
Abstract
Miscanthus, a member of the Saccharinae subtribe that includes sorghum and sugarcane, has been widely studied as a feedstock for cellulosic biofuel production. Here, we report the sequencing and assembly of the Miscanthus floridulus genome by the integration of PacBio sequencing and Hi-C mapping, resulting in a chromosome-scale, high-quality reference genome of the genus Miscanthus. Comparisons among Saccharinae genomes suggest that Sorghum split first from the common ancestor of Saccharum and Miscanthus, which subsequently diverged from each other, with two successive whole-genome duplication events occurring independently in the Saccharum genus and one whole-genome duplication occurring in the Miscanthus genus. Fusion of two chromosomes occurred during rediploidization in M. floridulus and no significant subgenome dominance was observed. A survey of cellulose synthases (CesA) in M. floridulus revealed quite high expression of most CesA genes in growing stems, which is in agreement with the high cellulose content of this species. Resequencing and comparisons of 75 Miscanthus accessions suggest that M. lutarioriparius is genetically close to M. sacchariflorus and that M. floridulus is more distantly related to other species and is more genetically diverse. This study provides a valuable genomic resource for molecular breeding and improvement of Miscanthus and Saccharinae crops.
Similar content being viewed by others


Main
The Saccharinae subtribe is within Andropogoneae tribe in the grass family (Poaceae). It is comprised of the Saccharum complex, the interspecific breeding group inclusive of Saccharum (sugarcane) and Miscanthus, and the Sorghinae subtribe, which has been merged into subtribe Saccharinae recently1. In this group, Miscanthus, Sorghum and Saccharum are widely studied for their favourable characteristics for bioenergy production2,3,4,5. In addition to their capability for C4 photosynthesis, they require low nitrogen for growth and can regrow after harvest. In sorghum, photosynthetically fixed carbon is mainly stored in the form of starch in grain and cellulose/hemicellulose in leaves and stems. Sugarcane stores most fixed carbon as free sugars in stems and cellulose/hemicellulose in stems and leaves, while abundant cellulose/hemicellulose as the main form of fixed carbon is predominantly stored in both stems and leaves of Miscanthus. Briefly, Miscanthus can yield ~30 t ha−1 dry biomass, comprising 41–46% cellulose and 29% hemicellulose on average6,7 and sorghum can produce ~15–25 t ha−1 dry biomass, comprising 26–29% cellulose and 20–26% hemicellulose on average8,9, whereas commercial sugarcane yields 39 t ha−1 dry biomass comprising 14–29% cellulose and 9–18% hemicellulose5,10. Thus, among these three species, Miscanthus provides the most cellulose/hemicellulose per unit area for bioenergy production. Moreover, with increasing demand for food there is a corresponding increase in the demand for agricultural land for crop production. Miscanthus, which can grow robustly on marginal lands such as saline or infertile lands, is considered to be the most promising energy crop species compared with sorghum and sugarcane4,6,11.
Polyploidization is among the strongest drivers of angiosperm evolution, contributing to the speciation and emergence of valuable traits in Saccharinae especially sugarcane and Miscanthus12,13,14. Miscanthus and sugarcane share a common ancestor, which split from the sorghum clade before their divergence. Following a single whole-genome duplication (WGD) event and chromosomal rearrangement, Miscanthus has a basic chromosome number of 19 (2n = 2x = 38 or 2n = 4x = 76), whereas sugarcane has a basic chromosome number of eight (Saccharum spontaneum) or ten (S. officinarum); these numbers were ultimately attained after two or more rounds of polyploidization accompanied by extensive genome rearrangement12,15. In general, both sugarcane and Miscanthus are categorized as neopolyploids, having undergone WGD within the last 5 million years14. Unlike in palaeopolyploids, a variety of evolutionary processes such as the activation of genes or retrotransposons, gene loss, gene silencing and subfunctionalization of genes remain ongoing13,14. Thus, Saccharinae may serve as a tractable system for studying potential impacts of genome duplication, subsequent subgenome divergence, genome rearrangement and extensive gene fractionation.
In monocots, members of the cellulose synthase (CesA) family and seven cellulose synthase-like (Csl) families, including CslA, CslC, CslD, CslE, CslF, CslH and CslJ, are generally believed to mediate the synthesis of cellulose and the hemicellulose polysaccharides respectively16,17,18,19,20. In the CesA lineage, OsCesA4, OsCesA7 and OsCesA9 are mainly required for cellulose synthesis in the secondary cell wall of rice, whereas OsCesA1, OsCesA3 and OsCesA8 are strongly co-expressed in tissues typical of primary cell walls21,22, with the function of OsCesA5/OsCesA6 being partially redundant with that of OsCesA3. Because of their great importance in cell wall synthesis and bioenergy production, gene expression and phylogeny of all genes belonging to the CesA/Csl superfamily have been extensively studied since their identification16,17,18,21,22,23,24,25. In Miscanthus, however, only a handful of CesA genes were identified and analysed on the basis of transcriptome sequencing and quantitative polymerase chain reaction (qPCR), and the comprehensive genome-wide identification and expression profiling of CesA/Csl genes superfamily are still lacking26,27.
The reference genomes of sorghum and sugarcane (S. spontaneum) have been previously reported15,28,29, given their economic importance as major sources of food and livestock feed. However, despite it being one of the most promising bioenergy crops, Miscanthus still does not have a high-quality reference genome available. Here, we report the genome assembly of M. floridulus—the chromosome-scale reference genome for the genus Miscanthus. The assembly of the M. floridulus genome and comparative genomic analysis of this genome with those of sorghum and sugarcane, provide an opportunity to systematically understand the evolutionary history of the Saccharinae group, the diversification of the Miscanthus clade and the underlying genetic basis for the high cellulose content of Miscanthus.
Results
Genome sequencing and assembly
Due to the highly heterozygous nature of the M. floridulus genome, we first performed an initial genome survey to determine genome size and composition via Illumina sequencing data. The k-mer analysis (k = 17; Supplementary Fig. 1) revealed a heterozygosity rate of 1.89% and an estimated genome size of 2,462.35 megabases (Mb) (Supplementary Table 1), which was slightly smaller than the previous estimation of 2,596.59 Mb on the basis of cytometry data30. In addition, repeat sequences were estimated to constitute 73.42% of the genome.
We then combined four different technologies for the de novo assembly of the M. floridulus genome: Illumina short-read sequencing, PacBio long-read sequencing, 10x Genomics technology and Hi-C technology. After the initial assembly was achieved via FALCON with PacBio data, FALCON-Unzip was applied to the initial assembly to produce a phased diploid assembly31, which included primary contigs and haplotigs to address the issue of genomic heterozygosity. After phased polishing was performed, FALCON-Phase combined the primary contigs and haplotigs together with Hi-C data to extend the phasing between the unzipped haplotig blocks32: that is, the heterozygous regions of the genome as bubbles in the assembly graphs, resulting in the generation of two new and better phased assemblies (Supplementary Note 1). One of the phased assemblies was chosen for further polishing with PacBio reads and Illumina short reads, and yielded an assembly consisting of 4,522 contigs with an N50 length of 820,435 base pairs (bp). The phased assembly was then integrated together with the 10x Genomics data to create a hybrid assembly consisting of 2,803 scaffolds totalling 2.68 gigabases (Gb) with an N50 length of 1,644,988 bp. At the final step, a Hi-C-based physical map (Supplementary Fig. 2) was used to assemble 19 pseudochromosomes that anchored 2.44 Gb of the genome, accounting for 91.03% of the whole-genome assembly (Supplementary Table 2). A high-density genetic map comprising 3,799 single-nucleotide polymorphisms (SNPs) was used to verify the Hi-C assembly, providing evidence supporting the consistency between the two methods in terms of both chromosomal assignment and order (Supplementary Fig. 3). Additionally, the Hi-C assembly was verified by Bionano optical mapping data. The scaffolds assembled by the resulting Bionano optical maps covered 91.40% of the assembled genomes (Supplementary Table 3) and were highly consistent with the Hi-C assembly with only 186 conflicts identified between the Hi-C assembled genomes and Bionano optical maps (Supplementary Data 1). These conflicts were subsequently corrected manually (Supplementary Fig. 4).
The completeness of the assembled M. floridulus genome was validated using the core eukaryotic genes mapping approach (CEGMA)33 and benchmarking universal single-copy orthologues (BUSCO)34. CEGMA analysis revealed that the assembled genome covered 224 (90%) complete gene models of the 248 core eukaryotic genes (CEGs) and partially covered 14 additional CEG models (Supplementary Table 4). When BUSCO was used, among 1,440 conserved genes in our assembly, 1,378 complete single-copy plant orthologues (96%) were recalled (Supplementary Table 5). The draft assembly was further evaluated by mapping short reads to the genome assembly, which revealed a mapping rate and genome coverage of 98.4% and 99.8%, respectively (Supplementary Table 6). In addition, long terminal repeat retrotransposons assembly index (LAI), a reference-free genome metric for assessment of the assembly of repeat sequences35, was used to evaluate the continuity of the genome assembly. This analysis resulted in an LAI assembly score of 9.32, which is close to the quality of a reference genome according to the classification system35. Collectively, these results validated the high level of completeness and reliability of our M. floridulus genome assembly.
Genome annotation
In total, we identified 63.6% of the genome as transposable elements (TEs), which were categorized as long terminal repeat retrotransposons (LTR-RTs) (55.8%), non-LTR-RTs (1.2%) and DNA transposons (6.6%) (Supplementary Table 7). Among the LTR-RTs, Gypsy elements (31.3%) were much more abundant than Copia elements (14.2%). In the M. floridulus genome, similar to those of other species, TEs tend to accumulate in intergenic regions particularly recombination-suppressed pericentromeric regions. In addition, the latest LTR-RT proliferation occurred within the last 4 million years (Supplementary Fig. 5).
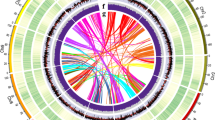
A set of 76,913 genes was annotated with high confidence using a comprehensive strategy that combined de novo gene prediction, protein-based homology searches and transcriptome-based predictions, and the results showed that protein-coding genes were distributed mainly on the ends of chromosome arms (Fig. 1). Functional annotations of all predicted genes resulted in the assignment of putative functional annotations for 71,637 (93.14%) genes. Comparison among Zea mays, Oryza sativa, Sorghum bicolor, S. spontaneum and M. floridulus revealed that a core set of 13,539 gene families were shared among all five grass genomes and that 2,219 gene families were unique to M. floridulus (Supplementary Fig. 6). Gene ontology (GO) enrichment analysis showed that these M. floridulus-specific genes were enriched in GO categories such as postembryonic development, phosphoprotein phosphatase activity, regulation of root development, defence response to oomycetes and other basic molecular functions (Supplementary Table 8). Comparison among the three Saccharinae species revealed that they shared 18,995 gene families (Supplementary Fig. 7) and GO enrichment analysis notably revealed enrichment for the GO term ‘xyloglucan metabolic process’ (Supplementary Table 9), echoing the common view that these grasses are highly promising biofuel feedstocks3.

Tracks indicate the following: gene density (a); TE density (b); gene expression level (TPM; the tracks from outermost to innermost indicate root, stem, leaf and inflorescence respectively) (c); SNP/Indels density from the resequencing data of the 75 accessions (outer track: SNP density; inner circle: Indel density) (d). Chr, chromosome. The outer circle represents the chromosome length of M. floridulus, with units in Mb.
Genome synteny and the evolutionary history of Saccharinae
Given that the annotated genome sequences of both sorghum and sugarcane are currently available15,28,29, we performed genomic synteny analyses among the three species of Saccharinae. We identified 2:1 and 2:4 syntenic depth ratios for alignments between M. floridulus–S. bicolor (Fig. 2a) and M. floridulus–S. spontaneum (Fig. 2b), respectively, thereby suggesting that the ancestor of Miscanthus had experienced one round of WGD since its divergence from sorghum. In addition, we concurrently identified the two subgenomes of Miscanthus (Supplementary Fig. 8). Furthermore, as revealed by a previously constructed genetic map of M. sinensis36, the ancestor of Sbchr7 was split around the centromere region into two segments. These two segments joined together at their telomere ends and formed a new, larger segment, which was then translocated into the centromeric region of the ancestral homoeologous chromosome of Sbchr4 to form MfChr8 (Fig. 2c and Supplementary Table 10). MfChr8 presented the lowest gene density in the second region connecting the split segments of ancestors of Sbchr4 and Sbchr7, suggesting that this region was most probably the newly evolved centromere of MfChr8 (Fig. 2c and d). This fusion resulted in the reduction from two sets of ten duplicated chromosomes produced by WGD to 19 chromosomes. In addition, Sbchr4 carried an inversion (Fig. 2c) specific to the sorghum lineage that was absent in both Miscanthus and sugarcane, which is consistent with previous observations15.

a, A set of two homoeologous chromosomes aligned to a single sorghum chromosome except MfChr7, MfChr13 and MfChr8, among which MfChr7 aligned to Sbchr4, MfChr13 aligned to Sbchr7 and MfChr8 aligned to Sbchr4 and Sbchr7. b, Alignment of M. floridulus chromosomes with S. spontaneum chromosomes. c, The split chromosome homoeologous to Sbchr7 were inserted into the chromosome homoeologous to SbChr4 in M. floridulus. The red triangle indicates the centromere of MfChr8. Note that there is an inversion in Sbchr4 compared with MfChr7, MfChr8 (blue lines) and SsChr4ABCD15. d, Gene count along the MfChr8 chromosome using 2 Mb as a window and 200 kb as a shift. Dashed lines indicate the joint regions linking the split chromosome segments homoeologous to Sbchr7 and Sbchr4 and the third region (blue dashed lines) corresponds to the new evolved centromere of MfChr8.
Previous studies have proposed that Miscanthus and Saccharum share a common allopolyploid ancestor that diverged from the Sorghum lineage ~3.8–4.6 million years ago (Ma)12. However, a recent study demonstrated that S. spontaneum experienced two rounds of WGD autopolyploidization, which occurred during only a brief interval of time15. To elucidate the genomic changes that occurred preceding and following the divergence of these three lineages, we calculated the Ks (synonymous substitutions per site) values between all homoeologous gene pairs in the intergenomic or intragenomic synteny blocks and plotted the Ks distributions of all the syntelogues. This analysis revealed that the Saccharum–Sorghum divergence (Ks = 0.079) and Miscanthus–Sorghum divergence (Ks = 0.072) occurred at approximately the same time (Fig. 3a). Although the Miscanthus–Saccharum divergence (Ks = 0.058) and the divergence of the Miscanthus subgenomes (Ks = 0.051) occurred at a similar time, the divergence of Miscanthus subgenomes occurred approximately half-a-million years later. Since the two rounds of S. spontaneum autopolyploidization occurred within a relatively narrow timeframe, only one Ks peak was detected to represent the two events. Compared with the Ks peak value for syntelogues of Miscanthus–Saccharum, the peak Ks value (Ks = 0.012) for syntelogues between the S. spontaneum subgenomes was very small, indicating that the autopolyploidization event within the Saccharum lineage occurred much later than the Miscanthus–Saccharum divergence.

a, Distribution of synonymous nucleotide substitutions between inter- or intraspecies. The lines with different colours represents the Ks distribution of syntelogues between two species or subgenomes of same species. b, A schematic species tree outlining the evolutionary history of Saccharinae group. The dark green explosion shapes show WGD events and light blue triangles indicate genome rearrangement events. A time line (leftmost) is shown in Ma and a Ks line (rightmost) is also displayed. Chromosome numbers of each state of a plant are shown along branches. (1) Time of divergence of Miscanthus and Saccharum with Sorghum. (2) Time of divergence of Misanthus with Saccharum. (3) Time of divergence of Miscanthus subgenomes. (4) Time of autopolyploidization of the ancestor of S. spontaneum. The plot was based on our data and analyses plus part of the results from a previous paper15.
Given that the Miscanthus subgenomes diverged from each other at a similar time as each of them diverged from Saccharum, which type of polyploidization the Miscanthus genome has undergone remains unclear. Previous studies suggested that the subgenomes of allopolyploids tend to exhibit subgenome dominance after genome rediploidization13,14,37,38,39. To determine if such dominance exists in Miscanthus and, if so, which subgenome predominated over the other, we first analysed gene losses in the synteny blocks between the two Miscanthus subgenomes through comparisons with their syntenic blocks in Sorghum. The results (Supplementary Table 11) showed that the average gene loss was ~25.8%, with no significant difference in overall gene loss between the two sets of homoeologous chromosomes (that is, two subgenomes; binomial test, P = 0.11) except for MfChr14-Sbchr8-MfChr15 (18.9% versus 58.3%). Notably, MfChr8, the fused chromosome derived from the ancestral Sbchr4 and Sbchr7 chromosomes, appeared to have lost fewer genes than did MfChr7 (20.9% versus 27.1%) and MfChr13 (19.2% versus 23.5%). These observations suggest that the Miscanthus subgenomes did not experience biased gene loss.
We then compared the gene expression levels of the syntelogues between the two subgenomes of Miscanthus (Supplementary Note 2) and found that there was no significant difference in the average expression level between genes in any two homoeologous chromosomes in any of the sampled tissues (Supplementary Fig. 9), including the fused chromosome MfChr8 and its non-fused chromosome homoeologues MfChr7 or MfChr13. These observations, together with unbiased gene losses from the two subgenomes suggest a lack of substantial global subgenome dominance in Miscanthus.
On the basis of previous reports, our own findings and the assumption that the synonymous substitution rate for coding genes is 6.5 × 10−9 synonymous substitutions yr−1 for grasses12,15,36,40, we were able to deduce the evolutionary history of the Saccharinae clade (Fig. 3b). The sorghum lineage diverged from the common ancestor of Miscanthus and Saccharum between 5.5 and 6.1 Ma, which then diverged from each other between 4.3 and 4.6 Ma. The ancestor of Saccharum then maintained a diploid state (2n = 20) for ~3.0 Myr, during which the ancestor of S. spontaneum underwent genome rearrangement and chromosome number reduction that resulted in a diploid state of 2n = 16. Approximately 1.0 Ma, the ancestor of S. spontaneum experienced the first round of autopolyploidization. Further genome rearrangement and a second round of WGD occurred sequentially and shortly after the first WGD. All these genomic changes followed by another slight genome rearrangement led to the speciation of S. spontaneum. Alternatively, two rounds of WGD occurred in the last 4 Myr and presumably gave rise to the basic chromosomes specific to the modern S. officinarum. In contrast, the ancestor of Miscanthus was most likely to experience an autopolyploidization event shortly after its divergence from the ancestor of Saccharum. When the tetraploid Miscanthus ancestor underwent the rediploidization process, chromosome fusion and gene losses occurred, contributing to further evolution towards the present forms of the Miscanthus genome.
Cellulose synthases in Miscanthus
Miscanthus, which has relatively high cellulose/hemicellulose content, is promising for use as a bioenergy feedstock3,4,6,11. To explore the molecular basis underlying this valuable trait, we identified the members of the CesA/Csl gene superfamily (except CslA/CslC; Methods) critical for cellulose/hemicellulose synthesis16,18,19,20 in the three lineages of Saccharinae. There are 35 CesA/Csl genes in S. bicolor (Supplementary Data 2), 77 in M. floridulus (Supplementary Data 3) and 109 in S. spontaneum (Supplementary Data 4). Phylogenetic analysis of the identified CesA/Csl proteins from the three Saccharinae species and rice revealed six groups, corresponding to six subfamilies of the rice CesA/Csl proteins (Supplementary Fig. 10). In light of these results, a phylogenetic tree of the CesA subfamily was then constructed and ten groups were identified with CesA3/CesA5 belonging to a same group and CesAX forming a new group not present in rice (Fig. 4a and Supplementary Note 3). On the basis of the synteny analysis and genomic coordinates, the CesA/Csl genes generally followed a 1:2 ratio in each single synteny block in S. bicolor versus two corresponding synteny blocks in M. floridulus, with a few exceptions. For example, there were five CslH genes on MfChr11 and only two CslH genes on MfChr12 syntenic to three tandem CslH genes on SbChr6 (Fig. 4b), suggestive of gene duplication and gene family expansion of the CslH subfamily in M. floridulus.

a, Maximum likelihood phylogenetic tree of CesA genes from rice, sorghum, M. floridulus and S. spontaneum. Different colour ranges correspond to different CesA groups. Numbers at tree nodes represent bootstrap support values (1,000 replications). b, The syntenic relationship of CslH genes on Sbchr6, MfChr11, MfChr12 and Ss5ABCD. Grey lines indicate the homoeologous genes between any two syntenic regions. Note that there are 3, 5, 1, 3, 4, 2 and 2 homoeologues on the syntenic regions from Sbchr6, MfChr11, MfChr12, Ss5A, Ss5B, Ss5C and Ss5D, respectively. c, Heatmap of the expression level of CesA/Csl genes in different tissues (root, growing stem, mature leaf and inflorescence) of M. floridulus. The expression levels were shown as log2(TPM + 0.5). Only genes with average TPM > 1 across all tissues were shown as indicated in Supplementary Data 3.
We then obtained the gene expression data of CesA/Csl for different tissues of mature M. floridulus plants from the RNA sequencing (RNA-seq) data (Supplementary Note 2). In general, the members of all CesA subfamilies except CesA10 and CesA11, including CesA1/3/5/6/8 which are expressed predominantly in primary cell walls and CesA4/7/9 which are responsible mainly for cellulose synthesis in secondary cell walls, were expressed at the highest level in growing stems of all collected tissues (Fig. 4c). With respect to Csls, one or several members of the CslD, CslE, CslF and CslH subfamilies were also highly expressed in growing stems and leaves (Fig. 4c), suggesting that active hemicellulose synthesis is occurring in these tissues. According to the histogram of expression level of all expressed genes (average transcripts per million mapped reads (TPM) > 1) in the stems (Supplementary Fig. 11), most expressed CesAs (11 of 17 genes) can be ranked among the top most expressed genes, suggestive of highly active cellulose synthesis; the same was true for several Csls in the stems (one CslD and two CslFs) and leaves (one CslH) (Fig. 4c). Taken together, these observations hint that Miscanthus had a strong ability for cellulose/hemicellulose synthesis, which is in agreement with its promising use as a bioenergy feedstock.
Population structure and diversity of the Miscanthus genus
To better understand the genetic diversity and population structure of the genus Miscanthus, we resequenced 74 diploid and tetraploid accessions distributed across China and one triploid Miscanthus × giganteus. These accessions include plants from M. floridulus, M. sinensis, M. lutarioriparius and M. sacchariflorus, as well as interspecific hybrids between them (Supplementary Table 12). After read mapping (Supplementary Data 5), variant calling and strict filtering (Supplementary Note 4), we identified 6.20 million high-confidence variants, which included 5,985,647 SNPs, 92,900 insertions and 122,070 deletions, with an average of 2.54 variants per kb (Fig. 1).
Principal component analysis (PCA) revealed three main clusters centred on M. floridulus, M. sinensis and M. sacchariflorus, encompassing most species, while the outliers consisted primarily of hybrids, M. × giganteus and some accessions of M. sinensis and M. lutarioriparius (Fig. 5a). The PCA results were supported by the phylogenetic tree constructed based on the SNP data (Fig. 5b), which showed ambiguity in the distinction between M. lutarioriparius and M. sacchariflorus, regardless of their differences in ploidy. Furthermore, M. × giganteus and other hybrids clustered very close to M. sinensis and M. sacchariflorus, which is consistent with the results of admixture-based analysis (Fig. 5c and Supplementary Fig. 12; for other K-values, see Supplementary Fig. 13). Notably, several accessions from M. sinensis and one accession from M. lutarioriparius did not cluster within their labelled species, although they were observed to be morphologically similar with their respective labelled species. Taken together, these observations suggest that most hybrids were probably derived from M. sinensis and M. sacchariflorus, whereas M. lutarioriparius is genetically similar to M. sacchariflorus. In addition, the fixation index (FST) calculation of the different species showed that M. sacchariflorus and M. lutarioriparius were closely related, while M. floridulus was more distant from either of the two species (Supplementary Table 13). This increased phylogenetic distance from other accessions may be related to our observations of higher genetic diversity (higher π value) among the accessions of M. floridulus compared to other species (Supplementary Fig. 14), suggesting that the levels of diversity are generally low in the other three species, especially in M. sacchariflorus and M. lutarioriparius, although accessions of these two species were acquired from geographically distant locations (Supplementary Table 12).

a, Principal components (PCs) of accession variation. The percentage numbers in the brackets indicates the proportions of the two PCs. b, Bootstrapped tree of 75 Miscanthus accessions based on genetic distance. The tetraploid was labelled by the tail of the accession ID. c, ADMIXTURE plot for Miscanthus, showing the distribution of K = 3 genetic clusters with the smallest cross-validation error. Mg, M. × giganteus.
Discussion
After their divergence from the sorghum lineage, both Miscanthus and Saccharum underwent one or more rounds of independent WGD. Previous studies based on sequences from next-generation sequencing (NGS) of Miscanthus and expressed sequence tags (ESTs) sequences for S. officinarum suggested that an ancestor of these two genera also experienced a WGD before their evolutionary bifurcation. However, by identifying the paralogues in a recently released S. spontaneum reference genome and by plotting the distributions of their Ks values, we found that the Ks peak (0.012) of S. spontaneum was different from that of S. officinarum previously reported (0.04, on the basis of the So1-So2 paralogues). By using the high-quality reference genomes of S. spontaneum15 and M. floridulus, we found it implausible that S. spontaneum and Miscanthus shared a common tetraploid ancestor. Moreover, the difference in basic chromosome number between S. officinarum and S. spontaneum suggests that a very long time was needed between the divergence of these species and their ancestral divergence from Miscanthus; otherwise, there would not be high conservation of morphological and physiological traits of S. officinarum and S. spontaneum.
Subgenome dominance has often been reported in the genomes of allopolyploids especially in allopalaeopolyploids14,37,38,39. However, some recent allopolyploids, such as Brassica napus41, Capsella bursa-pastoris42 and Triticum aestivum43, display indistinguishable or slight subgenome dominance. In M. floridulus, only negligible subgenome dominance was detected, including insignificant differences in the ratio of gene loss between subgenomes and unbiased expression of syntelogues, both of which can make it difficult to draw a convincing inference of the nature of its polyploidy. On the basis of our present analyses, it is most likely that the ancestor Miscanthus underwent autopolyploidization. However, we can also suggest that the ancestor of Miscanthus may have experienced allopolyploidization at ~3.8–4.1 Ma or long after that time but the differentiation between the two subgenomes in M. floridulus, as observed in other allopolyploids41,42,43, appears to be undetectable due to the relatively short timeframe.
CesA/Csl genes play central roles in cellulose/hemicellulose synthesis, the speed and ability of which are positively correlated with their expression19,44,45. We measured their expression in different tissues of Miscanthus and found that, among all the expressed genes, most CesAs and a subset of Csls showed extremely high expression in growing stems, which are a major source of biomass in Miscanthus46. Since WGD led to the duplication of most CesA genes and since each of the duplicated CesA genes was still highly expressed, the combined expression level of CesAs from the same subfamily and for the same function would be much higher than that of single CesA, resulting in increased synthesis of cellulose, as evidenced by the final content of cellulose in Miscanthus. Nevertheless, to reveal the relationship of the expression and number of CesA/Csl with the total cellulose/hemicellulose content, more well-designed experiments combining genetics, comparative genomics and transcriptomics are needed because of their importance in biofuel production.
By resequencing the 75 accessions from the genus Miscanthus, we provided a valuable genetic resource for Miscanthus breeding and improvement. Through the PCA, phylogenetic and structure analyses, this study also established that M. sacchariflorus and M. lutarioriparius are genomically indistinguishable from each other, validating the recent taxonomic evaluation and molecular results favouring the subspecies status of M. lutarioriparius belonging to M. sacchariflorus47. Moreover, there were some disagreements between morphology-based classification and phylogenetic and structure analysis-based classification of several accessions from M. sinensis and M. lutarioriparius, indicating that there was some hybridization or introgression among them, similar with the introgression among different subpopulations of another important bioenergy crop switchgrass48. These findings highlight the broad ability of different Miscanthus accessions to hybridize and further suggest that the boundaries between species may be less clear than previously thought. Additionally, crosses between Miscanthus and any member of the Saccharinae are also feasible and the ‘miscanes’ hybrids of Saccharum and Miscanthus have been used for sugarcane improvement49. With the completion of the reference genomes of three of the most economically important members of the Saccharinae, together with new tools for molecular breeding and transgenic research, we can envision the generation of a new hybrid that carries all of the desirable traits for bioenergy production from each species, such as high cellulose content and cold resistance from Miscanthus, drought tolerance from sorghum and disease resistance from sugarcane3,4,6.
Methods
Plant materials
The M. floridulus accession (‘MF70’, originally from Hunan Province in China) sequenced for genome assembly and annotations was grown in the field in Shandong Province, China. Accessions of different Miscanthus species and hybrids were also grown at the same location. Detailed information about them is listed in Supplementary Table 12.
Genome sequencing
For PacBio single-molecule real-time (SMRT) sequencing, at least 10 μg of sheared DNA was used to construct 20-kb-insert-size libraries in accordance with the PacBio protocol. The libraries were then sequenced on a PacBio Sequel instrument. For the polishing of genome assembly and whole-genome resequencing using Illumina NGS, genomic DNA was extracted from young leaf tissue of the different accessions. Afterwards, 500-bp paired-end libraries were constructed using a NEBNext Ultra DNA Library Prep Kit and subsequently sequenced with the Illumina HiSeq 2500 platform.
10x Genomics library construction and sequencing
DNA sample was prepared, indexed and barcoded using a GemCode instrument (10x Genomics). Briefly, ~0.7 ng of 50-kb-long input DNA was used in GEM PCR together with 16-bp-long barcodes introduced in droplets. The droplets were then fractured and the intermediate DNA was purified and sheared to 500 bp for library construction. Libraries were subsequently sequenced via the Illumina HiSeq X Ten platform.
Hi-C library construction and sequencing
The construction and sequencing of four Hi-C libraries was performed by the Annoroad Gene Technology Company. Briefly, young leaves of M. floridulus (accession ‘MF70’) were fixed with formaldehyde and then lysed, after which the cross-linked DNA was digested with MboI enzymes overnight. The enzymes were inactivated and cohesive ends were filled in by adding biotin-labelled dCTP. After proximity ligation in blunt-end ligation buffer, the cross-linking was reversed and DNA was purified for Hi-C library construction. The final library was sequenced on an Illumina HiSeq 2500 platform in 150-bp paired-end mode.
Bionano optical genome maps construction
Bionano optical maps were generated using a Saphyr Genome Imaging Instrument in conjunction with DLE1 non-nicking enzyme (CTTAAG). Extracted high-molecular-weight DNA was nicked and labelled using the SaphyrPrep Kit, and the labelled DNA was then loaded for imaging into the Saphyr system (Bionano Genomics). Imaged molecules were subsequently assembled using Bionano Solve (v.3.3) and the Bionano alignment images of the assembly were visualized with Bionano Access (v.1.5.2, https://bionanogenomics.com/support/software-downloads/).
Genome survey
The short reads used for polishing the genome assembly were quality filtered, after which the genome size, repeat sequence ratio and genome heterozygous rate were calculated using the k-mer method via Jellyfish50. The k-mer value used for the genome survey was 17.
Genome assembly
Before assembly, PacBio reads that were longer than 7,000 bp were used as seed reads to correct read errors and generate consensus sequences; this was accomplished with the ‘daligner’ function in the main script of the FALCON assembler. After error correction produced highly accurate (up to 99.999%) consensus sequences, FALCON identified the overlaps between all the pairs of the pre-assembled error-corrected reads. The read overlaps were used to construct a directed string graph by following Myers’ algorithm. After the initial assembly, FALCON-Unzip was used to produce primary contigs (p-contigs) and haplotigs31. After phased polishing, FALCON-Phase32 then combined the primary contigs and haplotigs with the Hi-C data to extend phasing between unzipped haplotig blocks and to generate two phased assemblies (Supplementary Note 1). One of the phased assemblies was further polished with PacBio reads by the consensus-calling algorithm Quiver2 (ref. 31). The Illumina reads were also used to correct the contigs via Pilon3 (ref. 51). Heterozygosity was then removed from the error-corrected contigs using purge_haplotigs together with the Illumina clean reads. Afterwards, with the default settings, BWA software52 was used to align the 10x Genomics data to the assembly and fill-in gaps. Scaffolding was performed by fragScaff with the barcoded sequencing reads and the three-stages option as follows: stage1 “-m 3000 -q 30”; stage2 “-C 5”; stage3 “-j 1 -u 3” (ref. 53). These processes ultimately yielded the final draft of the genome assembly. For Hi-C-assisted genome assembly, the Hi-C clean data were aligned to the preceding assembly using HiC-Pro54 and Bowtie2 (ref. 55) software. Only read pairs with both reads in the pair-aligned contigs were considered valid gene pairs for scaffolding. By the use of linkage information and restriction enzyme sites, a string graph was used to construct the scaffold graph with LACHESIS4 (ref. 56).
Linkage map construction
The mapping population comprised 116 F1 progeny generated from an interspecific cross between M. floridulus (MF70, 2n = 2x = 38) and M. sacchariflorus (MS62, 2n = 2x = 38). Genomic DNA was extracted from leaf tissue to construct restriction site-associated DNA sequencing (RAD-seq) libraries, which were then sequenced following standard protocols57. The generated reads were subsequently mapped to our genome assembly by BWA-MEM52 and variants were called by freebayes58 and then filtered by SnpSift59. A total of 6,472 SNPs were ultimately identified, including 5,827 parental testcross markers that were heterozygous only in M. floridulus (lm × ll) and 645 biparental markers that were heterozygous in both parents (hk × hk) according to cross-pollinator (CP) population type. A linkage map was constructed with JoinMap5 using the multipoint maximum likelihood mapping method60. A minimum independence logarithm of odds (LOD) score of 10 and a maximum recombination frequency of 0.4 were used to define linkage groups. In total, 3,799 SNPs clustered into 19 linkage groups covering 2,371.76 cM, with an average intermarker spacing of 0.65 cM. To validate the chromosome-scale assembly by Hi-C, the comparison of the genetic location and corresponding physical position of all SNP markers of the genetic map were assessed. In total, 92.74% of mapped markers were detected to be located at the same M. floridulus assembled chromosome. Of the markers, 5.35% were mapped equally well to homoeologous locations on the assembly (Supplementary Fig. 3).
Repeat annotation
Tandem repeats were extracted using TRF (http://tandem.bu.edu/trf/trf.html)61 by ab initio prediction. For TE annotations, a combined strategy based on de novo structural identification and on a homology-based alignment search for whole-genome repeats was applied. A de novo repetitive elements database was built by combining the results of LTR_FINDER (http://tlife.fudan.edu.cn/tlife/ltr_finder/)62 and RepeatScout63 (http://www.repeatmasker.org/), with those of RepeatModeler (http://www.repeatmasker.org/RepeatModeler), set to default parameters. All repeat sequences with lengths >100 bp and <5% ‘N’ gaps constituted the raw TE library. Repeat regions were extracted with a homology-based repeat prediction using the Repbase (http://www.girinst.org/repbase) database using RepeatMasker (http://www.repeatmasker.org/) software and its in-house scripts (RepeatProteinMask) with default parameters. A custom non-redundant library that was generated by a combination of Repbase-based search and a de novo TE library, which was processed by uclust, was subjected to RepeatMasker for final repeat identification.
Specifically, to investigate LTR-RTs in detail, highly confident intact LTR-RTs were identified using the LTR_retriever64 pipeline, which integrated the results from LTR_FINDER62 and LTRharvest65 and efficiently removed false positives. The insertion time was then estimated as T = K/2μ (where K is the divergence distance between the 5′ LTR and 3′ LTR of intact LTR-RTs and where μ is 1.38 × 10−8 for grasses66).
Gene model prediction and functional annotations
A combination of homology-based, ab initio and transcriptome-based gene prediction methods was used for protein-coding gene annotation. The protein sequences of six species (Brachypodium distachyon, O. sativa, S. bicolor, Setaria italic, S. spontaneum and Z. mays) were downloaded from the Ensembl plants database. The sequences were aligned to the genome using TBLASTN (v.2.2.26; E value ≤1 × 10–5), after which the matching proteins were aligned to homoeologous genome sequences for accurate spliced alignments via GeneWise (v.2.4.1)67, which was used to predict accurate gene structure within each protein region. For ab initio gene prediction, AUGUSTUS (v.3.2.3)68, GeneID (v.1.4)69, GeneScan (v.1.0)70, GlimmerHMM (v.3.04)71 and SNAP (https://github.com/KorfLab/SNAP) were used in our automated gene prediction pipeline. For genome annotation, RNA was extracted from five different tissues (root, leaf, stem, inflorescence and bud tissues) and mixed together equally for library construction. A de novo transcriptome assembly was then generated with Trinity (v.2.1.1)72, used for genome annotation and to optimize gene predictions. RNA-seq reads from different tissues were aligned to the genome using HISAT (v.2.0.4)73 with default parameters to identify exon regions and splice positions. The alignment results were then used as input by StringTie (v.1.3.3)73 with default parameters for genome-based transcript assembly. Finally, the non-redundant reference gene set was generated by merging genes predicted by the three methods above with EVidenceModeler (EVM; v.1.1.1)74 using PASA terminal exon support and including masked TEs as input for gene prediction.
Gene functions were assigned according to the best matches with Swiss-Prot protein sequences identified using BLASTP (with a threshold of E value ≤1 × 10–5). The motifs and domains were annotated using InterProScan (v.5.31)75 by searching against publicly available databases, including ProDom, PRINTS, Pfam, SMRT, PANTHER and PROSITE. The GO IDs for each gene were assigned according to the corresponding InterPro entry.
Further, transfer RNAs were predicted using the program tRNAscan-SE (http://lowelab.ucsc.edu/tRNAscan-SE/)76. Because ribosomal RNAs are highly conserved, we predicted rRNA sequences using BLAST with rRNAs from S. bicolor used as references. Other non-coding RNAs, including microRNAs and small nuclear RNAs, were identified by searching against the Rfam77 database via infernal software (http://infernal.janelia.org/)78 with the default parameters.
Genome assembly assessment
Both BUSCO34 (http://busco.ezlab.org/) analysis and the CEGMA33 (http://korflab.ucdavis.edu/Datasets/cegma/) pipeline were used to assess the completeness of the genome assembly and annotations. Further, the LAI, which was implemented in the LTR_retriever package, was also applied to assess the genome assembly quality35.
Gene family analysis
For gene family analysis, the online web server OrthoVenn2 (ref. 79) was used. Maize, sorghum, rice, S. spontaneum and M. floridulus protein sequences were used as inputs for online orthogroup clustering and annotations and GO enrichment.
Genome synteny
Protein sequences from M. floridulus, S. bicolor and S. spontaneum were blasted against themselves using BLASTP (E value <1 × 10–5) to identify intergenome synteny blocks. The BLAST results were subjected to c-score filtering (c-score = 0.99) and the Python version JCVI (https://github.com/tanghaibao/jcvi) of MCScan was used to generate dot plots. MCScanX80 was used to determine synteny blocks with default parameters and at least five genes were required to define a synteny block. To identify intragenome synteny blocks, protein sequences from two homoeologous chromosomes were extracted to query against each other and after c-score filtering by JCVI, MCScanX was used to determine synteny blocks with default parameters. The Ks value was calculated via KaKs_Calculator81 for each gene pair in the aligned blocks. The distributions of all Ks values were plotted via the R software and ggplot2 package82. The peak Ks values were converted to divergence time12 according to the formula T = Ks/2λ (T, time; λ, average substitution rate) by using an average substitution rate of 6.5 × 10−9 for grasses40 to infer speciation or WGD events that occurred during the evolutionary history.
Subgenome analysis
To analyse the gene loss of the two M. floridulus subgenomes, protein sequences of homoeologous chromosomes from M. floridulus and their corresponding homoeologous chromosome in S. bicolor were queried via BLAST against each other and synteny blocks were identified by MCScanX. Blocks shared by the three homoeologous chromosomes were then identified and syntelogues between any two of them were extracted for statistical analysis. For syntelogue expression analysis, RNA was extracted from leaf, growing stem, root and inflorescence tissues of mature M. floridulus plants and RNA-seq analysis was performed on these four tissues. There were three replicates for each tissue. The RNA-seq reads were quality filtered by fastp83 and then mapped to M. floridulus complementary DNA sequences by Salmon84. Meanwhile, TPM values were also calculated by Salmon. The expression of syntelogues across different tissues in each of homoeologous chromosome were retrieved and plotted via the R software.
Analysis of CesA/Csl genes
All genes from the three species S. bicolor, S. spontaneum and M. floridulus were first annotated using InterProScan. The CesA/Csl genes were then retrieved from the files generated from the Interproscan-based functional annotation with Pfam accession ID ‘PF03552’. Because there were only five and nine CslA/CslC genes (described by two Pfams ‘PF00535’ and ‘PF13632’) identified in S. bicolor and M. floridulus and these genes distantly diverged from other CesA/Csl genes16, those genes were excluded from the downstream analysis. The gene coordinates were retrieved from the GFF files of the three genomes and the expression level of CesA/Csl genes in M. floridulus were obtained from the RNA-seq results of the previous subgenome analysis. The expression of CesA/Csl genes were retrieved and heatmap was drawn using the pheatmap package (https://CRAN.R-project.org/package=pheatmap) of R software with values of log2(TPM + 0.5).
Resequencing and diversity analysis
DNA from different accessions of multiple Miscanthus species and hybrids was extracted for library construction and resequencing. The paired-end reads were quality filtered by fastp83 and subsequently mapped to the M. floridulus genome by BWA-MEM52. Via Picard, mapped reads were sorted and duplicated reads were marked. Uniquely mapped reads were then extracted from the sorted and duplicate-marked bam file with Sambamba85 using the parameter “-F ′′mapping_quality >= 30 and not (unmapped or secondary_alignment) and not ([XA] != null or [SA] != null)′′”. Then, SNPs and insertions/deletions (InDels) were called by freebayes58 using parameters “-standard-filters –min-coverage 4 –use-best-n-alleles 4 --genotype-qualities” and the polyploidy level of the different accessions were specified with “-A Miscanthus_freebayes_cnv_map.txt”. The variants were then filtered by a custom script to change low quality data (GQ < 30 or DP < 4 or DP > 50) to null and further filtered by SnpSift59 with the parameters “QUAL > 30 & NS >= 63 & (QUAL / AO > 10) & SAF > 0 & SAR > 0 & RPR > 1 & RPL > 1”. Afterwards, the multi-allelic genotype of polyploids were filtered and converted to bi-allelic genotype by our custom script and the remaining 6,200,617 variants were used in downstream analysis. PCA of the filtered variants was performed using the SMARTPCA program from EIGENSOFT (v.6.1.4)86. For phylogenetic analysis, SNPhylo87 software was used. Before tree construction, we filtered and pruned the SNPs with minor allele frequency (MAF) < 0.1, missing rate >0.1 and linkage disequilibrium (LD) threshold = 0.2. A total of 15,857 SNPs were ultimately selected for the SNPhylo pipeline. iTOL (v.5)88 was used to visualize the trees. For population structure analysis, the optimal ancestral population structure was estimated for the same variant set by using ADMIXTURE89 with ancestral population sizes K = 1–7 and choosing the population size with the smallest cross-validation error. On the basis of the K value, admixture analyses were then performed to infer population structure. Fixation index (FST) and π value (indicating genetic diversity) were calculated by VCFtools90.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The genome assembly and gene annotation have been deposited in the NCBI database under BioProject number PRJNA598249 and BioSample number SAMN13702268. Source data are provided with this paper.
Code availability
The custom codes used in this study are deposited in GitHub (https://github.com/sdysfhs319/Miscanthus_genome.git).
Change history
28 June 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41477-021-00968-0
28 June 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41477-021-00972-4
References
Paterson, A. H. Genomics of the Saccharinae (Springer, 2012).
Przybysz, K. et al. Production of sugar feedstocks for fermentation processes from selected fast growing grasses. Energies 12, 3129 (2019).
van der Weijde, T. et al. The potential of C4 grasses for cellulosic biofuel production. Front. Plant. Sci. 4, 107 (2013).
de Siqueira Ferreira, S., Nishiyama, M. Y., Paterson, A. H. & Souza, G. M. Biofuel and energy crops: high-yield Saccharinae take center stage in the post-genomics era. Genome Biol. 14, 210 (2013).
Waclawovsky, A. J., Sato, P. M., Lembke, C. G., Moore, P. H. & Souza, G. M. Sugarcane for bioenergy production: an assessment of yield and regulation of sucrose content. Plant Biotechnol. J. 8, 263–276 (2010).
Heaton, E. A., Dohleman, F. G. & Long, S. P. Meeting US biofuel goals with less land: the potential of Miscanthus. Glob. Change Biol. 14, 2000–2014 (2008).
Arnoult, S. & Brancourt-Hulmel, M. A review on Miscanthus biomass production and composition for bioenergy use: genotypic and environmental variability and implications for breeding. Bioenergy Res. 8, 502–526 (2014).
Rooney, W. L., Blumenthal, J., Bean, B. & Mullet, J. E. Designing sorghum as a dedicated bioenergy feedstock. Biofuels Bioprod. Biorefin. 1, 147–157 (2007).
Pimentel, L. D., Batista, V. A. P., Barros, A. F. D., Teófilo, R. F. & Dias, L. A. D. S. Chemical and bioenergetic characterization of sorghum agronomic groups. Pesqui. Agropecu. Trop. 47, 424–431 (2017).
Hoang, N. V. et al. High-throughput profiling of the fiber and sugar composition of sugarcane biomass. Bioenergy Res. 10, 400–416 (2016).
Lee, W.-C. & Kuan, W.-C. Miscanthus as cellulosic biomass for bioethanol production. Biotechnol. J. 10, 840–854 (2015).
Kim, C. et al. Comparative analysis of Miscanthus and Saccharum reveals a shared whole-genome duplication but different evolutionary fates. Plant Cell 26, 2420–2429 (2014).
Zhang, K., Wang, X. & Cheng, F. Plant polyploidy: origin, evolution, and its influence on crop domestication. Hortic. Plant J. 5, 231–239 (2019).
Cheng, F. et al. Gene retention, fractionation and subgenome differences in polyploid plants. Nat. Plants 4, 258 (2018).
Zhang, J. et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. 50, 1565–1573 (2018).
Little, A. et al. Revised phylogeny of the cellulose synthase gene superfamily: insights into cell wall evolution. Plant Physiol. 177, 1124–1141 (2018).
Yin, Y., Huang, J. & Xu, Y. The cellulose synthase superfamily in fully sequenced plants and algae. BMC Plant Biol. 9, 99 (2009).
Carpita, N. C. Update on mechanisms of plant cell wall biosynthesis: how plants make cellulose and other (1→4)-β-d-glycans. Plant Physiol. 155, 171–184 (2011).
Polko, J. K. & Kieber, J. J. The regulation of cellulose biosynthesis in plants. Plant Cell 31, 282–296 (2019).
Sarkar, P., Bosneaga, E. & Auer, M. Plant cell walls throughout evolution: towards a molecular understanding of their design principles. J. Exp. Bot. 60, 3615–3635 (2009).
Tanaka, K. et al. Three distinct rice cellulose synthase catalytic subunit genes required for cellulose synthesis in the secondary wall. Plant Physiol. 133, 73–83 (2003).
Wang, L. et al. Expression profiling and integrative analysis of the CESA/CSL superfamily in rice. BMC Plant Biol. 10, 282 (2010).
Vega-Sanchez, M. E. et al. Loss of cellulose synthase-like F6 function affects mixed-linkage glucan deposition, cell wall mechanical properties, and defense responses in vegetative tissues of rice. Plant Physiol. 159, 56–69 (2012).
Ermawar, R. A. et al. Distribution, structure and biosynthetic gene families of (1,3;1,4)-β-glucan in Sorghum bicolor. J. Integr. Plant Biol. 57, 429–445 (2015).
Jobling, S. A. Membrane pore architecture of the CslF6 protein controls (1-3,1-4)-β-glucan structure. Sci. Adv. 1, e1500069 (2015).
Hu, R. et al. Transcriptome analysis of genes involved in secondary cell wall biosynthesis in developing internodes of Miscanthus lutarioriparius. Sci. Rep. 7, 9034 (2017).
Zeng, X. et al. Differential expression patterns reveal the roles of cellulose synthase genes (CesAs) in primary and secondary cell wall biosynthesis in Miscanthus × giganteus. Ind. Crops Prod. 145, 112129 (2020).
Deschamps, S. et al. A chromosome-scale assembly of the sorghum genome using nanopore sequencing and optical mapping. Nat. Commun. 9, 4844 (2018).
Paterson, A. H. et al. The Sorghum bicolor genome and the diversification of grasses. Nature 457, 551–556 (2009).
Guo-te, D. et al. Estimation of genome size of Miscanthus floridulus. J. Plant Genet. Res. 14, 339–341 (2013).
Chin, C.-S. et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050 (2016).
Kronenberg, Z. N. et al. Extended haplotype phasing of de novo genome assemblies with FALCON-Phase. Preprint at bioRxiv https://doi.org/10.1101/327064 (2019).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 46, e126 (2018).
Swaminathan, K. et al. A framework genetic map for Miscanthus sinensis from RNAseq-based markers shows recent tetraploidy. BMC Genom. 13, 142 (2012).
Edger, P. P. et al. Subgenome dominance in an interspecific hybrid, synthetic allopolyploid, and a 140-year-old naturally established neo-allopolyploid monkeyflower. Plant Cell 29, 2150–2167 (2017).
Schnable, J. C., Springer, N. M. & Freeling, M. Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc. Natl Acad. Sci. USA 108, 4069–4074 (2011).
Zhao, M., Zhang, B., Lisch, D. & Ma, J. Patterns and consequences of subgenome differentiation provide insights into the nature of paleopolyploidy in plants. Plant Cell 29, 2974–2994 (2017).
Gaut, B. S., Morton, B. R., McCaig, B. C. & Clegg, M. T. Substitution rate comparisons between grasses and palms: synonymous rate differences at the nuclear gene Adh parallel rate differences at the plastid gene rbcL. Proc. Natl Acad. Sci. USA 93, 10274–10279 (1996).
Chalhoub, B. et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345, 950–953 (2014).
Douglas, G. M. et al. Hybrid origins and the earliest stages of diploidization in the highly successful recent polyploid Capsella bursa-pastoris. Proc. Natl Acad. Sci. USA 112, 2806–2811 (2015).
Consortium, I. W. G. S. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 345, 1251788 (2014).
Xie, L., Yang, C. & Wang, X. Brassinosteroids can regulate cellulose biosynthesis by controlling the expression of CESA genes in Arabidopsis. J. Exp. Bot. 62, 4495–4506 (2011).
Kim, W. C. et al. MYB46 directly regulates the gene expression of secondary wall-associated cellulose synthases in Arabidopsis. Plant J. 73, 26–36 (2013).
Bergs, M. et al. Comparing chemical composition and lignin structure of Miscanthus × giganteus and Miscanthus nagara harvested in autumn and spring and separated into stems and leaves. RSC Adv. 10, 10740–10751 (2020).
Clark, L. V. et al. Population structure of Miscanthus sacchariflorus reveals two major polyploidization events, tetraploid-mediated unidirectional introgression from diploid M. sinensis, and diversity centred around the Yellow Sea. Ann. Bot. 124, 731–748 (2019).
Lovell, J. T. et al. Genomic mechanisms of climate adaptation in polyploid bioenergy switchgrass. Nature 590, 438–444 (2021).
Chen, Y. & Lo, C. Disease resistance and sugar content in Saccharum–Miscanthus hybrids. Taiwan Sugar 36, 9–12 (1989).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://arxiv.org/pdf/1303.3997.pdf (2013).
Mostovoy, Y. et al. A hybrid approach for de novo human genome sequence assembly and phasing. Nat. Methods 13, 587 (2016).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Bio. 16, 259 (2015).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357 (2012).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119 (2013).
DaCosta, J. M. & Sorenson, M. D. Amplification biases and consistent recovery of loci in a double-digest RAD-seq protocol. PLoS ONE 9, e106713 (2014).
Garrison, E. & Marth, G. Haplotype-based variant detection from short-read sequencing. Preprint at https://export.arxiv.org/abs/1207.3907v2 (2012).
Ruden, D. M. et al. Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program, SnpSift. Front. Genet. 3, 35 (2012).
Van Ooijen, J. Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genet. Res. 93, 343–349 (2011).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Ou, S. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422 (2018).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 9, 18 (2008).
Ma, J., Devos, K. M. & Bennetzen, J. L. Analyses of LTR-retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Res. 14, 860–869 (2004).
Birney, E., Clamp, M. & Durbin, R. GeneWise and genomewise. Genome Res. 14, 988–995 (2004).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Blanco, E. & Abril, J. F. Computational gene annotation in new genome assemblies using GeneID. Methods Mol. Biol. 537, 243–261 (2009).
Lynn, A. M. et al. An automated annotation tool for genomic DNA sequences using GeneScan and BLAST. J. Genet. 80, 9–16 (2001).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Grabherr, M. G. et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 29, 644 (2011).
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667 (2016).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Zdobnov, E. M. & Apweiler, R. InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2001).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, D121–D124 (2005).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Xu, L. et al. OrthoVenn2: a web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 47, w52–w58 (2019).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
Zhang, Z. et al. KaKs_Calculator: calculating Ka and Ks through model selection and model averaging. Genomics Proteomics Bioinformatics 4, 259–263 (2006).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag, 2016).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417 (2017).
Tarasov, A., Vilella, A. J., Cuppen, E., Nijman, I. J. & Prins, P. Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034 (2015).
Patterson, N., Price, A. L. & Reich, D. Population structure and eigenanalysis. PLoS Genet. 2, e190 (2006).
Lee, T.-H., Guo, H., Wang, X., Kim, C. & Paterson, A. H. SNPhylo: a pipeline to construct a phylogenetic tree from huge SNP data. BMC Genomics 15, 162 (2014).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245 (2016).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Acknowledgements
We thank Z. Yi from the Hunan Agricultural University for providing partial materials for RNA sequencing. This research was supported by the National Natural Science Foundation of China to C.C. (grant nos. 31871267, 31271352 and 31071471), Shandong Agricultural Elite Variety Project to C.C. (grant nos. 2017LZN028 and 2019LZGC010) and Shandong First Class Discipline Construction Project for the Universities to C.C.
Author information
Authors and Affiliations
Contributions
C.C. designed and supervised this genome project. G.Z. and C.G. conducted the experiments. P.X., S.W., S.C., Y.H., Y.W., X.H., Y.Y., T.Y., X.X., X.Y., Q.L., W.W., W.L. and C.Z. collected and maintained plant materials and mapping populations. C.G. constructed the genetic map. C.G. and G.Z. conducted comparative genomic analysis. G.Z. and S. Deng processed the raw data and assembled and annotated the genome. G.Z. and Y.Z. studied WGDs and genome evolution and G.Z. analysed CesA/Csl genes. G.Z. wrote the manuscript. X.S., Z.W., R.M., S. Dong, J.M., X.Z. and C.C. revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Plants thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–13, Tables 1–14, Notes 1–4 and references.
Supplementary Data
Supplementary Data 1–5.
Source data
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, G., Ge, C., Xu, P. et al. The reference genome of Miscanthus floridulus illuminates the evolution of Saccharinae. Nat. Plants 7, 608–618 (2021). https://doi.org/10.1038/s41477-021-00908-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41477-021-00908-y
This article is cited by
-
Differential subgenome expression underlies biomass accumulation in allotetraploid Pennisetum giganteum
BMC Biology (2023)
-
A complete gap-free diploid genome in Saccharum complex and the genomic footprints of evolution in the highly polyploid Saccharum genus
Nature Plants (2023)
-
Investigation of genetic relationships within three Miscanthus species using SNP markers identified with SLAF-seq
BMC Genomics (2022)
-
Genomic Analyses of SUT and TST Sugar Transporter Families in Low and High Sugar Accumulating Sugarcane Species (Saccharum spontaneum and Saccharum officinarum)
Tropical Plant Biology (2022)
-
Advances in Sugarcane Genomics and Genetics
Sugar Tech (2022)
