity so often observed at the host–pathogen interface can compound these oxidative stresses by incapacitating the pathogens’ defense enzymes and increasing their susceptibility to ROS.17 While ROS-mediated DNA damage can kill microbial cells, it may also favor higher mutation rates which, when combined with the selective pressures at the host–pathogen interface, leads to great evolutionary potential.18
Other antimicrobial strategies employed by the host have similar mutagenic potential. For example, expression of antimicrobial peptide LL-37 has been described to increase mutation rates in invading pathogens. LL-37 is produced by host cells to disrupt bacterial cell membranes leading to cell lysis19 but recent studies have demonstrated that LL-37 can enter bacterial cells and induce mutation through erroneous replication by DNA polymerase IV.20 The increased mutation rates associated with LL-37 exposure in organisms like P. aeruginosa and Escherichia coli can even induce mutations involved in promoting resistance to the antibiotic rifampin.19
Another major stressor encountered by microorganisms infecting the human host is antibiotic treatment. Emergence of antibiotic resistance can be due to prolonged treatment, a major problem encountered in modern medicine. Current antibiotic treatments often fail when incomplete penetration of the drug into infectious sites creates exposure to subinhibitory concentrations and enables the emergence of partial resistance. Additionally, these subinhibitory concentrations of antibiotics can act as a signal for the microbial cells to alter their virulence and sometimes become more pathogenic.21 Such a microbial adaptation to stressors is not only detrimental to our ability to clear the infection, but it can also exacerbate the infection.22,23,24 This risk of adaptation to antibiotic treatment is greater in the presence of polymicrobial communities as a number of resistance genes counteracting many of the common microbially-derived antibiotics exists naturally within the microbial world. These resistance genes can sometimes be acquired by neighboring microbes. In fact, studies have shown that microbial genes conferring antibiotic-resistance in infected tissues can likely be acquired via horizontal gene transfer among co-infecting pathogens.25
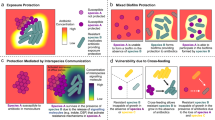
From the decades of research performed on studies of the host–pathogen interface, we now understand that invading microbial species experience a number of selective pressures in this environment produced by the host and/or nearby microbial species. The fact that some of these pressures can induce genetic mutation greater than the basal level of mutation that occurs during replication creates a breeding ground for adaptive mutations that can enable survival in a harsh environment (Fig. 1). Development of such pathoadaptive mutations are now being studied in detail.26,27 Interestingly, a large number of these mutations are witnessed only in chronic infections and it is now well-known that majority of chronic infections contain polymicrobial biofilms.28,29 Therefore, we expect that future research will uncover pathoadaptive traits promoting either competition or cooperation between microbial species to enable survival within host niches. In this review we seek to explore the idea that mutations at the host–pathogen interface might favor cooperative interactions between otherwise competitive species at the face of hostile host environment. Cooperative interactions can reduce individual biosynthesis cost, allow persistent colonization leading to establishment of polymicrobial chronic infections, and potentially increase virulence of the pathogens as a community (Fig. 2). This type of cooperation could be attributed to the ability of neighboring microbes to exploit each other’s mechanisms for survival at the host–pathogen interface.

Competitive interactions versus cooperative polymicrobial interactions at the host–pathogen interface. This figure compares the potential costs and/or benefits experienced by microbial cells when they are interacting competitively versus cooperatively with other species inside a host. a Microbial cells exhibiting a competitive lifestyle must not only expend biosynthetic costs for production of competitive molecules to combat surrounding pathogens, but they must also independently deal with the onslaught of host-associated harsh conditions such as immune cells, antibiotic treatment, and nutrient limitation. b Microbial cells exhibiting a more cooperative lifestyle do not expend energy on antimicrobials and therefore may have more energy to expend on molecules designed to combat the host-associated harsh conditions. Additionally, they might also reap the benefits of the secreted molecules produced by their neighboring microbes to achieve greater levels of survival at the host–pathogen interface
Evolution in lung infections
Biodiversity within lung infections
On the basis of standard culture techniques, lungs of healthy individuals were thought to be a sterile environment.30 However, the advent of metagenomic technologies has revealed that the healthy lung tissues contain microbial signatures from many species, mostly anaerobic bacteria like Haemophilus, Prevotella spp., and Veillonella spp., indicating that the healthy human lung might possess its own microbiome.31,32 The composition of this naturally occurring microbiota in healthy lung tissues is largely altered during infections.33,34 Difference in microbiome composition seen during different infections of the lung have been studied to understand their impact on host health. For example, an increase in population of microbes belonging to Pseudomonas, Haemophilus and Stenotrophomonas spp has been seen in the progression of chronic obstructive pulmonary disease.35,36 Chlamydia pneumoniae in lung tissues has been found in patients suffering from lung inflammations such as chronic bronchitis and asthma.37,38
Another disease that has been associated with dramatic shifts in the lung microbiome is cystic fibrosis. Cystic fibrosis is an autosomal recessive disorder caused by mutations in the cftr gene which encodes for the cystic fibrosis transmembrane regulator anion channel. Cystic fibrosis has many manifestations, one of which leads to accumulation of thick mucus in lung airways of the host and creates an environment ideal for microbial colonization.39 It was recently revealed that multiple microbial genera can occur in cystic fibrosis-associated lung infections as well. Different sequencing platforms have emphasized on different aspects of the microbial community composition in cystic fibrosis lungs. Some platforms detected presence of new species/strains in the samples whereas others identified the dominant species in the sample.40 Studies have shown that very early in life the lungs of cystic fibrosis patients tend to be colonized by common human commensals like Streptococcus, Veillonella and Actinomyces,41,42,43 but gradually they tend to be dominated by pathogenic species like, P. aeruginosa.44,45 Anaerobic bacterial species like Gemella spp.46 and fungal species like Candida albicans and Aspergillus fumigatus have also been identified in the lungs of cystic fibrosis patients.45 The composition of invading communities alters throughout the life of these patients and across different patients depending on host–pathogen and microbe-microbe interactions. While intervening host immune responses and antibiotic treatments can dramatically influence the microbial composition of infections,25 chronic infections can evolve stable and robust microbial systems that are often therapeutically resistant.47,48
Competitive interactions inside the host
While invading host tissues, pathogenic microbes face stresses presented by nearby microbes in the form of antimicrobial molecules and competition for nutrients and space. These nearby microbes can either be members of the natural host microbiota (the commensals) or other co-infecting pathogens. Nevertheless, overcoming these stresses are crucial for surviving and colonizing in host tissues and hence competitive interactions are very commonly observed in polymicrobial infections. For example, P. aeruginosa and S. aureus are commonly seen to co-colonize cystic fibrosis-associated lung airways.49 These microbes must compete for iron, which is sequestered by the host’s nutritional immunity. Therefore, iron depletion can act as a global regulatory signal for microbes such as P. aeruginosa to turn on genes for the synthesis of competitive molecules enabling the scavenging of this precious resource.50
In order to do so some microorganisms steal or “pirate” iron using siderophores of other pathogens, while others directly lyse cells to acquire the stored intracellular iron.8 Siderophores can be pirated through mutations in an organisms’ siderophore transport machinery to enable recognition of other siderophores. In order to prevent such activities, microorganisms often modify their siderophores structurally to increase specificity. Competing organisms can acquire mutations in their acquisition systems to recognize the new siderophore modifications giving rise to an evolutionary battle.2,8 A recent study also showed that under low iron conditions such as those occurring inside host peritoneum, pathogenic P. aeruginosa lyses S. aureus and S. pneumoniae cells to acquire their stored iron.51 Iron depletion in cystic fibrosis lungs also shows a similar phenomenon where antimicrobials such as 2-alkyl-4(1H)- quinolones are secreted by P. aeruginosa to suppress the growth of S. aureus.52
In cases of infections comprising P. aeruginosa and C. albicans, anti-microbials known as phenazines are produced by the bacterium which inhibit fungal biofilm formation by preventing the latter’s yeast to filament transition.53 Phenazines such as pyocyanin suppress growth of C. albicans on non-fermentable carbon sources by restricting respiration via limited oxygen availability. This interaction represents a competitive approach taken by P. aeruginosa via which it can utilize the products of fermentation for enhancing its growth and survival without actually having to produce them.54 However, polymicrobial interactions in the presence of phenazines do not always result in P. aeruginosa being the clear winner of the competition. For example, A. fumigatus possesses a unique ability to process and transform the P. aeruginosa-derived phenazines into unique molecules and can serve as interspecies signals.55 The presence of these signals activates a series of responses in A. fumigatus including oxidative stress responses as well as the induction of siderophore production, which might provide the fungus with its own competitive strategies against P. aeruginosa.55,56 Thus, a tug-of-war exists between microbial species to acquire essential nutrients. Interactions of such kind depict an ongoing molecular arms race within co-evolving pathogenic species.8
Individual adaptations to the lung environment
Despite the severe competitive pressure imposed by resource scarcity (such as transition metals) at the host–pathogen interface, a large number of the pathoadaptive mutations observed during chronic infections are not aimed at increasing microbial competitive strategies. Instead these adaptations are more focused on protective strategies such as evading the immune response and/or therapeutic intervention. This phenomenon indicates that the pressures faced as a result of the active immune response and antibiotic treatment may be even more influential to the evolution of pathogens than the pressures imposed by competing microbes. A plethora of adaptive changes have been described in S. aureus (Fig. 1), which is an early pathogenic colonizer of chronic lung infections.57,58 Biofilm formation,59 production of capsular polysaccharides,60 switching to small colony variant (SCV) phenotype,61 evolution of methicillin-resistant S. aureus strains (MRSA)62 and the recent emergence of vancomycin-resistant S. aureus strains (VRSA)63 are a few of such adaptations. These adaptive changes impart therapeutic resistance to S. aureus cells and facilitate recurrent infections in patients despite undertaking antibiotic treatments. This antibiotic resistance can be either due to acquisition of antibiotic-resistance genes as in the case of MRSA or due to absence of a proton motive force essential to pump antibiotics inside the microbe as in the case of SCVs.64 Biofilms are often associated with antibiotic tolerance simply due to limited diffusion through their thick biomass. Additionally, S. aureus can evolve mutations to survive in the presence of the competitive molecules produced by neighboring organisms such as P. aeruginosa.65
Many individual adaptations have also been observed in P. aeruginosa cells colonizing lung airways of adult cystic fibrosis patients66 (Fig. 1). For example, a study conducted whole-genome sequencing to compare gene sequences of two isolates of P. aeruginosa, one from an early (6 month) cystic fibrosis -infection sample and the other from a late (8 year) cystic fibrosis -infection sample. In total, 68 mutations were detected.67 Most mutations, in both the isolates, were present in the mexZ gene, a negative regulator of the MexXY-OprM multidrug efflux pump. Mutation in this gene confers resistance against aminoglycoside antibiotics like tobramycin.67 Another mutation was detected in the tyrosine catabolic pathway that leads to overproduction of pyomelanin, a molecule which provides resistance against oxidative stress.18,68 In other chronic cystic fibrosis lung isolates, mutations leading to overproduction of alginate have been commonly witnessed. Alginate is produced as a structural component of their biofilm extracellular polymeric substances (EPS), thus imparting antibiotic resistance and protection against ROS to microbial cells.69,70,71 A loss of function in the mucA gene leads to overproduction of alginate in P. aeruginosa.66,72 Some studies also show that LL-37, at sub-inhibitory levels, promotes polysaccharide production in organisms like Group A Streptococci, E. coli and P. aeruginosa.19,73 The range of microbial adaptive strategies to the harsh environment of the cystic fibrosis lung is wide, even within the same patient. For example, isolates of P. aeruginosa from different lobes of the same lung were found to have completely different antibiotic susceptibilities and virulence levels.74
Potential for evolution of cooperation to exploit intra- and interspecies adaptations
Microbial communities isolated from host tissues containing different strains and species exhibit diverse adaptive mutations. While the majority of these adaptations are likely to only benefit the adapted organism, some of these adaptations involve the secretion of molecules designed to make the host environment more hospitable (Figs 1 and 2). Theoretically, these secreted molecules could also be exploited by surrounding microorganisms (Fig. 2). For example, mixed communities of P. aeruginosa mucoid strains and nonmucoid revertants show enhanced resistance to both LL-37 and H2O2 produced by the host immune response. Alginate produced as a part of the EPS of mucoidy cells protects this community from LL-37 mediated killing and catalase produced by nonmucoidy cells protects the community from H2O2. The mucoid phenotype in P. aeruginosa is correlated with deteriorating cystic fibrosis lung infection and therefore this intraspecies mutualism indicates a selective benefit in co-existence.69,71 A study has shown that P. aeruginosa isolated from chronically infected CF patients exhibit parallel intraspecies mutualism where isogenic strains, auxotrophic for different nutrients, show complementation and reduced virulence.75
Similar intraspecies interactions have been seen in case of S. aureus. Growth-deficient mutants ΔhemB and ΔmenB have impaired growth and exhibit the SCV phenotype due to mutations in their heme and menaquinone biosynthetic pathways, respectively. These individual mutants “rescue” each other’s growth deficiencies via cross-feeding of heme and menaquinone when present together and achieve recovery of fitness and infectivity similar to the wild-type.76 Interestingly, this same study observed similar cooperative behavior occurring between S. aureus SCVs and other species isolated from cystic fibrosis infections, demonstrating that interspecies cooperation at the host–pathogen interface is possible.
In addition to the exchange of beneficial molecules, studies have demonstrated interspecies cooperation as a result of repression of competitive molecules produced by pathogenic microbes. An example of this type of cooperation is the reduction of pyocyanin production by multidrug resistant strains of P. aeruginosa isolated from chronically infected tissues. Pyocyanin, a bluish pigment, is a major virulence factor of P. aeruginosa and loss of it could enable this microbe to pursue non-competitive interactions with nearby species.77 In fact, it has been shown that the presence of calprotectin, an innate immune protein, in the host environment leads to a repression of pyocyanin and alkyl quinolones and promotes co-infection in the murine lung with S. aureus.78 This phenomenon is counterintuitive as the signal for repression of these molecules is zinc starvation induced by calprotectin and in this starved environment, an increase in the production of competitive molecules would be more expected. Mutations leading to alginate overproduction in P. aeruginosa also promotes co-infection with S. aureus by reducing production of the anti-staphylococcal molecules by P. aeruginosa.79
Potential advantages conferred by decrease in competitive molecules might be associated with the ability of P. aeruginosa to exploit the anti-immunity factors that S. aureus can produce. For example, S. aureus cells can assist in colonization of host tissues by Gram-negative bacteria. Virulence factors of S. aureus such as α-toxin allows proliferation and dissemination of Gram-negatives such as P. aeruginosa and Klebsiella pneumoniae by counteracting components of the human immune response.80 S. aureus also facilitates survival of lasR mutants of P. aeruginosa commonly found in cystic fibrosis patients by detoxifying surrounding nitric oxide released by host immune cells.81 In addition to S. aureus conferring benefits to P. aeruginosa, there are also instances of the reciprocal interaction. For example, protection of cystic fibrosis isolates of S. aureus cells from multiple antibiotics have also been reported recently.82 These cells were extracted from patients co-infected with P. aeruginosa. When present together, 4-hydroxy-2-heptylquinoline-N-oxide (HQNO) produced by P. aeruginosa cells inhibited respiration in S. aureus. While the presence of HQNO and associated reduction in respiration reduces the growth rate of S. aureus, it also protects the S. aureus cells from aminoglycosidic antibiotics.82 HQNO is produced by P. aeruginosa in its natural habitat primarily to slow down the growth of competitor cells in the vicinity83,84 but in host tissues the presence of HQNO may actually provide a benefit to S. aureus by promoting antibiotic tolerance.
This potential benefit of co-infection may extend past S. aureus and P. aeruginosa polymicrobial communities. For example, S. aureus cells have also been known to become tolerant to vancomycin in presence of the fungus, C. albicans.85 When present together S. aureus uses C. albicans cells as a substratum to form biofilms upon an existing biofilm. Also, the extracellular DNA component of C. albicans’s EPS promotes stability of C. albicans-S. aureus dual-biofilms and augments resistance of the former to anti-fungal molecules such as miconazole.86 In case of Streptococcus agalactiae, facilitation of growth via respiration by Lactococcus lactis has been studied as another example of interspecies cooperation. L. lactis synthesizes demethylmenaquinone in presence of heme which can be used by S. agalactiae to activate respiratory metabolism and enhance virulence in human blood.87 Respiration has been shown to be crucial in S. agalactiae’s virulence and thus, infections caused by S. agalactiae worsen in presence of L. lactis. Thus, we see there is significant potential for microbial cooperation among otherwise competing microbial species when present in polymicrobial contexts such as those observed in chronically infected host tissues. This phenomenon indicates that maybe tensions at the host–pathogen interface are driving an evolutionary shift in these species to become cooperative as opposed to being competitive for securing a long-term infection as a community. This type of cooperation can be corroborated by the study that showed that P. aeruginosa can protect members of the polymicrobial consortium of the cystic fibrosis lung namely, S. aureus, Inquilinus limosus and Stenotrophomonas maltophilia from antibiotics such as ciprofloxacin.88
Considering such discoveries, we hypothesize that due to the presence of the intensified pressures at the host–pathogen interface, the host versus microbe molecular arms race takes precedence over the natural microbe versus microbe race and opens doors to symbiotic or cooperative interactions with fellow pathogens and/or commensals. This phenomenon could be mediated by the fact that cooperative interactions can aid microbes to survive by joining forces against the host immune system and waging the battle together by exploiting shared resources, instead of expending energy in the production of antimicrobial compounds against each other (Fig. 2).
Applicability to other host niches
While this review is mostly focused on polymicrobial interactions within the lung, similar adaptations are likely to occur in other host-relevant niches. Microbial cooperation has been well characterized as occurring in human oral cavities and is likely also occurring in the context of chronic wound infections. Co-evolution has been very well depicted in the communities shaping these two host environments. The following sections describe instances where microorganisms of the oral cavity or the chronic wound environment display cooperation over competition.
Oral cavity
The healthy human oral cavity harbors as many as 200 different bacterial species89,90 along with viruses and fungi. These species form inter-connected networks to survive in the lotic environment of the mouth. Cooperation among these microbes is seen in terms of co-adhesion or co-aggregation of cells that attach to each other for establishing colonization onto oral surfaces like the gumline, tooth enamel and the sub-gingival cavities. Early colonizers such as Streptococcus gordonii, Propionibacterium acnes, Haemophilus parainfluenzae and Prevotella loescheii initiate colonization by binding to complementary salivary receptors such as sialylated mucins, proline-rich proteins and α-amylase present on the tooth surface. These cells coaggregate with late colonizers such as Aggregatibacter actinomycetemcomitans and Treponema denticola to promote polymicrobial biofilm formation.91,92 Sometimes, intermediary species even “bridge” the early and later colonizers. For example, Fusobacterium nucleatum expresses at least one adhesin that recognizes early colonizer Streptococci spp. and a galactose-specific lectin that interacts with late colonizer, Porphyromonas gingivalis.
Although a large portion of the oral microbiome consist of commensals, a subset of these are opportunistic pathogens.93 These pathogens can cause oral cavity infections such as periodontitis that have been widely studied for cooperative interactions among causative microorganisms.94 P. gingivalis, T. denticola and Tannerella forsythia are known to be the primary causative pathogens of oral periodontitis95 though recently many anaerobic bacterial species have been discovered to be associated as well.93 Periodontitis results from dysbiosis that changes the microbial composition of the oral cavity from a healthy to a diseased state.96 In particular, P. gingivalis has been revealed as a keystone pathogen driving the dysbiosis associated with chronic periodontitis through the production of a suite of virulence factors capable of benefiting itself as well as the surrounding diseased community.97 Additionally, P. gingivalis is known to release eDNA that can be up taken by microorganisms inducing adaptive mutations in them.98 This process can make neighboring pathogens immune to known antibacterial molecules and increase pathogenicity of the community. Other studies have revealed that periodontal pathogens such as F. nucleatum and A. actinomycetemcomitans generate short-chain fatty acids (SCFAs) such as butyric acid which aid in disease progression possibly via facilitating pathogen colonization.99,100,101 SCFAs induce ROS production and inflammation in the gingival tissues leading to growth inhibition of gingival epithelial cells that serves as a barrier against pathogenic invasion and impair healing.99,100
Synergistic interactions between early and late colonizers have been shown to enhance virulence of opportunistic oral pathogens such as A. actinomycetemcomitans. For example, during a co-culture of A. actinomycetemcomitans and the oral commensal S. gordonii, the latter increased the virulence of the former by providing it with l-lactate, a carbon source necessary for establishing robust infection in the gum subgingival crevice of human oral cavity.102 l-lactate is primarily produced by S. gordonii cells and is made available to A. actinomycetemcomitans via metabolic cross-feeding. S. gordonii cells also produce inhibitory levels of H2O2 to trigger catalase mediated break-down by A. actinomycetemcomitans. Production of such ROS augments bioavailability of oxygen during an infection, allowing A. actinomycetemcomitans cells to shift from a primarily fermentative to a respiratory metabolism which in turn enhances its growth and persistence.103 Additionally, this catalase produced by A. actinomycetemcomitans protects anaerobic pathogen P. gingivalis from ROS in the oral cavity and aids it to colonize oral surfaces other than the dental pockets where oxygen is limited.104
In a study about interactions of oral pathogens, intergeneric co-aggregation was shown in mixed-species communities of Streptococci and Veillonellae spp. during formation of early dental plaque. Lactic acid produced by Streptococci is cross fed to Veillonellae cells, where it serves as a carbon source and establishes metabolic interaction between the two microbial species. Biofilm community obtained from a retrievable human enamel surface revealed two Streptococci, S. oralis and S. gordonii, and an uncultivated Veillonellae spp. as members of such mixed-species colonies.105 Another study that investigated the interactions between the fungus C. albicans and three commensal Streptococci spp. of the human oral cavity showed that Streptococci cells displayed poor biofilm formation on abiotic or mucosal surfaces by itself but formed robust biofilms in presence of the fungus. On the other hand, these Streptococcal spp., such as S. oralis, enhanced the ability of C. albicans to invade oral and esophageal mucosa.106 Thus, co-adhesion and co-aggregation facilitated metabolic interdependencies have been very well established in the human oral cavity. These dependencies play important roles in determining the chronology of colonization in the oral cavity.
Chronic wounds
Chronically infected sites such as chronic wounds are also likely to experience similar levels of microbial interactions as in the oral cavity. Chronic wounds can be an outcome of burns, surgical site infections, and/or diabetic foot ulcers which are often slow-healing or non-healing.107 A study of 30 human wounds revealed the presence of 106 different bacterial genera, large fractions of which were strict and facultative anaerobes. Such research indicates the presence of a diverse microbial community in chronically wounded tissues.108
Recent studies have shown enhanced antibiotic tolerance in polymicrobial communities of chronically wounded tissues. For example, one study showed that chronic wounds colonized with biofilms of three bacterial species- S. aureus, P. aeruginosa and Clostridium perfringens, were found to be more tolerant to treatment than single-species wound infection.109 Another study aimed at understanding interspecies interaction between S. aureus, P. aeruginosa, Enterococcus faecalis and Finegoldia magna under in vivo mouse wound conditions, showed that the presence of a microbial consortium enabled growth of the obligate anaerobe F. magna.110 As in other host-associated niches, microbial cooperation in the form of cross-feeding appears to play an important role in establishment of the wound polymicrobial community.111 For example, wound-associated S. aureus, E. coli, and K. pneumoniae can provide molecules such as heme, menaquinone, and succinate to pathogenic Prevotella and Porphryomonas spp.112 In another study, augmentation of E. coli biofilms in mouse wound infection model has been shown to occur as a result of ornithine production by neighboring E. faecalis.111 Presence of ornithine modulates the surrounding environment of E. coli in ways that favor metabolic pathways leading to siderophore biosynthesis. In addition to nutrient complementation mediated by the surrounding microbial consortium, mice with mixed-species biofilm infections displayed wound healing impairment and increased antimicrobial tolerance as compared to mice infected with single species of these bacteria.113 Thus, synergistic interactions between different bacterial species in wounds confer the participating cells with benefits such as antibiotic tolerance and reduced interspecies competition. Additionally, the chronic wounds create an environment that promotes a reduction in competitive molecules produced by P. aeruginosa to enable this synergism. In addition to being able to suppress its antimicrobial arsenal in the presence of the innate immune protein calprotectin,78 P. aeruginosa exhibits reduced anti-staphylococcal activity in the presence of the abundant host protein albumin.114
Potential for therapeutic targeting
Infectious polymicrobial communities are often found to be more resistant to antibiotics than their mono-culture counterparts.82,88 Members of these polymicrobial communities are actively interacting with one another and building a network of interactions. These interactions are dictated by adaptive mutation which may initially seem detrimental but appear useful in context of the community. Here we have discussed several examples of microbial infections where we see how polymicrobial interactions and shifts in community composition modulate pathogenicity and antimicrobial susceptibility of the pathogens. Over the course of chronic infection, the microbial community composition tends to fluctuate, beginning with transient colonization and transforming into a highly pathogenic and persistent infection.115 An understanding of these shifts is important for designing therapeutics because of the metabolic differences exhibited by these communities. For example, in infections such as the ones caused by both P. aeruginosa and S. aureus, aminoglycosides might not be the most effective therapeutic due to the increased aminoglycoside resistance conferred to S. aureus by the presence of P. aeruginosa.82 In addition to being influenced by neighboring communities, microbial physiology is also driven by the overall community structure.116 This community structure introduces nutrient gradients within the community that drive physiological differentiation of microbes which can alter antibiotic susceptibility.117 Efforts are being made to better understand the core metabolism of chronic infectious communities as well as the biogeochemical forces influencing microbial physiology in order to identify the most effective drug targets.34
While polymicrobial interactions may provide complications for traditional therapeutics, it is also possible that these interactions could provide new drug targets. For example, if the structure of the microbial community is important for its persistence, mechanisms that disrupt the biogeography of the infection could prove useful.116 Mechanisms to target and disrupt structural components of biofilms are currently being pursued by many research groups.118,119,120 Additionally, if these complex microbial communities are relying on secreted shared resources for survival, targeting of these resources might severely impact both the producer microbes as well as beneficiary microbes in the vicinity.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
Ma, L., Terwilliger, A. & Maresso, A. W. Iron and zinc exploitation during bacterial pathogenesis. Metallomics 7, 1541–1554 (2015).
Hood, M. I. & Skaar, E. P. Nutritional immunity: transition metals at the pathogen-host interface. Nat. Rev. Microbiol. 10, 525–537 (2012).
Weinberg, E. D. Iron availability and infection. Biochim Biophys. Acta. 1790, 600–605 (2009).
Cassat, J. E. & Skaar, E. P. Iron in infection and immunity. Cell Host Microbe. 13, 509–519 (2013).
Saikia, S., Oliveira, D., Hu, G. & Kronstad, J. Role of ferric reductases in iron acquisition and virulence in the fungal pathogen Cryptococcus neoformans. Infect. Immun. 82, 839–850 (2014).
Li, K., Chen, W. H. & Bruner, S. D. Microbial siderophore-based iron assimilation and therapeutic applications. Biometals 29, 377–388 (2016).
Kalidasan, V., Joseph, N., Kumar, S., Hamat, R. A. & Neela, V. K. The ‘checkmate’ for iron between human host and invading bacteria: chess game analogy. Indian J. Microbiol. 58, 257–267 (2018).
Barber, M. F. & Elde, N. C. Buried treasure: evolutionary perspectives on microbial iron piracy. Trends Genet. 31, 627–636 (2015).
Hebert, G. A. & Hancock, G. A. Synergistic hemolysis exhibited by species of staphylococci. J. Clin. Microbiol. 22, 409–415 (1985).
Tomlin, K. L., Coll, O. P. & Ceri, H. Interspecies biofilms of Pseudomonas aeruginosa and Burkholderia cepacia. Can. J. Microbiol. 47, 949–954 (2001).
Bhargava, N., Sharma, P. & Capalash, N. Pyocyanin stimulates quorum sensing-mediated tolerance to oxidative stress and increases persister cell populations in Acinetobacter baumannii. Infect. Immun. 82, 3417–3425 (2014).
Valko, M. et al. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem Cell Biol. 39, 44–84 (2007).
Campbell, E. L. & Colgan, S. P. Neutrophils and inflammatory metabolism in antimicrobial functions of the mucosa. J. Leukoc. Biol. 98, 517–522 (2015).
Lemire, J., Alhasawi, A., Appanna, V. P., Tharmalingam, S. & Appanna, V. D. Metabolic defence against oxidative stress: the road less travelled so far. J. Appl. Microbiol. 123, 798–809 (2017).
Eason, M. M. & Fan, X. The role and regulation of catalase in respiratory tract opportunistic bacterial pathogens. Micro. Pathog. 74, 50–58 (2014).
Przybyszewska, J. & Zekanowska, E. The role of hepcidin, ferroportin, HCP1, and DMT1 protein in iron absorption in the human digestive tract. Prz. Gastroenterol. 9, 208–213 (2014).
Kehl-Fie, T. E. et al. Nutrient metal sequestration by calprotectin inhibits bacterial superoxide defense, enhancing neutrophil killing of Staphylococcus aureus. Cell Host Microbe. 10, 158–164 (2011).
Boles, B. R. & Singh, P. K. Endogenous oxidative stress produces diversity and adaptability in biofilm communities. Proc. Natl Acad. Sci. USA 105, 12503–12508 (2008).
Limoli, D. H. et al. Cationic antimicrobial peptides promote microbial mutagenesis and pathoadaptation in chronic infections. PLoS Pathog. 10, e1004083 (2014).
Limoli, D. H. & Wozniak, D. J. Mutagenesis by host antimicrobial peptides: insights into microbial evolution during chronic infections. Micro. Cell 1, 247–249 (2014).
Romero, D., Traxler, M. F., Lopez, D. & Kolter, R. Antibiotics as signal molecules. Chem. Rev. 111, 5492–5505 (2011).
Hoffman, L. R. et al. Aminoglycoside antibiotics induce bacterial biofilm formation. Nature 436, 1171–1175 (2005).
Bagge, N. et al. Pseudomonas aeruginosa biofilms exposed to imipenem exhibit changes in global gene expression and beta-lactamase and alginate production. Antimicrob. Agents Chemother. 48, 1175–1187 (2004).
Jorth, P. et al. Evolved aztreonam resistance is multifactorial and can produce hypervirulence in Pseudomonas aeruginosa. MBio. 8. https://doi.org/10.1128/mBio.00517-17 (2017).
Lim, Y. W. et al. Clinical insights from metagenomic analysis of sputum samples from patients with cystic fibrosis. J. Clin. Microbiol. 52, 425–437 (2014).
Bragonzi, A. et al. Pseudomonas aeruginosa microevolution during cystic fibrosis lung infection establishes clones with adapted virulence. Am. J. Respir. Crit. Care Med. 180, 138–145 (2009).
Lore, N. I. et al. Cystic fibrosis-niche adaptation of Pseudomonas aeruginosa reduces virulence in multiple infection hosts. PLoS ONE 7, e35648 (2012).
Mihai, M. M. et al. Microbial biofilms: impact on the pathogenesis of periodontitis, cystic fibrosis, chronic wounds and medical device-related infections. Curr. Top. Med. Chem. 15, 1552–1576 (2015).
Bjarnsholt, T. The role of bacterial biofilms in chronic infections. APMIS Suppl. 1–51. https://doi.org/10.1111/apm.12099 (2013).
Baughman, R. P., Thorpe, J. E., Staneck, J., Rashkin, M. & Frame, P. T. Use of the protected specimen brush in patients with endotracheal or tracheostomy tubes. Chest 91, 233–236 (1987).
Charlson, E. S. et al. Lung-enriched organisms and aberrant bacterial and fungal respiratory microbiota after lung transplant. Am. J. Respir. Crit. Care Med. 186, 536–545 (2012).
Hilty, M. et al. Disordered microbial communities in asthmatic airways. PLoS ONE 5, e8578 (2010).
Huang, Y. J. et al. The role of the lung microbiome in health and disease. A National Heart, Lung, and Blood Institute workshop report. Am. J. Respir. Crit. Care Med 187, 1382–1387 (2013).
Quinn, R. A. et al. Biogeochemical forces shape the composition and physiology of polymicrobial communities in the cystic fibrosis lung. MBio 5, e00956–00913 (2014).
Mammen, M. J. & Sethi, S. COPD and the microbiome. Respirology 21, 590–599 (2016).
Soler, N. et al. Airway inflammation and bronchial microbial patterns in patients with stable chronic obstructive pulmonary disease. Eur. Respir. J. 14, 1015–1022 (1999).
Shima, K., Coopmeiners, J., Graspeuntner, S., Dalhoff, K. & Rupp, J. Impact of micro-environmental changes on respiratory tract infections with intracellular bacteria. FEBS Lett. 590, 3887–3904 (2016).
Hahn, D. L., Azenabor, A. A., Beatty, W. L. & Byrne, G. I. Chlamydia pneumoniae as a respiratory pathogen. Front Biosci. 7, e66–e76 (2002).
Esther, C. R., Jr. et al. Mucus accumulation in the lungs precedes structural changes and infection in children with cystic fibrosis. Sci. Transl. Med. 11. https://doi.org/10.1126/scitranslmed.aav3488 (2019).
Hahn, A. et al. Different next generation sequencing platforms produce different microbial profiles and diversity in cystic fibrosis sputum. J. Microbiol. Methods 130, 95–99 (2016).
Li, J. et al. Data mining of lung microbiota in cystic fibrosis patients. PLoS ONE 11, e0164510 (2016).
Rabin, H. R. & Surette, M. G. The cystic fibrosis airway microbiome. Curr. Opin. Pulm. Med. 18, 622–627 (2012).
Beck, J. M., Young, V. B. & Huffnagle, G. B. The microbiome of the lung. Transl. Res. 160, 258–266 (2012).
Cox, M. J. et al. Airway microbiota and pathogen abundance in age-stratified cystic fibrosis patients. PLoS ONE 5, e11044 (2010).
McGuigan, L. & Callaghan, M. The evolving dynamics of the microbial community in the cystic fibrosis lung. Environ. Microbiol. 17, 16–28 (2015).
Carmody, L. A. et al. Changes in cystic fibrosis airway microbiota at pulmonary exacerbation. Ann. Am. Thorac. Soc. 10, 179–187 (2013).
Loesche, M. et al. Temporal stability in chronic wound microbiota is associated with poor healing. J. Invest Dermatol. 137, 237–244 (2017).
Fodor, A. A. et al. The adult cystic fibrosis airway microbiota is stable over time and infection type, and highly resilient to antibiotic treatment of exacerbations. PLoS ONE 7, e45001 (2012).
Nguyen, A. T. & Oglesby-Sherrouse, A. G. Interactions between Pseudomonas aeruginosa and Staphylococcus aureus during co-cultivations and polymicrobial infections. Appl. Microbiol. Biotechnol. 100, 6141–6148 (2016).
Vasil, M. L. & Ochsner, U. A. The response of Pseudomonas aeruginosa to iron: genetics, biochemistry and virulence. Mol. Microbiol. 34, 399–413 (1999).
Mashburn, L. M., Jett, A. M., Akins, D. R. & Whiteley, M. Staphylococcus aureus serves as an iron source for Pseudomonas aeruginosa during in vivo coculture. J. Bacteriol. 187, 554–566 (2005).
Nguyen, A. T., Jones, J. W., Ruge, M. A., Kane, M. A. & Oglesby-Sherrouse, A. G. Iron depletion enhances production of antimicrobials by Pseudomonas aeruginosa. J. Bacteriol. 197, 2265–2275 (2015).
Morales, D. K. et al. Control of Candida albicans metabolism and biofilm formation by Pseudomonas aeruginosa phenazines. MBio 4, e00526–e00512 (2013).
Hunter, R. C. et al. Phenazine content in the cystic fibrosis respiratory tract negatively correlates with lung function and microbial complexity. Am. J. Respir. Cell Mol. Biol. 47, 738–745 (2012).
Moree, W. J. et al. Interkingdom metabolic transformations captured by microbial imaging mass spectrometry. Proc. Natl Acad. Sci. USA 109, 13811–13816 (2012).
Zheng, H. et al. Redox metabolites signal polymicrobial biofilm development via the NapA oxidative stress cascade in Aspergillus. Curr. Biol. 25, 29–37 (2015).
Hirschhausen, N. et al. Extended Staphylococcus aureus persistence in cystic fibrosis is associated with bacterial adaptation. Int. J. /Med. Microbiol. 303, 685–692 (2013).
Kahl, B. C., Becker, K. & Loffler, B. Clinical significance and pathogenesis of Staphylococcal small colony variants in persistent infections. Clin. Microbiol Rev. 29, 401–427 (2016).
Donlan, R. M. & Costerton, J. W. Biofilms: survival mechanisms of clinically relevant microorganisms. Clin. Microbiol. Rev. 15, 167–193 (2002).
O’Riordan, K. & Lee, J. C. Staphylococcus aureus capsular polysaccharides. Clin. Microbiol. Rev. 17, 218–234 (2004).
Proctor, R. A. et al. Small colony variants: a pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat. Rev. Microbiol. 4, 295–305 (2006).
Jani, M., Sengupta, S., Hu, K. & Azad, R. K. Deciphering pathogenicity and antibiotic resistance islands in methicillin-resistant Staphylococcus aureus genomes. Open Biol. 7. https://doi.org/10.1098/rsob.170094 (2017).
Chang, S. et al. Infection with vancomycin-resistant Staphylococcus aureus containing the vanA resistance gene. N. Engl. J. Med. 348, 1342–1347 (2003).
von Eiff, C., Peters, G. & Becker, K. The small colony variant (SCV) concept–the role of staphylococcal SCVs in persistent infections. Injury 37(Suppl 2), S26–S33 (2006).
Noto, M. J., Burns, W. J., Beavers, W. N. & Skaar, E. P. Mechanisms of pyocyanin toxicity and genetic determinants of resistance in Staphylococcus aureus. J. Bacteriol. https://doi.org/10.1128/JB.00221-17 (2017).
Hogardt, M. & Heesemann, J. Microevolution of Pseudomonas aeruginosa to a chronic pathogen of the cystic fibrosis lung. Curr. Top. Microbiol. Immunol. 358, 91–118 (2013).
Smith, E. E. et al. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc. Natl Acad. Sci. USA 103, 8487–8492 (2006).
Rodriguez-Rojas, A. et al. Inactivation of the hmgA gene of Pseudomonas aeruginosa leads to pyomelanin hyperproduction, stress resistance and increased persistence in chronic lung infection. Microbiology 155, 1050–1057 (2009).
Hatch, R. A. & Schiller, N. L. Alginate lyase promotes diffusion of aminoglycosides through the extracellular polysaccharide of mucoid Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 42, 974–977 (1998).
Oliver, A. M. & Weir, D. M. The effect of Pseudomonas alginate on rat alveolar macrophage phagocytosis and bacterial opsonization. Clin. Exp. Immunol. 59, 190–196 (1985).
Pedersen, S. S., Kharazmi, A., Espersen, F. & Hoiby, N. Pseudomonas aeruginosa alginate in cystic fibrosis sputum and the inflammatory response. Infect. Immun. 58, 3363–3368 (1990).
Boucher, J. C., Yu, H., Mudd, M. H. & Deretic, V. Mucoid Pseudomonas aeruginosa in cystic fibrosis: characterization of muc mutations in clinical isolates and analysis of clearance in a mouse model of respiratory infection. Infect. Immun. 65, 3838–3846 (1997).
Gryllos, I. et al. Induction of group A Streptococcus virulence by a human antimicrobial peptide. Proc. Natl Acad. Sci. USA 105, 16755–16760 (2008).
Jorth, P. et al. Regional isolation drives bacterial diversification within cystic fibrosis Lungs. Cell Host Microbe 18, 307–319 (2015).
Qin, X. et al. Pseudomonas aeruginosa syntrophy in chronically colonized airways of cystic fibrosis patients. Antimicrob. Agents Chemother. 56, 5971–5981 (2012).
Hammer, N. D. et al. Inter- and intraspecies metabolite exchange promotes virulence of antibiotic-resistant Staphylococcus aureus. Cell Host Microbe 16, 531–537 (2014).
Frydenlund Michelsen, C. et al. Evolution of metabolic divergence in Pseudomonas aeruginosa during long-term infection facilitates a proto-cooperative interspecies interaction. ISME J. 10, 1323–1336 (2016).
Wakeman, C. A. et al. The innate immune protein calprotectin promotes Pseudomonas aeruginosa and Staphylococcus aureus interaction. Nat. Commun. 7, 11951 (2016).
Limoli, D. H. et al. Pseudomonas aeruginosa alginate overproduction promotes coexistence with Staphylococcus aureus in a model of cystic fibrosis respiratory infection. MBio 8. https://doi.org/10.1128/mBio.00186-17 (2017).
Cohen, T. S. et al. Staphylococcus aureus α toxin potentiates opportunistic bacterial lung infections. Sci. Trans. Med. 8, 329ra31 (2016).
Hoffman, L. R. et al. Nutrient availability as a mechanism for selection of antibiotic tolerant Pseudomonas aeruginosa within the CF airway. PLoS Pathog. 6, e1000712 (2010).
Hoffman, L. R. et al. Selection for Staphylococcus aureus small-colony variants due to growth in the presence of Pseudomonas aeruginosa. Proc. Natl Acad. Sci. USA 103, 19890–19895 (2006).
Machan, Z. A., Taylor, G. W., Pitt, T. L., Cole, P. J. & Wilson, R. 2-Heptyl-4-hydroxyquinoline N-oxide, an antistaphylococcal agent produced by Pseudomonas aeruginosa. J. Antimicrob. Chemother. 30, 615–623 (1992).
Heeb, S. et al. Quinolones: from antibiotics to autoinducers. FEMS Microbiol Rev. 35, 247–274 (2011).
Harriott, M. M. & Noverr, M. C. Candida albicans and Staphylococcus aureus form polymicrobial biofilms: effects on antimicrobial resistance. Antimicrob. Agents Chemother. 53, 3914–3922 (2009).
Kean, R. et al. Candida albicans mycofilms support Staphylococcus aureus colonization and enhances miconazole resistance in dual-species interactions. Front. Microbiol. 8, 258 (2017).
Rezaiki, L., Lamberet, G., Derre, A., Gruss, A. & Gaudu, P. Lactococcus lactis produces short-chain quinones that cross-feed Group B Streptococcus to activate respiration growth. Mol. Microbiol. 67, 947–957 (2008).
Magalhaes, A. P., Lopes, S. P. & Pereira, M. O. Insights into cystic fibrosis polymicrobial consortia: the role of species interactions in biofilm development, phenotype, and response to in-use antibiotics. Front. Microbiol. 7, 2146 (2016).
Kolenbrander, P. E., Palmer, R. J. Jr., Periasamy, S. & Jakubovics, N. S. Oral multispecies biofilm development and the key role of cell-cell distance. Nat. Rev. Microbiol. 8, 471–480 (2010).
Paster, B. J., Olsen, I., Aas, J. A. & Dewhirst, F. E. The breadth of bacterial diversity in the human periodontal pocket and other oral sites. Periodontol. 2000. 42, 80–87 (2006).
Ding, A. M., Palmer, R. J. Jr., Cisar, J. O. & Kolenbrander, P. E. Shear-enhanced oral microbial adhesion. Appl. Environ. Microbiol 76, 1294–1297 (2010).
Diaz, P. I. et al. Molecular characterization of subject-specific oral microflora during initial colonization of enamel. Appl Environ. Microbiol. 72, 2837–2848 (2006).
Paster, B. J. et al. Bacterial diversity in human subgingival plaque. J. Bacteriol. 183, 3770–3783 (2001).
Marsh, P. D. & Zaura, E. Dental biofilm: ecological interactions in health and disease. J. Clin. Periodo. 44(Suppl 18), S12–S22 (2017).
de Lillo, A., Booth, V., Kyriacou, L., Weightman, A. J. & Wade, W. G. Culture-independent identification of periodontitis-associated Porphyromonas and Tannerella populations by targeted molecular analysis. J. Clin. Microbiol. 42, 5523–5527 (2004).
Deng, Z. L., Szafranski, S. P., Jarek, M., Bhuju, S. & Wagner-Dobler, I. Dysbiosis in chronic periodontitis: Key microbial players and interactions with the human host. Sci. Rep. 7, 3703 (2017).
How, K. Y., Song, K. P. & Chan, K. G. Porphyromonas gingivalis: an overview of periodontopathic pathogen below the gum line. Front. Microbiol. 7, 53 (2016).
Ali Mohammed, M. M., Nerland, A. H., Al-Haroni, M. & Bakken, V. Characterization of extracellular polymeric matrix, and treatment of Fusobacterium nucleatum and Porphyromonas gingivalis biofilms with DNase I and proteinase K. J. Oral Microbiol. 5. https://doi.org/10.3402/jom.v5i0.20015 (2013).
Niederman, R., Buyle-Bodin, Y., Lu, B. Y., Robinson, P. & Naleway, C. Short-chain carboxylic acid concentration in human gingival crevicular fluid. J. Dent. Res. 76, 575–579 (1997).
Chang, M. C. et al. Butyrate induces reactive oxygen species production and affects cell cycle progression in human gingival fibroblasts. J. Periodontal Res. 48, 66–73 (2013).
Belibasakis, G. N., Mattsson, A., Wang, Y., Chen, C. & Johansson, A. Cell cycle arrest of human gingival fibroblasts and periodontal ligament cells by Actinobacillus actinomycetemcomitans: involvement of the cytolethal distending toxin. APMIS 112, 674–685 (2004).
Ramsey, M. M., Rumbaugh, K. P. & Whiteley, M. Metabolite cross-feeding enhances virulence in a model polymicrobial infection. PLoS Pathog. 7, e1002012 (2011).
Stacy, A., Fleming, D., Lamont, R. J., Rumbaugh, K. P. & Whiteley, M. A commensal bacterium promotes virulence of an opportunistic pathogen via cross-respiration. MBio 7. https://doi.org/10.1128/mBio.00782-16 (2016).
Zhu, B., Macleod, L. C., Newsome, E., Liu, J. & Xu, P. Aggregatibacter actinomycetemcomitans mediates protection of Porphyromonas gingivalis from Streptococcus sanguinis hydrogen peroxide production in multi-species biofilms. Sci. Rep. 9, 4944 (2019).
Chalmers, N. I., Palmer, R. J. Jr., Cisar, J. O. & Kolenbrander, P. E. Characterization of a Streptococcus sp.-Veillonella sp. community micromanipulated from dental plaque. J. Bacteriol. 190, 8145–8154 (2008).
Diaz, P. I. et al. Synergistic interaction between Candida albicans and commensal oral streptococci in a novel in vitro mucosal model. Infect. Immun. 80, 620–632 (2012).
James, G. A. et al. Biofilms in chronic wounds. Wound Repair Regen. 16, 37–44 (2008).
Dowd, S. E. et al. Survey of bacterial diversity in chronic wounds using pyrosequencing, DGGE, and full ribosome shotgun sequencing. BMC Microbiol. 8, 43 (2008).
Woods, J. et al. Development and application of a polymicrobial, in vitro, wound biofilm model. J. Appl. Microbiol. 112, 998–1006 (2012).
Dalton, T. et al. An in vivo polymicrobial biofilm wound infection model to study interspecies interactions. PLoS ONE 6, e27317 (2011).
Keogh, D. et al. Enterococcal metabolite cues facilitate interspecies niche modulation and polymicrobial infection. Cell Host Microbe 20, 493–503 (2016).
Bertesteanu, S. et al. Polymicrobial wound infections: pathophysiology and current therapeutic approaches. Int. J. Pharm. 463, 119–126 (2014).
Pastar, I. et al. Interactions of methicillin resistant Staphylococcus aureus USA300 and Pseudomonas aeruginosa in polymicrobial wound infection. PLoS ONE 8, e56846 (2013).
Smith, A. C. et al. Albumin inhibits Pseudomonas aeruginosa quorum sensing and alters polymicrobial interactions. Infect. Immun. 85. https://doi.org/10.1128/IAI.00116-17 (2017).
Conrad, D. et al. Cystic fibrosis therapy: a community ecology perspective. Am. J. Respir. Cell Mol. Biol. 48, 150–156 (2013).
Stacy, A., McNally, L., Darch, S. E., Brown, S. P. & Whiteley, M. The biogeography of polymicrobial infection. Nat. Rev. Microbiol. 14, 93–105 (2016).
Stewart, P. S. & Franklin, M. J. Physiological heterogeneity in biofilms. Nat. Rev. Microbiol. 6, 199–210 (2008).
Fleming, D., Chahin, L. & Rumbaugh, K. Glycoside hydrolases degrade polymicrobial bacterial biofilms in wounds. Antimicrob Agents Chemother. 61. https://doi.org/10.1128/AAC.01998-16 (2017).
Ren, Z. et al. Dual-targeting approach degrades biofilm matrix and enhances bacterial killing. J. Dent. Res. 98, 322–330 (2019).
Ren, Z. et al. Molecule targeting glucosyltransferase inhibits Streptococcus mutans biofilm formation and virulence. Antimicrob. Agents Chemother. 60, 126–135 (2016).
Acknowledgements
The work described in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R15GM128072. We would like to acknowledge the members of the Wakeman lab for critically reading the manuscript. We would also like to thank Moamen Elmassry for his valuable critique in putting together the article.
Author information
Authors and Affiliations
Contributions
J.B. and C.A.W. conceived the research focus for the review article. J.B. drafted the article and generated the figures. C.A.W. reviewed, edited, and revised the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Baishya, J., Wakeman, C.A. Selective pressures during chronic infection drive microbial competition and cooperation. npj Biofilms Microbiomes 5, 16 (2019). https://doi.org/10.1038/s41522-019-0089-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41522-019-0089-2
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
