
Abstract
Pulsed CO2 electroreduction (CO2RR) has recently emerged as a facile way to in situ tune the product selectivity, in particular toward ethanol, without re-designing the catalytic system. However, in-depth mechanistic understanding requires comprehensive operando time-resolved studies to identify the kinetics and dynamics of the electrocatalytic interface. Here, we track the adsorbates and the catalyst state of pre-reduced Cu2O nanocubes ( ~ 30 nm) during pulsed CO2RR using sub-second time-resolved operando Raman spectroscopy. By screening a variety of product-steering pulse length conditions, we unravel the critical role of co-adsorbed OH and CO on the Cu surface next to the oxidative formation of Cu-Oad or CuOx/(OH)y species, impacting the kinetics of CO adsorption and boosting the ethanol selectivity. However, a too low OHad coverage following the formation of bulk-like Cu2O induces a significant increase in the C1 selectivity, while a too high OHad coverage poisons the surface for C-C coupling. Thus, we unveil the importance of co-adsorbed OH on the alcohol formation under CO2RR conditions and thereby, pave the way for improved catalyst design and operating conditions.
Similar content being viewed by others


Introduction
Within the scope of reducing global CO2 emissions to limit climate change, carbon net-zero technologies have gained enormous interest. One promising technology is the CO2RR, which closes the carbon cycle by using renewable energies to transform CO2 back into useful chemicals and fuels1. Among the metals studied, only copper electrodes have the unique ability to produce the desired energy-dense alcohols and hydrocarbons such as ethanol and ethylene in significant amounts2. However, for further commercialization in high-current electrolyzers, Cu-based catalysts still suffer from a broad selectivity distribution, low activity, and low stability during long-term operation.
Several strategies have been developed to address these issues and to increase the product distribution toward C2+ products, which include tuning the catalyst structure and composition3,4,5,6,7 and modifying the electrolyte8,9,10. Additionally, the (initial) oxidation state of Cu plays a major role, particularly oxidized Cu species and oxide-derived Cu materials showed a major improvement in the selectivity and stability of the catalysts11,12,13,14,15,16. Thus, a simple way to regenerate the desired oxidation state of Cu in situ is by means of pulsed potential CO2RR, where an electrocatalytic potential alternates between an oxidizing and a CO2RR potential17. Hereby, the key pulse parameters are the applied cathodic and anodic potential, the pulse shape as well as the pulse length17.
By varying only the cathodic and anodic pulse lengths, while keeping the other pulse parameters constant, the catalytic properties of Cu electrodes could be enhanced17,18,19. In this way, the amount, as well as the type of Cu oxide species (Cu+, Cu2+) formed on the catalyst surface, was controlled20,21. In particular, distorted non-crystalline Cu(I)/Cu(II) domains were formed at short anodic pulses (below 2 s) as evidenced by operando X-ray absorption spectroscopy (XAS) and X-ray diffraction (XRD), which had been associated with an enhanced ethanol formation18. The dynamic alternation of the potential impacts also the restructuring (faceting and defects) and the roughening of the catalyst at longer anodic pulse lengths (above 1 s), which were observed to enhance the selectivity of either methane or ethylene18,20,22. However, the induced dynamics on the surface coverage of hydrogen (Had), hydroxide (OHad), and carbon monoxide (COad) are crucial to fully understand the underlying principle of selectivity changes via pulsed CO2RR. Several studies suggested that the application of the anodic pulse leads to higher OH coverage, while Had is removed due to the positive polarization of the electrodes19,21,23,24,25,26. The resulting higher OH coverage was proposed to stabilize the active COatop compared to the inactive CObridge species27,28,29, which then enhances the ethanol selectivity. The beneficial effect of a higher OH coverage and the resulting higher local pH was also linked to a change in the CO adsorption energy as well as a lower C–C coupling barrier30. While the CO binding configuration was previously tracked with operando surface-enhanced infrared absorption spectroscopy (SEIRAS)27,29, there is to date no experimental evidence for the postulated changes in the OH and CO coverages, and the selectivity-determining role of the adsorbed OH beyond CO dimerization stays unclear. Moreover, it remains challenging to determine the possible presence of surface Cu oxides due to the lack of surface-sensitive operando characterizations1. Thus, these tools are needed to gain a better mechanistic comprehension of pulsed CO2RR.
In this study, we, therefore, applied operando sub-second time-resolved surface-enhanced Raman spectroscopy (SERS), which is an ideal method to simultaneously track the changes in the surface oxidation state of Cu and the changes in the OHad and COad surface adsorbates as well as their surface coverage. In this way, we followed the impact of the applied anodic or cathodic pulse lengths during pulsed CO2RR. Thus, square-wave potential pulses were alternated between an electrocatalytic cathodic potential Ec at −1.0 V and an oxidizing anodic potential Ea at +0.6 V versus the reversible hydrogen electrode (RHE). Pre-reduced Cu2O nanocubes (NCs) served as catalysts, which exhibit tunable selectivities for valuable C2+ products such as ethanol or C1 products such as methane, depending on the selected pulse length18. In particular, we correlated the enhancement of ethanol observed to an increase of the OHad versus COad coverage and the formation of Cu-Oad and CuOx/(OH)y species under selected pulsed CO2RR conditions. Here, we demonstrate the impact of the adsorbates and surface species on the obtained selectivity trends in dependence on the applied pulse lengths to gain novel mechanistic insights in a sub-second time-resolved way.
Results
Time-dependent evolution of SERS
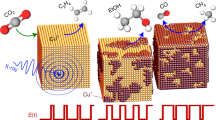
Figure 1a shows a scanning electron microscopy (SEM) image of the Cu2O NC (~20 nm in size) pre-catalysts deposited on a glassy carbon electrode. The pre-catalyst was first pre-reduced in an electrochemical operando Raman flow cell setup (Supplementary Note 1, Supplementary Figs. 1, 2), resulting in loss of the initial cubic shape and the growth of the particles (~30 nm in size, Fig. 1b). Subsequently, pulsed CO2RR with a selected cathodic pulse length tc and anodic pulse length ta was applied (Supplementary Table 1). For all pulsed CO2RR measurements in this study, Ec = −1.0 V and Ea = +0.6 V (vs. RHE, for all shown potentials in this study), and the electrolyte consists of 0.1 M CO2-saturated potassium bicarbonate (KHCO3) if not stated differently. We note that the morphology of the particles changed to larger structures after pulsed CO2RR, as observed in the SEM image in Fig. 1c. Figure 1d presents the applied potential and the measured current (top) during the symmetric pulses with tc = ta = 4 s and the corresponding normalized SERS signal intensities over the Raman shift (bottom) as a function of the time. For the normalization of the SERS spectra, the intensity values were modified to a mean of 0 and a variance of 1. We note that the optical properties of the working catalyst are changing under potential pulse conditions as the reduced Cu2O nanocubes transform into rough, porous nanostructures. Therefore, to account for different SERS enhancement at different pulse length conditions, the SERS bands in this study were only compared to each other within the same spectra to obtain intensity ratios (Supplementary Note 2). The periodic responses of the characteristic SERS bands in this spectroscopic region were fitted in Fig. 1e (with fit examples in Supplementary Fig. 3). The selected characteristic bands here are:
-
(I)
the COad vibrations on Cu, namely the Cu–CO rotation (COr) and stretching (COs) vibrations at 280 and 360 cm−1, where CO is considered to be adsorbed on Cu as shown with surface characterization methods31,32. The CO bands appear during each cathodic pulse as CO2RR intermediates and disappear during the anodic pulses6,33,34. The relative CO surface coverage (COcov) can be obtained by the intensity ratio of the two CO bands as COcov = Intensity (COs)/Intensity (COr). This relationship was derived in detail in our previous study through operando measurements in the presence of different CO concentrations and through DFT vibrational analysis for different CO coverages on Cu(100) surfaces33. However, the structural and morphological differences between the Cu(100) derived relationship to the oxide-derived Cu used in this work do not allow a direct comparison of the ratios and the CO coverage:
-
(II)
the OHad vibration on or close to Cu, which can appear during the cathodic pulse at 495 cm−1 and during the anodic pulse redshifted at 450 cm−1 if OH is adsorbed or close to the Cu surface via dipole-dipole interactions34,35,36:
-
(III)
the bands related to Cu2O on the surface at 410 cm−1 (multiphonon process), 530 cm-1 (Raman-allowed F2g mode), and 620 cm−1 (IR-allowed F1u mode), which appear during each anodic pulse due to the oxidation of Cu and disappear again during each cathodic pulse due to its reduction6,34,35,37. The additional presence of CuO or Cu(OH)2 as a minor component cannot be excluded due to similar band positions38.

a Ex situ SEM images of the as-prepared electrodes, b after potentiostatic CO2RR at -1.0 V for 1 h, and c after pulsed CO2RR at Ec = −1.0 V and Ea = 0.6 V with tc = ta = 4 s for 1 h. d Applied potential and current density over time with tc = ta = 4 s (top) and corresponding temporal evolution of the SERS signal intensity with marked Raman bands (bottom). e Applied potential over time and intensities of fits of selected characteristic Raman bands, namely the Cu–CO rotation (COr, 280 cm−1, dark blue), Cu–CO stretching (COs, 360 cm−1, light blue), Cu–OHad (OHad, 495 cm−1 at −1.0 V in turquoise and 450 cm−1 at +0.6 V in green) and the copper(I) oxide (Cu2O, sum of 530 and 620 cm−1 divided by two, orange) bands.
While the assignment of COad and Cu2O is straightforward, the assignment of OHad is still under debate, and some groups also assigned the band at 495 cm−1 to Cu–C and Cu–O species35,39. However, experiments in several previous studies with D2O showed a shift of the band position, which supports this assignment to chemisorbed OHad on Cu35,36,40,41. Furthermore, bands at ~500 and 700 cm−1, detectable during the electrochemical oxidation of Cu(111) and polycrystalline Cu surfaces, were linked by other groups to surface OH species through D2O experiments and DFT calculations42. Their DFT calculations propose that the 500 cm−1 band corresponds to a top-site OH stretching mode, possibly aligning with our scenario. In contrast, the 700 cm−1 band was attributed to the bending mode of free Cu–OH, a band mode that may be absent in our case due to co-adsorption with CO.
Additionally, a weak carbonate band at 1070 cm−1 was observed in some of the SERS spectra during the first 20 s, as shown in Supplementary Fig. 433,35,43.
Pulse length-dependent evolution of SERS
In order to follow the impact of the applied cathodic and anodic pulse duration on the catalyst structure and the adsorbates, we averaged the normalized operando SERS spectra of selected pulse conditions and fitted the characteristic bands over one pulse sequence, as shown in Fig. 2. The emphasis is placed on three pulse regimes that are typical for the highest ethanol (EtOH regime, Fig. 2a, d), ethylene/acetaldehyde (C2H4Ox regime, Fig. 2b, e), and C1 product selectivity (C1 regime, Fig. 2c, f). The EtOH regime is characterized by short anodic pulses (ta = 0.5 s), the C2H4Ox regime by intermediate anodic pulses (ta = 4 s), and in both cases, the cathodic pulses have an intermediate length of 4 s. In contrast, long anodic pulses (ta = 8 s) combined with short cathodic pulses (tc = 0.5 s) represent the C1 regime18. Thereby, the intensity of the COs compared to the COr band decreased with shorter ta and is the smallest in the EtOH regime. The OHad band is more intense during shorter ta (0.5 s, 4 s) in the EtOH/C2H4Ox regime, while no OHad bands, but stronger Cu oxide bands during long ta (8 s) and subsequent short tc (0.5 s) could be detected in the C1 regime. These observations are underlined by additional SERS data at different pulse lengths (Supplementary Figs. 5, 6). During the anodic pulses, the Cu2O band intensities increased with the duration of the anodic pulse, being comparably weak at ta = 0.5 s in the EtOH regime. Interestingly, there is also a contribution of Cu–Oad visible in the region of 606–626 cm−1 during the anodic pulse in the EtOH regime for anodic pulses shorter than 2 s (Supplementary Fig. 5a, b)42. Under these conditions, also intense OHad bands and additional bands at 370–380 and 520–540 cm−1 were detected, which can be attributed to disordered CuOx/(OH)y species35,40. Moreover, the carbonate band at 1070 cm-1 was predominantly observed at the onset of both the anodic and cathodic pulses, possibly linked to dynamic alterations of the electrical double layer. However, due to the low band intensities under these conditions, it is challenging to thoroughly analyze its characteristics further (Supplementary Figs. 5–7). To verify the reproducibility of the data the experiments of the main product regimes were repeated on freshly prepared electrodes showing similar spectra and trends (Supplementary Fig. 9).

a–c Normalized SERS spectra from bottom to top (as indicated with arrows) with highlighted characteristic SERS bands during pulsed CO2RR with varying pulse lengths at Ec = −1.0 V and Ea = +0.6 V and d–f intensities of fits of characteristic SERS bands averaged over one pulse sequence at selected pulse lengths. All averaged SERS spectra of c are shown in Supplementary Fig. 8. The data points in d–f represent the intensity fits of COr (280 cm−1, dark blue), COs (360 cm-1, light blue), OHad (495 cm−1 at −1.0 V in turquoise and 450 cm-1 at +0.6 V in green), Cu2O (sum of 530 and 620 cm−1 divided by two, orange) and Cu–Oad (610 cm−1, orange) bands. The red lines denote the exponential fits and serve as guides for the eye.
To obtain additional insights into the average surface coverage of the characteristic species and their corresponding adsorption/desorption and oxidation/reduction kinetics, we performed exponential fits of the Raman band intensities during the averaged pulse profiles (Fig. 2d–f, Supplementary Fig. 10, Supplementary Tables 2–4). In particular, the kinetics of the ad- and desorption of CO on Cu altered with the applied pulse lengths (Supplementary Table 3). For example, in the EtOH regime, the COs band intensity increased twice as fast during the cathodic pulse as it decreased during the anodic pulse. Additionally, the COs/COr band intensity ratio, which reflects the CO surface coverage, gradually and persistently escalated throughout the cathodic pulse (Supplementary Fig. 11). These findings suggest a change in the CO binding configuration during the cathodic pulse, which is expected to have implications for the catalytic function.
The differences in the CO binding configuration in terms of CO adsorption sites can be further evaluated by the C-O stretching vibration at higher wavenumbers (1900–2150 cm−1, Supplementary Figs. 12, 13). Particularly, the SERS spectra show the contribution of bridge CO (CObridge at 2030 cm−1) and two atop CO (COatop) bands, namely the linear low-frequency CO (COLFB at 2065 cm−1) and high-frequency CO (COHFB at 2095 cm−1) bands, as described in the literature29,44. The contribution of COatop sites, which are usually related to C–C coupling44, is more prominent in the EtOH and C2H4Ox regimes (Supplementary Fig. 13). Instead, the contribution of CObridge sites, which are usually considered inactive for C–C coupling27,29,44, increased significantly in the C1 regime. Nevertheless, we found high uncertainties during several pulse sequences owing to the constantly evolving surface reactions that lead to the dynamic nature of the C–O stretching vibrations at high Raman shifts34,44.
For the OHad species, in turn, it is not easy to extract the ad- and desorption behavior since the bands additionally shifted upon the potential switch and partially overlapped with the evolving Cu2O bands during the anodic pulse (Fig. 2a, b). The shift of the bands probably resulted from the Stark effect due to the change in the electric field, with a reasonable Stark tuning rate of ~28 cm−1/V45, rather than from a change in the bonding configuration of OHad, as also discussed in the literature where the potential was only changed by 0.2 V36. Furthermore, it is likely that OHad is an intermediate of the Cu2O formation during the anodic pulse.
The adsorption of oxygen on the Cu surface and the oxidation to Cu2O over the anodic pulse was, under all applied pulse conditions, slower than the corresponding desorption/reduction upon the application of the cathodic pulse (Supplementary Table 4). In the C1 regime, the amount of surface Cu2O continuously increased the fastest, which indicates a continuous growth of the oxide layer over the course of the anodic pulse. Moreover, the spectroscopic weights of the three deconvoluted Cu2O bands changed at different applied pulse lengths (Supplementary Table 5). The relative intensity contribution of the band at 410 cm−1 is stronger during longer cathodic pulses, which may have been influenced by the higher ratio of OHad36. On the other hand, the intensity of the 530 and 620 cm−1 bands contributes stronger for shorter cathodic pulses. Analyzing the time-dependent evolution of these bands in the C1 regime (Supplementary Fig. 14) revealed that the relative intensity of the band at 410 cm−1 was the highest in the first second of the anodic pulse, and its spectroscopic weight increased only slightly in the following. Also, the evolution of the intensity of the 620 cm-1 band suggests a temporary maximum in the initial phase of the anodic pulse. In contrast, the intensity of the 530 cm−1 band increased monotonously over the course of the anodic pulse. These changes might represent the formation/crystallization of the Cu2O layer terminating the Cu domains. In our previous work, we showed with operando XRD that over the course of an anodic potential pulse, crystalline Cu2O domains with a size of up to 3 nm were present at the end of a 10 s anodic pulse18. Additionally, operando XAS revealed a minor contribution of other Cu phases (e.g., Cu(II) compounds such as CuO/Cu(OH)2) in the initial phase of the electrochemical oxidation, which are likely highly disordered and thus absent in the XRD data. However, they cannot be directly correlated to a specific cationic Cu (surface) species based on SERS.
Correlations of selectivity, structure, and kinetics
To extract the link between the surface adsorbates and the catalytic function, Fig. 3 shows the change in the SERS band intensities (Supplementary Table 2) and in the Faradaic efficiencies (ΔFEs) during pulsed CO2RR in comparison to static CO2RR conditions at −1.0 V obtained from a variety of anodic and cathodic pulse lengths conditions18. We depicted the ΔFEs rather than the partial current densities of the products to compare with the qualitative changes in the relative spectroscopic weight of adsorbate-related bands from SERS. Figure 3b depicts the changes in the COcov during the cathodic part of pulsed CO2RR, which could be determined by the ratio of the COs and COr band intensities33. The CO surface coverage has been previously shown to be important for the C-C coupling step, and a higher CO coverage could be linked to the increase of C2+ products33. Under pulsed CO2RR, the determined CO coverage is almost unchanged in the C2H4Ox regime but decreased in the EtOH regime, while it increased in the C1 regime compared to static CO2RR conditions (Fig. 3a, b). Thus, the CO coverage does not seem to be the only crucial parameter determining the product selectivity under pulsed conditions.

Changes in product selectivities during pulsed CO2RR with Ec = −1.0 V, Ea = +0.6 V with respect to the anodic and cathodic pulse lengths after subtraction of the corresponding values under static CO2RR conditions at −1.0 V (indicated with Δ). a Change (Δ) of the sum of Faradaic efficiencies (ΔFEs) of the main C1 products (CO and CH4). b SERS intensity distribution of ΔCO coverage on Cu. c ΔFE of ethanol (EtOH). d Normalized SERS intensity distribution of ΔOHad. The selectivity data were taken from identical catalysts included in our previous publication18.
Therefore, we further investigated the role of OHad, where its beneficial effect was so far only hypothesized or suggested from (theoretical) studies and/or not quantified22,46,47. Fig. 3d presents the change of the normalized OHad intensities during the cathodic pulse (Supplementary Note 2). This map highlights the increased OHad intensities at shorter anodic pulse lengths (<5 s) in comparison to static CO2RR conditions, while the OHad intensities decreased at longer anodic pulse lengths. This fits to the change of the C1 products (Fig. 3a) for lower OH as well as to the enhanced EtOH selectivity at shorter anodic pulse lengths for higher OH intensities (i.e., coverage) (Fig. 3c).
In addition to the average band intensities, the kinetics of the intermediate formation are also expected to significantly influence the observed product generation due to the introduced variations by pulsing the potentials. Therefore, corresponding maps of the ad-/desorption of COad and Oad, as well as the oxidation and reduction of Cu2O depending on the pulse lengths condition, could be created by the use of the time constant of the exponential fits from Fig. 2 (Supplementary Figs. 15–17, Supplementary Tables 3, 4). In the EtOH regime, COr and COs vibrations developed faster than they vanished (Supplementary Fig. 15); thus, CO was rapidly available to form EtOH at these pulse lengths. Furthermore, the kinetics of the ad- and desorption of COs versus COr (Supplementary Fig. 16), which directly impacts the surface CO coverage, indicate faster kinetics of COs as compared to COr. This means that the rapidly available surface CO stems first mainly from COs, which has been related to the C–C coupling COatop in the literature33.
Moreover, even though we could not quantify the intensity of bands related to Cu oxides due their strong SERS sensitivity and missing normalization parameter, we can still follow the kinetic evolution of Oad at short anodic pulse lengths and surface Cu2O at long anodic pulse lengths (Supplementary Fig. 17). In particular, this demonstrates that Cu-Oad adsorbed kinetically quicker in the EtOH regime compared to the surface Cu2O in the C1 regime, which could be crucial for the rapid availability of oxygen species to form EtOH.
Hydroxide to carbon monoxide coverage ratio determines the products
To shift the focus from the applied pulse lengths, Fig. 4 directly plots the ΔFEs of selected products such as CO, C2H4, acetaldehyde and EtOH against the essential adsorbates previously identified, OHad and CO, which are reflected here by the change of their ratio. The changes were calculated with respect to stationary reaction conditions. The corresponding plots showing all the other products can be found in Supplementary Fig. 18. Figure 4 shows that the FEs of C2H4 and H2 under pulsed CO2RR were lower, irrespective of the surface adsorbate composition, while the selectivities toward CO, acetaldehyde, and EtOH were (mostly) increased. In fact, Fig. 4 can be divided into four different product regions depending on the OHad/CO ratio. The lowest OHad/CO ratio (-1, where no OHad is adsorbed) is characterized by the maximum of C1 products, such as CO and CH4. Increasing OHad/CO ratios (1–7.3) lead to the increased formation of C2+ products such as C2H4 and acetaldehyde. A further increase of the OHad/CO ratios (7.3–11.6) correlates with the highest ΔFE of EtOH, together with other minor alcohols, while the ΔFEs of CO, C2H4, and acetaldehyde decreased, highlighting the beneficial effect of enhanced OH coverage for the ethanol formation. Interestingly, the improvement of the CO2RR is not only seen in the FE of ethanol but also the partial current density increases at an optimal ratio of co-adsorbed CO and OH during CO2RR (Supplementary Fig. 19). However, at even higher ratios of OHad/COcov (>11.6) the FE of EtOH and ethylene started to decrease, while the FEs of H2 and C1 products such as CO and COO- (Supplementary Fig. 18a) increased. At these high ratios, the selectivities of EtOH and acetaldehyde were still slightly enhanced as compared to static CO2RR conditions18.

Correlations between the selectivity change ΔFE of selected products (CO, ethylene, acetaldehyde, and ethanol) and the Δ (OHad/COcov) under pulsed CO2RR conditions after subtracting the corresponding values under static CO2RR conditions at −1.0 V. The green star represents the change of the ethanol selectivity during pulsed CO2RR up to non-oxidizing potentials at Ea = 0 V.
We note that the decrease of the ethylene FE may be predominantly linked to irreversible (morphological) catalyst changes during the harsh working conditions, as shown in our previous work and will be further discussed later18.
In order to verify the dependence of the EtOH selectivity on the OHad concentration further, pulsed CO2RR up to non-oxidizing Ea = 0 V at tc/ta = 4 s/1 s was also measured (Supplementary Fig. 20, Supplementary Table 6). The slight enhancement of EtOH compared to the static conditions goes along with a slight enhancement in OHad versus COcov, as also indicated in Fig. 4 (green star), and supports the beneficial effect of OHad for the EtOH production. These findings highlight the crucial role of OHad and CO in the formation of EtOH.
Discussion
From the results discussed, Fig. 5 suggests the mechanism for pulsed CO2RR, where the periodic switching of the applied potential can change the Cu oxidation state and modify the adsorbate coverage of OH and CO with distinct kinetic behavior. Cu oxide-related species only formed during the anodic pulse and rapidly reduced again once the cathodic pulse was applied. At short anodic (ta < 2 s) and intermediate cathodic (tc = 4 s) pulse lengths, Cu-Oad vibrations and/or CuOx/(OH)y species were detected, which also correlate well with the observation of Cu(II) at short anodic pulses from operando XAS data in our prior study18. These CuOx/(OH)y species might be essential for the enhanced EtOH selectivity and are directly oxidized from Cu(0) to Cu(II) during the anodic pulse as no characteristic Cu2O bands could be detected. Instead, at longer anodic (ta > 2 s) and shorter cathodic pulses (tc ≤ 4 s), typical Cu2O bands appeared and reflected the growth and crystallization of Cu2O over the course of the anodic pulse. Larger amounts of bulk-like Cu2O, which were still present at the beginning of the cathodic pulse, led then to an enhancement in the methane selectivity linked to morphology changes, as also seen previously with XAS/XRD22.

Depiction of the processes observed during pulsed CO2RR with Ec = −1.0 V, Ea = +0.6 V of pre-reduced Cu2O NCs in dependence of the anodic and cathodic pulse lengths. COad, OHad, and Oad coverage are highlighted, and the oxidation state of Cu is indicated, which both contribute to different product selectivities during the cathodic pulse. The rate constant k of the CO ad- and desorption, as well as the Cu reduction and oxidation, are schematically specified by the lengths of the arrows to each other, where a longer arrow is correlated to a faster process.
Most importantly, the Cu redox transitions, the OH surface coverage, and the local pH seem to be intertwined. In general, the concentration of OH− close to and adsorbed on the catalyst surface is expected to increase during pulsed CO2RR at −1.0 V. Therein, OH- species are produced by the reduction of H2O during the cathodic pulse but are kept close to the catalyst surface due to the positive polarization of the Cu electrode during the anodic pulse. This is expected to increase the local pH during pulsed CO2RR compared to static CO2RR. The limited time at short ta values prevents the formation of an ordered Cu2O phase and favors the formation of the detected Cu–Oad and/or CuOx/(OH)y species. At longer oxidizing pulses, the low OH coverage values likely result from OHad/OH− consumption for the Cu2O formation (2 Cu + 2 OH− → 2 Cu–OH → Cu2O + H2O)22,24, leading to a decrease in the local pH during CO2RR, which is known to favor methane and CO production22. However, too high coverages of OHad start to block CO adsorption sites and prevent C–C coupling, which leads to an increase in C1 products and HER48.
It is important to note that the decrease of the ethylene and increase of the methane FE is linked to irreversible (morphological) catalyst changes caused by the pulsed reaction conditions (see ex-situ SEM images in Supplementary Fig. 21), which are especially prominent at longer anodic pulse length durations as also shown in our previous work18. Here, qualitative agreement to very high roughness and/or Cu dissolution of the Cu NCs could also lead to an increased population of low-coordinated sites, which are selective for hydrogen49. For example, sub-nanometer Cu clusters, likely formed under these continuous harsh redox cycles, were observed to be selective for methane formation50,51. This explains that under pulsed conditions, the highest CO coverage (without co-adsorbed OHad) was identified to enhance methane selectivities, while under less harsh stationary conditions, the highest CO coverage (usually with co-adsorbed OHad) was attributed to the highest C2+ product yield6,33. This suggests that under these pulsed reaction conditions, the C-C coupling is significantly hampered, leading to the reduction of the adsorbed CO to methane. To facilitate EtOH formation, the CO and OH coverage have to be balanced during pulsed CO2RR to enhance the protonation of the *CHxCO intermediates and/or hamper its O removal52, e.g., by a sufficient OH coverage blocking adjacent adsorption sites. Lower OH or higher CO coverages and, thus, more similar conditions as under static CO2RR shift the selectivity toward protonated C2+ products such as ethylene and acetaldehyde. Therefore, we hypothesize that increasing the interfacial availability of base (OH) sites under (acidic) CO2RR conditions might lead to even higher EtOH formation.
In the EtOH regime, we found faster kinetics of CO adsorbate formation, which are expected to facilitate CO–CO dimerization and, thus, decrease C1 product formation. The fast kinetics of the CO coverage in the EtOH regime might also be beneficial for the fast stabilization of OHad since COad has already been demonstrated to stabilize OHad by DFT calculations46. Furthermore, the fast kinetics of Cu–Oad in the EtOH regime compared to the formation of surface Cu2O in the C1 regime during the anodic pulse demonstrate the rapid availability of oxygen species with short anodic pulse durations that are needed for ethanol formation.
In conclusion, this study revealed the link between the changes in the adsorbate structure and composition and the catalytic function of oxide-derived Cu nanocatalysts during pulsed CO2RR by utilizing time-resolved operando SERS. By the implementation of sub-second time resolution, the development of characteristic adsorbates such as OH and CO could be tracked during each individual pulse. In this way, the observed ethanol enhancement could be attributed to an optimal OH and CO surface coverage, which was found to be selectivity-determining towards alcohols over hydrocarbons. Furthermore, these OH species had a significantly lower concentration during stationary CO2RR and were not present in noticeable amounts on Cu2O-derived working catalysts. Thus, only the intermittent formation of an OH/O-covered Cu surface triggers the continuous regeneration of the OH/CO-covered Cu catalyst during CO2RR. Furthermore, the preferable CO ad-/desorption kinetics were found to contribute to higher ethanol yields. On the contrary, OHad was found to be converted into bulk-like Cu2O species, which also led to the decrease of the near-surface pH and the formation of unfavorable C1 products, such as methane and CO, as well as tremendously different reduction mechanisms, not being able to stabilize the OH species. All in all, this study underlines the urgency of time-resolved surface-sensitive techniques such as operando SERS to understand the reaction mechanism of pulsed CO2RR in order to favor the desired product selectivities. Furthermore, we finally confirmed experimentally the importance of surface base (OH) sites within the CO2RR mechanistic framework which will pave the road for in-depth investigations and novel catalyst design approaches.
Methods
Catalyst preparation
Cu2O NCs were synthesized by a ligand-free method, as described in our previous study6. The reagents were purchased from Sigma Aldrich in ACS grade and used without further purification. 5 mL of a CuCl2*2 H2O solution (0.1 M) and 15 mL of a NaOH solution (0.2 M) were added to 200 mL of ultrapure water (>18 MΩ cm) at room temperature, and the solution was stirred for 5 min. Then, 10 mL of an l-ascorbic acid solution (0.1 M) was added to the mixture, and the solution was further stirred for 1 h. The solution was centrifuged and washed three times, twice with an ethanol-water mixture (1:1) and once with pure ethanol. The product was dried in a vacuum overnight, and the obtained powder was stored in the glove box.
To prepare the electrodes, 1 mg of the catalyst powder was dispersed in 0.5 mL of pure ethanol and ultrasonicated for 15 min to reach a concentration of Cu2O of 2 mg mL−1. For the operando Raman measurements, 31 µL of the dispersion were drop-casted on one side of a polished glassy carbon electrode (8 × 8 mm, Glassy Carbon SIGRADUR®, HTW) and dried at 60 °C for 5 min to obtain a Cu2O mass-loading of ~100 µg cm−2.
Electrolyte preparation
0.1 M KHCO3 (Alfa Aesar, 99.7–100.5%) was purified with a cation-exchange resin (Chelex 100 Resin, Bio-Rad) and saturated with CO2 (99.995%) for at least 15 min until a pH of 6.8 was reached.
Operando surface-enhanced Raman spectroscopy
Operando SERS was performed with a Raman spectrometer (Renishaw, InVia Reflex) coupled with an optical microscope (Leica Microsystems, DM2500M) together with a motorized stage for sample tracking (Renishaw, MS300 encoded stage). Calibration of the system was carried out by using a Si(100) wafer (520.5 cm-1). A near-infrared laser (Renishaw, RL785, λ = 785 nm, Pmax = 500 mW, grating 1200 and 1800 lines mm−1), as well as a HeNe laser (Renishaw, RL633, λ = 633 nm, Pmax = 17 mW, grating 1800 lines mm−1), were used as excitation sources. The backscattered light was Rayleigh-filtered and directed to a CCD detector (Renishaw, Centrus). For the operando measurements, the excitation source was focused on the surface of the sample, and Raman scattering signals were collected with a water immersion objective (Leica microsystems, ×63, numerical aperture of 0.9). The objective was protected from the electrolyte by a Teflon (FEP) film (Goodfellow, film thickness of 0.0125 mm), which was wrapped around the objective.
The electrochemical measurements were conducted at room temperature in a home-built spectro-electrochemical flow cell made of PEEK and controlled by a Biologic SP240 potentiostat (Supplementary Fig. 1). The cell was equipped with a leak-free Ag/AgCl reference electrode (LF-1-63, 1 mm OD, Innovative Instruments, Inc.) positioned close to the sample and a Pt counter electrode in the outlet of the flow. The working electrode with the catalyst drop-casted on glassy carbon was mounted from the bottom of the cell, and the area of the exposed catalyst was 0.25 mm2. The electrolyte (0.1 M KHCO3) was CO2-saturated (pH = 6.8) in its reservoir (V = 50 mL) outside of the Raman system and, from there, pumped through the cell with a peristaltic pump (PLP 380, Behr Labor-Technik).
The potentials in this manuscript were all converted to the RHE scale (\({E}({{{{{{\rm{vs.}}}}}}\; {{{{{\rm{RHE}}}}}}})={E}({{{{{{\rm{vs.}}}}}}\; {{{{{\rm{Ag}}}}}}}/{{{{{\rm{AgCl}}}}}})+0.242\,{{{{{\rm{V}}}}}}+0.059\,{{{{{\rm{V}}}}}}\times {{{{{{\rm{pH}}}}}}-{{{{{{\rm{iR}}}}}}}}\)) and corrected for iR drop as determined by electrochemical impedance spectroscopy.
The collection time of each spectrum depends on the applied electrochemical protocol. For the pulsed CO2RR experiments at the cathodic potential Ec = −1.0 V and the anodic potential Ea = +0.6 V, acquisition times between 0.1 and 0.8 s were used, depending on the cathodic and anodic pulse lengths tc and ta, respectively, to obtain at least three data points per pulse. The exact temporal resolutions for the low Raman shift region are given in Supplementary Table 1. To obtain a high time resolution, usually (if not stated differently), the static Raman mode in the region of 55–1272 cm−1 was applied together with the 785 nm laser and the 1200 lines mm−1 grating. In the region of 1700–2600 cm−1, the 633 nm laser and the 1800 lines mm−1 grating were used. The Raman data were first processed using the Renishaw WiRE 5.2 software to normalize the data and remove cosmic rays. Octave® scripts were written to combine the Raman and the electrochemical data, to fit characteristic Raman bands, and to average the Raman spectra. Averaged Raman spectra were obtained by averaging the Raman data points collected at the same times after the onset of each pulse cycle.
Ex situ scanning electron microscopy
Ex situ SEM images of the samples prior to and after the different electrocatalytic conditions were measured using an SEM (Apreo SEM, Thermo Fisher Scientific) with an in-lens secondary electron detector. The samples were deposited on glassy carbon and directly rinsed with ultrapure water (>18 MΩ cm) after each electrocatalytic CO2RR measurement to avoid electrolyte salt contamination on the sample.
Selectivity measurements
The main part of the selectivity measurements was already published in our previous work18 except for the new measurements acquired at Ec = −1.0 V, Ea = 0 V (versus RHE and iR drop corrected) and tc = 4 s, and ta = 1 s for a total duration of 4000 s. The measurements were carried out in an H-type cell equipped with an anion-exchange membrane (Selemion AMV, AGC) separating the cathodic and the anodic compartments and controlled by an Autolab (Metrohm) potentiostat. A leak-free Ag/AgCl reference electrode (LF-1, Alvatek) served as the reference electrode, a platinum gauze electrode (MaTecK, 3600 mesh cm−2) as the counter electrode, and Cu2O NCs deposited on carbon paper as the working electrode. The electrolyte was CO2-saturated 0.1 M KHCO3. Gas products were detected and quantified every 15 min by online gas chromatography (GC, Agilent 7890B), equipped with a thermal conductivity detector and a flame ionization detector. Liquid products were analyzed after each measurement with a high-performance liquid chromatograph (Shimadzu Prominence), equipped with a NUCLEOGEL SUGAR 810 column and a refractive index detector, and a liquid GC (Shimadzu 2010 plus), equipped with a fused silica capillary column and a flame ionization detector. The Faradaic efficiencies were calculated by taking only the cathodic part into account since there is no catalytic activity during the anodic pulse. Specifically, for the calculations of the Faradaic efficiencies of the gas products, the cathodic current was weighted by the term \(\frac{{t}_{c}}{{t}_{c}+{t}_{a}}\) to correct for effective time under CO2RR, while for the Faradaic efficiencies of the liquid products, just the cathodic charge was taken into account.
Data availability
Additional SERS data, fitting parameter, time constants and additional CO2RR data are given in the Supplementary Information. The raw SERS data (which require specialized software to process) are available from the corresponding authors on request.
References
Nitopi, S. et al. Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte. Chem. Rev. 119, 7610–7672 (2019).
Hori, Y.; Electrochemical CO2 Reduction on Metal Electrodes 89–189 (Springer, New York, 2008).
Mistry, H., Varela, A. S., Kühl, S., Strasser, P. & Roldan Cuenya, B. Nanostructured electrocatalysts with tunable activity and selectivity. Nat. Rev. Mater. 1, 16009–16022 (2016).
Jeon, H. S., Kunze, S., Scholten, F. & Roldan Cuenya, B. Prism-shaped Cu nanocatalysts for electrochemical CO2 reduction to ethylene. ACS Catal. 8, 531–535 (2018).
Jeon, H. S. et al. Operando insight into the correlation between the structure and composition of CuZn nanoparticles and their selectivity for the electrochemical CO2 reduction. J. Am. Chem. Soc. 141, 19879–19887 (2019).
Herzog, A. et al. Operando investigation of Ag-decorated Cu2O nanocube catalysts with enhanced CO2 electroreduction toward liquid products. Angew. Chem. Int. Ed. 60, 7426–7435 (2021).
Rüscher, M. et al. Tracking heterogeneous structural motifs and the redox behaviour of copper–zinc nanocatalysts for the electrocatalytic CO2 reduction using operando time resolved spectroscopy and machine learning. Catal. Sci. Technol. 12, 3028–3043 (2022).
Lum, Y., Yue, B., Lobaccaro, P., Bell, A. T. & Ager, J. W. Optimizing C–C coupling on oxide-derived copper catalysts for electrochemical CO2 reduction. J. Phys. Chem. C 121, 14191–14203 (2017).
Gao, D., Scholten, F. & Roldan Cuenya, B. Improved CO2 electroreduction performance on plasma-activated Cu catalysts via electrolyte design: halide effect. ACS Catal. 7, 5112–5120 (2017).
Singh, M. R., Kwon, Y., Lum, Y., Ager, J. W. & Bell, A. T. Hydrolysis of electrolyte cations enhances the electrochemical reduction of CO2 over Ag and Cu. J. Am. Chem. Soc. 138, 13006–13012 (2016).
Zhou, Y. et al. Stabilization of Cu+ via strong electronic interaction for selective and stable CO2 electroreduction. Angew. Chem. Int. Ed. 61, e202205832 (2022).
Mistry, H. et al. Highly selective plasma-activated copper catalysts for carbon dioxide reduction to ethylene. Nat. Commun. 7, 12123–12130 (2016).
Ren, D. et al. Selective electrochemical reduction of carbon dioxide to ethylene and ethanol on copper(I) oxide catalysts. ACS Catal. 5, 2814–2821 (2015).
De Luna, P. et al. Catalyst electro-redeposition controls morphology and oxidation state for selective carbon dioxide reduction. Nat. Catal. 1, 103–110 (2018).
Velasco-Vélez, J.-J. et al. The role of the copper oxidation state in the electrocatalytic reduction of CO2 into valuable hydrocarbons. ACS Sustain. Chem. Eng. 7, 1485–1492 (2019).
Dattila, F., Garcı́a-Muelas, R. & López, N. Active and selective ensembles in oxide-derived copper catalysts for CO2 reduction. ACS Energy Lett. 5, 3176–3184 (2020).
Casebolt, R., Levine, K., Suntivich, J. & Hanrath, T. Pulse check: potential opportunities in pulsed electrochemical CO2 reduction. Joule 5, 1987–2026 (2021).
Timoshenko, J. et al. Steering the structure and selectivity of CO2 electroreduction catalysts by potential pulses. Nat. Catal. 5, 259–267 (2022).
Kimura, K. W. et al. Controlled selectivity of CO2 reduction on copper by pulsing the electrochemical potential. ChemSusChem 11, 1781–1786 (2018).
Engelbrecht, A. et al. On the electrochemical CO2 reduction at copper sheet electrodes with enhanced long-term stability by pulsed electrolysis. J. Electrochem. Soc. 165, J3059–J3068 (2018).
Shiratsuchi, R. & Nogami, G. Pulsed electroreduction of CO2 on silver electrodes. J. Electrochem. Soc. 143, 582–586 (1996).
Jeon, H. S. et al. Selectivity control of Cu nanocrystals in a gas-fed flow cell through CO2 pulsed electroreduction. J. Am. Chem. Soc. 143, 7578–7587 (2021).
Le Duff, C. S., Lawrence, M. J. & Rodriguez, P. Role of the adsorbed oxygen species in the selective electrochemical reduction of CO2 to alcohols and carbonyls on copper electrodes. Angew. Chem. Int. Ed. 56, 12919–12924 (2017).
Kimura, K. W. et al. Selective electrochemical CO2 reduction during pulsed potential stems from dynamic interface. ACS Catal. 10, 8632–8639 (2020).
Kim, C., Weng, L.-C. & Bell, A. T. Impact of pulsed electrochemical reduction of CO2 on the formation of C2+ products over Cu. ACS Catal. 10, 12403–12413 (2020).
Arán-Ais, R. M., Scholten, F., Kunze, S., Rizo, R. & Roldan Cuenya, B. The role of in situ generated morphological motifs and Cu(i) species in C2+ product selectivity during CO2 pulsed electroreduction. Nat. Energy 5, 317–325 (2020).
Chou, T.-C. et al. Controlling the oxidation state of the Cu electrode and reaction intermediates for electrochemical CO2 reduction to ethylene. J. Am. Chem. Soc. 142, 2857–2867 (2020).
Iijima, G., Inomata, T., Yamaguchi, H., Ito, M. & Masuda, H. Role of a hydroxide layer on Cu electrodes in electrochemical CO2 reduction. ACS Catal. 9, 6305–6319 (2019).
Gunathunge, C. M., Ovalle, V. J., Li, Y., Janik, M. J. & Waegele, M. M. Existence of an electrochemically inert CO population on Cu electrodes in alkaline pH. ACS Catal. 8, 7507–7516 (2018).
Dinh, C.-T. et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 360, 783–787 (2018).
Roiaz, M. et al. Roughening of copper (100) at elevated CO pressure: Cu adatom and cluster formation enable CO fissociation. J. Phys. Chem. C 123, 8112–8121 (2019).
Gameel, K. M., Sharafeldin, I. M., Abourayya, A. U., Biby, A. H. & Allam, N. K. Unveiling CO adsorption on Cu surfaces: new insights from molecular orbital principles. Phys. Chem. Chem. Phys. 20, 25892–25900 (2018).
Zhan, C. et al. Revealing the CO coverage-driven C–C coupling mechanism for electrochemical CO2 reduction on Cu2O nanocubes via operando Raman spectroscopy. ACS Catal. 11, 7694–7701 (2021).
De Ruiter, J. et al. Probing the dynamics of low-overpotential CO2-to-CO activation on copper electrodes with time-resolved Raman spectroscopy. J. Am. Chem. Soc. 144, 15047–15058 (2022).
Moradzaman, M. & Mul, G. In situ Raman study of potential-dependent surface adsorbed carbonate, CO, OH, and C species on Cu electrodes during electrochemical reduction of CO2. ChemElectroChem 8, 1478–1485 (2021).
Niaura, G. Surface-enhanced Raman spectroscopic observation of two kinds of adsorbed OH− ions at copper electrode. Electrochim. Acta 45, 3507–3519 (2000).
Singhal, A. et al. Copper(I) oxide nanocrystals—one step synthesis, characterization, formation mechanism, and photocatalytic properties. Eur. J. Inorg. Chem. 2013, 2640–2651 (2013).
Deng, Y., Handoko, A. D., Du, Y., Xi, S. & Yeo, B. S. In situ Raman spectroscopy of copper and copper oxide surfaces during electrochemical oxygen evolution reaction: identification of CuIII oxides as catalytically active species. ACS Catal. 6, 2473–2481 (2016).
Li, J. et al. Electrokinetic and in situ spectroscopic investigations of CO electrochemical reduction on copper. Nat. Commun. 12, 3264–3274 (2021).
Zhao, Y. et al. Speciation of Cu surfaces during the electrochemical CO reduction reaction. J. Am. Chem. Soc. 142, 9735–9743 (2020).
Chan, H. Y. H., Takoudis, C. G. & Weaver, M. J. Oxide film formation and oxygen adsorption on copper in aqueous media as probed by surface-enhanced Raman spectroscopy. J. Phys. Chem. B 103, 357–365 (1999).
Bodappa, N. et al. Early stages of electrochemical oxidation of Cu(111) and polycrystalline Cu surfaces revealed by in situ Raman spectroscopy. J. Am. Chem. Soc. 141, 12192–12196 (2019).
Jiang, S., Klingan, K., Pasquini, C. & Dau, H. New aspects of operando Raman spectroscopy applied to electrochemical CO2 reduction on Cu foams. J. Chem. Phys. 150, 041718–041729 (2019).
An, H. et al. Sub-second time-resolved surface-enhanced Raman spectroscopy reveals dynamic CO intermediates during electrochemical CO2 reduction on copper. Angew. Chem. Int. Ed. 60, 16576–16584 (2021).
Chang, X. et al. Determining intrinsic stark tuning rates of adsorbed CO on copper surfaces. Catal. Sci. Technol. 11, 6825–6831 (2021).
Cao, Y. et al. Surface hydroxide promotes CO2 electrolysis to ethylene in acidic conditions. Nat. Commun. 14, 2387 (2023).
Sun, M., Staykov, A. & Yamauchi, M. Understanding the roles of hydroxide in CO2 electroreduction on a Cu electrode for achieving variable selectivity. ACS Catal. 12, 14856–14863 (2022).
Zhang, Q. et al. Regulated CO adsorption by the electrode with OH− repulsive property for enhancing C–C coupling. GreenChE 4, 331–337 (2023).
Reske, R., Mistry, H., Behafarid, F., Roldan Cuenya, B. & Strasser, P. Particle size effects in the catalytic electroreduction of CO2 on Cu nanoparticles. J. Am. Chem. Soc. 136, 6978–6986 (2014).
Jiang, K. et al. Effects of surface roughness on the electrochemical reduction of CO2 over Cu. ACS Energy Lett. 5, 1206–1214 (2020).
Akemann, W. & Otto, A. Vibrational modes of CO adsorbed on disordered copper films. J. Raman Spectrosc. 22, 797–803 (1991).
Hori, Y., Takahashi, R., Yoshinami, Y. & Murata, A. Electrochemical reduction of CO at a copper electrode. J. Phys. Chem. B 101, 7075–7081 (1997).
Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), project no. 327886311-SPP 2080. Additional financial support from the European Research Council under grant ERC-OPERANDOCAT (ERC-725915) was also greatly appreciated. A.H. and C.R. acknowledge support by the IMPRS for Elementary Processes in Physical Chemistry. We also thank Petrik Bischoff for his help in designing and manufacturing the operando flow cell. We thank Dr. Chao Zhan (FHI) for helpful discussions. Open access funding was provided by the Max Planck Society.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
A.H., A.B. and B.R.C. contributed to writing the manuscript. A.B. and B.R.C supervised the study. A.H. designed, planned, and analyzed all operando SERS experiments. A.H. and M.L.L. designed the operando SERS cell setup. A.H. and H.S.J. prepared the Cu2O NC samples. A.H. and C.R. performed the electrocatalytic CO2RR selectivity measurements. P.G. carried out the SEM measurements.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Juan Victor Perales-Rondón, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Herzog, A., Lopez Luna, M., Jeon, H.S. et al. Operando Raman spectroscopy uncovers hydroxide and CO species enhance ethanol selectivity during pulsed CO2 electroreduction. Nat Commun 15, 3986 (2024). https://doi.org/10.1038/s41467-024-48052-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-48052-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
