
Abstract
The differentiation of the stroma is a hallmark event during postnatal uterine development. However, the spatiotemporal changes that occur during this process and the underlying regulatory mechanisms remain elusive. Here, we comprehensively delineated the dynamic development of the neonatal uterus at single-cell resolution and characterized two distinct stromal subpopulations, inner and outer stroma. Furthermore, single-cell RNA sequencing revealed that uterine ablation of Pr-set7, the sole methyltransferase catalyzing H4K20me1, led to a reduced proportion of the inner stroma due to massive cell death, thus impeding uterine development. By combining RNA sequencing and epigenetic profiling of H4K20me1, we demonstrated that PR-SET7-H4K20me1 either directly repressed the transcription of interferon stimulated genes or indirectly restricted the interferon response via silencing endogenous retroviruses. Declined H4K20me1 level caused viral mimicry responses and ZBP1-mediated apoptosis and necroptosis in stromal cells. Collectively, our study provides insight into the epigenetic machinery governing postnatal uterine stromal development mediated by PR-SET7.
Similar content being viewed by others
Introduction
The uterus, which is derived from the Müllerian duct, serves as an essential organ for reproduction in mammals. Disturbances during uterine development exert ripple effects on uterine structure and functions in adulthood and give rise to infertility1.
In mice, the uterus is relatively rudimentary at birth (postnatal day (PND) 1), consisting of a single layer of luminal epithelium surrounded by undifferentiated mesenchyme. The emergence of endometrial glands through invagination and branching from PND5 to PND12, as well as the coordinated differentiation of the mesenchyme into endometrial stroma and myometrium, is considered two milestone events that occur during postnatal uterine development in rodents. In mice, the fundamental configuration of the uterus is established by PND152,3,4. While a variety of studies have identified many crucial genes that participate in the formation of uterine glands5,6,7,8,9, the dynamic process and underlying regulatory mechanisms of postnatal uterine stromal development remain largely elusive. A previous report proposed that a subluminal population of Misr2 (Müllerian inhibiting substance receptor 2)-positive cells functioned as progenitor cells, which further differentiated into two stromal subclusters according to single-cell RNA sequencing (scRNA-seq)10. However, the scRNA-seq analysis was performed at only one time point in this study and was incapable of providing a comprehensive landscape of stromal development.
Epigenetic machinery, including DNA methylation, histone modifications, and other modifications affecting chromatin accessibility and three-dimensional chromatin organization, plays a vital role in development and disease via controlling gene expression11. PR-SET7, an evolutionarily conserved histone methyltransferase, is the sole enzyme known to catalyze H4K20me1, which serves as the substrate for H4K20me2/312,13. In addition to its well-accepted canonical functions in cell cycle progression, DNA damage repair, and gene transcriptional regulation14,15,16,17,18,19, PR-SET7-mediated H4K20me1 was recently demonstrated to repress the intrinsic expression of endogenous retroviruses (ERVs), contributing to the maintenance of genomic stability20. Accumulating evidence has uncovered the indispensable roles of PR-SET7 and H4K20me1 in diverse physiological and pathological processes during the development and homeostasis of multiple organs and tissues21,22,23,24,25.
The pathophysiological significance of PR-SET7-mediated H4K20me1 in female reproduction has been reported. Trophoblast-specific deletion of Pr-set7 resulted in ERV derepression, viral mimicry responses, and necroptosis, which shed light on the potential etiology and pathogenesis of recurrent miscarriage20. Meanwhile, uterine PR-SET7 deficiency limited uterine epithelial population growth due to impaired DNA damage repair and severe epithelial cell apoptosis, which hampered gland formation in neonatal mice26. Nevertheless, the potential contributions of PR-SET7-mediated H4K20me1 to uterine stromal development have been neglected and remain ambiguous.
In the current study, we generate a dynamic transcriptomic atlas of uterine development in mice throughout the first two weeks after birth using scRNA-seq and describe the spatial localization, developmental trajectory, and unique characteristics of two stromal subsets, inner and outer stromal cells, in the neonatal uterus. In addition, we discover that uterine PR-SET7 deficiency disrupts the development of inner stromal cells and results in considerable cell death, leading to uterine atrophy. Utilizing a combination of bulk RNA sequencing (RNA-seq) and cleavage under targets and tagmentation (CUT&Tag) approaches, we elucidate that PR-SET7-mediated H4K20me1 epigenetically confines the aberrant activation of interferon responses and prevents ZBP1-mediated apoptosis and necroptosis in stromal cells, which sheds light on the pivotal role of PR-SET7-mediated H4K20me1 in calibrating interferon signaling to safeguard proper development of the endometrial stroma.
Results
The dynamic transcriptomic landscape of postnatal uterine development unraveled by scRNA-seq
The highly heterogeneous uterus is composed of various cell types and undergoes tremendous changes during development4,9,10. To achieve a comprehensive understanding of the dynamic process of postnatal uterine development, uterine tissues were collected on PND1, PND5, PND10 and PND15 and subjected to scRNA-seq (Fig. 1a). After quality control and batch effect correction, 13 distinct cell clusters were identified as visualized by uniform manifold approximation and projection (UMAP), including three stromal (Str) subpopulations, three epithelial (Epi) subpopulations, two myometrial (Myo) subpopulations, pericytes (Peri), mesothelial (Meso) cells, endothelial (Endo) cells, T cells and macrophages (Mac), according to well-documented marker genes (Fig. 1b–d and Supplementary Fig. 1a, b). The proportions of stromal, epithelial, and myometrial cells fluctuated wavily. Meanwhile, the percentages of pericytes, endothelial cells, and leukocytes increased gradually throughout the postnatal period (Fig. 1e), reflecting the maturation of the uterine vasculature and the establishment of a local immune microenvironment.

a Diagram illustrating the dynamic process of postnatal uterine development. b UMAP plot showing cell clusters in the neonatal uterus. c Spearman correlation of transcriptomic profiles of different cell populations. d Bubble plot indicating the expression of marker genes across different cell types. e Line chart showing the proportions of the major cell types at different time points. f–h Bar plot showing the stromal (f), epithelial (g), and myometrial (h) subset composition on PND1, PND5, PND10, and PND15. i–l UMAP visualization indicating the expression of Wnt16 (i), Htra3 (j), Foxa2 (k), and Acta2 (l) at different time points.
Stromal cells were the most abundant cell type in the neonatal uterus (Fig. 1b, e), consisting of Str_1 (which specifically expressed Wnt family member 16, Wnt16), Str_2 (which was characterized by relatively high level of HtrA serine peptidase 3, Htra3) and Str_3, a proliferative subset (Fig. 1d). The proportion of proliferative stromal cells gradually decreased, substantially dropping by PND15 (Fig. 1f), which coincided with the accomplishment of uterine development. In subsequent studies, we mainly put emphasis on Str_1 and Str_2. Both Str_1 and Str_2 already existed at birth. They seemed less differentiated since the expression levels of their marker genes were nearly undetectable at this time point. In the next two weeks, the expression levels of the marker genes of both Str_1 and Str_2 increased, and the expression pattern became more restricted (Fig. 1i, j), indicating the differentiation of stromal subpopulations.
The three epithelial subpopulations were defined as the luminal epithelial cells (Epi_1), a group of proliferative epithelial cells (Epi_2), and the glandular epithelial cells (Epi_3), based on the expression of tumor-associated calcium signal transducer 2 (Tacstd2), marker of proliferation Ki67 (Mki67) and forkhead box A2 (Foxa2), respectively (Fig. 1d). The formation of endometrial glands from PND5 to PND12 is a milestone event during uterine development3. Consistently, Foxa2-positive glandular epithelial cells appeared on PND10, and the number of these cells was significantly increased on PND15 (Fig. 1g, k).
The actin alpha 2 (Acta2)-positive uterine myometrium was subdivided into non-proliferative (Myo_1) and proliferative (Myo_2) myometrial cells, depending on Mki67 expression (Fig. 1d). The proportion of proliferative myometrial cells tended to increase from PND1 to PND10 (Fig. 1h), aligned with the progression of uterine myometrial development2. We also noticed myometrial cells with low expression level of Acta2 on PND1 before the morphological appearance of the myometrium2 (Fig. 1l), implying that the fates of these cells had already been determined at birth, and they remained in a less-differentiated state until the emergence of the myometrial layers.
Collectively, we depicted the dynamic process of uterine development after birth at single-cell resolution and demonstrated that although the uterine mesenchyme seemed homogeneous at birth, the less-differentiated stromal and myometrial cells had already undergone cell fate determination, earlier than previously thought.
Characterization of distinct stromal subpopulations in the neonatal uterus
To elucidate the localization of Str_1 and Str_2, single-cell resolution in situ hybridization on tissues (SCRINSHOT) was performed using specific probes for their respective marker genes, Wnt16 and Htra3. Wnt16-positive Str_1 was mainly distributed adjacent to the luminal epithelium, and this subpopulation was defined as the inner stromal cells (Fig. 2a). Meanwhile, Str_2, which highly expressed Htra3, was localized at the periphery of the endometrium, and these cells were defined as the outer stromal cells (Fig. 2b).

a, b SCRINSHOT analysis of Wnt16 (a) and Htra3 (b) in the uteri on PND1 and PND15. Dash lines represent the boundary between the epithelium and the stroma, as well as the stroma and the myometrium. Scale bar: 100 μm. c Visualization of RNA velocity analysis. d Heatmap showing the top-ranked regulons in inner stromal cells according to SCENIC analysis. e Violin plot displaying the expression level of Pgr in the inner and outer stroma throughout postnatal uterine development. f LacZ staining showing the expression pattern of Pgr in the uteri on PND1, PND5, PND10, and PND15. Scale bar: 100 μm. g IHC analysis of PR in the uteri on PND15. Dash lines represent the boundary between the stroma and the myometrium. Scale bar: 100 μm. h GO enrichment analysis of the highly expressed genes in the inner and outer stroma. Significance is based on over-representation analysis with clusterProfiler. i Gene expression clustering analysis and GO analysis of inner stromal cells at different time points. j, k CellChat analysis indicating the WNT (j) and BMP (k) signaling pathways among stromal cells and epithelial cells.
The inner and outer stroma displayed prominent differences in global gene expression (Supplementary Fig. 2a). To systematically decode the developmental trajectory of these stromal subclusters, we applied RNA velocity analysis and observed a potential differentiation orientation from outer stromal cells to inner stromal cells (Fig. 2c). In addition, single-cell regulatory network inference and clustering (SCENIC) analysis was performed to decipher the potential transcription factors governing the differentiation of inner stromal cells (Fig. 2d and Supplementary Fig. 2b). We found that the inner stromal cells gradually acquired progesterone receptor (PR) expression, which remained at a relatively low level in the outer stromal cells (Fig. 2e–g), suggesting the capability of inner stromal cells to better respond to progesterone in the adulthood.
To better depict the unique characteristics of the inner and outer stromal cells, genes that were highly and specifically expressed in these two subsets were selected and subjected to the gene ontology (GO) enrichment analysis. The outer stromal cells were involved in pathways related to extracellular matrix structure and collagen fibril organization (Fig. 2h), consistent with the RNA velocity result that this stromal subpopulation exhibited a less-differentiated state. On the other hand, the inner stromal cells particularly expressed genes relevant to WNT signaling, BMP signaling as well as mesenchyme and gland development (Fig. 2h), implying their potential roles in regulating the formation of uterine glands, as previously reported27. We noticed that the inner stromal cells highly expressed genes involved in chromatin remodeling and RNA processing on PND1 and PND5. On PND10, pathways related to WNT signaling and epithelial tube morphogenesis showed enrichment (Fig. 2i), synchronized with the emergence of uterine glands (Fig. 1g). Consistently, CellChat analysis revealed intimate interactions between inner stromal cells and glandular epithelial cells mediated by the WNT and BMP signaling pathways (Fig. 2j, k and Supplementary Fig. 2c). The expression of genes related to angiogenesis and leukocyte chemotaxis was significantly increased on PND10 and PND15, revealing the role of inner stromal cells in promoting these critical events (Fig. 2i and Supplementary Fig. 2d).
Overall, our scRNA-seq data provided detailed insights into the spatial localization, developmental trajectory, and unique features of different stromal subsets.
Uterine-specific deletion of Pr-set7 hampered stromal growth due to massive cell death
Our previous study focused mainly on the contribution of PR-SET7 to adenogenesis; however, we noticed that endometrial stromal growth was also impeded in the absence of PR-SET726. Therefore, in the present study, we intended to elucidate the physiological significance of PR-SET7-mediated H4K20me1 in uterine stromal development. We first assessed the spatiotemporal patterns of Pr-set7 expression and H4K20me1 modification in the neonatal uterus using in situ hybridization (ISH), immunofluorescence (IF) and western blotting (WB) (Fig. 3a–c). Pr-set7 and H4K20me1 signals were detected in all uterine cell types throughout the first two weeks after birth (Fig. 3a, b). The existence of H4K20me1 in the stromal compartment was further confirmed by co-staining with the stromal cell marker Wilms tumor gene 1 (WT1) (Fig. 3d).

a ISH analysis of Pr-set7 in the uteri on PND1, PND5, PND10, and PND15. Dash lines represent the boundary between the epithelium and the stroma. LE luminal epithelium, M mesenchyme, S stroma. Scale bar: 100 μm. b IF analysis of H4K20me1 in the uteri on PND1, PND5, PND10, and PND15. LE luminal epithelium, M mesenchyme, S stroma, GE glandular epithelium. Scale bar: 100 μm. c WB analysis of H4K20me1 in the uteri on PND1, PND5, PND10, and PND15. H3 served as a loading control. d Co-staining of H4K20me1 and WT1 in the uterus. LE luminal epithelium, S stroma. Dash lines represent the boundary between the epithelium and the stroma. Scale bar: 100 μm. e QRT-PCR analysis of Pr-set7 mRNA level in Pr-set7f/f (n = 3 mice) and Pr-set7d/d (n = 3 mice) uteri on PND5. The values were normalized to the Gapdh level. Data are presented as mean ± SEM. Two-tailed unpaired Student’s t-test. ***p = 0.0008. f WB analysis of H4K20me1 in Pr-set7f/f and Pr-set7d/d uteri on PND5, PND10 and PND15. H3 was used as a loading control. g IF analysis of H4K20me1 in Pr-set7f/f and Pr-set7d/d uteri on PND5. Scale bar: 100 μm. h IF analysis of VIMENTIN and α-SMA in Pr-set7f/f and Pr-set7d/d uteri on PND5, PND10 and PND15. Scale bar: 100 μm. i IF analysis of PDGFRα in Pr-set7f/f and Pr-set7d/d uteri on PND5, PND10 and PND15. Scale bar: 100 μm. j Fold change of stromal area in Pr-set7f/f (n = 5 mice) and Pr-set7d/d (n = 5 mice) uteri on PND5, PND10, and PND15. Data are presented as mean ± SEM. Two-tailed unpaired Student’s t-test. ***p = 0.0001 (PND10), ***p = 4e-15 (PND15). k TUNEL analysis of the Pr-set7f/f and Pr-set7d/d uteri on PND5, PND10 and PND15. Dash lines represent the boundary between the epithelium and the stroma, as well as the stroma and the myometrium. Scale bar: 100 μm. l Number of TUNEL+ stromal cells per section in the Pr-set7f/f (n = 5 mice) and Pr-set7d/d (n = 5 mice) uteri on PND5, PND10 and PND15. Data are presented as mean ± SEM. Two-tailed unpaired Student’s t-test. ***p = 4e-6 (PND5), ***p = 0.0002 (PND10), ***p = 5e-11 (PND15). Source data are provided as a Source Data file.
In order to unravel the potential role of PR-SET7 in uterine development after birth, mice with uterine-specific ablation of Pr-set7 (hereafter referred to as Pr-set7d/d) were generated by crossing Pr-set7 floxed mice (hereafter referred to as Prset7f/f) with Pgr-IRES-Cre mice. Specifically, the 7th exon of Pr-set7, which encodes the SET domain that is responsible for its methyltransferase activity, was deleted. According to LacZ staining, the Cre recombinase exhibited activity by PND5 in the uterus (Fig. 2f). On PND5, the Pr-set7 mRNA was efficiently deleted in the uterus according to quantitative reverse transcription-polymerase chain reaction (qRT-PCR) (Fig. 3e). Furthermore, the level of H4K20me1 was obviously declined upon Pr-set7 deletion, as revealed by WB (Fig. 3f) and IF (Fig. 3g).
Given that the murine uterus develops within two weeks after birth, we monitored the growth of the endometrial stroma throughout this pivotal period. As shown by IF staining of VIMENTIN and platelet-derived growth factor receptor alpha (PDGFRα), the area of the uterine stroma increased to more than three times its original size from PND5 to PND15 in Pr-set7f/f females. In contrast, the stromal area in the Pr-set7d/d group was even reduced (Fig. 3h–j).
It has been widely reported that PR-SET7 serves as a multifunctional switch for cell cycle and cell survival14. We thus surmised that the hampered development of the endometrial stroma in the Pr-set7d/d uterus resulted from aberrant cell proliferation or death. We examined the expression of several cyclins and cyclin-dependent kinase inhibitors (CKIs), among which p21 and p16 were markedly upregulated due to Pr-set7 deletion (Supplementary Fig. 3a, b). Nevertheless, the upregulated p21 was only detected in epithelial cells (Supplementary Fig. 3c), implying that the cell cycle progression was less affected in the Pr-set7d/d stroma. Consistently, the proliferation of stromal cells in the Pr-set7d/d uterus rivaled that in the Pr-set7f/f group, as visualized by immunohistochemistry (IHC) analysis of Ki67, proliferating cell nuclear antigen (PCNA) and phospho-histone H3 (p-H3) (Supplementary Fig. 3d–f). Subsequently, we evaluated stromal cell death using terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL). Barely any stromal cells in the Pr-set7f/f uterus exhibited TUNEL signals, while a large portion of the Pr-set7d/d stromal cells underwent cell death (Fig. 3k, l).
These findings proved that the limited growth of the endometrial stroma upon PR-SET7 loss was due to massive stromal cell death rather than impaired proliferation.
Uterine PR-SET7 deficiency led to a reduced number and derailed differentiation of inner stromal cells
To further delineate defective uterine stromal development due to Pr-set7 ablation, we compared stromal cell composition and gene expression between the Pr-set7f/f and Pr-set7d/d groups using scRNA-seq (Fig. 4a, b). An exaggerated decrease in the proportion of epithelial cells, especially glandular (Epi_3) epithelial cells, was observed in the Pr-set7d/d group (Fig. 4b), consistent with the previous study26. In addition, the proportion of inner stromal cells, which accounted for approximately 60% of total stromal cells in the Pr-set7f/f uterus, was reduced prominently to 40% upon PR-SET7 loss (Fig. 4b, c).

a Diagram illustrating uterine heterogeneity in Pr-set7f/f and Pr-set7d/d mice on PND15. b UMAP plot showing cell clusters in Pr-set7f/f and Pr-set7d/d uteri on PND15. c The percentage of stromal subsets in Pr-set7f/f and Pr-set7d/d uteri on PND15. d SCRINSHOT analysis of Wnt16 and Htra3 in the Pr-set7f/f and Pr-set7d/d uteri on PND15. Dash lines represent the boundary between the epithelium and the stroma, as well as the stroma and the myometrium. Scale bar: 100 μm. e IF analysis of PR in Pr-set7f/f and Pr-set7d/d uteri on PND15. Dash lines represent the boundary between the epithelium and the stroma, as well as the stroma and the myometrium. Scale bar: 100 μm. f IHC staining of PR in 4-week-old Pr-set7f/f and Pr-set7d/d uteri. Dash lines represent the boundary between the stroma and the myometrium. Scale bar: 100 μm. g Scatter plot showing the differentially expressed genes in the Pr-set7f/f and Pr-set7d/d inner and outer stroma. h GO functional analysis of the upregulated and downregulated genes in the inner and outer stromal cells due to PR-SET7 loss. Significance is based on over-representation analysis with clusterProfiler. i Violin plot indicating the expressions of Prdm1 and Gli1 in the Pr-set7f/f and Pr-set7d/d inner stroma. j UMAP visualization indicating the expression of Ihh in Pr-set7f/f and Pr-set7d/d uteri. k CellChat analysis showing the ncWNT signaling pathway among different cell clusters in Pr-set7f/f and Pr-set7d/d uteri.
Consistently, we observed significantly fewer Wnt16-positive inner stromal cells and more outer stromal cells that expressed Htra3 in the Pr-set7d/d uterus (Fig. 4d). The decline in the number of inner stromal cells was further verified by PR staining (Fig. 4e) since PR was expressed at a higher level in the inner stromal cells (Fig. 2e-g). Moreover, we unexpectedly discovered that PR-positive inner stromal cells vanished entirely in the uteri of 4-week-old Pr-set7d/d mice (Fig. 4f). Since more dead cells were observed underneath the luminal epithelium in the inner stroma in the absence of PR-SET7 (Fig. 3k), it was rational to speculate that the aberrantly decreased inner stromal subpopulation in the Pr-set7 d/d uterus was ascribed to massive cell death, which led to the complete disappearance of inner stromal cells before puberty.
In addition to the altered stromal subset composition, the transcriptional profiles of both inner and outer stromal cells displayed obvious differences between the Pr-set7f/f and Pr-set7d/d groups (Fig. 4g). Genes governing inner stromal differentiation and reproductive structure development, such as the transcription factors Prdm1 and Gli1, were markedly downregulated in the inner stromal cells compared to the outer stromal cells upon Pr-set7 deletion (Fig. 4h, i). Moreover, the pro-angiogenetic potential of inner stromal cells was also clearly suppressed (Fig. 4h), as visualized by downregulated expression of vascular endothelial growth factor a (Vegfa) and adrenomedullin (Adm) (Supplementary Fig. 4a). Meanwhile, the upregulated genes in the Pr-set7d/d inner stroma were particularly enriched in viral defense reactions and innate immune responses (Fig. 4h). These results suggested that uterine deletion of Pr-set7 interfered with the differentiation and homeostasis of inner stromal cells.
Postnatal uterine development relies on complex communications between the epithelium and the stroma4. Accompanied with the downregulation of Gli1 in the Pr-set7d/d inner stromal cells, the expression of Ihh, the upstream ligand of the Hedgehog signaling, vanished entirely in the epithelium (Fig. 4j). Furthermore, CellChat was applied to dissect out the complex interactions between different cell types in the Pr-set7f/f and Pr-set7d/d neonatal uteri. Both the number and the strength of cell-cell interactions were dramatically decreased in the Pr-set7d/d uterus (Supplementary Fig. 4b-c). The WNT and BMP signaling pathways, which are potentially implicated in gland formation (Fig. 2j, k), were completely abrogated between the inner stroma and the epithelium in the Pr-set7d/d uterus (Supplementary Fig. 4c). Stromal derived WNT5A, which activates the non-canonical WNT (ncWNT) signaling, has been reported to participate in the formation of glands27. We indeed observed a compromised ncWNT signaling pathway between inner stromal cells and epithelial cells in the Pr-set7d/d group (Fig. 4k).
Collectively, the deficiency of PR-SET7 resulted in a reduced number and compromised differentiation of inner stromal cells, which impaired proper epithelial-stromal interactions during uterine development.
Interferon responses were aberrantly elicited in the uterus due to PR-SET7 loss
We noticed that many of the upregulated genes in Pr-set7d/d inner stromal cells were enriched in immune activation and apoptotic signaling (Fig. 4h); thus, we conjectured that the massive cell death in the Pr-set7d/d inner stroma was related to aberrantly activated immune responses. Indeed, the expression of interferon stimulated genes (ISGs) was significantly increased in the inner stroma of the Pr-set7d/d uterus (Fig. 5a and Supplementary Fig. 5a). Moreover, CellChat analysis illustrated that several immunity-associated signaling pathways were remarkably activated upon Pr-set7 deletion (Supplementary Fig. 5b).

a UMAP visualization of Isg15 in the Pr-set7f/f and Pr-set7d/d uteri on PND15. The arrowhead indicates the expression of Isg15 in the inner stroma of the Pr-set7d/d uterus. b Volcano plot indicating the differentially expressed genes in the uteri after Pr-set7 deletion on PND5 and PND10. Significance is based on the negative binomial test with DESeq2. Blue and red dots represent downregulated and upregulated genes, respectively, while genes that were not significantly differentially expressed are shown in gray. c GO functional analysis of the upregulated genes due to PR-SET7 loss. Significance is based on over-representation analysis with clusterProfiler. d WB analysis of p-p65 and t-p65 level in Pr-set7f/f and Pr-set7d/d uteri on PND5 and PND10. β-ACTIN served as a loading control. e GSEA analysis indicating the enrichment of chemokine signaling pathway related genes in the Pr-set7d/d uteri compared with the Pr-set7f/f uteri. Significance is based on functional class scoring with clusterProfiler. NES, normalized enrichment score. f IF analysis of F4/80 in Pr-set7f/f and Pr-set7d/d uteri on PND5 and PND10. Dash lines represent the boundary between the epithelium and the stroma, as well as the stroma and the myometrium. Scale bar: 100 μm. g GSEA analysis assessing the enrichment of toll-like receptor signaling pathway associated genes in the Pr-set7d/d uteri compared with the Pr-set7f/f uteri. Significance is based on functional class scoring with clusterProfiler. NES, normalized enrichment score. h QRT-PCR analysis of Tlr7, Tlr8 and Tlr9 mRNA level in Pr-set7f/f (n = 3 mice) and Pr-set7d/d (n = 3 mice) uteri on PND5. The values were normalized to Gapdh level. Data are presented as mean ± SEM. Two-tailed unpaired Student’s t-test. **p = 0.0063 (Tlr7), ***p = 0.0004 (Tlr8), *p = 0.0421 (Tlr9). i Immunoblotting analysis of p-IRF3, IRF3, p-STAT1 and STAT1 in Pr-set7f/f and Pr-set7d/d uteri. GAPDH was used as a loading control. j QRT-PCR analysis of ISGs in Pr-set7f/f (n = 3 mice) and Pr-set7d/d (n = 3 mice) uteri on PND5. The values were normalized to Gapdh level. Data are presented as mean ± SEM. Two-tailed unpaired Student’s t-test. **p = 0.0085 (Ifitm6), *p = 0.0254 (Irf8), *p = 0.0461 (Isg15), *p = 0.0252 (Oas2), *p = 0.0139 (Zbp1). k Heatmap showing the differentially expressed ISGs between Pr-set7f/f and Pr-set7d/d groups on PND5 and PND10. l SCRINSHOT analysis of Tlr7, Isg15 and Zbp1 in Pr-set7f/f and Pr-set7d/d uteri. Magnified images represent the SCRINSHOT signals in the stromal area. Scale bar: 100 μm. Source data are provided as a Source Data file.
To further confirm these findings, the uteri of Pr-set7f/f and Pr-set7d/d mice were subjected to whole transcriptome sequencing. A total of 1347 and 2366 differentially expressed genes (fold change > =1.5, p < 0.05) were identified on PND5 and PND10, respectively (Fig. 5b). Specifically, 838 genes were upregulated and 509 genes were downregulated in the Pr-set7d/d uterus on PND5. On PND10, 1084 genes were upregulated, while 1282 genes were downregulated. GO enrichment analysis revealed that genes upregulated upon Pr-set7 deletion were mainly enriched in terms closely related to the activation of innate immune responses, including the response to virus, the production of cytokines and leukocyte migration (Fig. 5c).
The expression of genes involved in the NF-kappaB signaling, which serves as a crucial regulator of innate immunity and promotes the secretion of pro-inflammatory cytokines and chemokines, was significantly increased in the Pr-set7d/d uterus (Supplementary Fig. 6a). WB analysis further proved the activation of the NF-kappaB pathway resulting from PR-SET7 deficiency, as manifested by elevated level of phosphorylated p65 (Fig. 5d). In addition, an increase in the expression of genes involved in leukocyte chemotaxis was observed due to PR-SET7 loss (Supplementary Fig. 6a). Gene set enrichment analysis (GSEA) indicated activation of the chemokine signaling pathway (Fig. 5e). Furthermore, obvious infiltration and gathering of leukocytes, especially macrophages, were noticed in the stromal compartment of the Pr-set7d/d uterus, as visualized by CD45 and F4/80 staining (Fig. 5f and Supplementary Fig. 6b), implying an inflammatory environment in the stromal area.
As aforementioned, uterine PR-SET7 deficiency resulted in an exaggerated immune response to viral infection (Fig. 5c). Once viral infection takes place, the innate immune system of the host urges certain pattern recognition receptors (PRRs) to recognize microbe-derived pathogen associated molecular patterns (PAMPs, conserved microbial structures) or endogenous damage associated molecular patterns (DAMPs, tissue damage markers). These PRRs include toll-like receptors (TLRs), nucleotide oligomerization domain (NOD)-like receptors (NLRs), RIG-I-like receptors (RLRs) and so on28. Indeed, gene sets relevant to anti-viral pathways, including TLR signaling, NLR signaling and cytosolic DNA-sensing signaling, were remarkably enriched among the upregulated genes in the Pr-set7d/d uterus (Fig. 5g and Supplementary Fig. 6c), and qRT-PCR analysis further confirmed the elevated level of several TLRs (Fig. 5h).
Upon the recognition of viruses, PRRs activate downstream transcription factors, such as interferon regulatory factor 3 (IRF3) and 7 (IRF7), leading to the production of interferons, a group of cytokines responsible for anti-viral defense. By binding with specific receptors, interferons further drive the phosphorylation of signal transducer and activator of transcription 1 (STAT1) and 2 (STAT2), ultimately leading to the production of ISGs and the amplification of anti-viral responses29. We observed a prominent increase in the level of p-IRF3 and p-STAT1, demonstrating the activation of the interferon signaling pathway (Fig. 5i). In addition, the expressions of 56 ISGs, including interferon induced transmembrane protein 6 (Ifitm6), interferon regulatory factor 8 (Irf8), interferon stimulated gene 15 (Isg15), 2′−5′ oligoadenylate synthetase 2 (Oas2) and Z-DNA binding protein 1 (Zbp1), were significantly increased (fold change >= 1.2, p < 0.05) upon Pr-set7 deletion (Fig. 5j, k).
Since the abovementioned studies were implemented on the whole uterus, we subsequently aimed to investigate whether stromal cells experienced the hyperactivation of anti-viral defenses in the absence of PR-SET7. SCRINSHOT was applied to detect the localization of the upregulated TLRs and ISGs. In fact, significantly more signals for Tlr7, Isg15 and Zbp1 were observed in both the epithelial and inner stromal compartments (Fig. 5l), consolidating that inner stromal cell underwent excessive anti-viral immune activation in the absence of PR-SET7.
H4K20me1 has been widely reported to play an indispensable role in the repair of DNA damage15,18. Consistently, the ablation of Pr-set7 also led to considerable accumulation of DNA damage in the uterus. The p53 signaling pathway, which is elicited in response to DNA damage, was significantly activated in the Pr-set7d/d uterus (Supplementary Fig. 6d). Meanwhile, γH2A.X staining revealed more cells with DNA double strand breaks in the uterine stroma of Pr-set7d/d mutants compared to control mice (Supplementary Fig. 6e, f), which was consistent with the results of the TUNEL assay (Fig. 3k, l).
H4K20me1 restrained the expression of ISGs in both direct and indirect manners
According to published literature, H4K20me1 exerts either promotive or repressive effects on gene transcription16,30,31,32. To excavate the underlying cause of compromised inner stromal differentiation and overwhelming anti-viral responses upon Pr-set7 ablation, uterine tissues were subjected to CUT&Tag for H4K20me1 on PND5 and PND10. H4K20me1 modifications were predominantly distributed at gene promoters and introns (Fig. 6a–c).

a Pie chart displaying the genomic distribution of H4K20me1. b Profile plot showing the distribution of normalized H4K20me1 CUT&Tag signals. TSS, transcription start site; TES, transcription end site. c Heatmap indicating the distribution of normalized H4K20me1 CUT&Tag signals. TSS, transcription start site; TES, transcription end site. d Venn diagram showing the upregulated genes upon PR-SET7 loss (n = 1084) and genes with H4K20me1 modifications (n = 10578). e GO enrichment analysis of genes that were directly suppressed by H4K20me1. Significance is based on over-representation analysis with clusterProfiler. f Genome browser view of normalized H4K20me1 CUT&Tag signals as well as Pr-set7f/f and Pr-set7d/d RNA-seq signals on a portion of ISGs that were upregulated upon Pr-set7 ablation and possessed H4K20me1 occupancy. g Heatmap indicating the abnormally upregulated ERVs due to PR-SET7 abrogation on PND5 and PND10. h Profile plot showing Pr-set7f/f and Pr-set7d/d RNA-seq signals on the body region of MER70-int:ERVL:LTR. TSS, transcription start site; TES, transcription end site. i QRT-PCR analysis of MER70-int:ERVL:LTR level in Pr-set7f/f (n = 3 mice) and Pr-set7d/d (n = 3 mice) uteri on PND5. The values were normalized to Gapdh level. Data are presented as mean +/- SEM. Two-tailed unpaired Student’s t-test. ***p = 0.0002. j Profile plot showing normalized H4K20me1 CUT&Tag signals on the body region of MER70-int:ERVL:LTR. TSS, transcription start site; TES, transcription end site. k Genome browser view of normalized H4K20me1 CUT&Tag signals on MER70-int:ERVL:LTR copies. Source data are provided as a Source Data file.
Motif analysis indicated that gene promoters with H4K20me1 modifications possessed binding motifs for homeobox A10 (HOXA10) (Supplementary Fig. 7a), a critical mesenchymal transcription factor involved in female reproductive tract patterning33 and the specification of the inner stroma (Fig. 2d), suggesting the role of H4K20me1 in the transcriptional regulation of genes related to inner stromal development. Besides, H4K20me1 modifications were detected on the gene bodies of Prdm1 and Gli1 (Supplementary Fig. 7b), which were obviously downregulated in the Pr-set7d/d inner stromal cells (Fig. 4i).
Meanwhile, the promoters occupied by H4K20me1 exhibited motifs of interferon regulatory factor 4 (IRF4), reticuloendotheliosis oncogene (REL, NF-kappaB subunit) and IRF3 with a high probability (Supplementary Fig. 7a), implying the potential participation of H4K20me1 in modulating the transcription of innate immune related genes. The genes upregulated upon PR-SET7 loss were overlapped with genes modified by H4K20me1, and 242 genes directly repressed by H4K20me1 were identified (Fig. 6d). These genes were then subjected to GO analysis, and many of these genes were found to be associated with response to virus, leukocyte migration and activation of immune response (Fig. 6e). Therefore, we rationally speculated that the upregulated ISGs upon Pr-set7 deletion might be directly inhibited by H4K20me1 modifications. Indeed, combining CUT&Tag results with RNA-seq data, we observed that a portion of ISGs upregulated upon PR-SET7 deficiency were equipped with H4K20me1 modifications at promoters and gene bodies, including bone marrow stromal cell antigen 2 (Bst2), Zbp1 and Stat1 (Fig. 6f), suggesting that the expression of these genes was directly suppressed by H4K20me1. Nevertheless, we noticed that a subset of upregulated ISGs exhibited no obvious H4K20me1 occupancy (Supplementary Fig. 7c), indicating an alternative mechanism by which H4K20me1 influenced the transcription of these genes.
As described above, pathways in response to virus were incited in the Pr-set7d/d uterus. Given that viral infections rarely occur during postnatal uterine development, we hypothesized that the hyperactivated anti-viral reactions originated from the existence of ERVs. Under normal circumstances, ERVs are epigenetically silenced to maintain and ensure genome stability. The mechanisms by which ERVs are repressed include histone methylation (e.g., H3K9me3 and H4K20me3), DNA methylation and RNA methylation34. In order to verify that H4K20me1 was also involved in the silencing of ERVs, RNA-seq data were subjected to transposable element analysis, and a number of ERVs, including MER70-int:ERVL:LTR, were abnormally elevated upon PR-SET7 abrogation (Fig. 6g, h), which was also confirmed by qRT-PCR (Fig. 6i). Furthermore, CUT&Tag revealed H4K20me1 deposition on the gene body of MER70 as well as several other ERVs (e.g., LTR105_Mam:ERVL:LTR and MER68-int:ERVL:LTR) (Fig. 6j, k and Supplementary Fig. 7d).
Given the potential difficulty of short read sequencing in handling repetitive sequences, we noticed that the lengths of most ERVs we studied were shorter than 300 bp (Supplementary Fig. 8a), and different copies of the same ERV family are highly variable in sequences due to insertions, deletions and substitutions during evolution35. Since we performed paired-end 150 bp sequencing, the alignment of ERV sequences should be accurate. To further confirm this, we selected several ERV copies that were upregulated upon PR-SET7 loss according to our RNA-seq data (Supplementary Fig. 8b), amplified the full length of each copy from uterine stromal cell transcripts and performed Sanger Sequencing. The amplified full-length ERV copies were accurately mapped to ERV sequences downloaded from the RepeatMasker of the UCSC Genome Browser (Supplementary Fig. 8c), verifying the accuracy of our reads mapping.
In conclusion, these findings suggested that H4K20me1 directly restrained the transcription of certain ISGs; on the other hand, H4K20me1 indirectly prevents uncontrolled anti-viral immunity via mediating the silencing of ERVs.
Declined H4K20me1 level led to viral mimicry responses and culminated in ZBP1-mediated necroptosis and apoptosis
Considering that the neonatal uterus is comprised of multiple cell types, in order to reinforce the significance of PR-SET7 specifically in stromal cells, primary mouse endometrial stromal cells isolated from the postnatal uterus were cultured in vitro and treated with DMSO (served as the control) or UNC0379, a selective substrate-competitive inhibitor of PR-SET7. A dose of 5 μM UNC0379 was sufficient to dampen H4K20me1 level in cultured uterine stromal cells (Fig. 7a). Consistent with the in vivo evidence, we observed aberrantly elevated levels of ERVs (Fig. 7b). Since ERVs were previously reported to cause the accumulation of dsRNAs36, we performed dot blotting and IF analysis using the J2 antibody, which specifically recognizes dsRNAs. As expected, impairing PR-SET7 mediated H4K20me1 gave rise to a prominent increase in cytosolic dsRNA accumulation (Fig. 7c, d).

a WB analysis of H4K20me1 level in cultured endometrial stromal cells treated with DMSO (CTR) or UNC0379. GAPDH was used as a loading control. b QRT-PCR analysis of the mRNA level of ERVs in cultured uterine stromal cells treated with DMSO (CTR, n = 3 independent biological replicates) or UNC0379 (n = 3 independent biological replicates). The values were normalized to Gapdh level. Data are presented as mean ± SEM. Two-tailed unpaired Student’s t-test. **p = 0.0064 (MER70-int), ***p = 0.0002 (LTR105_Mam), ***p = 0.0008 (MER68-int). c IF analysis of J2 indicating dsRNA level in cultured endometrial stromal cells treated with DMSO (CTR) or UNC0379. Scale bar: 50 μm. d Dot blotting analysis of dsRNA level in cultured endometrial stromal cells treated with DMSO (CTR) or UNC0379. e Immunoblotting analysis of p-IRF3, IRF3, p-STAT1 and STAT1 in cultured endometrial stromal cells treated with DMSO (CTR) or UNC0379. GAPDH served as a loading control. f QRT-PCR analysis of the mRNA level of ISGs in cultured endometrial stromal cells treated with DMSO (CTR, n = 3 independent biological replicates) or UNC0379 (n = 3 independent biological replicates). The values were normalized to Gapdh level. Data are presented as mean ± SEM. Two-tailed unpaired Student’s t-test. ***p = 6e-5 (Bst2), **p = 0.0074 (Ifitm3), **p = 0.0032 (Isg15), ***p = 0.0001 (Oas2), **p = 0.0029 (Oasl2), **p = 0.0050 (Zbp1). g Annexin V/PI apoptosis analysis of cultured endometrial stromal cells treated with DMSO (CTR) or UNC0379. h–k Immunoblotting analysis of Cleaved CASPASE-3 (h), GSDMD (i), p-RIPK3, RIPK3, p-MLKL and MLKL (j), as well as ZBP1 (k) in cultured endometrial stromal cells treated with DMSO (CTR) or UNC0379. GAPDH served as a loading control. l ChIP-qRT-PCR analysis of H4K20me1 on Zbp1 locus. The data are representative of 3 independent biological replicates. Data are presented as mean ± SEM. Two-tailed unpaired Student’s t-test. ***p = 0.0003. m Annexin V/PI apoptosis analysis of cultured Zbp1+/− stromal cells treated with DMSO or UNC0379 and Zbp1−/− stromal cells treated with DMSO or UNC0379. n Percentage of cell death in Zbp1+/− stromal cells treated with DMSO (n = 3 independent biological replicates) or UNC0379 (n = 3 independent biological replicates) and Zbp1−/− stromal cells treated with DMSO (n = 3 independent biological replicates) or UNC0379 (n = 3 independent biological replicates). Data are presented as mean ± SEM. Two-tailed unpaired Student’s t-test. ***p = 0.0007, **p = 0.0023, *p = 0.0240. o Immunoblotting analysis of p-RIPK3, RIPK3, p-MLKL, MLKL, Cleaved CASPASE-3, ZBP1 and H4K20me1 in Zbp1+/− stromal cells treated with DMSO or UNC0379 and Zbp1−/− stromal cells treated with DMSO or UNC0379. GAPDH and H3 served as loading controls. Source data are provided as a Source Data file.
In the meanwhile, a viral mimicry cascade was ignited, as indicated by elevated p-IRF3 and p-STAT1 (Fig. 7e) as well as increased levels of ISGs, including Bst2, Isg15, Oas2, Zbp1, interferon induced transmembrane protein 3 (Ifitm3) and 2′−5′ oligoadenylate synthetase-like 2 (Oasl2) (Fig. 7f). Moreover, annexin V-FITC/PI co-staining evinced a greater ratio of cell death upon UNC0379 treatment (Fig. 7g and Supplementary Fig. 9). To further interrogate the cell death modality resulting from PR-SET7 inhibition, we examined markers of apoptosis (Fig. 7h), pyroptosis (Fig. 7i) and necroptosis (Fig. 7j), and found marked activation of both the apoptotic and necroptotic pathways, as revealed by elevated levels of cleaved CASPASE-3, p-RIPK3 (receptor-interacting protein kinase 3) and p-MLKL (mixed lineage kinase domain-like) (Fig. 7h, j).
In addition, to further consolidate our findings, we obtained primary uterine stromal cells from Pr-set7f/f mice and performed transient transfection with plasmids carrying the Cre recombinase. Despite the relatively low transfection efficiency in primary uterine stromal cells (Supplementary Fig. 10a–c), we also observed the activation of interferon responses and cell death (Supplementary Fig. 10d, e).
In the subsequent study, we intended to bridge the gap between the hyperactivated immune response and cell death, and eventually put emphasis on ZBP1. ZBP1, the expression of which is induced by the interferon signaling, is capable of detecting the left-handed conformation of nucleic acids (referred to as Z-form nucleic acids) either produced during viral infection or transcribed from ERVs through its Zα domains and recruiting RIPK3 via shared receptor-interacting protein homotypic interaction motifs (RHIMs), which culminates in diverse modes of cell death including apoptosis and necroptosis37. Declined H4K20me1 by either Pr-set7 deletion or UNC0379 treatment significantly upregulated the mRNA and protein levels of ZBP1 (Figs. 5j, l, and 7f, k). CUT&Tag revealed H4K20me1 occupancy at the promoter region of Zbp1 (Fig. 6f), and chromatin immunoprecipitation (ChIP)-qRT-PCR using a pair of primers targeting the promoter locus of Zbp1 further proved that Zbp1 was a direct target of H4K20me1 (Fig. 7l). To explore whether the increase in cell death upon UNC0379 administration was mediated by ZBP1, endometrial stromal cells obtained from the uteri of Zbp1+/− and Zbp1−/− mice were subjected to UNC0379 treatment. According to annexin V-FITC/PI co-staining and immunoblotting, cell apoptosis and necroptosis caused by PR-SET7 inhibition were remarkably attenuated by ZBP1 abrogation (Fig. 7m–o).
These findings highlighted the pivotal role of ZBP1 in the execution of cell death caused by abnormally activated ERVs and viral mimicry responses upon PR-SET7 inhibition.
Discussion
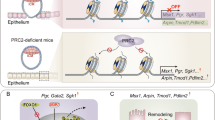
The spatiotemporal progression and potential regulatory machinery of postnatal uterine stromal development has been a long-standing enigma attributed to the scarcity of advanced approaches and appropriate genetic models. Here, we portrayed the transcriptomic atlas of the neonatal uterus throughout the postnatal development period at single-cell resolution. In line with the previous report10, two distinct stromal subpopulations were defined, and the spatial distribution, developmental trajectory, and potential functions of these two stromal subsets were dissected. Furthermore, utilizing mouse model with uterine specific deletion of Pr-set7, we demonstrated that PR-SET7-mediated H4K20me1 facilitated the proper development of the inner stromal subset via epigenetically limiting exaggerated interferon responses and consequent cell death (Fig. 8).

a Uterine Pr-set7 deletion resulted in impaired inner stromal development after birth. b PR-SET7-mediated H4K20me1 epigenetically restricted exaggerated interferon responses via both direct and indirect manners, protecting stromal cells from ZBP1-mediated apoptosis and necroptosis.
Accumulated evidence has reinforced the heterogeneity of the endometrial stroma/decidua in the adult uterus38,39,40,41. Through scRNA-seq, Saatcioglu et al. unraveled the heterogeneity of the neonatal uterine stroma, and hypothesized that a population of Misr2+ cells at birth (before PND2) represented uterine stromal progenitors and gave rise to two stromal layers by PND610. Since scRNA-seq was performed only on PND6 in this previous study, in order to provide a comprehensive and dynamic atlas of postnatal uterine stromal development, we carried out scRNA-seq analysis throughout the neonatal period (PND1-15). Our scRNA-seq data demonstrated that both stromal subsets already existed on PND1 in a less-differentiated state, and underwent further differentiation in the first two weeks after birth. Therefore, we present an alternative view that the fates of uterine stromal cells are already determined at birth depending on their “position codes” along the luminal-myometrial radial axis. The previous study identified two stromal subpopulations and their distributions in the neonatal uterus, but the characteristics of these two distinct subsets remained elusive. In order to fill this gap, we described the developmental trajectory of stromal subpopulations, transcription factors that potentially directed stromal differentiation, as well as the unique features of these two stromal subsets.
We observed an inequality of PR expression between the inner and outer stroma. The human endometrium is composed of the functional layer that experiences extensive proliferation, differentiation and decidualization in response to ovarian steroid hormones in each menstrual cycle, as well as the basal layer that remains relatively quiescent across the cycle42. Similarly in mice, stromal cells surrounding the implanting embryo transform into decidual cells under the influence of progesterone during pregnancy, leaving a thin layer of undifferentiated stroma adjacent to the myometrium43. Therefore, the relatively higher expression level of PR in the inner stroma implies its superior responsiveness to progesterone, which resembles the functional layer in humans, while the outer stroma is more analogous to the basal layer. Whether the inner and outer stroma in the neonatal uterus indeed represent the functional and basal layers that display diverging routes during the menstrual cycle/pregnancy due to differences in progesterone responsiveness deserves further exploration.
Postnatal uterine development requires the intimate crosstalk between the epithelium and the mesenchyme/stroma4. Tissue recombination experiments clearly indicated that the uterine mesenchyme/stroma directed and specified epithelial proliferation and differentiation, while the epithelium was necessitated to support stromal and myometrial development44,45,46. For instance, mice with deletion of Wnt7a, which is specifically expressed in uterine epithelial cells, not only failed in adenogenesis, but also exhibited severe stromal defects6. Meanwhile, uterine stromal WNT5A has been demonstrated to be essential for the formation of glands27. Our scRNA-seq analysis revealed that the inner stromal cells, which were found to be in close proximity to the luminal epithelium, highly expressed genes related to gland morphogenesis. The expression level of these adenogenesis-associated genes peaked on PND10, when the glandular buds experienced extensive branching and invasion into the stroma, which underscored the significance of stromal-epithelial interactions in the development of uterine glands. CellChat analysis further revealed that the WNT and BMP signaling pathways potentially mediated the communications between inner stromal cells and glandular epithelial cells.
Although the previous study proved that the failure of uterine adenogenesis upon Pr-set7 deletion resulted from limited epithelial population growth26, it cannot be excluded that the dysfunctional inner stroma also contributed to compromised gland development, and/or that certain detrimental factors were emitted from the dying inner stromal cells, hampering the survival and maintenance of epithelial cells. On the other hand, the derailed differentiation and severe cell death of inner stromal cells in the Pr-set7d/d uterus could partially arise from impaired epithelial functions and aberrant epithelial loss.
ERVs are evolutionary remnants originating from ancient retroviral infections during which the genetic materials of the viruses were integrated into the genome of host germ cells and transmitted across generations34. Despite the long-held misconception that ERVs are “junk DNA” without biological significance, emerging evidence has acknowledged their crucial contributions to various developmental events, such as embryogenesis and placentation, via functioning as regulatory elements (promoters or enhancers) and rewiring the transcriptional program47,48,49,50. However, aberrant transcription of ERVs disturbs genomic stability and is implicated in uncontrolled viral mimicry responses and even cell death51. Therefore, ERVs are tightly regulated by host surveillance mechanisms, including DNA methylation, histone modifications (e.g., H3K9me3 and H4K20me3) and RNA modifications (e.g., m6A), either individually or concurrently34. In addition to H3K9me3 and H4K20me3, a recent report demonstrated that PR-SET7-mediated H4K20me1 also participated in the transcriptional silencing of ERVs in trophoblast cells20, and we discovered a similar epigenetic regulatory mechanism in the neonatal uterus in the present study. Further investigations are required to interrogate whether H4K20me1 mediates the repression of ERVs alone or via the interplay with other epigenetic regulatory machinery during postnatal uterine development.
ZBP1 is an ISG and serves as a cytoplasmic sensor of double-stranded nucleic acids to further activate the innate immune system against viral infections52,53. Previous studies have confirmed a critical role of ZBP1 in programmed cell death. Particularly, ZBP1 interacts with RIPK3 through the RHIM domains, resulting in the oligomerization and autophosphorylation of RIPK3, which drives parallel CASPASE-8-dependent apoptosis and MLKL-dependent necroptosis54,55. Additionally, ZBP1 also induces the ignition of pyroptosis by activating NLRP356. Recently, the concept of PANoptosis, a multifaceted inflammatory cell death process involving the intricate interplay among apoptosis, necroptosis and pyroptosis, was proposed, and ZBP1 has been identified as a key upstream executor of PANoptosis57. Utilizing the primary culture of uterine stromal cells isolated from Zbp1−/− mice and the administration of the PR-SET7 inhibitor, we proved that the inhibition of PR-SET7 led to massive apoptosis and necroptosis, which were largely restored by Zbp1 deletion. However, the deficiency of ZBP1 alleviated but not eliminated cell death, suggesting the existence of alternative mechanisms contributing to the cell death induced by PR-SET7 inhibition.
Collectively, our study unearthed an unappreciated role of PR-SET7-mediated H4K20me1 in the regulation of postnatal uterine stromal subpopulation development via restricting hyperactivated interferon signaling and preventing uncontrolled cell death.
Methods
Animals
Neonatal (PND1-PND15) C57BL6 female mice were used in the present study. Pr-set7 floxed (Pr-set7f/f), Pgr-IRES-Cre, and Zbp1−/− transgenic mice used in the present study were constructed as previously described24,58,59. Mice with uterine specific deletion of Pr-set7 (Pr-set7d/d) were generated by crossing Pr-set7f/f mice with Pgr-IRES-Cre mice. All mice were housed in the animal care facility of Xiamen University (temperature: 22 ± 2 °C, humidity: 50–60%, 12 h light/dark cycle) according to the guidelines for the care and use of laboratory animals. All experimental procedures were approved by the Animal Care Committee of Xiamen University (XMULAC20170366). For the assessment of postnatal uterine development, Pr-set7f/f and Pr-set7d/d neonates were sacrificed, uteri of which were isolated and either frozen in liquid nitrogen or fixed in 10% neutral buffered formalin overnight depending on subsequent experimental requirements.
Primary uterine stromal cell culture
Uteri isolated from neonates were cut into small pieces and first digested using 25 mg/ml pancreatin (Sigma) and 6 mg/ml dispase (Gibco) for 10 min at 37 °C, and then incubated with 0.5 mg/ml collagenase (Sigma) for 30 min at 37 °C. After filtration, the obtained cells were plated and cultured in phenol red-free Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 Ham (Gibco) supplemented with 10% charcoal-stripped fetal bovine serum (Bioind). Adherent stromal cells were administrated with DMSO or 5 μM UNC0379 (MedChemExpress), or subjected to electrotransfection with plasmids carrying the CMV promoter-driven Cre recombinase.
ISH
After removal from −80 °C, frozen sections were warmed at 37 °C for 5 min, and then fixed in 4% paraformaldehyde for 1 h at room temperature. Slides were incubated with Digoxigenin-labeled probes overnight at 60 °C. Following hybridization, uterine sections were incubated with Anti-Digoxigenin-Alkaline phosphatase overnight at 4 °C. Signals were detected with Nitrotetrazolium blue chloride/5-Bromo-4-chloro-3-indolyl phosphate. Images were captured using NIS-Elements D 4.50.00 (Nikon). Probe for Pr-set7 was used in the present study, with detailed sequence information listed in Supplementary Table 1.
Immunostaining
For fixed specimens, uterine tissue was dehydrated using graded ethanol, xylene and paraffin. Paraffin sections were deparaffinized, rehydrated, subjected to heat-mediated antigen retrieval before antigen blocking. For frozen sections and cell slides, they were fixed in 4% polyformaldehyde for 30 min at room temperature before blocking. Slides were blocked with 1% bovine serum albumin, incubated with primary antibodies overnight at 4 °C and then either horseradish peroxidase-labeled secondary antibodies (Zhongshan Golden Bridge Biotechnology, 1:200) or fluorescent dye-conjugated secondary antibodies (Jackson ImmunoResearch, 1:200) for 1 h at room temperature. Images were captured using NIS-Elements D 4.50.00 (Nikon) or Zen 2.3 (Zeiss). Primary antibodies against PR (CST, 8757, 1:200), H4K20me1 (ABclonal, A2370, 1:200), WT1 (Santa Cruz, sc-393498, 1:200), VIMENTIN (Abcam, ab92547, 1:200), α-SMA (BioGenex, MU128-UC, 1:200), PDGFRα (CST, 3174, 1:200), F4/80 (CST, 70076, 1:200), J2 (SCICONS, 10010500, 1:500), p21 (ABclonal, A19094, 1:200), Ki67 (Abcam, ab15580, 1:100), PCNA (Santa Cruz, sc-7907, 1:200), p-H3 (CST, 9701, 1:500), CD45 (CST, 70257, 1:200) and γH2A.X (CST, 9718, 1:500) were employed in this study, with detailed information listed in Supplementary Table 2.
QRT-PCR
Total RNA was extracted from uterine tissue or cultured endometrial stromal cells using the TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. 1 μg RNA was reverse transcribed into cDNA. Expression level of target mRNAs was detected by quantitative real-time PCR analysis with SYBR Green (TAKARA). All assays were performed at least three times. Primers used in this study were listed in Supplementary Table 1.
WB
Proteins extracted from uterine tissue or cultured endometrial stromal cells were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis, and transferred to polyvinylidene difluoride membrane. The membrane with proteins was blocked with 5% skim milk for 1 h at room temperature, and incubated with primary antibodies overnight at 4 °C. In the subsequent procedure, the membrane was incubated with secondary antibodies (Zhongshan Golden Bridge Biotechnology, 1:5000) for 1 h at room temperature. Results were visualized using Supersignal West Pico (Thermo Scientific) according to the manufacturer’s instructions. Primary antibodies against H4K20me1 (ABclonal, A2370, 1:1000), H3 (Abmart, P30266, 1:5000), p65 (CST, 8242, 1:1000), p-p65 (CST, 3033, 1:1000), β-ACTIN (Bioworld, AP0060, 1:5000), IRF3 (CST, 4302, 1:1000), p-IRF3 (CST, 79945, 1:1000), STAT1 (CST, 9172, 1:1000), p-STAT1 (CST, 9167, 1:1000), GAPDH (Bioworld, AP0063, 1:5000), J2 (SCICONS, 10010500, 1:5000), RIPK3 (CST, 15828, 1:1000), p-RIPK3 (CST, 91702, 1:1000), MLKL (CST, 37705, 1:1000), p-MLKL (CST, 37333, 1:1000), Cleaved CASPASE-3 (CST, 9661, 1:1000), GSDMD (Abcam, ab219800, 1:1000), ZBP1 (Santa Cruz, sc-271483, 1:500) and p21 (ABclonal, A19094, 1:1000) were applied in this study, with detailed information listed in Supplementary Table 2.
SCRINSHOT
SCRINSHOT was performed as previously reported60. After removal from −80 °C, frozen sections were warmed at 45 °C for 3 min, and then fixed in 4% paraformaldehyde for 1 h at room temperature. After treated with the blocking solution (1× Ampligase Buffer, 0.05 M KCl, 20% Formamide deionized, 0.1 μM Oligo-dT, 0.2 μg/μl bovine serum albumin, 40 U/μl RNase Inhibitor, 10 μg/μl tRNA) for 30 min at room temperature, slides were incubated with specific Padlock probes for 20 min at 55 °C and then for 4 h at 45 °C. Following hybridization, uterine sections were subjected to probe ligation overnight at 25 °C using 0.5 U/μl SplintR (NEB, M0375) and rolling circle amplification overnight at 30 °C using 0.5 U/μl φ29 DNA polymerase (Vazyme, N106). Subsequently, sections were incubated with horseradish peroxidase-labeled detection oligos. Hybridization signals were amplified by the tyramide signal amplification system, and detected using Zeiss LSM 880 with Airyscan. Images were captured using Zen 2.3 (Zeiss). Padlock probes for Wnt16, Htra3, Tlr7, Isg15 and Zbp1 were used in the present study, with detailed sequence information listed in Supplementary Table 1.
TUNEL
The TUNEL assay was performed using the One Step TUNEL Apoptosis Assay Kit (Beyotime, C1086) according to the manufacturer’s protocol. Briefly, paraffin sections were deparaffinized, rehydrated and treated with 20 μg/ml proteinase K for 30 min at 37 °C, followed by the incubation of the TUNEL detection solution for 1 h at 37 °C in the dark. TUNEL signals were observed by Zeiss LSM 880 with Airyscan. Images were captured using Zen 2.3 (Zeiss).
LacZ staining
Uteri of Pgr-IRES-Cre;Rosa26-LacZ neonates on PND1, 5, 10 and 15 were collected, fixed in 0.2% paraformaldehyde overnight at 4 °C and then dehydrated in 30% sucrose overnight at 4 °C. Dehydrated tissue was mounted in optimal cutting temperature compound for sectioning. Frozen sections were fixed in 0.2% paraformaldehyde for 10 min at 4 °C, and stained with the LacZ staining solution (0.1 M phosphate buffer pH 7.3, 2 mM MgCl2, 0.01% sodium deoxycholate, 0.02% NP-40, 5 mM K3Fe(CN)6, 5 mM K4Fe(CN)6 and X-GAL) overnight at 37 °C. Images were captured using NIS-Elements D 4.50.00 (Nikon).
Dot blotting
1–2 μg total RNA extracted from cultured mouse endometrial stromal cells treated with DMSO or UNC0379 was dropped onto nitrocellulose membrane. Subsequently, the membrane was subjected to ultraviolet crosslinking, blocked with 5% skim milk for 1 h at room temperature and then incubated with primary antibody against J2 (SCICONS, 10010500, 1:5000) overnight at 4 °C. The next day, the membrane was incubated with secondary antibody (Zhongshan Golden Bridge Biotechnology, 1:5000) for 1 h at room temperature. Results were visualized using Supersignal West Pico (Thermo Scientific) according to the manufacturer’s instructions. Detailed information of the J2 antibody was listed in Supplementary Table 2.
Flow cytometry
Cultured mouse endometrial stromal cells treated with DMSO or UNC0379 were harvested and stained with Annexin V-FITC and propidium iodide (PI) in the dark for 20 min at room temperature according to manufacturer’s instructions of the Annexin V-FITC Apoptosis Detection Kit (Beyotime, C1062). After staining, cells were immediately examined by flow cytometry using Beckman CytoFLEX S (CytExpert 2.4). Flow cytometry data were further analyzed by the FlowJo 10.8.1 software. Each experiment was repeated three times.
RNA-seq
Total RNA was extracted from neonatal Pr-set7f/f and Pr-set7d/d uteri on PND5 and 10 using the TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. Purified RNA was subjected to RNA-seq using DNBSEQ-T7 (China, BGI).
RNA-seq data analysis
RNA-seq raw data were filtered to obtain clean data after quality control by Trim Galore (0.6.4). High-quality clean data were aligned to the mouse reference genome (GRCm38) using STAR (2.7.3) with default parameters. Differentially expressed genes were identified using the DESeq2 (1.30.1) package with the criterion of fold change greater than 1.5 and p value less than 0.05. The volcano plot and heatmap were performed using the ggplot2 (3.4.3) and pheatmap (1.0.12) package, respectively. The clusterProfiler (4.8.1) package was employed for GO and GSEA analysis.
CUT&Tag
Uterine tissue isolated from neonates on PND5 and 10 was cut into small pieces and first digested using 25 mg/ml pancreatin (Sigma) and 6 mg/ml dispase (Gibco) for 10 min at 37 °C, and then incubated with 0.5 mg/ml collagenase (Sigma) for 30 min at 37 °C. After filtration, the collected cells were counted, and 100 thousand cells were subjected to subsequent CUT&Tag assay using the NovoNGS CUT&Tag 4.0 High-Sensitivity Kit (Novoprotein, N259-YH01) according to manufacturer’s instructions. Briefly, cells were incubated with ConA magnetic beads for 10 min at room temperature, followed by primary antibody (anti-H4K20me1, ActiveMotif, 39727) incubation overnight at 4 °C, secondary antibody incubation for 1 h at room temperature, ChiTag transposome incubation for 1 h at room temperature, and then Tn5 tagmentation for 1 h at 37 °C. Next, DNA was extracted, amplificated and purified. The constructed library was sequenced using Illumina NovaSeq 6000. Detailed information of the primary antibody used in the study was listed in Supplementary Table 2.
CUT&Tag data analysis
CUT&Tag raw data were filtered to obtain clean data after quality control by Trim Galore (0.6.4). High-quality clean data were aligned to the mouse reference genome (mm10) using Bowtie2 (2.5.0). MACS2 (2.2.7.1) was applied for peak calling. Peak annotation was performed using the ChIPseeker (1.14.2) package. Heatmap and profile plot were obtained by deepTools (3.5.1).
ChIP-qRT-PCR
Cultured mouse endometrial stromal cells were fixed with 1% formaldehyde for 10 min at room temperature, which was terminated by 0.125 M glycine for 10 min on ice. Fixed cells were collected and lysed in Lysis Buffer 1 (50 mM HEPES pH 7.5, 1 mM EDTA, 140 mM NaCl, 0.5% NP-40, 10% glycerol, 0.25% Triton X-100), Lysis Buffer 2 (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 0.5 mM EGTA, 200 mM NaCl) and Lysis Buffer 3 (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 0.5 mM EGTA, 100 mM NaCl, 0.1% Sodium Deoxycholate, 0.1% N-lauroylsarcosine). All lysis buffers were supplemented with protease inhibitors cocktail (Roche) before use. Chromatin DNA was sheared to 500–1000 bp using the BioRuptor sonicator (Diagenode) and incubated with primary antibody against H4K20me1 (ActiveMotif, 39727) bound to protein A magnetic beads (Invitrogen) overnight at 4 °C. After washing with Low Salt Buffer (50 mM HEPES pH 7.9, 2 mM EDTA, 150 mM NaCl, 1% Triton X-100, 0.5% Sodium Deoxycholate), High Salt Buffer (50 mM HEPES pH 7.9, 2 mM EDTA, 500 mM NaCl, 1% Triton X-100, 0.5% Sodium Deoxycholate), LiCl Buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 250 mM LiCl, 0.5% NP-40, 0.5% Sodium Deoxycholate), TE Buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA) and elution with Elution Buffer (100 mM NaHCO3, 1% SDS), the protein-DNA complex was reversed overnight at 65 °C. Immunoprecipitated DNA and input DNA was purified and subjected to qRT-PCR. Detailed information of PCR primers and primary antibodies used in the study was listed in Supplementary Tables 1 and 2, respectively.
Single-cell RNA-seq
Uteri of Pr-set7f/f neonates on PND1, 5, 10, 15 and Pr-set7d/d neonates on PND15 were isolated and digested into single-cell suspensions, which were then loaded into microfluidic devices using the Singleron Matrix Single Cell Processing System (Singleron). Subsequently, libraries were constructed according to the protocol of the GEXSCOPE Single Cell RNA Library Kit (Singleron). The constructed library was sequenced using Illumina NovaSeq 6000.
Single-cell RNA-seq data analysis
Single-cell RNA-seq reads were processed by the CellRanger (4.0.0) pipeline with default and recommended parameters. The Seurat (4.3.0) toolkit was employed for quality control. High-quality cells (genes per cell >500 and the content of mitochondrial genes <5%) were subjected to subsequent analyses. 2000 highly variable genes were selected, and the top 50 principal components were calculated using the PCA function. The dimensionality reduction and cell clustering were performed using UMAP, and cell types were annotated according to well-known marker genes. Cell-cell interaction and communication analysis was performed using CellChat (1.5.0). For RNA velocity analysis, the BAM files were produced by default parameters of the CellRanger software. The velocyto.R package was used to calculate RNA velocity values for selected genes from each cell. Highly variably expressed genes computed by FindVariableFeatures of Seurat were further filtered out based on a cluster-wise expression, and the remaining highly variably expressed genes were selected as input for velocyto.R. Finally, The RNA velocity vectors were embedded in the UMAP plot.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 8.2.0 Software. All data were presented as mean ± SEM (standard error of mean). Each experiment included at least three independent samples. Comparisons between two groups were made by unpaired two-tailed Student’s t-test. P < 0.05 was considered to indicate statistical significance.
Statistics and reproducibility
All immunostaining, ISH, SCRINSHOT, WB, qRT-PCR, TUNEL, LacZ staining, dot blotting and flow cytometry experiments were repeated at least three times independently with similar results.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The single-cell RNA-seq, bulk RNA-seq and CUT&Tag data generated in this study have been deposited in the National Center for Biotechnology Information Sequence Read Archive under accession code PRJNA1046685. Source data are provided with this paper.
Code availability
Codes used in this study have been deposited at [https://github.com/dwb0211/PRSet7-scRNA].
References
Kobayashi, A. & Behringer, R. R. Developmental genetics of the female reproductive tract in mammals. Nat. Rev. Genet. 4, 969–980 (2003).
Brody, J. R. & Cunha, G. R. Histologic, morphometric, and immunocytochemical analysis of myometrial development in rats and mice: I. Normal development. Am. J. Anat. 186, 1–20 (1989).
Gray, C. A. et al. Developmental biology of uterine glands. Biol. Reprod. 65, 1311–1323 (2001).
Spencer, T. E., Hayashi, K., Hu, J. & Carpenter, K. D. Comparative developmental biology of the mammalian uterus. Curr. Top. Dev. Biol. 68, 85–122 (2005).
Bellessort, B. et al. Dlx5 and Dlx6 control uterine adenogenesis during post-natal maturation: possible consequences for endometriosis. Hum. Mol. Genet. 25, 97–108 (2016).
Dunlap, K. A. et al. Postnatal deletion of Wnt7a inhibits uterine gland morphogenesis and compromises adult fertility in mice. Biol. Reprod. 85, 386–396 (2011).
Jeong, J. W. et al. Foxa2 is essential for mouse endometrial gland development and fertility. Biol. Reprod. 83, 396–403 (2010).
Shelton, D. N. et al. The role of LEF1 in endometrial gland formation and carcinogenesis. PLoS ONE 7, e40312 (2012).
Spencer, T. E., Lowke, M. T., Davenport, K. M., Dhakal, P. & Kelleher, A. M. Single-cell insights into epithelial morphogenesis in the neonatal mouse uterus. Proc. Natl Acad. Sci. USA 120, e2316410120 (2023).
Saatcioglu, H. D. et al. Single-cell sequencing of neonatal uterus reveals an Misr2+ endometrial progenitor indispensable for fertility. eLife 8, e46349 (2019).
Xu, R. et al. Insights into epigenetic patterns in mammalian early embryos. Protein Cell 12, 7–28 (2021).
Fang, J. et al. Purification and functional characterization of SET8, a nucleosomal histone H4-lysine 20-specific methyltransferase. Curr. Biol. 12, 1086–1099 (2002).
Xiao, B. et al. Specificity and mechanism of the histone methyltransferase Pr-Set7. Genes Dev. 19, 1444–1454 (2005).
Beck, D. B., Oda, H., Shen, S. S. & Reinberg, D. PR-Set7 and H4K20me1: at the crossroads of genome integrity, cell cycle, chromosome condensation, and transcription. Genes Dev. 26, 325–337 (2012).
Dulev, S., Tkach, J., Lin, S. & Batada, N. N. SET8 methyltransferase activity during the DNA double-strand break response is required for recruitment of 53BP1. EMBO Rep. 15, 1163–1174 (2014).
Nishioka, K. et al. PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol. Cell 9, 1201–1213 (2002).
Tardat, M. et al. The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells. Nat. Cell Biol. 12, 1086–1093 (2010).
Tuzon, C. T. et al. Concerted activities of distinct H4K20 methyltransferases at DNA double-strand breaks regulate 53BP1 nucleation and NHEJ-directed repair. Cell Rep. 8, 430–438 (2014).
Wu, S. et al. Dynamic regulation of the PR-Set7 histone methyltransferase is required for normal cell cycle progression. Genes Dev. 24, 2531–2542 (2010).
Zhou, X. et al. Trophoblast PR-SET7 dysfunction induces viral mimicry response and necroptosis associated with recurrent miscarriage. Proc. Natl Acad. Sci. USA 120, e2216206120 (2023).
Driskell, I. et al. The histone methyltransferase Setd8 acts in concert with c-Myc and is required to maintain skin. EMBO J. 31, 616–629 (2012).
Malik, J., Lillis, J. A., Couch, T., Getman, M. & Steiner, L. A. The methyltransferase Setd8 is essential for erythroblast survival and maturation. Cell Rep. 21, 2376–2383 (2017).
Nikolaou, K. C. et al. Spontaneous development of hepatocellular carcinoma with cancer stem cell properties in PR-SET7-deficient livers. EMBO J. 34, 430–447 (2015).
Oda, H. et al. Monomethylation of histone H4-lysine 20 is involved in chromosome structure and stability and is essential for mouse development. Mol. Cell Biol. 29, 2278–2295 (2009).
Tanaka, H. et al. The SETD8/PR-Set7 methyltransferase functions as a barrier to prevent senescence-associated metabolic remodeling. Cell Rep. 18, 2148–2161 (2017).
Cui, T. et al. PR-Set7 deficiency limits uterine epithelial population growth hampering postnatal gland formation in mice. Cell Death Differ. 24, 2013–2021 (2017).
Mericskay, M., Kitajewski, J. & Sassoon, D. Wnt5a is required for proper epithelial-mesenchymal interactions in the uterus. Development 131, 2061–2072 (2004).
O’Neill, L. A. & Bowie, A. G. Sensing and signaling in antiviral innate immunity. Curr. Biol. 20, R328–R333 (2010).
Dalskov, L., Gad, H. H. & Hartmann, R. Viral recognition and the antiviral interferon response. EMBO J. 42, e112907 (2023).
Shoaib, M. et al. Histone H4 lysine 20 mono-methylation directly facilitates chromatin openness and promotes transcription of housekeeping genes. Nat. Commun. 12, 4800 (2021).
Nikolaou, K. C., Moulos, P., Harokopos, V., Chalepakis, G. & Talianidis, I. Kmt5a controls hepatic metabolic pathways by facilitating RNA Pol II release from promoter-proximal regions. Cell Rep. 20, 909–922 (2017).
Li, Z., Nie, F., Wang, S. & Li, L. Histone H4 Lys 20 monomethylation by histone methylase SET8 mediates Wnt target gene activation. Proc. Natl Acad. Sci. USA 108, 3116–3123 (2011).
Benson, G. V. et al. Mechanisms of reduced fertility in Hoxa-10 mutant mice: uterine homeosis and loss of maternal Hoxa-10 expression. Development 122, 2687–2696 (1996).
Wang, J., Lu, X., Zhang, W. & Liu, G. H. Endogenous retroviruses in development and health. Trends Microbiol. 32, 342–354 (2023).
Johnson, W. E. Origins and evolutionary consequences of ancient endogenous retroviruses. Nat. Rev. Microbiol. 17, 355–370 (2019).
Chen, Y. G. & Hur, S. Cellular origins of dsRNA, their recognition and consequences. Nat. Rev. Mol. Cell Biol. 23, 286–301 (2022).
Kuriakose, T. & Kanneganti, T. D. ZBP1: innate sensor regulating cell death and inflammation. Trends Immunol. 39, 123–134 (2018).
Kirkwood, P. M. et al. Single-cell RNA sequencing redefines the mesenchymal cell landscape of mouse endometrium. FASEB J. 35, e21285 (2021).
Li, R. et al. Spatial transcriptomic profiles of mouse uterine microenvironments at pregnancy day 7.5dagg†. Biol. Reprod. 107, 529–545 (2022).
Vento-Tormo, R. et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 563, 347–353 (2018).
Wang, W. et al. Single-cell transcriptomic atlas of the human endometrium during the menstrual cycle. Nat. Med. 26, 1644–1653 (2020).
Evans, J. et al. Fertile ground: human endometrial programming and lessons in health and disease. Nat. Rev. Endocrinol. 12, 654–667 (2016).
Liu, M. et al. Menin directs regionalized decidual transformation through epigenetically setting PTX3 to balance FGF and BMP signaling. Nat. Commun. 13, 1006 (2022).
Cunha, G. R. Stromal induction and specification of morphogenesis and cytodifferentiation of the epithelia of the Mullerian ducts and urogenital sinus during development of the uterus and vagina in mice. J. Exp. Zool. 196, 361–370 (1976).
Cunha, G. R., Young, P. & Brody, J. R. Role of uterine epithelium in the development of myometrial smooth muscle cells. Biol. Reprod. 40, 861–871 (1989).
Kurita, T., Cooke, P. S. & Cunha, G. R. Epithelial-stromal tissue interaction in paramesonephric (Mullerian) epithelial differentiation. Dev. Biol. 240, 194–211 (2001).
Fueyo, R., Judd, J., Feschotte, C. & Wysocka, J. Roles of transposable elements in the regulation of mammalian transcription. Nat. Rev. Mol. Cell Biol. 23, 481–497 (2022).
Goke, J. et al. Dynamic transcription of distinct classes of endogenous retroviral elements marks specific populations of early human embryonic cells. Cell Stem Cell 16, 135–141 (2015).
Lawson, H. A., Liang, Y. & Wang, T. Transposable elements in mammalian chromatin organization. Nat. Rev. Genet. 24, 712–723 (2023).
Mi, S. et al. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 403, 785–789 (2000).
Wang, R. et al. Gut stem cell necroptosis by genome instability triggers bowel inflammation. Nature 580, 386–390 (2020).
Jiao, H. et al. Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature 580, 391–395 (2020).
Maelfait, J. et al. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J. 36, 2529–2543 (2017).
Nogusa, S. et al. RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated apoptosis to protect against influenza A virus. Cell Host Microbe 20, 13–24 (2016).
Thapa, R. J. et al. DAI senses influenza a virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe 20, 674–681 (2016).
Kuriakose, T. et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 1, aag2045 (2016).
Lee, S. et al. AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature 597, 415–419 (2021).
Yang, C. F. et al. Sexually dimorphic neurons in the ventromedial hypothalamus govern mating in both sexes and aggression in males. Cell 153, 896–909 (2013).
Yang, D. et al. ZBP1 mediates interferon-induced necroptosis. Cell. Mol. Immunol. 17, 356–368 (2020).
Zhao, H. et al. Stromal cells-specific retinoic acid determines parturition timing at single-cell and spatial-temporal resolution. iScience 26, 107796 (2023).
Acknowledgements
We are grateful to Professor Danny Reinberg (New York University School of Medicine) and Professor Wei Mo (School of Medicine, Zhejiang University) for providing Pr-set7f/f and Zbp1−/− transgenic mice. This work was supported by the National Key Research and Development Program of China (2022YFC2702500 and 2021YFC2700302 to H.W.), National Natural Science Foundation of China (82288102 and 82030040 to H.W., 82222026 to S.K., 82122026 and 32171117 to W.D., 82301886 to H.B.).
Author information
Authors and Affiliations
Contributions
H.B., N.D., Y.J., G.L., Y.G., X.L., Y.T., and H.C. performed experiments. H.W., W.D., and S.K. designed the study. H.B., Y.S., L.Z., J.L., and W.D. analyzed data. H.B., Y.S., N.D., H.W., and S.K. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Francesco DeMayo, Marc Morgan, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bao, H., Sun, Y., Deng, N. et al. PR-SET7 epigenetically restrains uterine interferon response and cell death governing proper postnatal stromal development. Nat Commun 15, 4920 (2024). https://doi.org/10.1038/s41467-024-49342-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-49342-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
