
Abstract
Derivatives of free monocoordinated borylenes have attracted considerable interest due to their ability to exhibit transition-metal-like reactivity, in particular small molecules capture. However, such complexes are rare as the formation is either endergonic, or the resulting adduct is a transient intermediate that is prone to reaction. Here, we present the synthesis of two bis(N-heterocyclic carbene)-borylene complexes capable of capturing and functionalizing carbon dioxide. The capture and subsequent functionalization of CO2 by the bis(NHC)-disilylamidoborylene 1 is demonstrated by the formation of the bis(NHC)-isocyanatoborylene-carbon dioxide complex 3. Reversible capture of CO2 is observed using the bis(NHC)-mesitylborylene 2, and the persistent bis(NHC)-mesitylborylene-carbon dioxide adduct 4 can be stabilized by hydrogen bonding with boric acid. The reactions of 4 with ammonia-borane and aniline demonstrate that the captured CO2 can be further functionalized.
Similar content being viewed by others


Introduction
The conversion of carbon dioxide (CO2) into value-added chemicals has attracted much attention due to the increasing amount of anthropogenic CO2 in the atmosphere and consequent climatic problems1. Due to the high thermodynamic stability of CO2, reactive precious transition metal complexes have been developed to capture, activate, and transform CO2 into high-value chemical feedstocks, but some of these elements remain costly and susceptible to potential supply chain issues2,3,4,5. In this context, the development of sustainable alternatives that possess energetically accessible molecular orbitals to interact with CO2 is important.
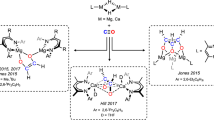
Boron derivatives with both strong electrophilic and nucleophilic characters were selected to examine their feasibility in transition-metal-like small-molecules activation. Braunschweig et al. showed that multiply bonded diboron compounds such as bis(NHC)-diborene (NHC = N-heterocyclic carbene) and bis(CAAC)-diboracumulene (CAAC = cyclic (alkyl)(amino)carbene) could activate CO2 via cycloaddition of B = B and partial B ≡ B bond with CO2, respectively (Fig. 1a)6. We further illustrated that the B = B double bond in an N-phosphinoamidinato NHC-diborene complex was capable of catalyzing hydroboration of CO2 with HBpin7. Kinjo et al. reported that the detached nucleophilic and electrophilic boron centres in a 6π-aromatic 1,3,2,5-diazadiborinine functioned like frustrated Lewis pairs (FLPs) to cooperatively activate CO28, which enabled the latter to undergo catalytic N-formylation with amines and HBpin (Fig. 1a)9. Wilson and Gilliard et al. showed that the CAAC ligand and boron anion in a 9-CAAC-9-borafluorene anion cooperatively activated two equivalents of CO2 to form a trioxaborinanone as a perceivable carbon monoxide releasing molecule (Fig. 1a)10. Wang and Mo et al. reported that the silylene ligand and borylene center in a distorted T-shaped bis(silylene)amidoborylene cooperatively activated CO2 and 9-BBN in hydroboration via a presumed 2-sila-4-boraoxetan-3-one intermediate (Fig. 1a)11.

a Carbon dioxide activation by various ambiphilic boron compounds reported previously. b Established structures of stable transition metal-carbon dioxide complexes and postulated analogues based on boron. c The first stable borylene-carbon dioxide adduct, bis(NHC)-isocyanatoborylene-carbon dioxide adduct 3.
In transition metal-mediated CO2 activation, the first step involves simple coordination of CO2 with a transition metal in the η2-CO2, η1-CO2-κC or η1-CO2-κO binding mode (Fig. 1b)12. Several stable transition metal complexes of CO2 such as Aresta’s [(Cy3P)2Ni(η2-CO2)]13, Herskowitz’s [(diars)2M(η1-CO2-κC)Cl] (M = Ir, Rh; diars = o-phenylenebis(dimethylarsine))14 and Gambarotta’s [(ONNO)V(OH)(η1-CO2-κO)] (ONNO= [2,4-Me2−2-(OH)C6H2CH2]2N(CH2)2NMe2)15 were isolated and structurally characterized. However, in boron metallomimetic chemistry, single-site-boron complexes of CO2 with the composition of B(η2-CO2), B(η1-CO2-κC) or B(η1-CO2-κO) are unknown. In the activation of CO2 by the 9-CAAC-9-borafluorene anion, bis(silylene)amidoborylene or bis(CAAC)-diboracumulene6,10,11, Density Functional Theory (DFT) calculations show that the nucleophilic boron center captures CO2 in the η1-CO2-κC or η2-CO2 binding mode (Fig. 1b). However, the reaction is either endergonic, or the resulting adduct is a transient intermediate that is prone to react with the electrophilic ligand backbone or the electrophilic boron center. In CO2 activation mediated by FLP of phosphine and borane16, the phosphine acts as a Lewis base to capture CO2 in the η1-CO2-κC binding mode while the electrophilic boron center stabilizes the captured CO2 via O-coordination17. In this context, a stable single-site-boron-carbon dioxide adduct is a highly sought-after compound not only for scientific curiosity, but also for a better understanding of how a single-boron center captures CO2 and enables the latter to further react with substrates for functionalization. In this paper, we report the synthesis of a bis(1,3,4,5-tetramethylimidazol-2-ylidene)-bis(trimethylsilyl)amidoborylene and -mesitylborylene and their complexes of CO2. The functionalization of the captured CO2 is also reported. DFT calculations were performed to elucidate electronic structures.
Results
Two equivalents of 1,3,4,5-tetramethylimidazol-2-ylidene (IMe) were reacted with RBBr2 [R = N(SiMe3)2 and mesityl (2,4,6-Me3C6H2)] and KC8 in toluene at room temperature to afford the bis(NHC)-disilylamidoborylene [(IMe)2B{N(SiMe3)2}] (1, Yield: 73%) and bis(NHC)-mesitylborylene [(IMe)2BMes] (2, Yield: 42%, Fig. 2a), respectively. They were both isolated as a red crystalline solid from the concentrated reaction mixture. The 11B{1H} NMR signals of 1 and 2 are 1.6 and −6.8 ppm, respectively. The molecular structures of compounds 1 and 2 obtained by X-ray crystallography show that the boron centers adopt a trigonal planar geometry (Fig. 2b, c). IMe ligands are tilted with respect to the boron centers. The B-CIMe bond lengths in compound 1 (1.508(2)−1.509(2) Å) and 2 (1.519(3)−1.520(3) Å) are almost equal. They are between the B-Cmesityl bond (1.588(5) Å) in 2 and B = C double bonds in methylene boranes (1.351–1.488 Å)18, indicating some multiple bond character in the B-CIMe bonds due to weak pB→pC π-back bonding. In addition, compound 1 has a gauche conformation with respect to the B1-N1 bond. The Si-N-B-C dihedral angle is 58(1)°. The B-Namido bond (1.5327(19) Å) in 1 is typical of a single bond. DFT calculations (M06-2X/Def2-TZVP) of compounds 1 and 2 show that the Highest Occupied Molecular Orbitals (HOMOs) are a dominant π-type lone pair orbital on the boron center forming π-back bonding with the vacant p orbital on IMe, while the Lowest Unoccupied Molecular Orbitals (LUMOs) are the empty p orbitals on the carbene centers (Fig. 2d, Supplementary Figs. 33, 34). Their HOMOs are of similar energy level (1: −3.801; 2: −3.865 eV), indicating that their nucleophilicity should be comparable. The Wiberg Bond Index (WBI) shows that the B-CIMe bonds in compounds 1 (WBI: 1.183–1.185) and 2 (WBI: 1.174–1.176) have weak double bond character with reference to the B-Cmesityl bond in compound 2 (WBI: 0.949). The WBI of the B-Namido bond in compound 1 is 0.789, which suggests that the B1-N1 bond has a single bond character. The Natural Population Analysis (NPA) charge of the boron center in compound 1 (0.251 e) is higher than that in compound 2 (−0.055 e) due to the inductive effect exerted by the disilylamido substituent. The trend is in line with the 11B{1H} NMR chemical shift of compounds 1 and 2.

a Synthetic routes to the bis(NHC)-borylenes 1 and 2. b The molecular structure of 1 obtained by X-ray crystallography. Thermal ellipsoids are shown at 50% probability. All hydrogen atoms are removed for clarity. Selected bond lengths (Å) and angles (deg): B1-C1 1.508(2), B1-C8 1.509(2), B1-N1 1.5327(19), C1-B1-C8 119.28(13), C1-B1-N1 120.63(13), C8-B1-N1 120.09(13). c The molecular structure of 2 obtained by X-ray crystallography. Thermal ellipsoids are shown at 50% probability. All hydrogen atoms are removed for clarity. Selected bond lengths (Å) and angles (deg): B1-C1 1.520(3), B1-C1A 1.519(3), B1-C8 1.588(5), C1A-B1-C8 120.99(15), C1-B1-C8 120.99(15), C1-B1-C1A 118.0(3). d HOMO (−3.865 eV) of 2 (isovalue 0.06) at M06-2X/def2-TZVP level of theory.
Synthesis of stable NHC-borylene complexes is a formidable challenge because the weak π-accepting property of NHC is insufficient to stabilize the Lewis ambiphilicity of the borylene centers. Robinson and Braunschweig et al. independently showed that NHC-borylene complexes are highly reactive, wherein dimerization or C-H bond activation often occurred19,20. In the case of bis(NHC)-borylene complexes where there are two weak π-accepting NHC ligands, it is important that the third ligand is strongly π-electronic withdrawing for the stabilization of the B lone pair of electrons. Braunschweig et al. reported a bis(NHC)-borylene analogue [(IiPr)2BCym] (Cym = (C5H4)Mn(CO)3, IiPr =:C{N(iPr)C(H)}2), wherein the lone pair of electrons in the presumed borylene center is stabilized by the B to Mn charge transfer via the Cym ligand leading to a borafulvenium or boratafulvene electronic structure21. Driess et al. reported a bis(NHC)-(isocyanide)-borylene cation [(IiPrMe)2B(CNR)]+ (IiPrMe =:C{N(iPr)C(Me)}2; R = cyclohexyl, tert-butyl), where excess electron density on the boron center delocalizes to the isocyanide ligand to afford a prominent boraketiminium resonance form22. As NHCs alone are insufficient to stabilize a borylene, strong π-accepting donors such as cyclic (alkyl)(amino)carbenes (CAACs)23,24, carbon monoxide or isocyanides25 were often used to extensively delocalize the boron lone pair of electrons in other stable bis(Lewis base)-borylene complexes. In contrast, compounds 1 and 2 are rare bis(NHC)-borylene complexes that do not need an extra π-electronic withdrawing substituent to stabilize the borylene centers. In addition, the weak π-acidity of NHC should preserve the nucleophilic character of borylenes for capturing CO2 and subsequently forming stable single-site-boron complexes of CO2.
Compound 1 was reacted with CO2 (1 bar) in toluene at room temperature, from which the bis(NHC)-isocyanatoborylene-carbon dioxide adduct [(IMe)2(OCN)B(η1-CO2-κC)] (3, Fig. 3) was isolated as a colorless crystalline solid (Yield: 80%). The 11B{1H} NMR signal (−16.0 ppm) supports that the boron center is four-coordinate. The molecular structure of 3 obtained by X-ray crystallography (Fig. 4a) shows that the B-CIMe (B1-C1: 1.640(3); B1-C8: 1.639(3) Å) and B-CCO2 (1.645(3) Å) bond lengths are typical of single bonds, while the identical C-O bond lengths of the captured CO2 (1.263(3) and 1.265(3) Å) indicate the presence of delocalized negative charge. The HOMO of 3 in DFT calculations shows the B-C σ orbital formed by the lone pair orbital on the B atom and the π* orbital on CO2, leading to a carboxylate anion electronic structure (Fig. 4b). For the formation of 3, it is proposed that the boron lone pair of electrons attack CO2 and the activated CO2 moiety in Int1 inserts into the N-Si bond of the disilylamido substituent to form a carbamate substituent in Int2. It further captures CO2 to form Int3, where the activated CO2 attacks the N-Si bond of the carbamate substituent to form the isocyanate and trimethylsilyl carboxylate substituent in Int4 and Me3SiO-. The latter reacts with the trimethylsilyl carboxylate substituent in Int4 to form compound 3 and O(SiMe3)2. DFT calculations (M06-2X/def2-TZVP/IEFPCM(toluene)) show that the mechanism is feasible.

Reaction of 1 with CO2 in toluene at room temperature. (DFT calculations, ΔG and ΔGǂ in the proposed mechanism are in kcal/mol and were calculated at the M06-2X/def2-TZVP/ IEFPCM(toluene) level of theory).

a The molecular structure of 3 obtained by X-ray crystallography with thermal ellipsoids shown at 50% probability. All hydrogen atoms are removed for clarity. Selected bond lengths (Å) and angles (deg): B1-C15 1.645(3), C1-B1 1.640(3), C8-B1 1.639(3), B1-N5 1.541(3), C8-B1-C1 106.55(17), C1-B1-N5 105.65(18), N5-B1-C8 111.13(17), O1-C15-O2 123.8(2). b HOMO (−6.329 eV) of 3 (isovalue 0.06) at M06-2X/def2-TZVP level of theory, showing the interaction of the lone pair orbital on the B and the π* orbital on CO2.
Compound 3 is the first stable borylene-carbon dioxide adduct26. The formation of 3 demonstrates that the boron center in a borylene can directly attack the carbon center of CO2, which is a result that has remained unattainable using electrophilic borane. The formation of the isocyanate substituent in compound 3 suggests that functionalization of captured CO2 should be feasible. In this context, compound 2 was used to mediate the functionalization of CO2 due to the presence of a spectated Mes substituent.
The reaction of compound 2 with CO2 (1 bar) in CD3CN at room temperature afforded the persistent bis(NHC)-mesitylborylene-carbon dioxide adduct [(IMe)2(Mes)B(η1-CO2-κC)] (4, 11B{1H} NMR: −15.7 ppm, Fig. 5a). When the reaction mixture was placed under reduced pressure or heated at 70 °C, compound 4 was instantaneously converted back into compound 2 as confirmed by 11B{1H} NMR spectroscopy, showing that the CO2 capture was reversible. Isolating compound 4 by recrystallization was not attained due to its instability.

a Synthesis of 4 and 4·B(OH)3 from 2. b Reaction of 4 with NH3BH3 in toluene at room temperature to form 5. (DFT calculations, ΔG and ΔGǂ in the proposed mechanism are in kcal/mol and were calculated at the M06-2X/def2-TZVP/ IEFPCM(toluene) level of theory). c Reaction of 4 with PhNH2 in toluene at room temperature to afford 6.
In carbon monoxide dehydrogenase, CO2 is captured by the nucleophilic nickel(0) and electrophilic iron(II) centers, and the activated CO2 substrate is further stabilized by hydrogen-bonding with aptly situated amino acid residues27. Based on this, an effective single-site catalyst motif, where ligands with pendant proton donors are used to coordinate with a transition metal for the activation of CO2 through the stabilization of transition metal-CO2 adduct by hydrogen-bonding, has been developed in biomimetic artificial CO2 reduction catalysis2,28. It is thus anticipated that the captured CO2 in compound 4 could be stabilized by hydrogen-bonding by mimicking carbon monoxide dehydrogenase. Boric acid B(OH)3 was used to react with compound 4 in THF at room temperature to afford compound 4·B(OH)3 (Fig. 5a), where the captured CO2 moiety was stabilized by hydrogen-bonding with B(OH)3. Compound 4·B(OH)3 was stable in solution under reduced pressure and was isolated as a colorless crystalline solid (Yield: 53%) from the concentrated reaction mixture. The 11B{1H} NMR spectrum of compound 4·B(OH)3 shows a signal at −15.8 ppm attributable to the mesityl-bonded boron center, which is comparable with that of compound 4. The molecular structure of compound 4·B(OH)3 obtained by X-ray crystallography shows that the C-O bonds (1.2643(14), 1.2790(14) Å) are unequal and the oxygen atoms point to two OH substituents of B(OH)3 (Fig. 6a). The O···H distances (O2···H5: 1.77 Å; O1···H3: 1.76 Å) indicate the presence of hydrogen bonding. The B-CIMe (B1-C1: 1.6532(16); B1-C8: 1.6514(17) Å) and B-CCO2 bonds (1.6810(17) Å) are typical of single bonds. The B-Cmesityl bond (1.6608(17) Å) in compound 4·B(OH)3 is significantly lengthened in comparison with that of compound 2, probably due to the steric congestion at the four-coordinate boron center.

a The molecular structure of 4·B(OH)3 obtained by X-ray crystallography. Thermal ellipsoids are shown at 50% probability. All hydrogen atoms except for those on boric acid are removed for clarity. Selected bond lengths (Å) and angles (deg): C1-B1 1.6532(16), C8-B1 1.6514(17), C15-B1 1.6608(17), C24-B1 1.6810(17), C24-O1 1.2790(14), C24-O2 1.2643(14), C1-B1-C8 114.24(9), C1-B1-C15 112.85(9), C8-B1-C15 106.30(9), O1-C24-O2 121.60(11), O1-C24-B1 119.45(10), O2-C24-B1 118.68(10). b The molecular structure of 5 obtained by X-ray crystallography. Thermal ellipsoids are shown at 50% probability. All hydrogen atoms except for those on the boron and formate are removed for clarity. Selected bond lengths (Å) and angles (deg): C1-B1 1.635(4), C8-B1 1.621(4), C15-B1 1.622(4), C24-O1 1.223(4), C24-O2 1.210(4), O1-C24-O2 133.6(4), C1-B1-C15 117.2(2), C1-B1-C8 108.4(2), C8-B1-C15 116.3(2). c The molecular structure of 6 obtained by X-ray crystallography. Thermal ellipsoids are shown at 50% probability. All hydrogen atoms except for those on the boron and carbamate are removed for clarity. Selected bond lengths (Å) and angles (deg): C1-B1 1.618(3), C4-B1 1.631(3), C3-B1 1.643(3), C9-O1 1.246(2), C9-O2 1.268(2), C9-N5 1.406(2), C1-B1-C4 111.65(15), C1-B1-C3 115.09(16), C3-B1-C4 111.65(15), O1-C9-O2 126.14(18), O1-C9-N5 119.53(17), O2-C9-N5 114.33(17).
Borylene-mediated hydrogenation of CO2 with H2 or transfer hydrogenation agents is unknown as yet. As the captured CO2 in compound 4·B(OH)3 is capable of interacting with the hydrogen atoms of B(OH)3, hydrogenation of the captured CO2 should be feasible. As such, NH3BH3 was used to undergo hydrogenation with compound 4 in toluene at 25 °C to form a formate [(IMe)2(Mes)BH](HCO2) (5, Yield: 51%, Fig. 5b), which was isolated as a colorless crystalline solid from the concentrated reaction mixture. The 11B NMR signal of −23.3 ppm (doublet) supports the formation of a B-H bond. The molecular structure of compound 5 obtained by X-ray crystallography shows that the boron center adopts a tetrahedral geometry (Fig. 6b), which is consistent with the upfield 11B NMR signal. The OCO skeleton is bent and the C-O bond lengths (1.223(4) and 1.210(4) Å) are shorter than those of 3, indicating the formation of a formate anion. It is proposed that the hydride from the -BH3 moiety of NH3BH3 attacks the carbon atom of the captured CO2, while the negatively charged oxygen atom of the captured CO2 in compound 4 abstracts a proton from the -NH3 moiety of NH3BH3 to form formic acid HC(O)OH and regenerate compound 2. The borylene center in compound 2 then activates the O-H bond of HC(O)OH to form compound 5. The feasibility of this proposed mechanism is demonstrated by DFT calculations (M06-2X/def2-TZVP/IEFPCM(toluene)). Compound 2 is the first borylene capable of mediating hydrogenation of captured CO2 by NH3BH3 in Lewis-acid-free conditions to form a formate derivative. With the aid of both sterically hindered Lewis acid and base, Stephan et al. showed that hydrogenation of CO2 with NH3BH3 was mediated by an FLP mechanism. In the reaction, the FLP of Mes3P and AlX3 (X = Cl, Br) activated CO2 to form [Mes3PC(OAlX3)2], which subsequently reacted with NH3BH3 and H2O to form methanol via a postulated intermediate [Mes3PH][(MeO)nAlX4-n]29. Instead of using NH3BH3, Dyson and Corminboeuf et al. reported that the FLP of tris(p-bromo)tridurylborane (tbtb) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) activated H2 under high pressure to form [DBU-H]+[H-tbtb]-, which reacted with CO2 via hydride transfer to afford the formate salt [DBU-H]+[HCOO]- and regenerate tbtb30. The molecular structure of the formate salt [DBU-H]+[HCOO]- is similar to that of compound 5.
Aromatic amines cannot react with CO2 at ambient conditions to form carbamic acids or carbamates31. Conversely, the captured CO2 in compound 4 could react with PhNH2 in toluene at room temperature to form the carbamate [(IMe)2(Mes)BH][PhN(H)CO2] (6, Yield: 72%, Fig. 5c), which was isolated as a colorless crystalline solid from the concentrated reaction mixture. The 11B NMR signal of compound 6 is −23.4 ppm (doublet) and its X-ray crystallographic data is consistent with compound 5 (Fig. 6c). The formation of compound 6 illustrates another example of further functionalization of the captured CO2 in compound 4.
Discussion
This work reports the synthesis of bis(NHC)-disilylamidoborylene 1 and bis(NHC)-mesitylborylene 2 through the reaction of two equivalents of IMe with RBBr2 (R = N(SiMe3)2, 2,4,6-Me3C6H2) and KC8. Compounds 1 and 2 are examples of rare bis(NHC)-borylene complexes that do not need an extra π-electronic withdrawing substituent to stabilize the borylene centers. The weak π-acidity of NHC preserves the nucleophilic character of the borylenes and enables the capture of CO2 in the form of the bis(NHC)-isocyanatoborylene-carbon dioxide adduct 3, which is the first stable single-site-boron complex of CO2. Reversible coordination with CO2 was also demonstrated using compound 2 to form the persistent bis(NHC)-mesitylborylene-carbon dioxide adduct 4, which was stabilized by hydrogen bonding with B(OH)3 to form compound 4·B(OH)3. Compound 4 was found to be able to undergo hydrogenation with NH3BH3 to form formate 5 and amination with PhNH2 to form carbamate 6, which demonstrates that the captured CO2 can be further functionalized.
Methods
General procedures
All operations were carried out under an inert atmosphere of argon gas by standard Schlenk techniques. The synthesis of the starting materials (TMS)2NBBr2 and MesBBr2 were adapted from published procedures, which can be found below. All other chemicals were purchased from Sigma-Aldrich and used directly without further purification. All solvents were dried over K metal or CaH2 prior to use. The 1H, 11B, 11B{1H}, 13C{1H}, and 29Si{1H} NMR spectra were recorded on a JEOL ECA 400 spectrometer or Bruker Avance III 400. The NMR spectra were recorded in deuterated solvents and the chemical shifts are relative to SiMe4 for 1H, 13C and 29Si; BF3.Et2O for 11B, respectively. The following abbreviations are used to describe signal multiplicities: s = singlet, d = doublet, m = multiplet, brs = broad singlet. Coupling constants J are given in Hertz (Hz). HRMS spectra were obtained at the Mass Spectrometry Laboratory in the Division of Chemistry and Biological Chemistry, Nanyang Technological University. Melting points were measured with an OptiMelt automated melting point system. Fourier transform infrared (FT-IR) spectra were recorded on a Bruker Alpha FT-IR spectrometer.
Synthesis of (TMS)2NBBr2 adapted from a published procedure32.
A hexane solution (2.5 M) of n-BuLi (8.0 mL, 20 mmol) was added dropwise into a hexane solution of hexamethyldisilazane (4.19 mL, 20 mmol) at −78 °C. The mixture was allowed to warm to room temperature and stirred for 4 h, then cooled to −78 °C, to which a hexane solution of BBr3 (1.90 mL, 20 mmol) was added dropwise. The mixture was gradually warmed to room temperature and stirred overnight. The resulting suspension was filtered, and all volatiles were removed in vacuo to give a yellow liquid. Distillation afforded (TMS)2NBBr2 as a colorless liquid in 32 % yield (2.12 g, 6.65 mmol).
Synthesis of MesBBr2 adapted from a published procedure33.
A toluene solution of BBr3 (0.95 mL, 10 mmol) was added dropwise into a toluene solution of mesitylcopper(I) (2.01 g, 10 mmol) at −78 °C. The mixture was stirred for 2 h at −78 °C before it was allowed to warm to room temperature and stirred overnight. The resulting suspension was filtered, and all volatiles were removed in vacuo to give a yellow liquid. Distillation afforded MesBBr2 as a colorless liquid in 72 % yield (2.08 g, 7.17 mmol).
Synthesis of 1
A toluene solution of (TMS)2NBBr2 (1.0 mmol) was added into a 100 mL Schlenk flask containing 1,3,4,5-tetramethylimidazolin-2-ylidene (2.0 mmol, 0.25 g) and KC8 (2.0 mmol, 0.27 g) at room temperature, following which, the reaction mixture was stirred for 8 h. The resulting bright red-purple suspension was filtered, and the filtrate was concentrated to 10 mL and kept for 3 days at room temperature to afford compound 1 as red block crystals (0.31 g) in 73 % yield. M.p.: 76 °C. 1H NMR (399.5 MHz, C6D6, 25 °C): δ 3.27 (s, 6 H, N-CH3), 2.41 (s, 6 H, N-CH3), 1.66 (s, 12 H, C-CH3), 0.44 (s, 18 H, N(Si(CH3)3)2). 11B{1H} NMR (128 MHz, C6D6, 25 °C): δ 1.6 (s). 13C{1H} NMR (101 MHz, C6D6, 25 °C): δ 121.3, 119.8 (C = C), 34.9, 34.8 (NCH3), 10.0, 9.3 (CH3), 4.7 (N(Si(CH3)3)). 29Si{1H} NMR (79.4 MHz, C6D6, 25 °C): δ −1.5 (s). HRMS (ESI): m/z calcd for C20H43BN5Si2: 420.3150 [(M + H)]+; found: 420.3157.
Synthesis of 2
A toluene solution of dibromo(2,4,6-trimethylphenyl)borane (MesBBr2) (1.0 mmol) was added into a 100 mL Schlenk flask containing 1,3,4,5-tetramethylimidazolin-2-ylidene (2.0 mmol, 0.25 g) and KC8 (2.0 mmol, 0.27 g) at room temperature, following which, the reaction mixture was stirred for 8 h. The resulting bright red suspension was filtered, and the filtrate was concentrated to 10 mL and kept for 3 days at room temperature to afford compound 2 as red block crystals (0.16 g) in 42 % yield. M.p.: 94 °C. 1H NMR (399.5 MHz, C6D6, 25 °C): δ 7.20 (s, 2 H, ArH), 2.75 (s, 6 H, N-CH3), 2.72 (s, 6 H, N-CH3), 2.57 (s, 6 H, Ar-CH3), 2.47 (s, 3 H, Ar-CH3), 1.72 (s, 6 H, C-CH3), 1.55 (s, 6 H, C-CH3). 11B{1H} NMR (128 MHz, C6D6, 25 °C): δ – 6.8 (s). 13C{1H} NMR (101 MHz, C6D6, 25 °C): δ 141.5, 130.9, 127.5 (Ar-C), 120.3, 118.8 (C = C), 34.9, 33.8 (NCH3), 25.1, 21.7 (Ar-CH3), 10.0, 9.2 (CH3). HRMS (ESI): m/z calcd for C23H36BN4: 379.3033 [(M + H)]+; found: 379.3035.
Synthesis of 3
A toluene solution of 1 (0.13 g, 0.3 mmol) in a Schlenk flask was degassed by a freeze–pump–thaw method. Then, CO2 (1 bar) was filled. The resulting solution changed from red-purple to colorless immediately. After 30 min of stirring, all volatiles of the resulting suspension were removed under vacuum to give 3 as a colorless solid (0.08 g) in 80% yield. Colorless crystals of 3 were isolated from the saturated acetonitrile solution. M.p.: 81 °C. 1H NMR (399.5 MHz, CD3CN, 25 °C): δ 3.52 (s, 12 H, N-CH3), 2.14 (s, 12 H, C-CH3). 11B{1H} NMR (128 MHz, CD3CN, 25 °C): δ −16.0 (br). 13C{1H} NMR (101 MHz, CD3CN, 25 °C): δ 126.7 (C = C), 33.2 (N-CH3), 8.9 (CH3). HRMS (ESI): m/z calcd for C16H25BN5O3: 346.2050 [(M + H)]+; found: 346.2056.
Synthesis of 4
A CD3CN solution of 2 (0.04 g, 0.1 mmol) in a J-Young NMR tube was degassed by a freeze–pump–thaw method. Then, CO2 (1 bar) was filled. The resulting solution changed from red to colorless immediately. 1H NMR (399.5 MHz, CD3CN, 25 °C): δ 6.68 (s, 2 H, ArH), 3.29 (s, 12 H, N-CH3), 2.18 (s, 3 H, Ar-CH3), 2.13 (s, 12 H, C-CH3), 1.96 (s, 6 H, Ar-CH3). 11B{1H} NMR (128 MHz, CD3CN, 25 °C): δ – 15.7 (s). 13C{1H} NMR (101 MHz, CD3CN, 25 °C): δ 144.4, 135.0, 130.2 (Ar-C), 126.4 (C = C), 34.1 (N-CH3), 24.3, 20.7 (Ar-CH3), 9.2 (CH3).
Synthesis of 4·B(OH)3
A THF solution of 2 (0.15 g, 0.4 mmol) in a 100 mL Schlenk flask was degassed by a freeze–pump–thaw method. Then, CO2 (1 bar) was filled. The reaction mixture was stirred for 30 min at room temperature. Boric acid B(OH)3 (0.03 g, 0.5 mmol) was then added into the colorless solution. After which, the reaction mixture was stirred for 2 h. The resulting suspension was filtered and concentrated to give compound 4·B(OH)3 as colorless crystals (0.11 g) in 53% yield. M.p.: 93 °C. 1H NMR (399.5 MHz, CD3CN, 25 °C): δ 6.71 (s, 2 H, ArH), 3.25 (s, 12 H, N-CH3), 2.18 (s, 3 H, Ar-CH3), 2.14 (s, 12 H, C-CH3), 1.92 (s, 6 H, Ar-CH3). 11B{1H} NMR (128 MHz, CD3CN, 25 °C): δ 19.7 (s, B(OH)3), – 15.8 (s, Ar-B). 13C{1H} NMR (101 MHz, CD3CN, 25 °C): δ 144.4, 134.8, 130.2 (Ar-C), 126.3 (C = C), 34.0 (N-CH3), 24.3, 20.7 (Ar-CH3), 9.2 (CH3). HRMS (ESI): m/z calcd for C24H39B2N4O5: 485.3107 [(M + H)]+; found: 485.3121.
Synthesis of 5
A toluene solution of 2 (0.15 g, 0.4 mmol) in a 100 mL Schlenk flask was degassed by a freeze–pump–thaw method. Then, CO2 (1 bar) was filled. The reaction mixture was stirred for 2 h at room temperature. Ammonia borane (NH3BH3) (0.012 g, 0.4 mmol) was then added into the colorless solution. After which, the reaction mixture was stirred for 4 h. The resulting suspension was filtered and concentrated to give compound 5 as colorless crystals (0.09 g) in 51% yield. M.p.: 262 °C. 1H NMR (399.5 MHz, CDCl3, 25 °C): δ 6.75 (s, 2 H, ArH), 3.28 (s, 12 H, N-CH3), 2.23 (s, 3 H, Ar-CH3), 2.22 (s, 12 H, C-CH3), 1.77 (s, 6 H, Ar-CH3). 11B NMR (128 MHz, CDCl3, 25 °C): δ – 23.3 (d, J = 84.0 Hz). 13C{1H} NMR (101 MHz, CDCl3, 25 °C): δ 167.7 (C = O), 141.8, 135.7, 129.3 (Ar-C), 126.5 (C = C), 32.6 (N-CH3), 23.4, 21.0 (Ar-CH3), 9.3 (CH3). HRMS (ESI): m/z calcd for C24H37BN4O2: 441.3037 [(M + H)]+; found: 441.3038.
Synthesis of 6
A toluene solution of 2 (0.15 g, 0.4 mmol) in a 100 mL Schlenk flask was degassed by a freeze–pump–thaw method. Then, CO2 (1 bar) was filled. The reaction mixture was stirred for 2 h at room temperature. Aniline (PhNH2) (0.037 g, 0.4 mmol) was then added into the colorless solution. After which, the reaction mixture was stirred for 4 h. The resulting suspension was filtered and concentrated to give compound 6 as colorless crystals (0.12 g) in 72 % yield. M.p.: 229 °C. 1H NMR (399.5 MHz, CDCl3, 25 °C): δ 7.37 (d, 1 H, ArH, 3JH-H = 7.6 Hz), 7.13-7.09 (m, 1 H, ArH), 7.06-7.02 (m, 1 H, ArH), 6.72 (s, 2 H, ArH), 6.72-6.62 (m, 2 H, ArH), 3.21 (s, 12 H, N-CH3), 2.19 (s, 3 H, Ar-CH3), 2.15 (s, 12 H, C-CH3), 1.72 (s, 6 H, Ar-CH3). 11B NMR (128 MHz, CDCl3, 25 °C): δ – 23.4 (d, J = 81.7 Hz). 13C{1H} NMR (101 MHz, CDCl3, 25 °C): δ 162.4 (C = O), 158.9, 146.5, 144.3, 141.7, 135.5, 129.3, 129.2, 128.2 (Ar-C), 125.9 (C = C), 118.5, 118.1, 116.6, 115.1(Ar-C), 32.4 (N-CH3), 23.3, 20.9 (Ar-CH3), 9.1 (CH3). HRMS (ESI): m/z calcd for C30H42BN5O2: 441.3037 [(M + H)]+; found: 441.3038.
Data availability
All data generated or analyzed during this study are included in this manuscript (and its Supplementary Information). Details about materials and methods, experimental procedures, characterization data, and NMR spectra are available in the Supplementary Information. The optimized cartesian coordinates are provided in the Source Data file. The structures of 1–6 in the solid state were determined by single-crystal X-ray diffraction studies and the crystallographic data for these structures have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers 2235472 (1), 2257718 (2), 2235466 (3), 2257719 (4.B(OH)3), 2307337 (5), 2307338 (6). These data can be obtained free of charge from via www.ccdc.cam.ac.uk/data_request/cif. All data are also available from corresponding authors upon request. Source data are provided with this paper.
References
Burkart, M. D., Hazari, N., Tway, C. L. & Zeitler, E. L. Opportunities and challenges for catalysis in carbon dioxide utilization. ACS Catal. 9, 7937–7956 (2019).
Cokoja, M., Bruckmeier, C., Rieger, B., Herrmann, W. A. & Kühn, F. E. Transformation of carbon dioxide with homogeneous transition-metal catalysts: a molecular solution to a global challenge? Angew. Chem. Int. Ed. 50, 8510–8537 (2011).
Sahoo, P. K., Zhang, Y. & Das, S. CO2-promoted reactions: an emerging concept for the synthesis of fine chemicals and pharmaceuticals. ACS Catal. 11, 3414–3442 (2021).
Klankermayer, J., Wesselbaum, S., Beydoun, K. & Leitner, W. Selective catalytic synthesis using the combination of carbon dioxide and hydrogen: catalytic chess at the interface of energy and chemistry. Angew. Chem. Int. Ed. 55, 7296–7343 (2016).
Wang, W.-H., Himeda, Y., Muckerman, J. T., Manbeck, G. F. & Fujita, E. CO2 hydrogenation to formate and methanol as an alternative to photo- and electrochemical CO2 reduction. Chem. Rev. 115, 12936–12973 (2015).
Stoy, A. et al. CO2 Binding and splitting by boron–boron multiple bonds. Angew. Chem. Int. Ed. 57, 5947–5951 (2018).
Fan, J., Mah, J.-Q., Yang, M.-C., Su, M.-D. & So, C.-W. A N-phosphinoamidinato NHC-diborene catalyst for hydroboration. J. Am. Chem. Soc. 143, 4993–5002 (2021).
Wu, D. et al. 1,3,2,5-Diazadiborinine featuring nucleophilic and electrophilic boron centres. Nat. Commun. 6, 7340 (2015).
Wu, D. et al. Electrostatic catalyst generated from diazadiborinine for carbonyl reduction. Chem 3, 134–151 (2017).
Wentz, K. E. et al. Activation of carbon dioxide by 9-carbene-9-borafluorene monoanion: carbon monoxide releasing transformation of trioxaborinanone to luminescent dioxaborinanone. J. Am. Chem. Soc. 144, 16276–16281 (2022).
Chen, X., Yang, Y., Wang, H. & Mo, Z. Cooperative bond activation and catalytic CO2 functionalization with a geometrically constrained bis(silylene)-stabilized borylene. J. Am. Chem. Soc. 145, 7011–7020 (2023).
Mascetti, J. Carbon Dioxide Coordination Chemistry and Reactivity of Coordinated CO2. In Carbon Dioxide as Chemical Feedstock (ed. Aresta, M.) 55–88 (Wiley-VCH, 2010).
Aresta, M., Nobile, C. F., Albano, V. G., Forni, E. & Manassero, M. New nickel–carbon dioxide complex: synthesis, properties, and crystallographic characterization of (carbon dioxide)-bis(tricyclohexylphosphine)nickel. Chem. Commun. 636–637 (1975).
Herskovitz, T. Carbon dioxide Coordination Chemistry. 3. Adducts of Carbon Dioxide with Iridium(I) Complexes. J. Am. Chem. Soc. 99, 2391–2392 (1977).
Viasus, C. J., Gabidullin, B. & Gambarotta, S. Linear end-on coordination modes of CO2. Angew. Chem. Int. Ed. 58, 14887–14890 (2019).
Ashley, A. E. & O’Hare, D. FLP-mediated activations and reductions of CO2 and CO. Top Curr Chem 334, 191–217 (2013).
Mömming, C. et al. Reversible metal-free carbon dioxide binding by frustrated lewis pairs. Angew. Chem. Int. Ed. 48, 6643–6646 (2009).
Berndt, A. Classical and nonclassical methyleneboranes. Angew. Chem. Int. Ed. 32, 985–1009 (1993).
Wang, Y. et al. A stable neutral diborene containing a BB double bond. J. Am. Chem. Soc. 129, 12412–12413 (2007).
Bissinger, P. et al. Generation of a Carbene-Stabilized Bora-borylene and its Insertion into a C–H Bond. J. Am. Chem. Soc. 133, 19044–19047 (2011).
Schmidt, U. et al. Tuneable Reduction of Cymantrenylboranes to Diborenes or Borylene-Derived Boratafulvenes. Chem. Commun. 56, 14809–14812 (2020).
Hadlington, T. J., Szilvási, T. & Driess, M. Striking Transformations of the Hydroborylene Ligand in a HB:→NiII Complex with Isocyanides and CO. Chem. Sci. 9, 2595–2600 (2018).
Kinjo, R., Donnadieu, B., Celik, M. A., Frenking, G. & Bertrand, G. Synthesis and Characterization of a Neutral Tricoordinate Organoboron Isoelectronic with Amines. Science 333, 610–613 (2011).
Braunschweig, H. et al. Main-Group Metallomimetics: Transition Metal-like Photolytic CO Substitution at Boron. J. Am. Chem. Soc. 139, 1802–1805 (2017).
Braunschweig, H. et al. Multiple Complexation of CO and Related Ligands to a Main-Group Element. Nature 522, 327–330 (2015).
Fan, J. et al. Tetrakis(N-heterocyclic Carbene)-Diboron(0): Double Single-Electron-Transfer Reactivity. J. Am. Chem. Soc. 145, 11669–11677 (2023).
Jeoung, J.-H. & Dobbek, H. Carbon Dioxide Activation at the Ni,Fe-Cluster of Anaerobic Carbon Monoxide Dehydrogenase. Science 318, 1461–1464 (2007).
Chapovetsky, A. et al. Pendant hydrogen-bond donors in cobalt catalysts independently enhance CO2 reduction. ACS Cent. Sci. 4, 397–404 (2018).
Ménard, G. & Stephan, D. W. Room temperature reduction of CO2 to methanol by Al-based frustrated lewis pairs and ammonia borane. J. Am. Chem. Soc. 132, 1796–1797 (2010).
Das, S., Turnell-Ritson, R. C., Dyson, P. J. & Corminboeuf, C. Design of frustrated lewis pair catalysts for direct hydrogenation of CO2. Angew. Chem. Int. Ed. 61, e202208987 (2022).
Mannisto, J. K. et al. Mechanistic insights into carbamate formation from CO2 and amines: the role of guanidine–CO2 adducts. Cata. Sci. & Technol. 11, 6877–6886 (2021).
Neilson, R. H., Li, B. L. & Goodman, M. A. Synthesis and Steroechemistry of Some Alkyl[bis(trimethylsilyl)amino]boranes. Inorg. Chem. 23, 1368–1371 (1983).
Jäkle, F. & Sundararaman, A. A comparative study of base-free arylcopper reagents for the transfer of aryl groups to boron halides. J. Organomet. Chem. 681, 134–142 (2003).
Acknowledgements
This work was supported by the Ministry of Education Singapore, AcRF Tier 1 (RG72/12) and A*STAR MTC Individual Research Grants (M21K2c0117) (C.-W.S.). C.-S.W. and M.-D.S. acknowledge the National Center for High-Performance Computing of Taiwan for generous amounts of computing time and the Ministry of Science and Technology of Taiwan for the financial support. We also thank Prof. Weng Kee Leong and Zhen Xuan Wong (NTU) for their assistance in Fourier transform infrared spectroscopy, as well as Dr. Yongxin Li (NTU) for his assistance in the X-ray crystallographic measurements and analysis.
Author information
Authors and Affiliations
Contributions
J.F. and A.-P.K. performed the synthetic experiments and spectroscopic characterizations. C.-S.W. and M.-D.S. did the theoretical calculations. C.-W.S. conceived and supervised the study, and drafted the manuscript with assistance from J.F. and A.-P.K. All authors contributed to discussion.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Hao Wang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fan, J., Koh, AP., Wu, CS. et al. Carbon dioxide capture and functionalization by bis(N-heterocyclic carbene)-borylene complexes. Nat Commun 15, 3052 (2024). https://doi.org/10.1038/s41467-024-47381-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-47381-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
