
Abstract
Precisely modulating the Ru-O covalency in RuOx for enhanced stability in proton exchange membrane water electrolysis is highly desired. However, transition metals with d-valence electrons, which were doped into or alloyed with RuOx, are inherently susceptible to the influence of coordination environment, making it challenging to modulate the Ru-O covalency in a precise and continuous manner. Here, we first deduce that the introduction of lanthanide with gradually changing electronic configurations can continuously modulate the Ru-O covalency owing to the shielding effect of 5s/5p orbitals. Theoretical calculations confirm that the durability of Ln-RuOx following a volcanic trend as a function of Ru-O covalency. Among various Ln-RuOx, Er-RuOx is identified as the optimal catalyst and possesses a stability 35.5 times higher than that of RuO2. Particularly, the Er-RuOx-based device requires only 1.837 V to reach 3 A cm−2 and shows a long-term stability at 500 mA cm−2 for 100 h with a degradation rate of mere 37 μV h−1.
Similar content being viewed by others

Introduction
Renewable-driven water electrolysis is widely recognized as a promising and sustainable route to scalable production of green hydrogen1,2,3,4. Proton exchange membrane water electrolysis (PEMWE) technology shows great potential on account of its high intermittent compatibility, low Ohmic resistance, high current density, low operating pressure, and limited side reactions5,6,7. However, the widespread application of PEMWE is obstructed by the lack of efficient and cost-affordable electrocatalysts for acidic oxygen evolution reaction (OER)8,9. OER with four proton-coupled electron transfer necessitates a high overpotential due to its sluggish kinetics, thereby decreasing the operating efficiency of PEMWE10,11. Besides, strong oxidative bias and extremely acidic corrosion collectively challenge the durability of available OER electrocatalysts for high-current-density operation12.
Currently, iridium (Ir)-based catalysts, e.g. IrO2, remain the only practical choice for anode electrocatalysts in PEMWE due to their well balance in durability and activity13,14. However, the spreading of PEMWE is drastically limited by the availability of Ir, which eventually leads to severe Ir shortage15,16. In this context, numerous efforts have been devoted to searching for alternatives to Ir, of which the most promising one is identified to be Ru-based oxides for their intrinsic high activity. Nevertheless, Ru-based OER catalysts always suffer from insufficient stability at industrial current density, which hampers their deployment in PEMWE for green hydrogen industry17. The instability of Ru-based anode originates from the over-oxidation of Ru species, generating soluble RuO4 under OER potentials18,19,20. Despite considerable efforts on stabilizing Ru for OER electrocatalysis, a significant performance gap remains to meet the industrial requirements21,22,23. To fill this gap, it would be more efficient to establish a fundamental and manageable strategy for stabilizing Ru-based OER electrocatalysts, instead of the conventional trial-and-error approach.
The stability of nanocrystalline RuO2-based catalysts is closely tied to the covalency of Ru–O bonds6. Weakening the Ru–O bond covalency can localize O 2p and Ru 3d orbitals below Fermi level, inhibiting lattice oxygen’s participation in OER and the formation of oxygen vacancies, thereby preventing excessive overoxidation of Ru species into soluble RuO4 during OER24. Meanwhile, excessively low Ru–O covalency is detrimental as it makes Ru be easily leached, leading to the direct demetallation of surface Ru and the subsequent structural degradation of RuO223. Prior studies focused on modulating the electronic structure of RuO2 using 3d, 4d, and 5d metal substrates/dopants6,21,25. However, as d orbital locates the outermost of transition metals, it is susceptible to external influences of crystal field and coordination environment26,27,28. For instance, the introduction of consecutive elements such as Ni (3d84s2), Cu (3d104s1), and Zn (3d104s2) into RuOx results in the transformation of their valence electrons into 3d8, 3d10, and 3d10, respectively20,29,30. Such external influences render it be extremely challenging to modulate the Ru–O covalency in a precise and continuous manner.
Herein, we reason that lanthanide (Ln)-group elements with the 4f orbital buried under 5s/p can minimize external influences and consequently enable precise and continuous tuning of Ru–O covalency for durable OER electrocatalysis. Density functional theory (DFT) calculations were conducted on Ln-RuOx systems, showing that the resultant 4d−2p−4f hybridization induces a continuously varying Ru–O covalency for dictating OER performance. Benefiting from an optimal Ru–O covalency, Er-RuOx was screened out as it demonstrated the largest formation energy of the lattice oxygen and Ru vacancy. The operando characterizations confirm the critical role of Er dopants in stabilizing the Ru–O structure for improved OER durability. Moreover, the up-shifted \({d}_{{z}^{2}}\)-state energy level (−0.855 eV) of Er-RuOx, relevant to the benchmark RuO2 (−2.171 eV), results in less electronic occupancy in the antibonding states and a stronger *OH adsorption, as validated by methanol molecular probe experiments, thereby significantly boosting the catalytic activity of Er-RuOx. This work validates a novel Ln-regulating approach for precisely and continuously modulating Ru–O covalency, aiding to more economic affordability of PEMWE in a hydrogen economy scenario.
Results
Lanthanide-regulating Ru–O covalency as OER descriptor
Ln elements offer a flexible avenue for fine-tuning and optimizing the electrocatalytic performance of catalysts due to the shielding effect of 5s/5p orbitals, gradient-filled 4f orbital electron configuration, rich electronic energy levels, and the ability to accommodate various coordination numbers. The gradient orbital coupling of Ru, O, and Ln shows promise for enhancing the OER performance based on group theory-directed symmetric analysis31,32. For [RuO6], the orbital coupling of valence 4d, 5s, and 5p orbitals with sp-mixing orbitals of coordinated O atoms produces metal-oxygen (M-O) bonding states composed of a1g, t1u, eg, with their corresponding (M-O)* antibonding states a1g*, t1u*, eg*, and t2g non-bonding terms. For [LnO6], the orbital coupling between Ln-4f and O-2p is contributed by t1u and t2u with their antibonding t1u* and t2u* terms, and a2u non-bonding terms. Thanks to the σ conjunction with O-p orbitals, the gradient orbital coupling of Ru–O–Ln can be formed (Fig. 1a), resulting in more flexible electronic interactions for electrocatalytic adaptation.

a The qualitative molecular orbital diagram obtained from [RuO6] and [LnO6]. b The ΔGO vacancy and (c) ΔGRu vacancy as a function of -ICOHP for Ln-RuOx. The upshift values of ICOHP indicate lower Ru–O covalency. d The reaction paths on Er-RuOx and RuO2 at 1.23 V. e Volcano plot for different electrocatalysts and corresponding structures.
We first carried out the DFT calculations with Ru–O–Ln configuration constructed based on the rutile RuO2(110) (Supplementary Figs. 1–4). The Bader charge analysis shows electron transfers from Ln to Ru, reducing the charge of Ru sequentially from 1.52 (RuO2) to 1.50 (Tm-RuOx), 1.48 (Er-RuOx), and 1.22 (Ho-RuOx) (Supplementary Fig. 5), which confirms the d-p-f orbital hybridization in Ln-RuOx. Through the analysis of bonding and antibonding orbital filling, the crystal orbital Hamilton population (COHP) and integrated COHP (ICOHP) calculations results demonstrate that the introduction of Ln can weaken the Ru–O bonding state occupancy from −1.614 eV (RuO2) to −1.523 eV (Ho-RuOx), −1.573 eV (Er-RuOx), and −1.574 eV (Tm-RuOx) (Supplementary Fig. 6). The fine tuning of Ru–O covalency originates from the shielding effect of 5 s/5p orbitals for Ln elements with gradient-filled 4 f orbital electron configuration.
To evaluate the stability of lattice oxygen and Ru, we calculated the formation energy of the lattice oxygen (ΔGO vacancy) and Ru vacancy (ΔGRu vacancy), which are utilized together to assess the stability of the electrocatalysts. As Ln dissolution would take place during OER process33, defective structures containing Ln vacancies were constructed. In the presence of Ln vacancies, Er-RuOx exhibits the highest ΔGO vacancy (0.33 eV), surpassing RuO2, Ho-RuOx, and Tm-RuOx by 0.29, 0.02, and 0.12 eV, respectively (Fig. 1b). Specifically, the regulation of Ru–O covalency leads to a modified ΔGRu vacancy, increasing from 2.58 eV in RuO2 to 3.49, 3.78, and 3.44 eV for Ho-RuOx, Er-RuOx, and Tm-RuOx, respectively (Fig. 1c). Considering the ΔGO vacancy and ΔGRu vacancy in combination, the stability of Ln-RuOx follows the volcanic-like trend as a function of Ru–O covalency. This in turn verifies our proposed design principle that the fine tuning of Ru–O covalency by Ln regulates stability.
To further investigate the mechanism by which Ln regulated the OER activity, we calculated the Gibbs free energy of oxygen intermediates during OER, yielding the theoretical overpotential for Ho-RuOx, Er-RuOx, Tm-RuOx, and RuO2. As shown in Fig. 1d and Supplementary Figs. 7 and 8, the potential determining step (PDS) for RuO2 is the evolution from *O to *OOH, with a calculated overpotential of 0.53 V, while the overpotential decreases to 0.48, 0.46, and 0.50 V for Ho-ErOx, Er-RuOx, and Tm-RuOx, respectively. The theoretical overpotential of various electrocatalysts are depicted in Fig. 1e employing a three-dimensional volcano-shaped plot that delineates the free energy difference between *O and *OH intermediates. Benefitting from the optimized Ru–O covalency, Er-RuOx demonstrates an enhanced *OH-binding strength (Supplementary Fig. 9) and provides near-optimal free energies for each intermediate, thus leading to the low theoretical OER overpotential. For the samples with both lower (Ho-RuOx, Tm-RuOx) and higher (RuO2) Ru–O covalency, the free energy increase of PDS is observed. Ru–O covalency of the catalysts reflects the Ru-*OH bonding interaction, suggesting an optimal Ru–O covalency that is neither too weak nor too strong is favorable for OER.
Overall, the incorporation of Ln can continuously optimize the Ru–O covalency within a narrow range, thereby controlling the dissolution kinetics of RuO2-based catalysts and influencing the energy barriers of key reaction steps.
Materials synthesis and characterization
As Er-RuOx theoretically possesses high OER activity and stability, it is selected for further investigation. To experimentally verify the predictions above, the Er-RuOx catalyst was prepared by a one-pot glucose-blowing method. Pure RuO2 was also synthesized employing the same method for comparison. The scanning electron microscopy (SEM) images (Supplementary Fig. 10) manifest that Er-RuOx possesses a porous sheet-like structure with a specific surface area (SBET) of 64.78 m2 g−1 (Supplementary Fig. 11). Furthermore, the porous sheets are composed of a large number of small nanoparticles (Supplementary Fig. 12). The X-ray diffraction (XRD) patterns reveal that Er-RuOx possesses a rutile-type structure with no distinct diffraction peaks related to ErOx (Supplementary Fig. 13), suggesting the successful incorporation of Er atoms into RuO2. High-resolution transmission electron microscopy (HRTEM) image shows Er-RuOx nanosheets consist of small nanoparticles with sizes around 5–10 nm, which are composed of the (110) facet-dominated RuO2 (Supplementary Fig. 14).
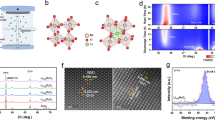
Furthermore, we utilized the aberration-corrected transmission electron microscopy (AC-TEM) to reveal the fine atomic-scale structure of the Er-RuOx catalyst. The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images (Fig. 2a, d) show the clear edge of the Er-RuOx grain. Figure 2b and Supplementary Fig. 15 display the surface plots of Er-RuOx, allowing the depiction of the intensity of the atomic columns. The atomic configuration of Er-RuOx can be clearly seen from the atomic HAADF-STEM images along the [001] and [110] zone axes (Fig. 2c, e), consistent with that of pure RuO2. The energy-dispersive spectra (EDS) line scan in Fig. 2f was recorded from the HAADF-STEM image of Er-RuOx (Fig. 2d, the green line OO’). The O, Ru, and Er signals are detected and recorded in the line scan profile. Especially, the signals resulting from Er only appears in the regions of Ru signal, further indicating that Er was successfully introduced into RuO2. The relatively weak signal intensity of Er element in contrast to that of Ru reveals the low Er content, matching with the result of EDS spectra (Supplementary Fig. 16). The element mapping images further demonstrate the coexistence and atomic-scale distribution of Ru and Er atoms (Fig. 2g–q). Er atoms are distributed within the catalyst, with a preference for surface localization, which contributes to the electrocatalytic performance as the Ru–O covalency can be regulated as discussed above.

a HAADF-STEM and b surface plot of Er-RuOx along [001] zone axes, scale bar: 2 nm. c Atomic STEM image along [001] zone axes and structural model (inset) of Er-RuOx, scale bar: 2 Å. d HAADF-STEM (scale bar: 2 nm) and (e) atomic STEM images (scale bar: 2 Å) of Er-RuOx along [110] zone axes. f EDS line scan of O, Ru, and Er signal recorded from the green line OO’ in (d). HAADF-STEM image and corresponding element mapping images of Er-RuOx along the (g–k) [110] and (l–p) [001] zone axes, scale bar: 2 nm. q Schematic illustration of Er-RuOx.
Electrocatalytic performance in three-electrode configurations and PEMWE devices
The OER performance of Er-RuOx and the control samples were measured in 0.5 M H2SO4 solution in a three-electrode system. As exhibited in Fig. 3a, Er-RuOx represents superior OER activity to commercial and home-made RuO2. Excitingly, the required overpotential to reach current density of 10 mA cm−2 is only 200 ± 8 mV on Er-RuOx, 87 ± 5 and 77 ± 7 mV lower than those of commercial RuO2 and home-made RuO2, respectively. Furthermore, when normalized to the electrochemical active area, the catalytic activity of Er-RuOx remains better than that of commercial RuO2 (Supplementary Fig. 19). The Er-RuOx catalyst also demonstrates a decreased Tafel slope of 45 mV dec−1 compared to commercial RuO2 (105 mV dec−1) (Fig. 3b), suggesting the boosted reaction kinetics.

a OER polarization curves of home-made RuO2, Er-RuOx and commercial RuO2 with iR-corrected (95%), where R was measured to be 1.1 ± 0.1 Ω. b Tafel plots of home-made RuO2, Er-RuOx and commercial RuO2. c Polarization curves of Er-RuOx and commercial RuO2 before (solid line) and after (dash line) 30,000 CV cycles. d The CP curves of Er-RuOx and commercial RuO2 at 10 mA cm−2. e Schematic diagram of the PEMWE electrolyzer. f Polarization curves of the Er-RuOx and commercial RuO2-based PEMWE at 80 °C. g The CP curves of Er-RuOx-based PEMWE electrolyzer operated at 200, 500, and 1000 mA cm−2, respectively.
The stability of the as-prepared catalysts was investigated by cyclic voltammetry (CV) between 1 V and 1.45 V vs. RHE. Er-RuOx demonstrates a much smaller attenuation than commercial RuO2 after 30,000 cycling tests (Fig. 3c). The concentration of dissolved Ru after 30,000 CV cycles for Er-RuOx was measured to be 13.7 ppb, which was much lower than that of commercial RuO2 (40.9 ppb). These results suggest that the incorporation of Er suppresses the dissolution of RuOx. In addition, the catalytic durability was evaluated by chronopotentiometry (CP) at 10 mA cm−2 (Fig. 3d), demonstrating a more pronounced stability advantage of Er-RuOx over commercial RuO2. In detail, the required overpotential of commercial RuO2 increased by 674 mV after 73 h stability test at 10 mA cm−2, which was ~35.5 times higher than that of Er-RuOx, verifying the beneficial role of Er on catalytic stability. The OER performance of Ho-RuOx and Tm-RuOx was shown in Supplementary Figs. 22–24, being in good agreement with DFT prediction. As shown in Supplementary Tables 6 and 7, both the activity and stability of Er-RuOx were higher than those of the recently reported Ru-based electrocatalysts7,22,23,25, confirming the economic efficiency of Er-RuOx.
To investigate the application potential of Er-RuOx for water electrolysis, we constructed a PEMWE device (Nafion 117 membrane) using Er-RuOx and commercial Pt/C as the anode and cathode catalyst, respectively (Fig. 3e). The current-voltage curves (without iR compensation) in Fig. 3f clearly demonstrate the superior water electrolysis performance of the Er-RuOx-based PEMWE in comparison to the commercial RuO2||Pt/C PEMWE device. Specifically, the Er-RuOx-based electrolyzer (at 80 °C) required only 1.590, 1.713, and 1.837 V to reach an industrial current density of 1, 2, and 3 A cm−2, respectively, outperforming the PEMWE using the other state-of-the-art RuOx-based catalysts (Supplementary Table 8). Moreover, the PEMWE employing the Er-RuOx catalyst achieves an efficiency of approximately 80% at 1 A cm−2 (Supplementary Fig. 26), with an estimated cost of about US$ 0.85 per kg of H2, which is significantly below the US Department of Energy (DOE)’s target of US$ 2 per kg of H234. The stability of the Er-RuOx||Pt/C PEMWE device was evaluated at 200, 500, and 1000 mA cm−2, respectively, and no significant activity decay was observed over the device after a 100-h stability test for each condition (Fig. 3g). The degradation rate at 500 mA cm−2 is mere 37 μV·h−1, demonstrating the application potential of Er-RuOx for green hydrogen production.
Origin of the excellent performance of Er-RuOx
X-ray photoelectron spectroscopy (XPS) was conducted to determine the chemical composition and valence states of the as-prepared catalysts. In the Ru 3p XPS spectrum (Supplementary Fig. 27), the peaks at 463.7 and 463.1 eV can be attributed to Ru3+ and Ru4+, respectively35. Compared with RuO2, the Ru4+ ratio in Er-RuOx is lower than that in RuO2, indicating a lower oxidation state of Ru in Er-RuOx. To explore the interaction of Er and Ru, X-ray absorption spectroscopy (XAS) was utilized to probe the atomic and coordination environment. The X-ray absorption near-edge structure spectra (XANES) of Ru K-edge (Fig. 4a) reveal that the absorption threshold position of Er-RuOx is higher than that of the Ru foil but lower than that of RuO2. The calculated oxidation state of Ru in Er-RuOx is 3.80 (Supplementary Fig. 28), consistent with the results of XPS and Bader charge. Moreover, the Ru K-edge Fourier transform extended X-ray absorption fine structure (FT-EXAFS) spectrum of Er-RuOx (Fig. 4b) displays a peak located at ~1.98 Å, assigned to the Ru–O bonds. This peak exhibits a positive shift towards longer interatomic distances in comparison to that of RuO2 because of the slightly weakened Ru–O covalency (Supplementary Table 9), matching well with theoretical calculation predictions. The Wavelet transform analysis also demonstrates that Ru–O coordination in Er-RuOx is similar to that of RuO2, while no Ru–Ru bond is detected in Er-RuOx, aligning with the FT-EXAFS results (Fig. 4g–i).

a Ru K-edge XANES spectra of Ru foil, RuO2, and Er-RuOx. b EXAFS spectra of Ru foil, RuO2, and Er-RuOx. In situ Raman spectra obtained under various applied potential on (c) RuO2 and (d) Er-RuOx. e Ru K-edge XANES spectra of Er-RuOx with different applied bias. f The variation trend of Ru oxidation states in RuO2 and Er-RuOx under different potentials. WT-EXAFS of (g) Ru foil, (h) RuO2, and (i) Er-RuOx.
We then conducted the in situ Raman spectroscopy measurement (Supplementary Fig. 29) to elucidate the structure evolution during OER. Raman spectra shows the nanocrystalline nature of RuO2 and Er-RuOx (Fig. 4c, d), with two major peaks located at ~430 and 600 cm−1, corresponding to the Eg and A1g, respectively. As the potential increases from open circuit potential (OCP) to 1.8 V vs. RHE, Er-RuOx maintains a nearly constant Raman shift, indicating the stability of Ru–O configurations. In contrast, a ~ 10 cm−1 red shift is observed in RuO2, suggesting the shrinkage in Ru–O bonding length during OER.
Furthermore, Ru K-edge XANES of Er-RuOx and RuO2 were collected with applied potential rise from 1.3 to 1.7 V vs. RHE to investigate the changes in local Ru atomic structure and chemical coordination. As presented in Fig. 4e, f and Supplementary Fig. 30, Er-RuOx and RuO2 exhibit significant differences in the variation of Ru oxidation sates, especially at high bias. In detail, when the applied voltage is 1.3 V, the oxidation state of Ru in Er-RuOx increases from 3.80 to 4.16, followed by a negligible change when potential further increased to 1.7 V. In contrast, the oxidation states of Ru in RuO2 steadily increases to 4.36 as the applied bias varies from 1.3 to 1.7 V, showing no sign of stabilization. The result suggests that the stable structure of Er-RuOx can be maintained at high oxidation potentials, whereas RuO2 undergoes significant structural evolution. The Ru 3d peak in Er-RuOx XPS spectrum after stability test is slightly positive-shift (Supplementary Fig. 31), indicating that the surface of Er-RuOx is oxidized to a higher valence state, consistent with the XANES results. TEM images after the stability test confirm that the crystalline structure of Er-RuOx (Supplementary Fig. 32) is well preserved, indicating no significant structure deterioration occurred under the strong oxidative bias.
Adsorption behavior analysis of Er-RuOx with oxygen intermediates
To rationalize the improved OER performance on Er-RuOx, the adsorptions of Er-RuOx and RuO2 to oxygen intermediates were evaluated. As shown in Fig. 5a, Er-RuOx exhibits a lower d-band center of Ru (−1.945 eV) than that of the pure RuO2 (−1.900 eV), indicating that the 4f-2p-4d orbital hybridization slightly changes the electronic environment of Ru d orbitals. Further, the \({d}_{{z}^{2}}\) orbitals can be hybridized into σ and σ* with *OH intermediates. The Ru \({d}_{{z}^{2}}\)-state energy level (−0.855 eV) of Er-RuOx up-shifts to the Fermi level relative to that of the RuO2 (−2.171 eV) (Fig. 5b), thereby resulting in less electronic occupancy in the antibonding states and stronger *OH adsorption (Fig. 5c). As shown in Fig. 5d, more charge accumulation toward *OH verifies a stronger electron transfer behavior between Er–O–Ru and *OH, confirming the strengthened *OH adsorption.

a The calculated PDOS of Ru d, O p, and Er f orbitals of Er-RuOx. b The PDOS of Ru \({d}_{{z}^{2}}\) orbital of RuO2 and Er-RuOx. c Schematic diagram of orbital hybridization between \({d}_{{z}^{2}}\) orbital of RuO2 and Er-RuOx and the *OH bonding orbitals. d Charge density difference of *OH-adsorbed Er-RuOx (left) and RuO2 (right). Yellow and blue iso-surfaces represent charge accumulation and depletion, respectively. Polarization curves of (e) RuO2 and (f) Er-RuOx in 0.5 M H2SO4 solution with (dashed lines) and without (solid lines) 1 M methanol. g Current difference between the polarization curves in 0.5 M H2SO4 solution with and without 1.0 M methanol for Er-RuOx and RuO2. ATR-SEIRAS analysis of (h) commercial RuO2 and (i) Er-RuOx.
The strengthened *OH adsorption was also experimentally verified using methanol as a probe. The methanol oxidation reaction (MOR) follows a well-established mechanism, in which methanol molecules tend to nucleophilically attack the electrophilic *OH. As a result, MOR is more active on surfaces with stronger *OH adsorption36. When 1.0 M methanol was introduced into the 0.5 M H2SO4 solution, the current densities of Er-RuOx and RuO2 showed a substantial increase compared to those before the addition of methanol, attributed to the methanol electrooxidation (Fig. 5e, f). The difference in current densities induced by MOR, which was directly proportional to the number of charges transferred, was quantified by calculating the filled area between the curves. The bigger current difference observed between the MOR and OER over Er-RuOx than that of RuO2 suggested its stronger MOR competition reaction, verifying the enhanced *OH adsorption on Er-RuOx (Fig. 5g).
To acquire the information of oxygen intermediates for a more comprehensive mechanistic understanding, the in situ attenuated total reflection-surface enhanced infrared absorption spectroscopy (ATR-SEIRAS) was performed using a home-made electrochemical cell. The ATR-SEIRAS of both RuO2 and Er-RuOx were measured between 1000 and 1400 cm−1 from 1.3 V to 1.7 V vs. RHE. As shown in Fig. 5h, a potential-dependent peak at 1211 cm−1 for RuO2, corresponding to the stretching vibration of *OOH species18, became more prominent as the potential increased from 1.5 V to 1.7 V. For Er-RuOx, the peak of *OOH species was emerged at 1.4 V (Fig. 5i), in accordance with the lower PDS of *O to *OOH (Fig. 1d). Meanwhile, the emergence of *OOH species indicated that the adsorbate evolution mechanism pathway dominated O2 generation over Er-RuOx.
Discussion
In summary, we demonstrate that precisely tuning of the Ru–O covalency in RuOx can be achieved by introducing Ln elements through d-p-f orbital hybridization. Benefiting from the optimized Ru–O covalency, Er-RuOx is screened out and exhibits the high catalytic stability, significantly outperforming pristine RuO2 by orders of magnitude. The operando characterizations indicate that the oxidation state of Ru in Er-RuOx initially increases as the applied potential increases and then remains nearly constant. In contrast, the oxidation state of Ru in RuO2 continuously rises with no sign of stabilization. DFT calculations and methanol molecular probe experiments validate the stronger *OH adsorption on Er-RuOx relative to that on RuO2, thus leading to the enhanced OER activity. The PEMWE (Nafion 117 membrane) employing Er-RuOx as the anode catalyst requires only 1.837 V to reach 3 A cm−2 and exhibit long-term stability at 500 mA cm−2 for 100 h with a degradation rate of mere 37 μV h−1. This study provides a principled catalyst design framework for the precise Ru–O covalency control, thereby guiding the development of ruthenium-based catalysts suitable for practical implementation in PEMWE systems.
Methods
Materials
Ruthenium chloride (RuCl3, 99.5%) was purchased from Beijing InnoChem Science & Technology Co., Ltd. Holmium nitrate pentahydrate (Ho(NO3)3·5H2O, 99.99%), erbium nitrate pentahydrate (Er(NO3)3·5H2O, 99.9%) and thulium nitrate hexahydrate (Tm(NO3)3·6H2O, 99.9%) was purchased from Shanghai Macklin Biochemical Co., Ltd. Urea (AR) and glucose (AR) were obtained from Beijing Tong Guang Fine Chemicals Company. Commercial Pt/C (70% Pt) was obtained from Johnson Matthey Company. Nafion 117 membrane (thickness: 0.18 mm) was obtained from DuPont Co. All reagents were used without further purification.
Synthesis of Ln-RuOx and RuO2
In the typical synthesis of Ln-RuOx, 40 mg RuCl3 and a certain amount of Ln(NO3)3 were added into 5 mL deionized water with 1 g urea and 5 g glucose (keeping the atomic ratio of Ln:Ru at 1:10). The mixture was stirred until a homogeneous solution was attained. Subsequently, it was subjected to heating at 140 °C for 8 h in an oven to form a porous foam, and then annealed at 500 °C for 10 h in the air to obtain Ln-RuOx. For comparison, pure RuO2 was also prepared without the addition of Ln(NO3)3.
Electrochemical measurements
All electrochemical measurements were conducted with the CHI 760E electrochemical workstation employing a three-electrode cell at room temperature. The as-prepared catalysts (2 mg) were dispersed in a mixture of 980 μL isopropanol and 20 μL Nafion D-521 (5 wt%) solution. After ultrasonication for 1 h, the homogeneous ink was carefully dropped onto the carbon paper (1 cm2) to obtain the working electrode with a desirable loading of 0.5 mg cm−2. The reference electrode was Hg/Hg2SO4, calibrated in a three-electrode system in which Pt wires served as both working electrode and counter electrode, and H2-saturated 0.5 M H2SO4 solution was employed as the electrolyte. Subsequently, CV was measured at a scan rate of 1 mV s−1. The average potential at which the current crosses zero was determined as the thermodynamic potential relative to Hg/Hg2SO433. All potentials were calibrated to RHE by the equation: E (vs. RHE) = E (vs. Hg/Hg2SO4) + 0.707 V. The graphite rod was used as the counter electrode. The polarization curves were performed at a scan rate of 5 mV s−1 in 0.5 M H2SO4 solution (pH ≈ 0.3). All linear sweep voltammetry (LSV) curves measured in three-electrode cell were iR-corrected (95%) unless otherwise stated, where R was measured to be 1.1 ± 0.1 Ω. During electrolyte preparation, 13.6 mL sulfuric acid (98%) was added to a beaker containing a suitable volume of deionized water, followed by adjustment of the volume to 500 mL in a volumetric flask and shake to ensure thorough mixing. The electrolyte is freshly prepared and promptly utilized. Electrochemical impedance spectroscopy was performed at 1.485 V vs. RHE in the frequency range from 10−2 Hz to 105 Hz. CVs at various scan rates (namely, 10, 20, 30, 40, and 50 mV s−1) were performed to calculate the electrochemical active surface area (ECSA), which was proportional to the double layer capacitance (Cdl). Assuming that the specific capacitance of a flat surface was ~40 μF for 1 cm2 of real surface area, then the ECSA was estimated as: ECSA =\(\,\frac{{C}_{{dl}}({mF}{{cm}}^{-2})}{40\mu F{{cm}}^{-2}{per}{{cm}}_{{ECSA}}^{2}}\).
PEMWE tests
The PEMWE was assembled with Er-RuOx or commercial RuO2 as anode, with a loading of ~3 mg cm−2. Commercial Pt/C (70 wt% Pt) was utilized as the cathode catalyst (~0.5 mgPt cm−2). Pt-coated Ti fiber was used as gas diffusion layers. Nafion 117 was used as the proton exchange membrane (PEM), which was sequentially treated with H2O2 and 0.5 M H2SO4 at 80 °C for 1 h. The anode and cathode catalyst ink were separately prepared by dispersing the catalyst in a mixture of Nafion (5%), isopropanol, and distilled water. All the cathode ink was sprayed onto the PEM. As for anode, half of catalyst ink was sprayed onto the PEM, while the remaining half was sprayed onto the surface of gas diffusion layer. The catalyst-loaded PEM and gas diffusion layers were then hot-pressed together at 120 °C for 2 min under a pressure of 2 MPa to fabricate the membrane electrode assembly (MEA), which was sandwiched by two Ti bipolar plates to complete a PEMWE device. Each Ti bipolar plate featured a serpentine flow channel with 1 cm × 1 cm reactive area. The PEMWEs were operated at 80 °C utilizing distilled water as the electrolyte, which was delivered to the anode by a peristaltic pump. All measurements in PEMWEs were recorded without iR-correction.
In situ ATR-SEIRAS measurements
The in situ ATR-SEIRAS measurement was carried out on Bruker 70 V Fourier-transform infrared (FTIR) spectrometer. The measurement featured a spectral resolution of 8 cm−1, with 64 interferograms co-added for each spectrum. The preparation of working electrodes comprised of two steps: firstly, an ultra-thin Au film was chemical deposited on the Si crystal to enhance the IR signal and facilitate electron conduction; secondly, the catalyst ink was dropped onto the Au film with a loading of 0.1 mg cm−2. The Si crystal loaded with catalyst was placed onto a spectro-electrochemical three-electrode cell. Ag/AgCl electrode and Pt wire served as the reference and counter electrodes, respectively. The 0.1 M HClO4 solution was used as the electrolyte. All SEIRAS spectra were obtained during the LSV test.
DFT calculations
The calculations were performed employing the density functional theory as implemented in the Vienna ab initio simulation package (VASP)37. The exchange-correlation function was described using the generalized gradient approximation (GGA)38 parameterized by the Perdew–Burke–Ernzerhof (PBE). The cut-off energy for the plane wave basis was set to 450 eV. RuO2(110) surface (Supplementary Fig. 1) was modeled by a 2 × 2 × 2 supercell (with 55 O atoms and 24 Ru atoms), in which the top three atomic layers are allowed to relax. As for Ln-RuOx, two Ru atoms in the RuO2(110) model was substituted by Ln atoms (Supplementary Figs. 2–4 and Supplementary Data 1), leading to a chemical composition of 8.3 at percentage Ln and 91.7 at percentage Ru. A vacuum spacing of 20 Å was set along the z-direction to prevent the interaction between the slab and its periodic motif. The termination of RuO2(110) and Ln-RuOx has all Ru filled with oxygen but one coordinatively unsaturated site Ru unfilled, which is the active site for intermediates adsorption. The Monkhorst-Pack method was used for sampling the Brillouin zone with a 3 × 3 × 1 mesh. The geometry relaxation and convergence criteria for the electronic structure were 0.05 eV/Å and 5 × 10−5 eV, respectively. The free energies of the reaction steps were calculated by the equation: \({\varDelta G}_{{ads}}=\,{\varDelta E}_{{ads}}+\,{\varDelta E}_{{ZPE}}-T\varDelta S\), where \({\varDelta E}_{{ads}}\) is the adsorption energy of intermediates, and T is temperature. \({\varDelta E}_{{ZPE}}\) and ΔS represent the energy variance in zero-point energy and entropy, respectively. When calculating the formation energy of Ru vacancies, the lost Ru atoms originate from unsaturated surface coordination sites, consistently from the same position across all models. In the calculation of oxygen vacancy formation energy, both Ln-RuOx and RuO2(110) lose oxygen atoms surrounding unsaturated Ru coordination sites on the surface. ICOHP and Bader charge represent the values at active site.
Data availability
The data that support the conclusions of this study are available within the paper and Supplementary information. Source data are provided with this paper.
References
Chong, L. et al. La- and Mn-doped cobalt spinel oxygen evolution catalyst for proton exchange membrane electrolysis. Science 380, 609–616 (2023).
Seh, Z. W. et al. Combining theory and experiment in electrocatalysis: insights into materials design. Science 355, eaad4998 (2017).
Guo, J. et al. Direct seawater electrolysis by adjusting the local reaction environment of a catalyst. Nat. Energy 8, 264–272 (2023).
Li, F. & Baek, J.-B. Active site engineering accelerates water electrolysis. Nat. Synth. 3, 558–559 (2024).
Shi, Z. et al. Enhanced acidic water oxidation by dynamic migration of oxygen species at the Ir/Nb2O5-x catalyst/support interfaces. Angew. Chem. Int. Ed. 61, e202212341 (2022).
Shi, Z. et al. Customized reaction route for ruthenium oxide towards stabilized water oxidation in high-performance PEM electrolyzers. Nat. Commun. 14, 843 (2023).
Liu, H. et al. Eliminating over-oxidation of ruthenium oxides by niobium for highly stable electrocatalytic oxygen evolution in acidic media. Joule 7, 558–573 (2023).
Li, L., Wang, P., Shao, Q. & Huang, X. Recent progress in advanced electrocatalyst design for acidic oxygen evolution reaction. Adv. Mater. 33, 2004243 (2021).
Wang, Q. et al. Long-term stability challenges and opportunities in acidic oxygen evolution electrocatalysis. Angew. Chem. Int. Ed. 62, e202216645 (2023).
Ge, S. et al. A robust chromium-iridium oxide catalyst for high-current-density acidic oxygen evolution in proton exchange membrane electrolyzers. Energy Environ. Sci. 16, 3734–3742 (2023).
Liu, R.-T. et al. Recent advances in proton exchange membrane water electrolysis. Chem. Soc. Rev. 52, 5652–5683 (2023).
Chen, F.-Y., Wu, Z.-Y., Adler, Z. & Wang, H. Stability challenges of electrocatalytic oxygen evolution reaction: from mechanistic understanding to reactor design. Joule 5, 1704–1731 (2021).
Dang, Q. et al. Iridium metallene oxide for acidic oxygen evolution catalysis. Nat. Commun. 12, 6007 (2021).
Chen, S. et al. Reconstructed Ir-O-Mo species with strong Brønsted acidity for acidic water oxidation. Nat. Commun. 14, 4127 (2023).
Gao, J., Tao, H. & Liu, B. Progress of nonprecious-metal-based electrocatalysts for oxygen evolution in acidic media. Adv. Mater. 33, 2003786 (2021).
Li, L. et al. Electrochemically modifying the electronic structure of IrO2 nanoparticles for overall electrochemical water splitting with extensive adaptability. Adv. Energy Mater. 10, 2001600 (2020).
Cao, L. et al. Dynamic oxygen adsorption on single-atomic ruthenium catalyst with high performance for acidic oxygen evolution reaction. Nat. Commun. 10, 4849 (2019).
Yao, Y. et al. Engineering the electronic structure of single atom Ru sites via compressive strain boosts acidic water oxidation electrocatalysis. Nat. Catal. 2, 304–313 (2019).
Zagalskaya, A. & Alexandrov, V. Role of defects in the interplay between adsorbate evolving and lattice oxygen mechanisms of the oxygen evolution reaction in RuO2 and IrO2. ACS Catal. 10, 3650–3657 (2020).
Wu, Z.-Y. et al. Non-iridium-based electrocatalyst for durable acidic oxygen evolution reaction in proton exchange membrane water electrolysis. Nat. Mater. 22, 100–108 (2023).
Wang, Y. et al. Unraveling oxygen vacancy site mechanism of Rh-doped RuO2 catalyst for long-lasting acidic water oxidation. Nat. Commun. 14, 1412 (2023).
Galyamin, D. et al. Active and durable R2MnRuO7 pyrochlores with low Ru content for acidic oxygen evolution. Nat. Commun. 14, 2010 (2023).
Jin, H. et al. Safeguarding the RuO2 phase against lattice oxygen oxidation during acidic water electrooxidation. Energy Environ. Sci. 15, 1119–1130 (2022).
Miao, X. et al. Quadruple perovskite ruthenate as a highly efficient catalyst for acidic water oxidation. Nat. Commun. 10, 3809 (2019).
Zhu, W. et al. Direct dioxygen radical coupling driven by octahedral ruthenium-oxygen-cobalt collaborative coordination for acidic oxygen evolution reaction. J. Am. Chem. Soc. 145, 17995–18006 (2023).
Li, L. et al. Spin-polarization strategy for enhanced acidic oxygen evolution activity. Adv. Mater. 35, 2302966 (2023).
Li, L. et al. Compensating electronic effect enables fast site-to-site electron transfer over ultrathin RuMn nanosheet branches toward highly electroactive and stable water splitting. Adv. Mater. 33, 2105308 (2021).
Zheng, X. et al. Ru-Co pair sites catalyst boosts the energetics for the oxygen evolution reaction. Angew. Chem. Int. Ed. 61, e202205946 (2022).
Su, J. et al. Assembling ultrasmall copper-doped ruthenium oxide nanocrystals into hollow porous polyhedra: highly robust electrocatalysts for oxygen evolution in acidic media. Adv. Mater. 30, 1801351 (2018).
Zhang, D. et al. Construction of Zn-doped RuO2 nanowires for efficient and stable water oxidation in acidic media. Nat. Commun. 14, 2517 (2023).
Wang, X. et al. Engineering 3d-2p-4f gradient orbital coupling to enhance electrocatalytic oxygen reduction. Adv. Mater. 34, 2206540 (2022).
Wang, X. et al. Spin-selective coupling in Mott-Schottky Er2O3-Co boosts electrocatalytic oxygen reduction. Small Methods 7, 2300100 (2023).
Li, L. et al. Optimizing the electronic structure of ruthenium oxide by neodymium doping for enhanced acidic oxygen evolution catalysis. Adv. Funct. Mater. 33, 2213304 (2023).
Bender, G. & Dinh, H. N. HydroGEN: Low-Temperature Electrolysis (LTE) and LTE/Hybrid Supernode (No. NREL/PR-5900-659). (National Renewable Energy Lab.(NREL), 2020).
Hu, Y. et al. Single Ru atoms stabilized by hybrid amorphous/crystalline FeCoNi layered double hydroxide for ultraefficient oxygen evolution. Adv. Energy Mater. 11, 2002816 (2021).
Tao, H. B. et al. A general method to probe oxygen evolution intermediates at operating conditions. Joule 3, 1498–1509 (2019).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Acknowledgements
S.J.G. acknowledge the fundings from National Natural Science Foundation of China (Nos. 52333010, 52025133, 22309004, 52261135633, 52303363, 52302207, 22205010, 22305010, 22105007), National Key R&D Program of China (No. 2022YFE0128500), China National Petroleum Corporation-Peking University Strategic Cooperation Project of Fundamental Research, the Beijing Natural Science Foundation (No. Z220020), New Cornerstone Science Foundation through the XPLORER PRIZE, CNPC Innovation Found (No. 2021DQ02-1002), China National Postdoctoral Program for Innovative Talents (No. BX20220009), China Postdoctoral Science Foundation (Nos. 2022M720225, 2023M730029, 2022M710187, 2023M730051, 2020M670018) and Yunnan Fundamental Research Projects (grant NO. 202401AT070370). This work was carried out with the support of 1W1B beamline at Beijing Synchrotron Radiation Facility. The authors thank the photoemission photoendstations BL14W1 in the Shanghai Synchrotron Radiation Facility (SSRF) for the help with characterizations.
Author information
Authors and Affiliations
Contributions
S.J.G. conceived and supervised the project. M.C.L. guided and supervised the whole research. L.L. and G.W.Z. performed the experiments, collected and analyzed the data. C.H.Z., F.L., H.L. and D.W.W. participated in the PEMWE tests. Y.J.T., Y.H., Y.X.L. and C.S.S. helped with the Operando ATR-SEIRAS measurements. L.Y.Z., Q.Z.H., R.J.Z. and N.Y. participated in part of basic experiments. L.L. wrote the manuscript. All authors took part in the discussion of data and gave comments on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, L., Zhang, G., Zhou, C. et al. Lanthanide-regulating Ru-O covalency optimizes acidic oxygen evolution electrocatalysis. Nat Commun 15, 4974 (2024). https://doi.org/10.1038/s41467-024-49281-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-49281-2
This article is cited by
-
Operando identification of the oxide path mechanism with different dual-active sites for acidic water oxidation
Nature Communications (2024)
-
Durable ruthenium oxide catalysts for water oxidation reaction
Science China Chemistry (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
