
Abstract
Skeletal modifications enable elegant and rapid access to various derivatives of a compound that would otherwise be difficult to prepare. They are therefore a powerful tool, especially in the synthesis of natural products or drug discovery, to explore different natural products or to improve the properties of a drug candidate starting from a common intermediate. Inspired by the biosynthesis of the cephalotane natural products, we report here a single-atom insertion into the framework of the benzenoid subfamily, providing access to the troponoid congeners — representing the reverse of the proposed biosynthesis (i.e., a contra-biosynthesis approach). Computational evaluation of our designed transformation prompted us to investigate a Büchner–Curtius–Schlotterbeck reaction of a p-quinol methylether, which ultimately results in the synthesis of harringtonolide in two steps from cephanolide A, which we had previously prepared. Additional computational studies reveal that unconventional selectivity outcomes are driven by the choice of a Lewis acid and the nucleophile, which should inform further developments of these types of reactions.
Similar content being viewed by others
Introduction
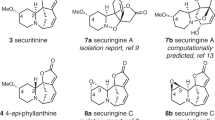
The total synthesis of natural products remains an active area of research in chemical synthesis1,2,3. In cases where many congeners of a family of natural products are targeted for synthesis, it is often more efficient to prepare a late-stage intermediate that can be diversified to access the entire collection4,5. In some instances, such late-stage diversification approaches have closely mimicked the biosynthetic pathway to the targeted molecules. For example, congeners of terpenoid secondary metabolites often arise from oxidation or oxygenation reactions that are effected by tailoring P450 enzymes in what has come to be referred to as the oxidase phase6,7. This general approach has been adopted to great effect in preparing many terpenoids8,9,10,11. In our laboratory, we have applied the late-stage diversification approach to the syntheses of members of the longiborneols12,13, the phomactins14,15, the diterpenoid alkaloids16,17, and more recently, cephalotane natural products such as the cephanolides and ceforalides (e.g., 1 and 2) that were prepared from pentacycle 3 (Fig. 1A)18,19.

A Benzenoid subfamily and our previous work. B Troponoids and non-aromatic seven-membered ring congeners. C Biosynthesis of the cephalotanes and our strategy employing a single-atom insertion. [O]: oxidation.
The cephanolides20 and ceforalides21 are structurally related to harringtonolide (4), first isolated in 1978 from C. harringtonia (Fig. 1B)22. This natural product has been shown to possess interesting bioactivity, including antiviral and antineoplastic activity23,24. The key difference between these structures is that the cephanolides and ceforalides bear an arene A-ring or oxidized variant thereof (hence our reference to these compounds as the benzenoid congeners), whereas 4 possesses a tropone A-ring. Over the last half-decade, a large number of additional troponoids and non-aromatic seven-membered A-ring cephalotane congeners have been isolated25, including the fortalpinoids (e.g., 5)26, mannolides (e.g., 6)27, and cephinoids (e.g., 7)28. While syntheses of these latter classes of cephalotanes are beginning to appear29,30,31, harringtonolide remains a popular synthetic target32,33,34. Biosynthetically, it is proposed that the benzenoids might be derived from the troponoid subfamily (e.g., 8, Fig. 1C) through a 6π electrocyclization to arrive at the corresponding cyclopropanone (9), which, following a Baeyer–Villiger type oxidation and aromatization, would give the benzenoid type I framework (10). Subsequent decarboxylation and oxidation events would then yield a variety of other congeners bearing the benzenoid type II and III frameworks (11 and 12)20,35. This proposed cephalotane biosynthesis, which relies on a net one-carbon deletion inspired us to explore a contra-biosynthetic approach employing single-atom insertion to prepare the troponoids from the benzenoid subfamily36.
Strategies to achieve such single-atom skeletal edits to access privileged scaffolds continue to emerge and draw the interest of the synthesis community37. Because nitrogen-containing heteroaromatics are the most commonly occurring structural motifs in pharmaceuticals and agrochemicals38,39, many current methods for skeletal editing have relied on the intrinsic reactivity of aza-heterocycles. In our planned approach, we saw an opportunity to highlight skeletal editing of carbocyclic arenes through ring expansion (and ultimately, also ring contraction) to access families of natural products. The challenge of effecting a skeletal change in complex sp3-rich polycyclic structures with multiple functionalities offered opportunities to develop new methods. Here, we report the realization of the benzenoid-to-troponoid conversion of the cephalotanes, culminating in a two-step synthesis of harringtonolide from cephanolide A. Notably, the success of our studies was guided by valuable insights gained through computational analysis of the key ring expansion reaction.
Results and discussion
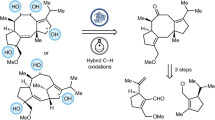
Harringtonolide (4) has been synthesized by the groups of Mander32, Tang33, and Zhai34 using highly innovative approaches. In particular, Mander’s approach32, which relied on an intramolecular Büchner reaction40,41, was highly inspirational to our planned benzenoid-to-troponoid conversion for the synthesis of 4 (Fig. 1C, Approach A). However, this approach was uniformly unsuccessful even following an extensive survey of reaction conditions (Supplementary Table 1)42,43,44,45,46,47,48. Given the limitations of our attempted intermolecular cycloadditions, we decided to investigate different ring expansion approaches that relied on the reactivity of carbonyl groups (Fig. 1C, Approach B). We were drawn to oxidative dearomatizations of phenols to provide quinols49, which could be followed by a Büchner–Curtius–Schlotterbeck (BCS) reaction50,51,52 to afford the desired tropone moiety (Fig. 2A)53,54,55,56,57,58,59. In general, the BCS reaction has been well-explored and established using saturated ketones60. To the best of our knowledge, there were no reports employing p-quinol derivatives such as 16 as substrates when we carried out these studies. However, during the review of our work, related studies appeared61,62,63. We postulated that a BCS reaction using 16 would undergo ring expansion to 18 via 17. An elimination of the alkoxy group in 18 would yield the desired tropone (19).

A Büchner–Curtius–Schlotterbeck reaction and our hypothesis. B Computational study performed at the ωB97X-D/def2-TZVPP(SMD=CH2Cl2)//ωB97X-D/def2-SVP(SMD=CH2Cl2) level of theory to evaluate the feasibility of tropone formation. LA: Lewis acid, p-: para-.
Reaction design and experimental investigation
To evaluate the feasibility of this tropone synthesis, we have undertaken computational studies as outlined in Fig. 2B64. In the context of the synthesis of harringtonolide (4), we envisioned using quinol methylether 20, which would undergo a one-carbon insertion by a BCS ring-expansion via 21, followed by tautomerization of 22 and loss of methanol (see 22’) to afford 4. We first began our calculations at the several levels of theory (Supplementary Fig. 6A) (Quantum chemical calculations were performed with Gaussian 16 rev. C.01 for geometry optimizations and ORCA 5.0.4 for single-point energy corrections; see the Supplementary Information for full computational details and references.)65,66,67,68 by modeling the reaction of 20 with CH2N2 in the presence of BF3•OEt2, which represents one of the most commonly employed conditions for these types of reactions50. We theorized that the Lewis acid likely binds to the carbonyl lone-pair of 20 away from the α-Me group, as shown in 20’-A. At this stage, two diastereoselective additions of CH2N2 are possible, leading to adducts 21 or 21’, respectively, in which the convex adduct 21 (via TS1; ∆G‡ = 11.7 kcal/mol) is marginally favored by 0.6 kcal/mol. The formation of the tropone ring by ring expansion of 21 is energetically feasible via TS2-A/B (∆G‡ = 6.5 kcal/mol), leading to two constitutional isomers (22 and 23). We also found the possibility of intramolecular oxygen replacement via TS2-C to give rise to epoxide 24. Overall, the C–C migration (TS2) was calculated to be product-determining, wherein a Curtin–Hammett scenario is one of many possibilities to account for our observations.
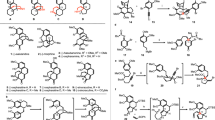
Given the promising preliminary computational results, we commenced our investigation of the planned BCS reaction by preparing p-quinol derivative 20 (Fig. 3A). Treatment of cephanolide A (1), which was prepared by a modified 12-step sequence (Supplementary Fig. 1), with Kita oxidative dearomatization conditions69,70 afforded 20 in 55% yield. Based on our preliminary calculations, we initially attempted conditions using CH2N2 in the presence of BF3•OEt2 for the tropone formation (Table in Fig. 3A). Unfortunately, these conditions were ineffective and led primarily to the recovery of the starting material (entry 1). Likely, CH2N2 was not nucleophilic enough to react with the carbonyl group of 20 and decomposed under the conditions. Therefore, we turned to other diazomethane equivalents and first examined TMSCHN2 (2.0 equiv) in the presence of BF3•OEt2 (1.2 equiv). To our delight, conducting the reaction at –78 °C yielded tropone 4 but in only 9% isolated yield along with a 57% yield of 25 (a 1:6.3 ratio; entry 2). We also found that using 3.0 equiv of TMSCHN2 at –60 °C, 20 was fully consumed to give 4 in 19% yield and 25 in 70% yield (a 1:3.7 ratio; entry 3). To increase the selectivity for the formation of 4, we then screened a range of Lewis acids (entries 4–9). As a result, we found that AlCl3 (3.0 equiv) along with 5.0 equiv of TMSCHN2 converted 20 to a 37% yield of 4 and 45% yield of 25 (a 1:1.2 ratio; entry 7). Overall, these conditions proved to be optimal (see Supplementary Tables 2 and 3 for full details). Of note, the conversion of 20 to harringtonolide (4) represents the shortest synthesis of this natural product reported to date (14 steps from commercially available material). The selectivity outcome, unexpected based on our preliminary DFT calculations with CH2N2 (entry 3), as well as the improved ratio obtained using AlCl3 (entry 7), led us to undertake additional calculations to gain more insight into the selectivity of this reaction.

A Optimization table for the synthesis of harringtonolide. B Selectivity for the nucleophilic attack of TMSCHN2 on the two prochiral faces of substrate 20-[B]. C Potential energy surface for the reaction between TMSCHN2 and 20-[LA]; All calculations were performed at the ωB97M-V/def2-TZVPP(SMD=CH2Cl2)//ωB97X-D/def2-SVP(SMD=CH2Cl2) level of theory. PIDA: phenyliodine(III) diacetate, TMS: trimethylsilyl, TS: transition state.
Computational studies
With some experimental results in hand, we performed benchmarking computational studies to rationalize the observed selectivity using a range of computational protocols. Based on our computational benchmarking, we found that ωB97M-V/def2-TZVPP(SMD=CH2Cl2)//ωB97X-D/def2-SVP(SMD=CH2Cl2) level of theory most accurately reproduced the empirically observed selectivity. A revised PES has also been calculated for the reaction of 20 with CH2N2 described in Fig. 2B (Supplementary Fig. 6B). Our calculations showed that the attack of TMSCHN2 should occur on the si-face of 20-[LA] — favored by 1 kcal/mol in the case of the BF3-activated substrate (Fig. 3B). However, two possible orientations of the attacking nucleophile are possible, leading to either 26a or 26b. Rearrangement of 26a would yield 27a or 28a, whereas 26b would lead to 27b or 28b. For the computed scenario with BF3•OEt2 as the Lewis acid at –60 °C (Fig. 3C), TS1a-[B] was found to have a 0.5 kcal/mol higher barrier compared to TS1b-[B]. In this case, we believe that the energy difference between TS1a-[B] and TS1b-[B] accounts for the observed distribution of products, which compares favorably with the empirical observation (i.e., the ratio of 4/25 = 1:3.7, which corresponds to a ~ 0.6 kcal/mol difference). With AlCl3 as a Lewis acid, there is no difference in stability between TS1a-[Al] and TS1b-[Al], consistent with our observed ratio (4/25 = 1:1.2). Overall, these computational results show that in the BCS reaction using TMSCHN2, the addition of TMSCHN2 (TS1) to the Lewis acid-bound p-quinol derivative is the selectivity-dictating step.
To gain deeper insight into the impact of the choice of Lewis acid on the reaction outcome, we conducted a comprehensive analysis of the product-determining TSs for both the BF3•OEt2 and AlCl3-mediated systems (Fig. 4). In the case of the minor pathway via TS1a-[B], we observed a C–C bond distance of 2.18 Å between the nucleophilic carbon of TMSCHN2 and the adjacent carbonyl group in an eclipsed orientation. This unexpected, eclipsed orientation of the incoming substituents along the forming C–C bond can be attributed to favorable dispersive interactions between the highly polarizable TMS group and the carbonyl group, as evidenced by the non-covalent interaction (NCI) isosurfaces71. In addition, in the case of TS1b-[B], which features a similar C–C bond distance of 2.19 Å, we observed a staggered orientation of the substituents, with the TMS group placed in close proximity to the BF3 Lewis acid, which sits in the plane of the carbonyl group. This change in orientation from eclipsed to staggered is driven by the favorable interactions between the partially negatively charged fluoride atoms and the electropositive silicon atom, located within 3.26 Å. As such, the preferential reactivity via TS1b-[B] can be attributed to favorable electrostatic and dispersive interactions between the TMS group and the Lewis acid in the case of BF3•OEt2.

Effect of the Lewis acid on the product-determining TS of the reaction demonstrated for BF3•OEt2 and AlCl3. The relatively small BF3•OEt2 (left) lies in the plane of the carbonyl group, leading to TS1b-[B] as the favored TS, whereas the larger Lewis acid AlCl3 (right) rotates out of the plane and therefore both TS are equally present. TS: transition state.
When we conducted a similar analysis on the same two competing TSs (i.e., TS1a-[Al] and TS1b-[Al]), using AlCl3 as the Lewis acid, some significant structural differences emerged. Firstly, the forming C–C bonds between TMSCHN2 and the substrate were found to be 0.1 Å longer, which is consistent with earlier TSs, indicating the lower activation energy barriers in this case compared to using BF3 (as shown in Fig. 3). However, a more significant change was observed in the orientation of the AlCl3 group, which moved out of co-planarity with the carbonyl group due to increased steric demand. This fundamental structural alteration eliminates any favorable interaction between the Lewis acid and the TMS group in TS1b-[Al] and promotes the reorientation of the TMS group to a more favorable eclipsed position, similar to that observed in TS1a-[Al]/[B]. Consequently, this loss of favorable non-covalent interactions destabilizes TS1b-[Al], resulting, overall, in better selectivity toward the desired product 27. Finally, in a preliminary study, we have shown that the tropone formation can be extended to other substrates (Supplementary Fig. 2).
In conclusion, we have shown that an oxidative dearomatization and ring expansion starting from cephanolide A accomplishes a benzenoid-to-troponoid ring expansion to afford harringtonolide. To gain insight into the regioselectivity-determining factors in the ring expansion reaction, we have carried out extensive computational studies. These calculations have unveiled the unique effects of the different Lewis acids in establishing secondary interactions with TMSCHN2 which significantly affect the regioselectivity by changing the relative energies of the different transition structures. The extension of the ring expansion transformation described here to other quinol derivatives are provided in Supplementary Fig. 2. Future studies will focus on the application of the Büchner–Curtius–Schlotterbeck transformation to other natural product classes.
Methods
General considerations
Commercial reagents and solvents were purchased from Fisher Scientific, Acros Organics, Alfa Aesar, and/or Sigma Aldrich, and used without additional purification. Diazomethane (CH2N2) was generated using an Aldrich® diazomethane-generator with System 45TM. MeCN and MeOH were sparged with argon and dried by passing through alumina columns using argon in a Glass Contour solvent purification system. DCM was freshly distilled over calcium hydride under a N2 atmosphere before each use. Reaction progress was monitored by thin-layer chromatography (TLC) on Macherey-Nagel TLC plates (60 Å, F254 indicator). TLC plates were visualized by exposure to ultraviolet light (254 nm), and/or stained by submersion in aqueous potassium permanganate solution (KMnO4), p-anisaldehyde, or phosphomolybdic acid stain and heating with a heat gun. Organic solutions were concentrated under reduced pressure on a Heidolph temperature-controlled rotary evaporator equipped with a dry ice/isopropanol condenser.
Oxidative dearomatization
To a solution of cephanolide A (1) (25.0 mg, 83.8 μmol, 1.0 equiv) in MeCN/MeOH (1:1 v/v, 838 µL, 0.1 M) was added phenyliodine(III) diacetate (PIDA; 32.4 mg, 101 μmol, 1.2 equiv) at 0 °C under a N2 atmosphere. After stirring at room temperature for 5 h, the reaction mixture was quenched with sat. aq. NaHCO3 (2 mL), diluted with H2O (3 mL) and extracted with DCM (3 × 5 mL). The combined organic phase was dried over Na2SO4 and concentrated in vacuo. The resulting residue was purified by silica gel flash column chromatography (hexanes/EtOAc = 2:1), yielding methyl-ceforalide H (20) (15.2 mg, 46.3 μmol, 55%) as a colorless solid.
Ring-expansion
A flame-dried vial with a magnetic stir bar was transferred to a glovebox and charged with AlCl3 (12.2 mg, 91.4 µmol, 3.0 equiv). The vial was sealed with a septa cap and removed from the glove box. The vial was evacuated and backfilled with N2 three times and cooled to –60 °C. Freshly distilled DCM (50 µL) was added, and the suspension was stirred at –60 °C for 5 min. A solution of methyl-ceforalide H (20) (10.0 mg, 30.5 µmol, 1.0 equiv) in freshly distilled DCM (250 µL) was added and stirred at –60 °C for 10 min to give a grayish suspension. TMS-diazomethane (0.2 M, prepared from a 2.0 M solution in hexanes diluted with freshly distilled DCM, 760 µL, 152 µmol, 5.0 equiv) was added over 2 min resulting in a yellowish solution. The mixture was stirred at –60 °C for 3 h and quenched with sat. aq. NaHCO3 (500 µL). The suspension was diluted with H2O (2 mL) and extracted with DCM (3 × 3 mL). The combined organic layers were dried over anhydrous Na2SO4, and the solvent was removed under reduced pressure. The residue was purified by preparative TLC (hexanes/EtOAc = 1:3), yielding harringtonolide (4) (3.5 mg, 11.3 µmol, 37%) as a colorless solid and iso-harringtonolide (25) (4.3 mg, 13.9 µmol, 46%) as a colorless solid.
Computational methods
The range-separated dispersion corrected ωB97X-D density functional65 was used in conjunction with the double-zeta valence polarized def2-SVP basis set67, to optimize the geometry of all stationary points. Additional single points energy correction was carried out with the newer generation meta-augmented range separated density functional ωB97M-V9 that employs the Vydrov and van Voorhis VV10 dispersion correction72, together with the triple-zeta valence polarized def2-TZVPP basis set. The VV10 dispersion corrected family of functionals developed by the Head-Gordon group have been demonstrated to be one of the most robust functionals for assessment of main group thermochemistry and for describing non-covalent interactions (Quantum chemical calculations were performed with Gaussian 16 rev. C.01 for geometry optimizations and ORCA 5.0.4 for single-point energy corrections; see the Supplementary Information for full computational details and references.). All calculations included the integral equation formalism variant of the polarizable continuum model (IEF-PCM), with the SMD solvation model to account for solvation effects (solvent = dichloromethane)68. Conformational sampling was performed manually. Gaussian16 version C.01 was employed for all density functional theory (DFT) geometry optimization calculations, using the default ultrafine pruned (99,590) grid for numerical integration of the exchange-correlation functional and its derivatives (Quantum chemical calculations were performed with Gaussian 16 rev. C.01 for geometry optimizations and ORCA 5.0.4 for single-point energy corrections; see the Supplementary Information for full computational details and references.). Single point corrections were carried our using ORCA 5.0.473. Vibrational frequency calculations were used to verify that stationary points were either minima or first-order saddle points on the corresponding potential energy surface. Additional intrinsic reaction coordinate (IRC) calculations were performed to ensure that the transition state structures connected to their appropriate initial and final geometries74. The computed thermochemistry data were further corrected following Grimme’s quasi-harmonic (QHA)75 model for entropy with a frequency cut-off value of 100.0 cm−1 using the GoodVibes76 program at 213.15 K (−60° C). In addition, GoodVibes applied 1 M standard concentration corrections to all individual calculations to account for reactions in solution (i.e., change in standard concentration from 1 atm to 1 M)77. XYZ coordinate files were also generated using GoodVibes.
Data availability
The data supporting the findings of this study are available within the paper and its Supplementary Information. Detailed information for reaction conditions, compound characterization data, computational data, and crystallographic data are in the Supplementary Information. The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Center (CCDC), under deposition numbers 2293695 (20) and 2293696 (25). These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif. All data are available upon request from the corresponding authors.
References
Nicolaou, K. C., Vourloumis, D., Winssinger, N. & Baran, P. S. The art and science of total synthesis at the dawn of the twenty-first century. Angew. Chem. Int. Ed. 39, 44–122 (2000).
Wender, P. A. & Miller, B. L. Synthesis at the molecular frontier. Nature 460, 197–201 (2009).
Hoffmann, R. W. Natural product synthesis: changes over time. Angew. Chem. Int. Ed. 52, 123–130 (2013).
Li, L., Chen, Z., Zhang, X. & Jia, Y. Divergent strategy in natural product total synthesis. Chem. Rev. 118, 3752–3832 (2018).
Kanda, Y., Ishihara, Y., Wilde, N. C. & Baran, P. S. Two-phase total synthesis of taxanes: tactics and strategies. J. Org. Chem. 85, 10293–10320 (2020).
Maimone, T. J. & Baran, P. S. Modern synthetic efforts toward biologically active terpenes. Nat. Chem. Biol. 3, 396–407 (2007).
Ishihara, Y. & Baran, P. S. Two-phase terpene total synthesis: historical perspective and application to the taxol® problem. Synlett 12, 1733–1745 (2010).
Chen, K. & Baran, P. S. Total synthesis of eudesmane terpenes by site-selective C–H oxidations. Nature 459, 824–828 (2009).
Jørgensen, L. et al. 14-Step Synthesis of (+)-Ingenol from (+)−3-Carene. Science 341, 878–882 (2013).
Kawamura, S., Chu, H., Felding, J. & Baran, P. S. Nineteen-step total synthesis of (+)-phorbol. Nature 532, 90–93 (2016).
Chu, H., Smith, J. M., Felding, J. & Baran, P. S. Scalable synthesis of (−)-thapsigargin. ACS Cent. Sci. 3, 47–51 (2017).
Lusi, R. F., Sennari, G. & Sarpong, R. Total synthesis of nine longiborneol sesquiterpenoids using a functionalized camphor strategy. Nat. Chem. 14, 450–456 (2022).
Lusi, R. F., Sennari, G. & Sarpong, R. Strategy evolution in a skeletal remodeling and C–H functionalization-based synthesis of the longiborneol sesquiterpenoids. J. Am. Chem. Soc. 144, 17277–17294 (2022).
Kuroda, Y. et al. Isolation, synthesis, and bioactivity studies of phomactin terpenoids. Nat. Chem. 10, 938–945 (2018).
Leger, P. R., Kuroda, Y., Chang, S., Jurczyk, J. & Sarpong, R. C–C bond cleavage approach to complex terpenoids: development of a unified total synthesis of the phomactins. J. Am. Chem. Soc. 142, 15536–15547 (2020).
Marth, C. J. et al. Network-analysis-guided synthesis of Weisaconitine D and Liljestrandinine. Nature 528, 493–498 (2015).
Kou, K. G. M. et al. A unifying synthesis approach to the C18-, C19-, and C20-diterpenoid alkaloids. J. Am. Chem. Soc. 139, 13882–13896 (2017).
Haider, M., Sennari, G., Eggert, A. & Sarpong, R. Total synthesis of the cephalotaxus norditerpenoids (±)-cephanolides A–D. J. Am. Chem. Soc. 143, 2710–2715 (2021).
Sennari, G. et al. Unified total syntheses of benzenoid cephalotane-type norditerpenoids: cephanolides and ceforalides. J. Am. Chem. Soc. 144, 19173–19185 (2022).
Fan, Y.-Y. et al. cephanolides A–J, cephalotane-type diterpenoids from Cephalotaxus sinensis. J. Nat. Prod. 80, 3159–3166 (2017).
Ge, Z.-P. et al. Cephalotane-type Norditerpenoids from Cephalotaxus fortune var. alpina. Chin. J. Chem. 40, 1177–1184 (2022).
Buta, J. G., Flippen, J. L. & Lusby, W. R. Harringtonolide, a plant growth inhibitory tropone from Cephalotaxus harringtonia (Forbes) K. Koch. J. Org. Chem. 43, 1002–1003 (1978).
Sun, N.-J., Xue, Z., Liang, X.-T. & Huang, L. Studies on the structure of a new antitumor agent – hainanolide. Acta Pharm. Sin. 14, 39–43 (1979).
Kang, S. Q., Cai, S. Y. & Teng, L. Antiviral effect of hainanolide. Acta Pharmacol. Sin. 16, 867–868 (1981).
Jiang, C. et al. Progress in structure, synthesis and biological activity of natural Cephalotane Diterpenoids. Phytochemistry 192, 112939 (2021).
Ge, Z.-P. et al. 17-nor-Cephalotane-type diterpenoids from Cephalotaxus Fortunei. J. Nat. Prod. 82, 1565–1575 (2019).
Ni, G. et al. Mannolides A–C with an intact diterpenoid skeleton providing insights on the biosynthesis of antitumor Cephalotaxus Troponoids. Org. Lett. 18, 1880–1883 (2016).
Ni, L. et al.Bioactive norditerpenoids from Cephalotaxus fortunei var alpina and C. lanceolata. Phytochemistry 151, 50–60 (2018).
Ren, Z. et al. Total synthesis of (+)‐3‐Deoxyfortalpinoid F, ( + )‐Fortalpinoid A, and (+)‐Cephinoid H. Angew. Chem. Int. Ed. 60, 18572–18576 (2021).
Ao, Q., Zhang, H., Zheng, J., Chen, X. & Zhai, H. Asymmetric total synthesis of (+)‐Mannolide C. Angew. Chem. Int. Ed. 60, 21267–21271 (2021).
Wang, H. et al. Asymmetric total synthesis of Cephalotaxus Diterpenoids: Cephinoid P, Cephafortoid A, 14-epi-Cephafortoid A and Fortalpinoids M–N, P. J. Am. Chem. Soc. 145, 16988–16994 (2023).
Frey, B., Wells, A. P., Rogers, D. H. & Mander, L. N. Synthesis of the unusual diterpenoid tropones hainanolidol and harringtonolide. J. Am. Chem. Soc. 120, 1914–1915 (1998).
Zhang, M., Liu, N. & Tang, W. Stereoselective total synthesis of hainanolidol and harringtonolide via oxidopyrylium-Based [5 + 2] cycloaddition. J. Am. Chem. Soc. 135, 12434–12438 (2013).
Zhang, H.-J. et al. Total synthesis of the Diterpenoid (+)-Harringtonolide. Angew. Chem. Int. Ed. 55, 11638–11641 (2016).
Xu, J.-B. et al. Cephalotanins A-D, four norditerpenoids represent three highly rigid carbon skeletons from Cephalotaxus Sinensis. Chem. Eur. J. 22, 14648–14654 (2016).
Wu, X. et al. Semi-synthesis of harringtonolide derivatives and their antiproliferative activity. Molecules 26, 1380 (2021).
Jurczyk, J. et al. Single-atom logic for heterocycle editing. Nature Synthesis 1, 352–364 (2022).
Taylor, R. D., MacCoss, M. & Lawson, A. D. G. Rings in drugs: miniperspective. J. Med. Chem. 57, 5845–5859 (2014).
Vitaku, E., Smith, D. T. & Njardarson, J. T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals: miniperspective. J. Med. Chem. 57, 10257–10274 (2014).
Ye, T. & McKervey, M. A. Organic synthesis with α-diazo carbonyl compounds. Chem. Rev. 94, 1091–1160 (1994).
Reisman, S. E., Nani, R. R. & Levin, S. Büchner and beyond: arene cyclopropanation as applied to natural product total synthesis. Synlett 17, 2437–2442 (2011).
Fleming, G. S. & Beeler, A. B. Regioselective and enantioselective intermolecular Buchner ring expansions in flow. Org. Lett. 19, 5268–5271 (2017).
Su, J., Hu, X., Huang, H., Guo, Y. & Song, Q. Difluorocarbene enables to access 2-fluoroindoles from ortho-vinylanilines. Nat. Commun. 12, 4986 (2021).
Reber, K. P., Gilbert, I. W., Strassfeld, D. A. & Sorensen, E. J. Synthesis of (+)-lineariifolianone and related cyclopropenone-containing sesquiterpenoids. J. Org. Chem. 84, 5524–5534 (2019).
Ciamician, G. L. & Dennstedt, M. Über die einwirkung des chloroforms auf die kaliumverbindung pyrrols. Ber. Dtsch. Chem. Ges. 14, 1153–1163 (1881).
Ma, D., Martin, B. S., Gallagher, K. S., Saito, T. & Dai, M. One-carbon insertion and polarity inversion enabled a pyrrole strategy to the total syntheses of pyridine-containing lycopodium alkaloids: complanadine A and lycodine. J. Am. Chem. Soc. 143, 16383–16387 (2021).
Holmberg-Douglas, N., Onuska, N. P. R. & Nicewicz, D. A. Regioselective arene C–H alkylation enabled by organic photoredox. Catalysis. Angew. Chem. Int. Ed. 59, 7425–7429 (2020).
Ciamician–Dennstedt Reaction. Comprehensive organic name reactions and reagents; John Wiley & Sons, Inc: 2010; 646–648. https://doi.org/10.1002/9780470638859.conrr143.
Roche, S. P. & Porco, J. A. Dearomatization strategies in the synthesis of complex natural products. Angew. Chem. Int. Ed. 50, 4068–4093 (2011).
Büchner, E. & Curtius, T. Synthese von ketonsaureanthern aus aldehyden und diazoessigäther. Chem. Ber. 18, 2371–2377 (1885).
Schlotterbeck, F. Transformation of aldehydes into ketones by means of diazomethane. Chem. Ber. 40, 1826–1827 (1907).
Büchner–Curtius–Schlotterbeck Reaction. Comprehensive organic name reactions and reagents; 567–569 (John Wiley & Sons, Inc. 2010).
Liu, N., Song, W., Schienebeck, C. M., Zhang, M. & Tang, W. Synthesis of naturally occurring tropones and tropolones. Tetrahedron 70, 9281–9305 (2014).
Sennari, G., Hirose, T., Iwatsuki, M., Ōmura, S. & Sunazuka, T. A concise total synthesis of puberulic acid, a potent antimalarial agent. Chem. Commun. 50, 8715–8718 (2014).
Sennari, G. et al. Antimalarial troponoids, puberulic acid and viticolins; divergent synthesis and structure-activity relationship studies. Sci. Rep. 7, 7259 (2017).
Bemis, C. Y. et al. Total synthesis and computational investigations of sesquiterpene-tropolones ameliorate stereochemical inconsistencies and resolve an ambiguous biosynthetic relationship. J. Am. Chem. Soc. 143, 6006–6017 (2021).
Murelli, R. P., Berkowitz, A. J. & Zuschlag, D. W. Carbocycloaddition strategies for troponoid synthesis. Tetrahedron 130, 133175 (2023).
Umekubo, N. & Yokoshima, S. Total syntheses of malettinins C and E. Org. Lett. 25, 4530–4533 (2023).
Combs, J. et al. Enantioselective synthesis of anhydrogukulenin A C2-acetate. Preprint at ChemRxiv. Cambridge: Cambridge Open Engage; (2023); This content is a preprint and has not been peer-reviewed. https://doi.org/10.26434/chemrxiv-2023-8dj1k.
Candeias, N. R., Paterna, R. & Gois, P. M. P. Homologation reaction of ketones with diazo compounds. Chem. Rev. 116, 2937–2981 (2016).
Our work was published in ChemRxiv on February 6, 2024 https://doi.org/10.26434/chemrxiv-2024-v0670.
On March 25, 2024 during peer review of our work, another paper describing related chemistry appeared, seeShao, H. et al. Bio-inspired total synthesis of cephalotaxus diterpenoids and their structural analogues. Angew. Chem. Int. Ed., e202402931 (2024).
In addition, another manuscript on this topic was uploaded as a preprint on February 14, seeZhang, Z.-A. et al. Total synthesis of chephanolide A and harringtonolide: A unified strategy connecting benzenoid and troponoid cephalotaxus diterpenoids. Preprint at ChemRxiv. Cambridge: Cambridge Open Engage; https://doi.org/10.26434/chemrxiv-2024-rp8s5 (2024).
Sakai, T., Ito, S., Furuta, H., Kawahara, Y. & Mori, Y. Mechanism of the regio- and diastereoselective ring expansion reaction using trimetheylsilyldiazomethane. Org. Lett. 14, 4564–4567 (2012).
Chai, J.-D. & Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 10, 6615–6620 (2008).
Mardirossian, N. & Head-Gordon, M. wB97M-V: A combinatorially optimized, range-separated hybrid, meta-GGA density functional with VV10 nonlocal correlation. J. Chem. Phys 144, 214110 (2016).
Weigend, F. & Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305 (2005).
Marenich, A. V., Cramer, C. J. & Truhlar, D. G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 113, 6378–6396 (2009).
Tamura, Y., Yakura, T., Tohma, H., Kikuchi, K. & Kita, Y. Hypervalent iodine oxidation of p-Alkoxy- and related phenols: A facile and efficient synthesis of p-quinones. Synthesis 1989, 126–127 (1989).
Kita, Y., Yakura, T., Tohma, H., Kikuchi, K. & Tamura, Y. A synthetic approach to discorhabdin alkaloids: hypervalent iodine oxidation of p-substituted phenol derivatives to azacarbocyclic spirodienones. Tetrahedron Lett. 30, 1119–1120 (1989).
Contreras-Garcia, J. et al. NCIPLOT: A program for plotting noncovalent interaction regions. J. Chem. Theory Comput. 7, 625–632 (2011).
Vydrov, O. A. & Van Voorhis, T. J. Nonlocal can der Waals density functional: the simpler the better. J. Chem. Phys. 133, 244103 (2010).
Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2, 73–78 (2012).
Fukui, K. The path of chemical reactions – the IRC approach. Acc. Chem. Res. 14, 363–368 (1981).
Grimmer, S. Supramolecular binding thermodynamics by dispersion-corrected density functional theory. Chem. Eur. J. 18, 9955–9964 (2012).
Luchini, G., Alegre-Requena, J. V., Funes-Ardoiz, I. & Paton, R. S. GoodVibes: automated thermochemistry for heterogeneous computational chemistry data. F1000Research 9, 291 (2020).
Bryantsev, V. S., Diallo, M. S. & Goddard, W. A. III Calculation of solvation free energies of charged solutes using mixed cluster/continuum models. J. Phys. Chem. B 112, 9709–9719 (2008).
Acknowledgements
S.W. is grateful to the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, WI 5665/1-1) for a postdoctoral scholarship. G.S. thanks the Uehara Memorial Foundation for a postdoctoral fellowship. K.E.G. thanks the NSF for a graduate research fellowship (DGE 1752814). K.A. is grateful to the JSPS Overseas Challenge Program for Young Researchers and the Satomi Foundation for support of a visiting student stay at UC Berkeley. We thank Dr. Hasan Celik and UC Berkeley’s NMR facility in the College of Chemistry (CoC-NMR) for spectroscopic assistance. Instruments in CoC-NMR are supported in part by NIH S10OD024998. We are grateful to Dr. Nicholas Settineri (UC Berkeley) for X-ray crystallographic studies. This work utilized the Alpine HPC resource, which is jointly funded by the University of Colorado Boulder, the University of Colorado Anschutz, and Colorado State University, and the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) through allocation TG-CHE180056. The authors thank the NSF under the CCI Center for Computer-Assisted Synthesis (CHE-2202693) and the National Science Foundation (CHE-18566228) for support.
Author information
Authors and Affiliations
Contributions
The initial experiments were carried out by G.S. and S.W. planned the project and designed the experiments. The experimental work was performed by S.W., G.S., K.E.G. and K.A. The computational studies were conducted by M.V.P. The initial draft was written by G.S. with input from all authors. S.W., G.S., M.V.P., K.E.G., R.S.P. and R.S. were involved in editing and finalizing the manuscript. R.S.P. and R.S. supervised the project and secured funding.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wiesler, S., Sennari, G., Popescu, M.V. et al. Late-stage benzenoid-to-troponoid skeletal modification of the cephalotanes exemplified by the total synthesis of harringtonolide. Nat Commun 15, 4125 (2024). https://doi.org/10.1038/s41467-024-48586-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-48586-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
