
Abstract
DNAzymes – synthetic enzymes made of DNA — have long attracted attention as RNA-targeting therapeutic agents. Yet, as of now, no DNAzyme-based drug has been approved, partially due to our lacking understanding of their molecular mode of action. In this work we report the solution structure of 8–17 DNAzyme bound to a Zn2+ ion solved through NMR spectroscopy. Surprisingly, it turned out to be very similar to the previously solved Pb2+-bound form (catalytic domain RMSD = 1.28 Å), despite a long-standing literature consensus that Pb2+ recruits a different DNAzyme fold than other metal ion cofactors. Our follow-up NMR investigations in the presence of other ions — Mg2+, Na+, and Pb2+ – suggest that at DNAzyme concentrations used in NMR all these ions induce a similar tertiary fold. Based on these findings, we propose a model for 8–17 DNAzyme interactions with metal ions postulating the existence of only a single catalytically-active structure, yet populated to a different extent depending on the metal ion cofactor. Our results provide structural information on the 8-17 DNAzyme in presence of non-Pb2+ cofactors, including the biologically relevant Mg2+ ion.
Similar content being viewed by others


Introduction
Nature has assigned the role of biological catalysts to proteins and to a lesser extent RNA molecules. In spite of this state of affairs conserved throughout biology, artificially engineered DNA catalysts (DNA enzymes; DNAzymes) have emerged in the 1990s1,2. The more inert chemical nature and inherent stability of such enzymes makes them most appealing for industrial and biomedical applications. DNAzymes whose catalytic activity is related to RNA processing (site-specific cleavage, ligation, phosphorylation, etc.) generate the most promise in the fields of biotechnology and medicine3. RNA-cleaving DNAzymes especially have already been widely applied as biosensors for targets, as diverse as, metal ions4 up to whole bacterial cells5. However, ever since their discovery, the most anticipated use of RNA-cleaving DNAzymes is mRNA-level control in vivo. Unfortunately, DNAzyme application in therapy remains as of yet an unfulfilled promise, due to factors such as low concentration of their cofactors -- divalent metal ions — in cells or the difficulty to achieve multiple catalytic turnover in these conditions6,7. Rationally redesigning DNAzyme molecules to amend for these shortcomings is currently a very difficult task, as our knowledge regarding their molecular mode of action remains limited. Only a deep structural and functional understanding of the most prominent RNA-cleaving DNAzymes can pave the path towards their successful therapeutic usage.
The 8-17 DNAzyme (Fig. 1A) is an archetypical RNA-cleaving DNA enzyme, first reported in 19972 and later repeatedly rediscovered in the presence of different metal ion cofactors8. Its constant reappearance in in vitro selections performed under very diverse conditions makes it the most versatile and pertinent DNAzyme identified to date. While originally discovered in the presence of Mg2+2, it was shown capable of using many other divalent ions as cofactors with the following trend of relative catalytic rates8: Pb2+ » Zn2+ » Mn2+ ≈ Co2+ > Cd2+ > Ni2+ > Mg2+ ≈ Ca2+ > Sr2+ ≈ Ba2+. 8–17 is also the most thoroughly investigated DNAzyme in terms of structure, sequential requirements, and reactivity8. Structural studies using techniques such as FRET9,10 and CD spectroscopy11 uncovered a puzzling peculiarity in 8–17 DNAzyme’s mode of action (Fig. 1D), by suggesting that it is capable to carry out its catalytic role in two distinct folded states, selected based on the cofactor present. Both techniques concluded that Pb2+ binds directly to the apo form of the enzyme without substantially changing its open structure, and the catalytic reaction proceeds from this state. In contrast, in case of other cofactors - most prominently Zn2+ and Mg2+ - binding is accompanied by a large-scale conformational rearrangement within the DNAzyme towards a more compact structure and only this distinct fold supports catalysis with these cofactors. Thus, it appeared that in order to understand the 8–17 DNAzyme not one, but two catalytic mechanisms had to be elucidated, yet both of them long remained elusive in the absence of high resolution structural data8.

A consensus structure, (B) construct used in the crystallographic study13, (C) construct used in this NMR investigation, (D) current paradigm for 8–17 DNAzyme interplay with divalent metal ion cofactors, (E) the tertiary fold observed in Pb2+-bound crystallographic structure, (F) the proposed catalytic mechanism.
While attempts at high-resolution structural characterization of DNAzyme molecules have long remained unsuccessful, recent years have brought major progress in the field. The first crystal structure of an RNA-ligating DNAzyme 9DB1 was reported in 201612. This was followed by the crystal structures of the apo and Pb2+ bound forms of 8–17 DNAzyme13 (sharing the same overall fold; Fig. 1E) and most recently by the NMR structure of the 10–23 DNAzyme14. This last report has actually brought results that fulfill the promise of structure-informed DNAzyme engineering by proposing a single-atom substitution that rendered 10–23 several times more active14. Similarly, the crystal structures of 8–17 DNAzyme have reinvigorated the efforts to deeper understand its molecular mode of action. Based on the crystallographic report13 and several follow-up experimental15,16,17,18 and computational19,20 studies a consensus regarding the catalytic mechanism in the presence of Pb2+ started to emerge (Fig. 1F). First, the DNAzyme locks the nucleophile (2’OH group of rN18) and the scissile phosphate into a conformation that enables the in-line attack of the deprotonated nucleophile on the phosphorus atom13. Such a catalytic strategy is referred-to as α-catalysis21,22. In the crystal structure of 8–17 DNAzyme the entire tertiary fold appears to be stabilizing this particular conformation, with two substrate-binding arms containing the residues N1 and N18 forced into in a V-like orientation through co-axial stacking with two short helices present within the catalytic domain (Fig. 1E). The presence of one of those helices (Helix 4 in Fig. 1E) and the pseudo-knot it introduces within the catalytic domain, were only first made apparent in the crystallographic structure, leading to a previously unexpected intricate and rigid fold. Second, guanosine residue G14 was identified13,15 as the general-base that facilitates the deprotonation of the nucleophile (γ-catalysis). Third, the metal ion was in-turn proposed to assist in the protonation of the O5’ leaving group (δ-catalysis) and/or to electrostatically stabilize the negatively charged transition state (β-catalysis). The Pb2+ ion may also be implicated in the disruption of non-productive hydrogen bonds formed by the nucleophile in the pre-catalytic state20. Thus, 8-17 is now believed to utilize an intricate combination of different catalytic strategies to achieve its high RNA cleavage rates in the presence of Pb2+, with its active site following the L-platform architecture found in multiple RNA-cleaving nucleic acid enzymes23.
However, it remains unclear to what extent these mechanisms are relevant for the 8–17 DNAzyme catalysis in the presence of other divalent metal ions cofactors, more important for activity in vivo, due to the global structural change that the latter impose on the DNAzyme. Some similarities are already known to be present, e.g., G14 was shown15 to act as a general base also in the presence of Mg2+, but no complete mechanistic model can be proposed without a high resolution structural information on the DNAzyme fold that these ions induce.
In the current study we set out to solve the solution structure of the 8–17 DNAzyme in the presence of Zn2+ and to characterize its interactions with this ion. A shortened, unimolecular construct of 8–17 DNAzyme (Fig. 1C) facilitating NMR structural studies is designed, validated for unaltered catalytic activity, and characterized by NMR. Most surprisingly, while pronounced spectral changes are indeed observed upon Zn2+ titration, confirming ion-induced folding, the high-resolution structure that we solve is very similar from the previous crystallographic ones in terms of both the fold of the catalytic domain and the positioning of substrate-binding arms.
To understand this unexpected result, we confirm by NMR that moderately high (0.2–0.4 M) concentrations of Na+ induce the same tightly folded structure as Zn2+, as was already suggested previously11. Given significant Na+ concentrations present in the crystallization conditions13, we propose that the previous crystal structures captured the highly compacted structure usually associated with ions such as Zn2+ or Mg2+ instead of the hypothesized open apo form (Fig. 1D). Moreover, most unexpectedly, NMR experiments performed in presence of Pb2+ reveal that even this cofactor induced 8–17 DNAzyme global folding at NMR sample concentrations (>1 mM). Based on these results, reinforced by a series of activity assays and comparisons with previous literature we bring forward and discuss the idea that Pb2+ might carry out its catalytic role in the same folded state as other ions, even if at most often employed Pb2+ concentrations the population of this state might remain low.
Moreover, this study brings important details regarding divalent ion binding by the 8–17 DNAzyme with the identification of the Zn2+ binding site responsible for the ion-induced folding. However, given the position of this site away from the scissile phosphate, we conclude that it has to be of purely structural importance and another Zn2+ ion must be required for catalysis. While our current attempts at capturing also this second catalytic Zn2+ site were not successful, the results presented herein establish a much more intricate picture of 8–17 DNAzyme’s interactions with divalent ions with distinct structural and catalytic metal-binding sites being involved, similarly to what was previously proposed for several naturally occurring ribozymes24,25,26,27,28.
Results
Construct optimization and validation
To facilitate a high-resolution NMR study, an 8–17 DNAzyme construct of the shortest length is highly desired. While the catalytic core of 8–17 does not tolerate the deletion of any nucleotides, with the exception of N15.08 (Fig. 1A), the substrate binding arms, usually containing over a dozen base pairs each, can potentially be significantly shortened without disrupting the DNAzyme’s structure. The reduced propensity for hybridization between the DNAzyme and substrate strands can be counteracted by connecting the two into a single unimolecular construct, preferably with the ultra-stable GAA triloop29,30.
Following this general template, a series of 8–17 constructs with different arm sequences were designed and evaluated by NMR in the presence of Zn2+, as described in detail in the Supplementary Note 1 and Supplementary Fig. 1. Throughout this optimization the catalytic domain was kept unaltered with respect to the crystallographic study to facilitate a structural comparison, with the exception that A15.0 was deleted. As is a common approach in structural studies of DNAzymes the tested constructs were synthetized in all-DNA form to abolish self-cleavage and only the selected variant, presented in Fig. 1C and referred to as 8–17_short from now on, was also obtained in a cleavable form and tested for catalytic activity. Cleavage rate constants of 0.453 min−1, 0.728 min−1 and 0.063 min−1 were measured (Supplementary Fig. 2a) in the presence of 700 µM Zn2+, 50 µM Pb2+ and 50 mM Mg2+, respectively (at pH 6.0). In the same conditions the rate constants measured for bimolecular full-length 8-17 construct used in the crystallographic study13 (Supplementary Fig. 2b) were 0.246 min−1, 0.488 min−1 and 0.100 min−1, showing that 8–17_short retained catalytic capabilities comparable to longer, bimolecular DNAzyme variants. The activities of the two constructs were also studied as functions of the three divalent metal ion concentrations (Supplementary Fig. 3).
The shortened construct was tested for Zn2+-induced folding by NMR and CD spectroscopies. The results of the CD-monitored titration with Zn2+ are presented in Fig. 2A. They show gradual and saturable spectral changes in response to the metal ion. The effect is much more pronounced here than originally reported11, probably due to a smaller B-helical contribution from the DNAzyme’s arms. The spectral changes in function of Zn2+ concentration can be fitted to a 1:1 interaction model, yielding an apparent Kd value of ≈15 μM (previous literature reported 52 μM9, 18.9 μM10 and 3.1 μM11 for different full-length 8–17 constructs). Given such an apparent Kd, the binding of Zn2+ to 8–17_short should be practically quantitative at NMR sample concentrations (>1 mM). A Zn2+ titration monitored by 1D 1H-NMR presented in Fig. 2B confirms this conclusion. At least two spectral forms can be observed in the imino and methyl regions of the spectrum when no Zn2+ is present. As the first equivalent of Zn2+ is titrated many of the peaks gradually disappear (marked with red stars in Fig. 2), until only one spectral form remains. The folding is also accompanied by the gradual appearance and sharpening of new resonances, like the one observed at around 0.6 ppm in Fig. 2B. Additional equivalents of Zn2+ give rise only to subtle movements of some peaks, indicating that the folding transition is already saturated. Similar results were obtained when experiments were repeated at pH 7 (Supplementary Fig. 4), confirming that the lower pH used did not perturb the structural transition. Overall, our shortened construct turned out to behave very similarly to longer 8–17 variants in terms of both the catalytic activity and Zn2+-induced folding. From now on, when referring to specific residues in 8–17_short, we will use the numbering specific to this construct (Fig. 1C) adding whenever relevant the standard numbering of the residues of the catalytic domain (Fig. 1A) in parenthesis in bold e.g., G29(G14).

A monitored by CD spectroscopy and (B) monitored by 1D 1H-NMR. NMR resonances belonging to an initially present second spectral form are marked by red stars. The titration was performed in 10 mM sodium cacodylate buffer which originally contained no additional salts. Source data are provided as a Source Data file.
NMR structure of 8–17_short in presence of Zn2+ is similar to Pb2+-bound crystal structure
While one equivalent of Zn2+ is enough to saturate the folding transition at NMR concentrations, the folded structure is not particularly thermally stable in these conditions. For this reason, for NMR structure determination a sample containing 2 eq. of Zn2+ as well as 200 mM of NaCl to increase the ionic strength was used, for which good quality spectra could be recorded up to 35 °C. NMR spectral assignments were obtained using standard approaches (see Methods) and confirmed through isotope labeling of key residues within the catalytic domain (Supplementary Fig. 5). A detailed description of observed structurally relevant NMR spectral features, including the set of measured long-range NOEs, can be found in the Supplementary Note 2 complemented by Supplementary Figs. 6 and 7. The NMR structure of 8–17_short in the presence of Zn2+ was determined through a restrained molecular dynamics protocol described in Methods, with the structure determination statistics provided in Supplementary Table 1. The fold of the catalytic domain is very well defined in the NMR structural bundle (RMSD = 0.99 Å), with a bit more uncertainty regarding the mutual positioning of the substrate-binding arms, resulting in a RMSD of 1.83 Å for the entire structure.
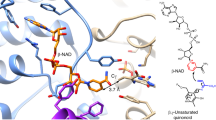
Apart from the three helices presented in Fig. 1A, the structure (Fig. 3; the whole NMR ensemble presented in Fig. 4) also features the G22(G7)-C28(C13), A21(A6)-G29(G14) and G6(rN18)-A30(N15) base pairs (Fig. 3B), that were previously only observed in the crystal structures of the apo and Pb2+-bound forms of the enzyme. These base pairs are crucial for the formation of the L-platform23 tertiary fold in the catalytic domain and for positioning G29(G14) base to act as a general base during catalysis. The G22-C28 pairing is directly visible in the NMR spectra through strong NOE contacts between the exchangeable protons of the two bases. On the other hand, sheared G:A base pairs rely on hydrogen bonding between groups that are difficult to directly observe by NMR (amine groups of G and A residues), but their presence is required to satisfy a set of long range NOE contacts between nucleobase and sugar protons of the residues involved (listed in Supplementary Table 2). The tertiary fold of the DNAzyme is defined by the co-axial stacking of helices 1–4 and 2–3 (Fig. 3A) which fixes the entire structure into a V-like shape with the interhelical angle between the two substrate binding arms averaging 83° ± 11° for different conformers within the NMR bundle.

A the overall fold, (B) geometric details of Helix 4.

Main panel uses a single conformer from the NMR bundle for clarity, while the insert shows the entire bundle.
Overall, the catalytic domain fold that we observe in the presence of Zn2+ is very similar to the one reported previously in the crystal structures, with the average catalytic domain RMSD of the members of our NMR bundle to the crystal structure equal to 1.28 Å (which is just beyond the uncertainty of the bundle itself). When the phosphate group of T27(N12), the only moiety whose position in the crystal structure appears to fall outside the range of uncertainty of the NMR bundle upon visual inspection is omitted the RMSD further drops to 1.18 Å. Also, the interhelical angle between substrate-binding arms encountered in the crystal (≈75°) is close to what we observe in our NMR structure. The degree of similarity between the two structures can be appreciated in Fig. 4.
Mg2+ and Na+ induce the same overall 8–17_short fold as Zn2+
Previous literature suggests that other divalent ions, such as Mg2+ as well as high concentrations of monovalent ions, like Na+, induce a folding transition in the 8–17 DNAzyme, akin to that promoted by Zn2+9,10,11. We decided to verify whether such behavior can also be observed for our 8–17_short construct. We have started the investigation by performing CD- and NMR-monitored titrations of 8–17_short with MgCl2 and NaCl, presented in Fig. 5. The induced spectral changes are indeed qualitatively similar to what was observed for Zn2+ (Fig. 2), yet require several equivalents of Mg2+ to saturate even at NMR concentrations (Fig. 5B; apparent Kd ≈ 90 μM from CD measurements) and even orders of magnitude more metal ion is needed in case of Na+ (Fig. 5A; apparent Kd of Na+ ≈90 mM), consistent with the literature9,10,11. To confirm that tertiary folds induced by these ions and Zn2+ are indeed the same, we have performed NMR resonance assignments and analyzed the NOE patterns for 8–17_short in the presence of either 7.5 mM Mg2+ or 400 mM NaCl. While the lower spectral quality made accurate integration of many peaks difficult and thus precluded structure calculation in these conditions, the observed patterns of long-range NOE connectivities and characteristic chemical shifts was qualitatively the same as for Zn2+ (as described in detail in Supplementary Note 3 and illustrated in Supplementary Fig. 8). One can thus conclude that Mg2+ and Na+ indeed induce the same overall DNAzyme fold as Zn2+. To retroactively exclude the possibility that the structure determined for sample containing both Zn2+ and 200 mM Na+ was dictated by the monovalent ion rather than by Zn2+, we have also performed the 2D-NMR analysis for a sample containing only Zn2+ without added NaCl. Once again, the same pattern of long-range NOEs was observed (see Supplementary Note 3, Supplementary Table 2 and Supplementary Fig. 8), confirming that one equivalent of Zn2+ alone is indeed enough to induce the folded structure.

A titrations with Na+ (B) titrations with Mg2+. For each ion the folding was monitored by CD spectroscopy (top) and 1D 1H-NMR (bottom). NMR resonances belonging to an initially present second spectral form are marked by red stars. The titration was performed in 10 mM sodium cacodylate buffer which originally contained no additional salts. Source data are provided as a Source Data file.
Even Pb2+ ions induce global folding at NMR concentrations
As control experiments we have also performed NMR and CD titrations with Pb2+, the only activating ion not shown to induce 8–17 DNAzyme folding9,10,11. Surprisingly even in this case we observed extensive spectral changes in both techniques. The CD titration (Fig. 6A) initially followed a pattern similar to what was observed for the other ions (Figs. 2 and 5) with a gradual decrease of the band around 280 nm. Yet, starting at around 2 equivalents Pb2+ and before the initial structural transition could be saturated, a new spectral band started to appear centered at around 320 nm. This second transition saturated at around 10–15 equivalents Pb2+. Similarly, in the first part of the NMR titration (Fig. 6B) we have once again observed a transition from multiple spectral forms to only one, alike of what was described above for Zn2+, Mg2+ and Na+. Unlike the CD titration this transition saturated at around 2 eqivalents of Pb2+, due to much higher concentrations of interaction partners used in NMR, as demonstrated by the virtual disappearance of the peaks marked with red stars in Fig. 6B. The addition of the additional equivalents of Pb2+ finally resulted in the appearance of a small fraction of a distinct structural form (with a distinct set of imino proton resonances located around 12 ppm; green stars in Fig. 6B), probably equivalent to the one responsible for the 320 nm band in CD. To confirm this assumption we have performed another NMR titration (Fig. 6C) at a DNA concentration more akin to what we used for CD spectroscopy. In this case when 10 equivalents of Pb2+ was reached the structural form with imino resonances around 12 ppm (green stars in Fig. 6C) became the dominant one, in accordance with the CD data. Thus overall, the addition of Pb2+ ions to 8–17_short causes two consecutive structural transitions: (1) one from the initial ensemble of structures to a single folded state that becomes the only form present at around 2–3 equivalents of Pb2+ at NMR concentrations (Fig. 6B) and (2) a second transition to a different folded state at higher Pb2+-to-DNA molar ratios. This second transition is accompanied by the disappearance of imino proton resonances of Watson-Crick base-pairs (Fig. 6C), including those from the DNAzyme’s arms, and thus we would like to argue that it is a reflection of the known tendency of Pb2+ ions to destabilize helical folds at higher molar ratios by direct coordination of nucleobases, especially guanines31. The rather early onset of this transition for 8–17_short may be related to its very short and G-rich helices. This argument is presented in detail in the Supplementary Discussion 1 (strengthened by additional experimental data presented in Supplementary Figs. 9 and 10), culminating in a suggestion that the structure formed is a Pb2+-stabilized G-quadruplex. Here we will instead focus on the fold resulting from the initial transition. A set of 2D NMR spectra was recorded and analyzed for a sample containing 2.33 equivalents of Pb2+ for which only a single structural form was present. Similarly to the previous samples containing only a single type of ion (Na+, Mg2+ and Zn2+) some of the NMR resonances were broadened making the accurate integration of their NOESY cross-peaks difficult. Still a fairly complete resonance assignment was possible (Supplementary Data 1 and 2) and a significant number of long-range NOESY connectivities was observed within the catalytic domain (Supplementary Table 2). Both these long-range NOE contacts and non-standard chemical shifts were very similar to the ones observed in all the samples discussed thus far, as described in detail in the Supplementary Note 3, which is only possible if the overall fold of the molecules is conserved throughout the studied conditions. Thus, at least for our 8–17_short construct, Pb2+ appear to induce the same overall 8–17 DNAzyme fold as other metal ions at NMR concentrations.

(A) monitored by CD spectroscopy and (B) monitored by 1D 1H-NMR at millimolar DNA concentrations, (C) monitored by 1D 1H-NMR at submilimolar DNA concentrations. NMR resonances belonging to an initially present second spectral form are marked by red stars while those to the form appearing at higher concentrations of Pb2+ with green ones. The titration was performed in 10 mM sodium cacodylate buffer which originally contained no additional salts. Source data are provided as a Source Data file.
Control experiments with full-length 8-17 DNAzyme
As the just described folding of the 8–17_short DNAzyme by Pb2+ ions is at odds with the current state of knowledge about this system, we have repeated the CD and NMR-monitored titrations with all four metal ions for a more standard 8−17 DNAzyme variant – the bimolecular, full-length construct used previously for the crystallographic study. Surprisingly, also for this construct our CD titrations revealed qualitatively similar spectral changes for all four metal ions — a saturable decrease of intensity of the CD band around 280 nm, although with somewhat different end-points for the different metal ions (Fig. 7). This discrepancy with the previous results11 cannot be attributed to difference in experimental conditions, but is rather the effect of specific DNAzyme sequences being studied, as when we repeated the CD experiments using DNAzyme variants used in reference 11 we obtained the same results as the original authors (only marginal CD spectral changes upon Pb2+ titration, Supplementary Fig. 11). We are currently conducting a more systematic study using different 8−17 variants to shed more light on this issue in the future, yet the results presented in Fig. 7 show that at least the crystallographic 8−17 DNAzyme construct undergoes structural changes upon Pb2+ titration. The NMR titrations (Supplementary Fig. 12) show less spectacular spectral changes due to significant spectral crowding in 1D NMR severely obstructing the observations, nevertheless the changes that can be observed are very similar among all studied metal ions — gradual shifts of the same two methyl proton resonances as well as the appearance of a new imino resonance around 13.1–13.2 ppm and a highly shielded proton resonance around 0.9 ppm (Supplementary Fig. 12).

CD-monitored titrations with four different metal ions: (A) Zn2+, (B) Na+, (C) Mg2+ and (D) Pb2+. Source data are provided as a Source Data file.
Are the global folds assumed by 8–17 DNAzyme in presence of Zn2+ and Pb2+ the same?
It is a long-standing literature consensus that Pb2+ binds to the open, apo form of 8−17 DNAzyme without affecting its original conformation, while Zn2+ binding triggers a global structural rearrangement towards a more compact fold (Fig. 1D). Despite this, the solution structure of Zn2+-bound 8−17 DNAzyme that we report here is practically indistinguishable from the Pb2+-bound and apo crystal structures, in terms of catalytic domain fold and substrate-binding arms orientation (Fig. 4). Clearly, for some reason, all these structural studies have captured the same state of the enzyme, yet which one is it — the apo one or the ion-induced one? We wish to argue that it is the latter given that our NMR and CD titrations clearly indicate that the structure we solved was only formed upon Zn2+ addition (Fig. 2). The appearance of such ion-induced fold in crystals formed in presence of Pb2+ and even in those without any divalent ions would of course be highly unexpected, yet we believe that the bulk of experimental results presented in the current work allows us to propose a way to reconcile these results.
Regarding the apo state, our 1D NMR spectra recorded before addition of any metal ions (e.g., in Fig. 2B, bottom) show the enzyme to be present as a mixture of different conformations, which then gradually collapse into a single state upon metal ion titration. The complexity of such mixture precludes any detailed structural characterization of the different states forming it, however, as some monovalent ions are always present to compensate the negative charge of DNA, this conformational ensemble must contain a fractional population of the folded state (Fig. 8, top). When a biomolecule is dynamic/polydisperse, crystallization often selects only a single among the forms present to construct the crystal lattice, usually the more structured, rigid one. We thus find it plausible that the tightly folded, ion-induced structure was selected from the conformational ensemble of 8−17 DNAzyme during crystallizations. To strengthen this argument, one can notice that monovalent ions that also promote folding were present in crystallization drops, in quantities which could make the folded state abundant. The protein AsfvPolX, crystalized in complex with 8–17 DNAzyme, was supplied in 300 mM NaCl13 (final concentration not provided) and two out of three crystallization conditions also contained 200 mM NH4+. The presence of what we argue to be the compact form of the DNAzyme in the Pb2+-containing crystals could also be rationalized using the same argument (conformational selection when forming the crystal lattice). However, on top of that our Pb2+ titrations (Figs. 6 and 7) show that Pb2+ ions themselves can at higher concentrations — as used for both NMR and crystallographic structure determination studies — induce the compact fold of the DNAzyme.

The top part of the scheme represents the conformational ensemble of the apo- form of the enzyme, while the effects of Pb2+ and Zn2+ ions are depicted below in the left and right panels, respectively. The occupation state of structural and catalytic metal binding sites is also shown in each case for the well-folded DNAzyme molecules. When the catalytic site is filled the DNAzyme is activated, as marked by the black, highlighted scissors.
This finding itself may be the most unexpected result of our investigation, as previous FRET and CD studies have shown no signs of Pb2+-induced folding. These studies were of course performed at different DNAzyme concentrations, using different sequence variants and sample conditions, including much higher metal-to-DNA molar ratios. In the Supplementary Discussion 2 we discuss how these differences might have prevented the observation of Pb2+-induced folding. Here, we would like to bring forward one significant difference between Pb2+ and other activating ions that is apparent in our dataset and that could explain why Pb2+-induced folding was not considered in previous studies of the DNAzyme. Namely, in case of both Zn2+ and Mg2+ we observe a several orders of magnitude wide discrepancy between the metal ion concentrations needed to induce DNAzyme folding and those needed to saturate its catalytic activity (Table 1). Concentrating on Zn2+, for the 8–17_short the apparent Kd for Zn2+ binding estimated from the CD-titration is 15 µM, while the Kd derived from catalytic activity measurements is instead around 4 mM (Supplementary Figure 3). Similarly mismatched Kd values were also obtained for the full-length construct: 23 µM based on CD data and 3 mM based on activity assays (Table 1). Indeed, similar discrepancies are present throughout the available literature between the Zn2+ affinities estimated using 8–17 DNAzyme folding (always micromolar)9,10,11 and those derived from the Zn2+ concentration dependence of the measured rate constants kobs (always millimolar)9,32.
This conundrum may suggest that at least two Zn2+ (or Mg2+) ions are involved in specific interactions with 8–17, one causing its folding (binding with micromolar Kd values) and another one activating the DNAzyme for cleavage (and having a millimolar affinity). A similar conclusion was already suggested in the first paper reporting ion-induced folding of 8–17 DNAzyme9, yet for some reason did not find much reflection in later literature. The affinity discrepancy between these two sites leads to a situation in which at Zn2+ concentrations when cleavage rates become appreciable (hundreds of µM, Supplementary Fig. 3) the DNAzyme is already fully folded (Fig. 8, right branch).
On the other hand for lead ions the dissociation constants estimated using CD and activity assays appear to be much more similar (the CD-derived values cannot be compared to previous literature, as Pb2+-induced folding has not been reported before, while the activity-derived ones are consistent with previous reports within order of magnitude)9. In such a case the Pb2+ induced activity could already be high at metal concentrations like dozens of µM for which the DNAzyme is only partially folded into the active state (Fig. 8, left branch). Even the presence of only a fraction of the active folded state at a given time can give rise to the high final cleavage yields observed at these Pb2+ concentrations, because as the active state is gradually consumed and ions are released the dynamic equilibrium between the different DNAzyme conformations would work to repopulate it.
Thus, in our opinion it may be reasonable to argue that the explanation for the unexpected coincidence of the Zn2+-induced solution structure and the Pb2+-bound crystal one is that the two ions actually use the same folded state for catalysis. The main difference between the two ions would be the extent to which the active state is populated at metal concentrations capable of inducing appreciable catalytic activities — the Zn2+-induced activity occurs only at concentrations at which the DNAzyme is already fully folded, while Pb2+ starts to activate the DNAzyme even without significantly increasing the population of the folded state in the conformational ensemble of the apo state.
Formulated in terms of metal-DNA interactions, this hypothesis would argue for the existence of two distinct structural and catalytic binding sites. Ions such as Zn2+ and Mg2+ would have a much higher affinity of the structural site and thus saturate it preferentially, only starting to occupy the catalytic site at higher molar excesses (Fig. 8, right branch). Pb2+ on the other hand would either have the two affinities much closely aligned (Fig. 8, left branch) or use only a single binding site to achieve both DNAzyme activation and folding.
While this proposal provides a different interpretation of the available ion-induced folding data9,10,11 to the one currently accepted in the literature – a single active conformation populated to a different extent with Pb2+ compared to other ions, in place of the postulate of two different active states –, we believe that it is not directly at odds with previous experimental observations (see Supplementary Discussion 2 for a more thorough discussion).
If our hypothesis was true, then it should be possible to enhance Pb2+-induced activity at its typical concentrations (dozens-to-hundreds µM) by the addition of other metal ions — like Mg2+ – in quantities that are not enough to induce appreciable activity themselves, but are capable of significantly increasing the population of the folded active state for Pb2+ to use. On the other hand, if Mg2+ and Pb2+ use different global folds of the DNAzyme for catalysis then similar Mg2+ additions should inhibit the Pb2+-induced activity by transforming the DNAzyme into a fold that Pb2+ cannot use. Thus, to test our structural proposal we have measured the cleavage rates for two variants of 8–17 DNAzyme in presence of 50 µM of Pb2+ alone and alongside increasing additions of Mg2+, up to 500 µM (these concentrations of Mg2+ by themselves induce very little cleavage even after 2 h). For both variants we have indeed observed a gradual increase in activity alongside increased Mg2+ doping (Supplementary Fig. 13), as our one active state hypothesis would predict. Moreover, a similar observation was recently published regarding the effect of Na+ ions on 8-17 DNAzyme catalytic activity in the presence of Pb2+18. However, it has to be noted that the authors of that paper assigned the observed effect to a direct involvement of Na+ ions in Pb2+-induced cleavage reaction proposed by a previous MD study19, rather than to a structural shift.
Identification of the structural Zn2+ binding site
In the NMR experiments metal ions such as Zn2+ can be only observed indirectly through their influence on chemical shifts of the atoms within the macromolecule they bind to (chemical shift perturbations – CSPs). If the metal ion binds without affecting the overall structure of the macromolecule then the localization of CSP carry information about where the binding occurs. The possibility of inducing the folded state of 8–17_short using an ion with orders of magnitude weaker affinity (Na+) opens the possibility of studying Zn2+ binding using this principle. When Zn2+ is titrated to the Na+-stabilized DNAzyme (Fig. 9) it displaces the monovalent ion from the binding site(s), without changing the global structure, only affecting the local chemical environment of atoms close to the binding site. As seen in Fig. 9a, b, among the imino and aromatic protons the highest CSPs are recorded for the residues G7(N1) and T17(T2) forming the G:T base pair, adjacent to the cleavage site, suggesting Zn2+ interaction at this site. When all the measured CSPs (base, sugar, phosphate) are visualized on the structure (Fig. 9c) the shifts adjacent to the G:T base pair remain dominant, yet some moderate CSPs can be seen all throughout the catalytic domain. Their appearance could be related either to some local structural differences between the Zn2+ and Na+-stabilized structures or to the fact that with Na+ alone the population of the folded structure was not 100%. Interestingly, only a relatively small CSP of 0.09 ppm was measured for the 31P atom of the scissile phosphate, suggesting no direct Zn2+ interaction with this group.

A spectral changes in the imino region of 1D 1H-NMR spectrum upon Zn2+ titration, (B) spectral changes in the aromatic region of 1H-13C HSQC, (C) measured CSPs visualized on the 3D structure, color coded from 0.00 [ppm] (white) to ≥0.25 [ppm] (red). Source data are provided as Supplementary Data files.
Subsequent titration of two additional equivalents of Zn2+ yielded much smaller CSPs in the vicinity of the G:T base pair (Fig. 9A, B), confirming near-saturation of this binding site after just one equivalent. Instead, two regions of moderate CSPs appear, probably corresponding to additional weaker interaction sites (CSPs visualized on the structure in Supplementary Fig. 14b). One of them was located in the vicinity of residues C23(Y8) and G24(N9) (Helix 3 and its capping residue) and the other in one of the substrate-binding arms. The location of these additional sites, on the far peripheries of the molecule, makes any involvement of the metal bound there in the catalytic process highly unlikely. Similar titrations were also performed for the sample already structured by one equivalent of Zn2+, yet containing no added NaCl. In this case again, additional equivalents of Zn2+ induced some moderate CSPs around the two above-mentioned weak binding sites, but not around the G:T base pair (Supplementary Fig. 14c). This confirms that in these conditions this binding site is already saturated and thus it is the one responsible for the Zn2+-induced folding (the structural site introduced above).
While the CSPs themselves cannot reveal the exact geometry of this interaction, their pattern suggests that Zn2+ is interacting with the Hoogsteen edge of the G7(N1) residue which forms the G:T base pair. First, both the imino and aromatic protons of G7(N1) experience strong CSP, while for the thymidine only the imino proton is affected, with almost no CSP on the H6 and methyl protons (Fig. 9C). Second, the only other aromatic proton with a strong CSP belongs to G8, which directly stacks with G7(N1), further locating the binding site on this strand of the helix. Given that guanosine’s Hoogsteen edge is the strongest interaction site for Zn2+ on single nucleotide level33, these CSP data are most readily explained by the binding to G7(N1). As to why Zn2+ interaction with such a site could facilitate the DNAzyme’s folding, one can notice that in our structure the Hoogsteen edge of G7 directly faces the phosphate group of T27(N12) (the N7 atom of G7 is located 5.5–8.0 Å from the phosphorus atom in our structural bundle). Electrostatic repulsion between these two entities might represent a major obstacle for folding that is removed when a divalent cation binds and shields this repulsive interaction. The influence of the Zn2+ ion on the local environment of T27(N12) phosphate is reflected in the 0.25 ppm 31P CSP, the highest among all phosphate groups in the molecule. The involvement of G7(N1)-N7 and T27(N12)-P in Zn2+ binding was tested by studying the folding of 8–17_short variants containing either the 7-deaza or phosphorothioate modifications at the respective sites (see Supplementary Fig. 15 and Supplementary Note 4). For the 7-deaza-G7(N1) construct a fivefold decrease in binding affinity was observed, while for the T27(N12) phosphorothioate modification the decrease was two- to fourfold, depending on the method of data fitting (see Supplementary Note 4). These Kd changes confirm the involvement of these two moieties in the interactions, yet their magnitudes may point towards second sphere interactions, rather than direct metal coordination. On the other hand, the Zn2+ ion bound at this site almost certainly does not directly interact with the scissile phosphate, as (1) the 31P CSP of this group is small throughout our titrations and (2) the phosphorothioate substitution at this site does not influence observed ion binding (Supplementary Fig. 15). Available literature data provides strong evidence that the catalytically-required Zn2+ ion should have a direct interaction with the scissile phosphate, through its Rp oxygen atom17. This reinforces the idea that the strong site we observe is not the catalytic one. The lack of CSPs in the direct vicinity of the scissile phosphate indicate that this catalytic site was not appreciably populated in our titrations, even though Zn2+ concentrations of up to 4.5 mM were reached (Fig. 9 and S13). This might be either due to other competing sites of similar affinity (a factor which does not play such an important role during kinetic experiments performed at much lower DNA concentrations and thus many-fold excess of the metal) or to some distortion of the catalytic site in the all-DNA construct used for structural studies (the lack of the 2’OH at residue G6(rN18) might alter both the local conformation and electrostatic potential).
Discussion
The crystal structures of the 8–17 DNAzyme13 provided much momentum for the in-depth studies of its catalytic mechanism, as described in the Introduction. However, due to the consensus that the available structures represented the open/apo state of the enzyme (Fig. 1B), all the conclusions of these studies were believed to only be directly applicable to Pb2+-driven catalysis. With our current finding that the DNAzyme fold induced by Zn2+ and other ions is the same as one found in the crystals and follows the L-platform23 architecture, many of these conclusions can be extended to catalysis in the presence of other divalent metal ion cofactors.
First, the V-like overall fold of the DNAzyme, believed to orient the 2’OH group of rN18 (Fig. 2B) for in-line attack on the scissile phosphate (α-catalysis), is retained in the Zn2+-stabilized structure suggesting that this mechanism of catalysis is relevant in presence of all divalent metal ions. Second, with G14 (G29 in our construct) nucleobase sharing its position in both the crystal and solution structures, its role as general base appears to also be conserved regardless of the metal ion present. This conclusion is supported by the previous literature, as the role of G14 as the general base was already proven experimentally also in the presence of Mg2+15. On the other hand, much less can be inferred about the structural aspects of catalytic involvement of divalent ions other that Pb2+, as our current study did not manage to locate the weak (Kd > 1 mM)9 catalytically relevant Zn2+ binding site. Given the same DNAzyme structure, this interaction site might actually be similar to the one occupied by Pb2+ in the crystal, however, with somewhat different interaction geometry to allow for the direct coordination of the scissile phosphate17. Interestingly, MD simulations preformed previously for the Pb2+-bound crystal structure featured a first sphere contact with Rp oxygen even for this metal19, indicating that such a geometry is at least electrostatically favorable. Establishing whether other activating metal ions utilize such a binding mode requires further experimental or computational investigations. Nevertheless, the structural data presented here sets the stage for a more in-depth mechanistic characterization of 8–17 DNAzyme catalysis in the presence of non-Pb2+ cofactors, such as Mg2+, and eventual design of its more in vivo active variants.
Methods
Sample preparation
All the all-DNA constructs (including the 7-deaza-dG and phosphorothioate modified ones), as well as their cleavable, fluorescein labeled DNA/RNA hybrid counterparts for kinetic experiments were purchased from Metabion GmbH. The site-specifically 13C/15N labeled samples were synthesized in house using phosphoramidites purchased from Silantes GmbH. Before usage, the all-DNA constructs were purified from residual organic cation detrimental for NMR measurements (triethyl amine, remaining after HPLC) by centrifugation on Amicon filters in the excess of 10 mM cacodylate buffer (pH 6.0) that was used during all subsequent experiments. The cleavable constructs were instead used directly as provided by the supplier to avoid any premature cleavage during a similar pretreatment.
Kinetic assays
Kinetic assays were conducted to measure the catalytic activities for (1) the truncated 8–17_short construct and (2) a reference full-length bimolecular 8–17 DNAzyme construct from the crystallographic study (Supplementary Figures 2 and 3) in the presence of Zn2+, Mg2+ and Pb2+. Both constructs were 3′-FAM labeled and contained a single RNA residue at the cleavage site. The dried samples purchased from Metabion were dissolved in the reaction buffer (10 mM sodium cacodylate buffer, pH 6.0) to 40 μM concentrations and used without further purification to minimize premature cleavage. In case of the bimolecular variant the FAM-labeled substrate and DNAzyme strands were used in 1:1 concentration ratio. Kinetic assays were performed at 25 °C. The reactions were initiated by adding the appropriate amounts of metal ions from stock solutions. Aliquots were collected at appropriate time points and quenched using a stop buffer containing 0.5 M EDTA and 8 M Urea. Samples were resolved by electrophoresis on a 15% urea gel and scanned on a Amersham Typhoon phosphorimager. The extent of reaction at each time point was quantified using ImageQuantTL software, version 10.2. The observed rate constants \({k}_{{obs}}\) were obtained by fitting the observed fraction cleaved (\(y\)) against time (\(t\)) to the equation: \(y={y}_{0}+A\left(1-{e}^{-{k}_{{obs}}t}\right)\). The reaction was repeated three times in each of the tested conditions presented in Supplementary Fig. 2 and the error bars represent the standard deviations of the fraction cut at each time point between these repetitions. The dependencies of the measured \({k}_{{obs}}\) values on metal ion concentrations (Supplementary Fig. 3) were fitted using a 1:1 interaction model to estimate the apparent Kd values for metal ion binding. For this figure the \({k}_{{obs}}\) values at each metal concentration were derived in the same manner, however only for the 8–17_short interaction with Mg2+ the experiments were done in triplicate, for the other 5 series each condition was tested only once (due to excellent reproducibility observed between the three replicates in the 8–17_short-Mg2+ series that was performed as the first).
CD spectroscopy
The circular dichroism spectra (CD) were recorded using a JASCO J815 spectropolarimeter equipped with a Peltier temperature controller and operating on JASCO Spectra Manager software version 2.09.11. Cuvettes with a path length of 0.1 cm were used. Spectra were collected in the range between 215 and 320 nm, as a sum of seven repetitions at 20 °C and the buffer baseline was subtracted from each spectrum. The sample concentrations were adjusted to always give rise to absorbance values close to 1. This resulted in 40 μM samples for 8–17_short (similar to what was used in ref. 11) and around half that concentration for the full-length constructs. CD spectra were expressed in the units of molar ellipticity Δϵ [cm2 mmol−1], without normalization by the number of residues in the molecule.
NMR spectroscopy
The NMR spectra were primarily collected using a 700 MHz Bruker Avance III spectrometer equipped with a QCI-P CryoProbe using the Topspin 3.6.5 software. Only the 31P-observed HP-COSY spectra were recorded on a 500 MHz Bruker Avance III spectrometer with a room-temperature BBO probe using Topspin 3.6.2, instead. The resonance assignment of non-exchangeable protons and phosphorus atoms was achieved using standard procedures34,35, through the analysis of NOESY (with water suppression through excitation sculpting36 and 80 or 150 ms mixing times), TOCSY37, HC-HSQC38 and HP-COSY39 spectra recorded in 100% D2O at 20 °C and 35 °C when possible. The exchangeable protons were assigned using NOESY spectra (with WATERGATE water suppression40 and 80 or 250 ms mixing times) measured in 90% H2O/10% D2O at 5, 12.5, and 20 °C. The full range of sample conditions used are provided in Supplementary Data 1 and 2. All the spectra were analyzed in NMRFAM-Sparky41 version 1.470. The resonance assignments were gathered in Supplementary Data 1 and 2, while snippets of assigned spectra can be found in Supplementary Figs. 7 and 8. All the reported chemical shift perturbations (CSPs) were calculated by simple subtraction of 1H chemical shifts measured at different points of the titrations.
Restraint generation and NMR structure calculations
The structure determination of 8–17_short was driven by the classical NOE-derived distance restraints and dihedral angle restraints. The distance restraints between non-exchangeable protons were extracted from the peak volumes in NOESY spectra recorded in D2O with and 150 ms mixing time. The NOESY cross-peaks were classified as strong (1.8–3.0 Å), medium (2.0–4.0 Å), weak (2.2–5.0 Å) and very weak (2.4–6.0 Å), using fixed distances (H5-H6 in cytidines and H2’-H2” in deoxyriboses) as reference. Any peak that was not reliably integrable due to partial overlap was assigned to the broadest 1.8–7.0 Å category. The distance restraints between exchangeable protons were derived from the NOESY spectra recorded in H2O (150 ms mixing) using a similar procedure, with the mean volume of the imino-amino contacts of guanosines used as reference. Regarding dihedral angle restraints, the backbone dihedral angles α and ζ were restrained to exclude the trans rotamer for all residues with 31P chemical shift within the standard range. Moreover, for residues for which the P(n)-H4’(n-1) cross peak was observable the β and γ dihedral angles were restrained to 180° and 60°, respectively (±60°). The χ dihedral angle was in turn restrained to the anti orientation for all residues based on the intensities of H1′-H6/H8 NOESY cross-peaks. For the residues with JH3’-H4’ ≈ 0 Hz sugar puckers were restrained to the C2’-endo conformation. Hydrogen bond restraints for G-C base pairs were imposed based on the observation of strong NOE cross-peaks between specific cytidine NH2 and guanosine imino protons. For the C22-G28 base pair the assignment of the imino proton was confirmed using site-specific isotopic labeling (Supplementary Fig. 5a). Overall, a total of 365 NOE, 80 hydrogen bond, and 156 torsion angle (including 21 sugar pucker) restraints were obtained.
The structure calculations were performed using a two-step procedure, involving (1) initial folding from random extended conformations, in implicit solvent (generalized Born model) (2) refinement of the best pre-folded structures in explicit solvent. Both stages were executed in the SANDER module of the AMBER 18 molecular dynamics suite of programs42. All calculations used the parm99bsc0χOL15 force field43 with SPC/E water model and Joung-Cheatham parameters for monovalent cations44. Long-range electrostatics were calculated using the particle mesh Ewald method with the nonbonded cutoff set to 8 Å. The covalent bonds were constrained using SHAKE and the integration time step was set to 2 fs. The Langevin thermostat with collision frequency 1.0 ps-1 was used to control the temperature and Berendsen barostat for constant pressure simulation. In the first step a total of 200 structures were calculated using a simulated annealing protocol designed to enhance conformational sampling by significantly reducing the strength of the electrostatic and van der Waals components of the force field during the high temperature stage. Best among these structures were then solvated and refined using a much milder simulated annealing scheme in explicit solvent. In this final step 200 structures were calculated and then 20 were selected based on their reproduction of the experimental NMR data (Supplementary Table 1). All structure visualizations presented in this work were prepared in Pymol Molecular Graphics System, Version 1.6, Schrödinger, LLC.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Atomic coordinates and the list of experimental restraints for the reported NMR structure have been deposited with the Protein Data bank under accession code 8OR8 while the chemical shifts have been deposited at the BMRB under the numbers 34805 https://doi.org/10.13018/BMR34805 and 52355 https://doi.org/10.13018/BMR52355. The CD spectra and cleavage assays (gel scans) generated in this study are provided in the Source Data file. The raw NMR data used in this study are available in the Zenodo database under https://doi.org/10.5281/zenodo.11047570. This study also makes use of the previously published crystallographic structure: 5XM8 for structural comparison. Source data are provided with this paper.
References
Breaker, R. R. & Joyce, G. F. A DNA enzyme that cleaves RNA. Chem. Biol. 1, 223–229 (1994).
Santoro, S. W. & Joyce, G. F. A general purpose RNA-cleaving DNA enzyme. Proc. Natl Acad. Sci. USA 94, 4262–4266 (1997).
Silverman, S. K. Catalytic DNA: scope, applications, and biochemistry of deoxyribozymes. Trends Biochem. Sci. 41, 595–609 (2016).
Zhou, W., Saran, R. & Liu, J. Metal sensing by DNA. Chem. Rev. 117, 8272–8325 (2017).
Zhang, W., Feng, Q., Chang, D., Tram, K. & Li, Y. In vitro selection of RNA-cleaving DNAzymes for bacterial detection. Methods 106, 66–75 (2016).
Wang, Y., Liu, E., Lam, C. H. & Perrin, D. M. A densely modified M2+-independent DNAzyme that cleaves RNA efficiently with multiple catalytic turnover. Chem. Sci. 9, 1813–1821 (2018).
Wang, Y., Nguyen, K., Spitale, R. C. & Chaput, J. C. A biologically stable DNAzyme that efficiently silences gene expression in cells. Nat. Chem. 13, 319–326 (2021).
Schlosser, K. & Li, Y. A versatile endoribonuclease mimic made of DNA: characteristics and applications of the 8-17 RNA-Cleaving DNAzyme. ChemBioChem 11, 866–879 (2010).
Kim, H.-K. et al. Metal-dependent global folding and activity of the 8-17 DNAzyme studied by fluorescence resonance energy transfer. J. Am. Chem. Soc. 129, 6896–6902 (2007).
Kim, H. K., Rasnik, I., Liu, J., Ha, T. & Lu, Y. Dissecting metal ion–dependent folding and catalysis of a single DNAzyme. Nat. Chem. Biol. 3, 763–768 (2007).
Mazumdar, D. et al. Activity, Folding and Z-DNA Formation of the 8-17 DNAzyme in the presence of monovalent ions. J. Am. Chem. Soc. 131, 5506–5515 (2009).
Ponce-Salvatierra, A., Wawrzyniak-Turek, K., Steuerwald, U., Höbartner, C. & Pena, V. Crystal structure of a DNA catalyst. Nature 529, 231–234 (2016).
Liu, H. et al. Crystal structure of an RNA-cleaving DNAzyme. Nat. Commun. 8, 2006 (2017).
Borggräfe, J. et al. Time-resolved structural analysis of an RNA-cleaving DNA catalyst. Nature 601, 144–149 (2021).
Cepeda-Plaza, M., McGhee, C. E. & Lu, Y. Evidence of a General Acid–Base Catalysis Mechanism in the 8–17 DNAzyme. Biochemistry 57, 1517–1522 (2018).
Cortés-Guajardo, C., Rojas-Hernández, F., Paillao-Bustos, R. & Cepeda-Plaza, M. Hydrated metal ion as a general acid in the catalytic mechanism of the 8–17 DNAzyme. Org. Biomol. Chem. 19, 5395–5402 (2021).
Moon, W. J., Huang, P.-J. J. & Liu, J. Probing metal-dependent phosphate binding for the catalysis of the 17E DNAzyme. Biochemistry 60, 1909–1918 (2021).
Parra-Meneses, V., Rojas-Hernández, F. & Cepeda-Plaza, M. The role of Na+ in catalysis by the 8–17 DNAzyme. Org. Biomol. Chem. 20, 6356–6362 (2022).
Ekesan, Ş. & York, D. M. Dynamical ensemble of the active state and transition state mimic for the RNA-cleaving 8–17 DNAzyme in solution. Nucleic Acids Res. 47, 10282–10295 (2019).
Ganguly, A., Weissman, B. P., Piccirilli, J. A. & York, D. M. Evidence for a catalytic strategy to promote nucleophile activation in metal-dependent RNA-cleaving ribozymes and 8-17 DNAzyme. ACS Catal. 9, 10612–10617 (2019).
Breaker, R. R. et al. A common speed limit for RNA-cleaving ribozymes and deoxyribozymes. RNA 9, 949–957 (2003).
Bevilacqua, P. et al. An ontology for facilitating discussion of catalytic strategies of RNA-cleaving enzymes. ACS Chem. Biol. 14, 1068–1076 (2019).
Gaines, C. S., Piccirilli, J. A. & York, D. M. The L-platform/L-scaffold framework: a blueprint for RNA-cleaving nucleic acid enzyme design. RNA 26, 111–125 (2020).
Nakano, S., Proctor, D. J. & Bevilacqua, P. C. Mechanistic characterization of the HDV genomic ribozyme: assessing the catalytic and structural contributions of divalent metal ions within a multichannel reaction mechanism. Biochemistry 40, 12022–12038 (2001).
Lee, T.-S., Radak, B. K., Harris, M. E. & York, D. M. A two-metal-ion-mediated conformational switching pathway for HDV ribozyme activation. ACS Catal. 6, 1853–1869 (2016).
Skilandat, M., Rowinska-Zyrek, M. & Sigel, R. K. O. Secondary structure confirmation and localization of Mg2+ ions in the mammalian CPEB3 ribozyme. RNA 22, 750–763 (2016).
Liu, Y., Wilson, T. J. & Lilley, D. M. J. The structure of a nucleolytic ribozyme that employs a catalytic metal ion. Nat. Chem. Biol. 13, 508–513 (2017).
Mir, A. et al. Two divalent metal ions and conformational changes play roles in the hammerhead ribozyme cleavage reaction. Biochemistry 54, 6369–6381 (2015).
Hirao, I. et al. Most compact hairpin-turn structure exerted by a short DNA fragment, d(GCGAAGC) in solution: an extraordinarily stable structure resistant to nucleases and heat. Nucleic Acids Res. 22, 576–582 (1994).
Yoshizawa, S., Kawai, G., Watanabe, K., Miura, K. & Ichiro, H. GNA trinucleotide loop sequences producing extraordinarily stable DNA Minihairpins. Biochemistry 36, 4761–4767 (1997).
Billet, B. et al. Aptamer switches regulated by post-transition/transition metal ions. Angew. Chem. Int. Ed. 60, 12346–12350 (2021).
Brown, A. K., Li, J., Pavot, C. M.-B. & Lu, Y. A lead-dependent DNAzyme with a two-step mechanism. Biochemistry 42, 7152–7161 (2003).
Sigel, R. K. O. & Sigel, H. A stability concept for metal ion coordination to single-stranded nucleic acids and affinities of individual sites. Acc. Chem. Res. 43, 974–984 (2010).
Varani, G., Aboul-ela, F. & Allain, F. H.-T. NMR investigation of RNA structure. Prog. Nucl. Magn. Reson. Spectrosc. 29, 51–127 (1996).
Wijmenga, S. S. & van Buuren, B. N. M. The use of NMR methods for conformational studies of nucleic acids. Prog. Nucl. Magn. Reson. Spectrosc. 32, 287–387 (1998).
Hwang, T. L. & Shaka, A. J. Water suppression that works. excitation sculpting using arbitrary wave-forms and pulsed-field gradients. J. Magn. Reson. Ser. A 112, 275–279 (1995).
Bax, A. & Davis, D. G. MLEV-17-based two-dimensional homonuclear magnetization transfer spectroscopy. J. Magn. Reson. 65, 355–360 (1985).
Schleucher, J. et al. A general enhancement scheme in heteronuclear multidimensional NMR employing pulsed field gradients. J. Biomol. NMR 4, 301–306 (1994).
Sklenář, V., Miyashiro, H., Zon, G., Todd Miles, H. & Bax, A. Assignment of the 31P and 1H resonances in oligonucleotides by two-dimensional NMR spectroscopy. FEBS Lett. 208, 94–98 (1986).
Piotto, M., Saudek, V. & Sklenář, V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2, 661–665 (1992).
Lee, W., Tonelli, M. & Markley, J. L. NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327 (2015).
Case, D. A. et al. Amber 2018, University of California, San Francisco. https://ambermd.org/doc12/Amber18.pdf (2018).
Zgarbová, M. et al. Refinement of the Cornell et al. nucleic acids force field based on reference quantum chemical calculations of glycosidic torsion profiles. J. Chem. Theory Comput. 7, 2886–2902 (2011).
Joung, I. S. & Cheatham, T. E. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B 112, 9020–9041 (2008).
Acknowledgements
This work was supported by the National Science Center (Poland) under grant agreement: [UMO-2018/31/D/ST4/01467] to W.A. The calculations were performed at Poznań Supercomputing and Networking Center. The authors would like to thank Dr Karolina Zielińska for help in preparing the manuscript figures.
Author information
Authors and Affiliations
Contributions
W.A. designed the research. W.A. and Z.G. supervised the research. J.W., A.P., E.P., and K.P. collected the data, W.A., J.W., and A.P. analyzed the data, W.A. wrote the manuscript with comments and contributions from all other authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Solen Ekesan and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wieruszewska, J., Pawłowicz, A., Połomska, E. et al. The 8-17 DNAzyme can operate in a single active structure regardless of metal ion cofactor. Nat Commun 15, 4218 (2024). https://doi.org/10.1038/s41467-024-48638-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-48638-x
This article is cited by
-
The dynamic world of the 8–17 DNAzyme
Nature Communications (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
