
Abstract
Inositol 1,4,5-trisphosphate receptors (IP3Rs) are endoplasmic reticulum Ca2+ channels whose biphasic dependence on cytosolic Ca2+ gives rise to Ca2+ oscillations that regulate fertilization, cell division and cell death. Despite the critical roles of IP3R-mediated Ca2+ responses, the structural underpinnings of the biphasic Ca2+ dependence that underlies Ca2+ oscillations are incompletely understood. Here, we collect cryo-EM images of an IP3R with Ca2+ concentrations spanning five orders of magnitude. Unbiased image analysis reveals that Ca2+ binding does not explicitly induce conformational changes but rather biases a complex conformational landscape consisting of resting, preactivated, activated, and inhibited states. Using particle counts as a proxy for relative conformational free energy, we demonstrate that Ca2+ binding at a high-affinity site allows IP3Rs to activate by escaping a low-energy resting state through an ensemble of preactivated states. At high Ca2+ concentrations, IP3Rs preferentially enter an inhibited state stabilized by a second, low-affinity Ca2+ binding site. Together, these studies provide a mechanistic basis for the biphasic Ca2+-dependence of IP3R channel activity.
Similar content being viewed by others

Introduction
Inositol-1,4,5-trisphosphate receptors (IP3Rs) are large, tetrameric cation channels that serve as the primary intracellular calcium (Ca2+) release channels in nonexcitable cells. Expressed in the endoplasmic reticulum (ER), IP3Rs mediate the flow of Ca2+ from the ER into the cytoplasm and other cellular compartments where Ca2+ contributes to the regulation of cell division1, differentiation2, metabolism3, migration4,5, and cell death6. Consequently, dysregulation of IP3Rs is associated with numerous pathologies including cancer7,8,9, neurological10,11, cardiac12, and immune13 diseases. IP3R activation requires nanomolar cytosolic Ca2+ and the second messenger IP3, whose production is stimulated by receptor tyrosine kinase and G protein-coupled receptor signaling pathways14,15,16,17,18,19,20. Notably, IP3Rs are inhibited by micromolar cytosolic Ca2+ concentrations, resulting in a biphasic dependence on Ca2+ for channel activity. The recursive nature of IP3R regulation by its permeant ion results in the emergent phenomenon of Ca2+ oscillations in cells. The Ca2+ dependence of both activation and inhibition are further modified by the concentration of IP3 as well as ATP, ER Ca2+ and numerous protein interaction partners21,22,23. In this manner, IP3Rs integrate multiple upstream signals to tune the frequency and amplitude of Ca2+ oscillations that encode regulatory information for diverse cellular processes such as mitochondrial oxidative metabolism24, gene expression25, lymphocyte activation26 and neuronal development27.
Structural snapshots of IP3Rs have revealed the overall architecture of the channel and how IP3 and Ca2+ can stabilize conformational changes28,29,30,31,32,33,34. These studies revealed that IP3Rs possess a transmembrane domain that resembles other 6 transmembrane (6TM) ion channels such as voltage-gated ion channels and TRP channels, as well as a large cytosolic domain (CD) that contains all of the known regulatory ligand-binding sites, including two Ca2+ binding sites, and shares some homology with the Ryanodine Receptor (RyR)35. When both Ca2+ binding sites are occupied, the pore remains closed regardless of IP3 binding status29. In contrast, a recent structure suggests that the pore opens when only one of the Ca2+ binding sites is occupied in the presence of IP333. However, many additional conformations have been resolved whose functional corollaries remain unclear. More broadly, the conformational landscape that enables IP3Rs to pivot from activation to inhibition to generate Ca2+ oscillations remains unknown. Here, we collect electron cryomicroscopic images of human type 3 IP3R (hIP3R3) vitrified in a broad range of Ca2+ concentrations and treat particle abundance as a proxy for the relative free energy of each state to establish high-resolution thermodynamic models of IP3R activation and inhibition, which combined with cellular Ca2+ imaging elucidates the structural basis for IP3R-generated Ca2+ oscillations.
Results
Structural Ca2+ titration reveals conformational landscape of hIP3R3
To elucidate the mechanisms by which IP3 and Ca2+ together activate the channel, and high Ca2+ concentrations inhibit the channel, we collected transmission electron cryomicroscopic (cryo-EM) images of purified human type 3 IP3 receptors (hIP3R3) prepared with saturating (200 µM) IP3, saturating (1 mM) ATP, and five concentrations of nominal free Ca2+ spanning a range from 1 nM to 10 µM (Fig. 1a and Supplementary Fig. 1). Our cryo-EM conditions correspond to a range where electrophysiological analyses predict that hIP3R3 displays a biphasic relationship between Ca2+ concentration and channel open probability. To track the Ca2+-dependence of the IP3R conformational landscape in an unbiased manner, we merged these datasets and performed image processing in aggregate (Supplementary Fig. 2 and Supplementary Table 1).

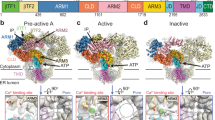
a Schematic for cryo-EM Ca2+ titration of hIP3R3. b–f C4-symmetrized composite cryo-EM density maps viewed from the cytosol (left) and the side (right) with structural heuristics (top-right corner) and ligand binding sites (bottom insets for IP3, CD Ca2+, JD Ca2+, and ATP) for the resting (b), preactivated (c), preactivated+Ca2+ (d), activated (e), and inhibited states (f). Insets in b-f are colored by domain: BTF1 (purple), BTF2 (blue), ARM1 (light blue), CLD (cyan), ARM2 (green), ARM3 (yellow), JD (orange), and TMD (red). Insets in (b) are contoured at 4, 8, 9 and 4 σ thresholds for the IP3, CD Ca2+, JD Ca2+, and ATP sites, respectively. Insets in (c–e) are contoured at 5, 7, 9 and 4 σ thresholds for the IP3, CD Ca2+, JD Ca2+, and ATP sites, respectively. Insets in (f) are contoured at 1, 9, 15 and 6 σ thresholds for the IP3, CD Ca2+, JD Ca2+, and ATP sites, respectively.
Using hierarchical classification without imposing symmetry, we resolved five major states for hIP3R3 at resolutions up to 2.5 Å to which we initially assigned C4 symmetry (Supplementary Table 2). By relaxing our assumption of C4 symmetry and computing latent representations of the conformational heterogeneity present in the remaining classes using 3D variability analysis (3DVA)36, we were also able to reconstruct discrete low-abundance intermediates, including several that are asymmetric. Following classification, we improved the interpretability of the reconstructions by performing symmetry expansion and local refinements that were subsequently merged into composite reconstructions.
Due to overlapping ligand-binding profiles of the major states and several minor states, we established a heuristic describing four features of the channel that facilitate comparisons between the states as well as with existing IP3R structures. The features that comprise the heuristic are the beta-trefoil (BTF) ring, armadillo repeat domain 2 (ARM2), the juxtamembrane domain (JD) ring and the pore (Fig. 1b–f). The most predominant of these features is the conformation of the cytosolic BTF ring, which adopts either an intact tetrameric ring structure that stabilizes the entire cytosolic domain (CD), or a disrupted state in which the CDs of the four protomers are decoupled and highly dynamic. Second is the conformation of the peripheral ARM2 domain, which can be either extended away from the rest of the CD or retracted. Third is the JD ring, located at the interface between the CD and the transmembrane domain (TMD) and can adopt either an intact ring structure or a disrupted, open conformation. Last is the pore, which can either be closed or open.
In the first of the major states, the BTF ring is intact, ARM2 is extended, the JD ring is intact, and the pore is closed (Fig. 1b, Supplementary Fig. 3 and Supplementary Table 2). As this state resembles previously published ligand-free states of IP3Rs in various detergents (PDB: 3JAV, 6DQJ, 6MU2, 6UQK, 7LHF) and lipid environments (PDB: 7LHE)28,29,30,31,32, we assigned this conformation as a resting state. Two similar minor states were also present that share the overall conformation of the resting state but differ slightly in the conformation of the TMD with much weaker density for the peripheral S1-S4 domain (Supplementary Fig. 4 and Supplementary Table 3). Due to the increased conformational heterogeneity of the TMD in these states, we assigned them as labile resting state 1 and labile resting state 2.
The second and third major states also have intact BTF and JD rings and a closed pore, but their ARM2 domains adopt the retracted conformation, where ARM2 is rotated towards the central linker domain (CLD) (Fig. 1c–d, Supplementary Figs. 5 and 6 and Supplementary Table 2). Differentiating these two states is the presence of a non-protein density occupying the previously identified JD Ca2+ binding site that we assigned as a bound Ca2+ ion. A fourth state shares the intact BTF ring and retracted ARM2 domain with the second and third states, but its pore is open, and its JD ring is disrupted (Fig. 1e, Supplementary Fig. 7 and Supplementary Table 2). Based on the open conformation of the pore, we assigned the fourth state as an activated state. This activated state is similar to a recently published structure of hIP3R3 with its pore in an open configuration (PDB: 7T3T)33. As the second and third states differ from the activated state only in their closed pores and intact JD rings, we assigned them as a preactivated state and a preactivated+Ca2+ state, respectively. The preactivated state, with its intact BTF ring and retracted ARM2 domain and closed pore, is similar to previously reported structures of IP3Rs in the presence of IP3 (PDB: 6DQV, 7T3P, 7T3Q, 7T3R)33,37, but the preactivated+Ca2+ state has not been previously described.
In addition to the four-fold symmetric resting and preactivated states, we also resolved classes with asymmetric CDs. In these classes, either one, two or three of the ARM2 domains adopt the retracted conformation (see below and Supplementary Table 4). Together, these classes represent a continuum of states between the resting state, where all four ARM2 domains are extended, and the preactivated state, where all four ARM2 domains are retracted, a finding we previously reported for channels in the presence of IP3 (PDB: 6DQN, 6DQS, 6DQZ, 6DR0, 6DQV)29. While we were able to resolve structures for these states, we observed significant continuous heterogeneity among these asymmetric classes. Therefore, we combined these particles into an ensemble that we call the resting-to-preactivated transitions for quantification. We also observed classes with asymmetric features in the JD and TMD that otherwise resembled the resting or preactivated states. The pore in these classes has undergone movements that result in either two-fold pseudosymmetric (~C2) or four-fold pseudosymmetric (~C4) dilations compared to the closed states (see below, Supplementary Fig. 8 and Supplementary Table 5). We will refer to the classes with extended ARM2 domains as resting TMD transitions and those with retracted ARM2 domains as preactivated TMD transitions.
In the fifth major state, the BTF ring is disrupted, ARM2 is retracted, the JD ring is intact, and the pore is closed (Fig. 1f, Supplementary Fig. 9 and Supplementary Table 2). A minor population of particles sharing these features was also identified in which the channels were organized into higher-order assemblies containing two or more tetrameric channels (see below, Supplementary Fig. 10 and Supplementary Tables 3, 10, and 11). Notably, the interactions that mediate the assemblies are the only distinguishing feature between these two states. Otherwise, the channels adopt similar conformations. These two BTF ring disrupted states are reminiscent of previously published Ca2+-bound hIP3R3 structures (PDB: 6DRC, 6DR2, 6DRA, 7T3U)29,33, where BTF ring disruption confines IP3-mediated conformational changes to the CD, so we assigned the major state as an isolated inhibited state and the minor state as a higher-order assembly of inhibited states.
Ligand dependence of hIP3R3 conformations
To evaluate the relationship between ligand occupancy and conformational state, we inspected the cryo-EM maps and identified densities in the resting, preactivated, preactivated+Ca2+, activated and inhibited states consistent with an IP3 bound at the BTF2-ARM1 interface and with a Zn2+ and an ATP bound in the JD of all five C4 symmetric major states (insets in Fig. 1b–f). Density for IP3 is also present in the asymmetric subclasses that belong to the resting-to-preactivated transitions (Supplementary Fig. 11), indicating that the 200 µM IP3 concentration used for vitrification was sufficient to saturate the nM-affinity binding site38, and that asymmetry of the ARM2 conformations did not arise from substoichiometric IP3 binding. The IP3-binding site is best resolved in the resting state where Arg568 on ARM1 coordinates the 1-phosphate of IP3 conferring a specific orientation to IP3 in this pocket as predicted by mutagenesis39. Arg266 and Arg270 on BTF2, and Arg503, Lys507, Arg510, and Lys569 on ARM1 complete the positively charged binding site to coordinate IP3 (PDB: 1N4K, 3T8S, 3UJ0)40,41,42. As observed previously29, IP3 can bind the channel via two modes (Supplementary Fig. 12). Comparing the resting state to a previously published ligand-free state (PDB: 6DQJ), IP3 binding results in a contraction of the IP3-binding pocket through movement of a loop (Leu265-Ser278) on BTF2 (Supplementary Fig. 12). Conversely, in the ARM2 retracted states, ARM1 tilts towards IP3 to contract the ARM1-BTF2 interface. Notably, IP3 is coordinated by the same residues in both binding modes (insets in Fig. 1b–f).
The Zn2+ ion bound in the JD is coordinated by a C2H2 zinc-finger fold formed by Cys2538, Cys2541, His2558, and His2563, where it has been observed in other IP3R structures28 (insets in Fig. 1b–f). The adenine base of the nearby ATP is buried in a hydrophobic cavity that was recently identified as an ATP-binding site that is structurally conserved with RyRs (Supplementary Fig. 13; PDB: 7T3P, 5TAP)33,43. Specificity for adenine bases21,44,45,46 is imparted through the primary amine of the base forming interactions with the backbone carbonyl oxygen of His2558 and thiolate of Cys2538. The triphosphate moiety of ATP extends away from the JD with clear densities corresponding to the α and β phosphates, which are directly coordinated by Lys2152 and Lys2560, respectively (Fig. 1b–f). The γ-phosphate is poorly resolved and does not form direct interactions with the channel. Taken together, the coordination of ATP is consistent with both ATP and ADP having greater potentiating effects on IP3Rs over AMP21,44,45.
In contrast to the saturating conditions for IP3 and ATP, our buffers sampled a range of Ca2+ concentrations that span the reported apparent affinities for both activation and inhibition of IP3Rs, suggesting that we might resolve a range of Ca2+ occupancies among the major states. To assess the Ca2+-dependence of each conformation, we first inspected the cryo-EM density near the previously identified JD and CD Ca2+ binding sites29 (Fig. 1b–f). In both the resting and preactivated states, no density peaks consistent with a bound Ca2+ ion were observed at either the JD or CD binding sites (insets in Fig. 1b–c). In the preactivated+Ca2+ state, we observed a density peak that we assigned as a Ca2+ in the JD site while the CD site was unoccupied (inset in Fig. 1d). The Ca2+-binding profile of the activated state is the same as the preactivated+Ca2+ state, with an occupied JD site and an empty CD site (inset in Fig. 1e). Only in the inhibited state did we observe densities corresponding to Ca2+ in both sites (inset in Fig. 1f). In the three JD Ca2+-bound states, the backbone of Thr2581 from the JD and side chains of Glu1882, Glu1946, and Gln1949 from ARM3 coordinate Ca2+ (Fig. 1d–f and below). The CD Ca2+, observed exclusively in the inhibited state, is coordinated by the backbone of Arg743 from the CLD and side chain of Glu1125 and backbone of Glu1122 from ARM2 (Fig. 1f and below). Outside of the CD and JD sites, no densities consistent with bound Ca2+ ions could be identified in any of the maps. Taken together with our previous analyses of hIP3R3 in saturating Ca2+29, these data are consistent with the JD and CD sites being the primary Ca2+ binding sites in IP3Rs. Thus, in addition to their distinct global conformations, the five major states display defining ligand-binding properties. The resting and preactivated states, which bind IP3, ATP, and Zn2+, but not Ca2+, differ in how they coordinate IP3. In addition to IP3, ATP, and Zn2+, a single Ca2+ ion is bound to each protomer of the preactivated+Ca2+ and activated states, while two Ca2+ ions are bound to each protomer of the inhibited state.
Ca2+ perturbs the energetic landscape of hIP3R3
Single-particle cryo-EM analysis of vitrified samples represents a near equilibrium assessment of their conformational landscape, allowing one to infer relative conformational free energy from the number of particles that populate specific structural classes47. Therefore, by analyzing the effects of Ca2+ on the relative abundance of each hIP3R3 conformation or ensemble, we can assess how Ca2+ biases the energetic landscape of the channel to favor activation at intermediate concentrations and favor inhibition at high concentrations (Fig. 2 and Supplementary Table 7). Furthermore, the Ca2+-dependent conformational landscape can provide additional confidence in the assignment of functional correlates to the observed conformational states. For example, the abundance of the putative resting state, which closely resembles the ligand-free state and shows no evidence of bound Ca2+ ions, is negatively correlated with the concentration of Ca2+. At low Ca2+, 45.2% of the particles adopt the resting state whereas this percentage drops to 0.7% at high Ca2+. Together, the two labile resting states follow a similar pattern, starting at 20.4% of the particles at 1 nM and falling to 1.2% at 10 µM. The ensemble of resting TMD transitions, comprised of the ~C2 and ~C4 states, is also similar, starting at 6% at 1 nM and falling to 2.4% at 10 µM.

a Relative percent abundance of the five major states and the ensembles of minor states. b Aggregate abundances of all states across the Ca2+ titration. Graphs are colored by state: resting (grey), resting labile (magenta), resting TMD transitions (purple), resting-to-preactivated (blue), preactivated (cyan), preactivated+Ca2+ (green), preactivated TMD transitions (yellow), activated (sand), higher-order inhibited (orange), and isolated inhibited (red).
We observed two distinct inhibited states – an isolated inhibited state and an assembled inhibited state in which several inhibited tetramers form higher-order assemblies (Fig. 1f and below, Supplementary Figs. 9 and 10). Although the states are structurally very similar with disrupted BTF rings, they have distinct abundance profiles with respect to Ca2+ concentration (Fig. 2a). The abundance of the isolated inhibited channels is the inverse of the resting state i.e. positively correlated to Ca2+ concentration, increasing monotonically to a maximum of 74.5% at 10 µM. The assembled inhibited state follows the same pattern at low Ca2+ concentrations, increasing from 5.9% at 1 nM to a maximum of 20.1% at 100 nM. However, higher Ca2+ concentrations do not have any additional effect as the abundance of the assembled inhibited state plateaus between 17.6% and 20.1%. Although the structures of the tetramers in the higher-order assemblies are very similar to the isolated inhibited tetramers, their divergent Ca2+-dependence suggests that they are distinct states and that formation of higher-order assemblies may represent an alternative mechanism for achieving an inhibited state, as we will discuss later.
In contrast to the resting-like states and the inhibited states, the distribution of the preactivated-like and activated states exhibit biphasic Ca2+ dependencies, achieving their maximum abundance at intermediate Ca2+ concentrations (Fig. 2a). Starting with the ensemble of resting-to-preactivated transitions, which achieve a maximum of 10.5% at 10 nM, the profiles of the preactivated, preactivated+Ca2+, the ensemble of ~C2 and ~C4 preactivated TMD transitions, and the activated state are shifted rightward to progressively higher Ca2+ concentrations. Apart from the activated state, the maximum abundance achieved by these states also decreases in a progressive manner, consistent with these states being progressively higher energy intermediates along a reaction coordinate extending from the resting state to the activated state. This continuum of inter-convertible states also provides a rationale for why the ensemble of resting-to-preactivated transitions and the preactivated state display a clear correlation with Ca2+ despite not showing evidence of binding Ca2+ themselves.
The abundance profile of the activated state agrees with decades of single-channel electrophysiological analyses of IP3Rs, showing a biphasic open probability in the presence of saturating IP3 and ATP with maximal activity occurring in the high nM Ca2+ range (Fig. 2a)19. Moreover, the Ca2+-dependent conformational landscape of IP3Rs resolves a bipartite mechanism for this biphasic relationship between Ca2+ concentration and channel open probability. At low Ca2+ IP3Rs must escape a low-energy ARM2 extended resting state in order to activate by binding Ca2+ at the high-affinity JD site. At high Ca2+, IP3Rs preferentially enter a low-energy inhibited state stabilized by a second Ca2+ ion binding to the low-affinity CD site.
The JD Ca2+ site is essential for Ca2+ oscillations
The multimodal regulation of IP3Rs, including activation and feedback inhibition by Ca2+, produces IP3R-dependent Ca2+ oscillations in cells48,49,50,51. Structurally, we observe that Ca2+ binding at the JD can occur in the putative activated state, while Ca2+ binding at the CD site occurs only in the inhibited states (Fig. 3a–e). To assess the roles of these sites in producing cellular Ca2+ oscillations and to attempt to establish a functional corollary to the conformational states obtained through the structural Ca2+ titration, we employed a fluorescence-based Ca2+ imaging assay that monitors Ca2+ oscillations in cells. We first incubated HEK293T cells lacking all three IP3R isoforms (IP3R-null) with Cal-520-AM, a fluorogenic calcium-sensitive dye, and then stimulated intracellular IP3 generation by adding carbachol to the bath solution (Fig. 4a)52. Saturating carbachol concentrations (100 µM) were added to cells to minimize potential stimulus-dependent effects on the IP3R response in cells53. Consistent with earlier reports52, no detectable changes in cytosolic Ca2+ were observed in IP3R-null cells (Supplementary Fig. 14). Conversely, Ca2+ oscillations of two or more peaks were observed in cells transiently expressing hIP3R3, indicating that the construct used for structural analysis expresses a functional channel (Fig. 4b). We assessed the temporal characteristics of the carbachol-stimulated Ca2+ spikes in cells by aligning the initial peak of each normalized cellular trace that produced an oscillatory response (Fig. 4c). For IP3R-null transiently expressing wild-type hIP3R3, the mean slope of the rising phase at the half-maximal intensity was 0.103 ± 0.015 Fluorescencenorm sec-1. Traces were also analyzed to determine the number of peaks observed in cells showing oscillatory responses following carbachol stimulation, with cells expressing wild-type hIP3R3 having a median of 4 peaks/cell (Fig. 4d). Finally, to calculate the time between successive Ca2+ spikes (inter-spike interval), we extracted traces from segmented cells, then smoothed and adjusted the baseline to automatically identify peaks. For wild-type hIP3R3 the mean inter-spike interval was 21.7 seconds, which is within the range of times measured for endogenous IP3R-mediated cytosolic Ca2+ 54,55.

a Side view of the activated state highlighting domain architecture on the left protomer and ligand binding sites on the right protomer. Front and rear protomers removed for clarity. b–c Superpositions of the JD Ca2+ binding site in the Ca2+-free states (b) and Ca2+-bound states (c). d–e Superpositions of the CD Ca2+ binding site in the Ca2+-free states (d) and the Ca2+-bound state (e). f–i Superpositions of the ARM3-JD interface aligned by the JD for transitions from resting to preactivated (f), preactivated to preactivated+Ca2+ (g), preactivated+Ca2+ to activated (h), and activated to inhibited (i). Magenta arrows highlight movements of the proximal and distal regions of the JD between states.

a Schematic describing Cal-520-AM fluorescence-based Ca2+ imaging assay and data analysis. b, e, h Representative z-score normalized Cal-520-AM fluorescence traces recorded from cells expressing hIP3R3 (b), CD mutant (c, E1125Q) and JD mutant (h, E1882Q+E1946Q) in an IP3R-null background following stimulation by carbachol. N = 3 biologically independent samples. c, f, i Aligned first peak of every oscillatory trace (thin lines) normalized to 1 for hIP3R3 (c), CD mutant (f, E1125Q) and JD mutant (i, E1882Q+E1946Q). Bold line represents mean and dashed lines represent 95% confidence interval. d, g, j, Peak count distributions for all oscillatory traces observed for hIP3R3 (d), CD mutant (g, E1125Q) and JD mutant (j, E1882Q+E1946Q). Individual points represent mean and error bars represent S.E.M. Source data are provided as a Source Data file.
Having established metrics that describe the carbachol-induced Ca2+ oscillations of wild-type hIP3R3, we next examined the effects of perturbing the Ca2+-binding sites. We transiently expressed hIP3R3 with mutations to the JD site (Glu1882Gln+Glu1946Gln), the CD site (Glu1125Gln) or both sites (Glu1125Gln+Glu1882Gln+Glu1946Gln) in IP3R-null cells. Robust Ca2+ oscillations were observed in cells expressing the CD mutant (Fig. 4e–g). While the mean rising phase was similar to wild-type hIP3R3 (Fig. 3f, Supplementary Table 8), the mean inter-spike interval was nearly half (59%) at 12.7 seconds (Supplementary Fig. 14, Supplementary Table 8), suggesting that perturbing the CD site alters gating of hIP3R3. As the CD site is exclusively occupied in the inhibited states, our structural and functional analyses are consistent with Ca2+ binding at the CD site contributing to channel inhibition.
Unlike cells expressing wild-type channels or the CD mutant, we did not observe oscillatory responses in cells expressing either the JD mutant (Fig. 4h–j) or the JD/CD double mutant (Supplementary Fig. 14). Instead, we observed a single slow non-oscillatory event in both mutants that did not resemble the events seen in cells expressing the wild-type channel. The mean slope of the rising phase was 3.7 times slower for cells expressing the JD mutant and 3.0 times slower for cells expressing the JD/CD double mutant than those of cells expressing wild-type hIP3R3 (Fig. 3c, i and Supplementary Fig. 14). Therefore, although perturbations to the JD site do not abolish IP3R-mediated Ca2+ release, consistent with recent electrophysiological analyses showing diminished activity of JD site mutants56, the JD site is essential for ensuring the fidelity of agonist-evoked cytosolic Ca2+ oscillations in cells.
Binding of the JD Ca2+ ion has distinct effects on channel conformation
Although Ca2+ binding to the JD site is required for Ca2+ oscillations in cells, it is also occupied in the closed preactivated+Ca2+ and the closed inhibited states. To gain insights into how Ca2+ binding can stabilize these three distinct conformations, we aligned the JD of the five major states (Fig. 3f–i) to visualize the progressive changes to the JD Ca2+ binding site during activation and inhibition. The pair-wise comparisons reveal that large changes to the ARM3-JD interface occur exclusively during the transition from the preactivated+Ca2+ to activated state: the JD-distal region of ARM3 rotates 6 Å towards the JD while the JD-proximal region shifts upwards 2 Å back to its resting state position (Fig. 3h). The changes that occur during the other transitions are more subtle. For example, the transitions from resting to preactivated and from preactivated to preactivated+Ca2+ are each accompanied by 1 Å downward movements of the JD-proximal part of ARM3 (Fig. 3f–g). Binding a second Ca2+ at the CD site also results in a minimal rearrangement of the ARM3-JD interface, with both the distal and proximal regions of ARM3 moving down 1 Å during the transition from the activated to inhibited state (Fig. 3i). Surprisingly, despite the large global conformational differences between the preactivated+Ca2+, activated and inhibited states, the configuration of the residues that form the JD site are nearly identical. The JD binding site appears to adopt only two conformations, a Ca2+-free expanded conformation in the resting and preactivated states and a Ca2+-bound contracted conformation in the preactivated+Ca2+, activated and inhibited states (Fig. 3d–e). Furthermore, we only observe stable occupancy of the JD site in the ARM2 retracted states, suggesting that the IP3-stabilized movement of ARM2 increases the affinity for Ca2+. Allosteric coupling between Ca2+ and IP3 binding is consistent with biochemical experiments suggesting that Ca2+ binding can increase the affinity for IP357,58, and kinetic experiments showing IP3 binding exposes a high-affinity Ca2+ binding site59. In summary, although Ca2+ binding to the JD site stabilizes a single, distinct Ca2+-bound conformation of the binding site, the effect of Ca2+ binding on channel conformation at the global level can be varied and is influenced by the presence of other ligands.
IP3 primes channel activation through a cooperative process involving ARM2 retraction
Activation of IP3Rs requires that all four IP3 binding sites be intact52, suggesting that a coordinated IP3-mediated conformational change must occur prior to pore opening. Our previous analysis revealed that the transition between ARM2 extended and ARM2 retracted states is both IP3-mediated, with the retracted state only being resolved in the presence of IP3, and cooperative, with the four-fold symmetric extended or retracted conformations being substantially favored over the asymmetric states as opposed to a binomial distribution29. We therefore hypothesized that the IP3 binding mode of a protomer can be sensed by its neighbors and that this communication may underlie the requirement for four intact IP3 binding sites. To evaluate the relationships between a single protomer and its neighbors, we performed symmetry expansion, focused refinement, and 3DVA on the CD of a single protomer, which includes the uniformly occupied IP3 binding site and ARM2, for the resting-to-preactivated ensemble (Fig. 5a and Supplementary Table 4). By calculating reconstructions for particles segmented along the primary dimension of variability, we can visualize the progression of one protomer (labeled b in Fig. 5b–g) from the ARM2 extended conformation resolved in the resting state to the ARM2 retracted conformation of the preactivated state. In the most extended ARM2 position of the central protomer, ARM2b forms two interactions with the counterclockwise protomer (labeled a), one with ARM1a, and a second with BTF1a (Fig. 5b). The transition of protomer a to the ARM2 retracted state is accompanied by a contraction of the ARM1-BTF2 interface around IP3. A consequence of this contraction is that ARM1a is pulled away from ARM2b, disrupting one of ARM2b’s interprotomer interactions (Fig. 5c). The diminished association with the neighboring protomer results in a more dynamic state for ARM2b, which manifests in weaker averaged density at its distal end (Fig. 5d). The increased flexibility of ARM2b destabilizes its remaining interprotomer interaction with BTF1a and allows it to transiently disengage from BTF1a and rotate towards CLDb to adopt the retracted conformation. In the retracted conformation, ARM2b establishes a new interprotomer interface with BTF1a (Fig. 5e). ARM2b retraction results in a tilt of ARM1b away from ARM2 on the clockwise protomer and the entire progression repeats, enabling a cascade around the tetramer that primes the JD site for Ca2+ binding (Fig. 5f–g).

a Unsharpened 5 Å low-pass filtered cryo-EM density of five states showing a range of ARM2 positions between the resting state (left) and preactivated state (right). The three intermediates are derived from the resting-to-preactivated transitions (see Supplementary Table 4). Magenta arrows highlight movements of ARM2 compared to the preceding panel. b–g Unsharpened 5 Å low-pass filtered cryo-EM density along a trajectory of protomer b from the extended state to the retracted state (see Supplementary Table 4). b ARM2b forms two interactions with the adjacent protomer in the extended state. IP3 bound to adjacent protomer a is highlighted. c The first movement is the displacement of ARM1a away from ARM2b. d Further displacement of ARM1a away is accompanied by a destabilization of the distal end of ARM2b. e ARM2b is repositioned into the retracted conformation near CLDb where ARM2b can contact BTF1a as ARM2a continues to move towards IP3a. f Once ARM2b adopts the retracted conformation, ARM1b can move towards the bound IP3 of protomer b, repeating the progression. This process results in torsion of the CLDb. g The movements reach their extremes in the retracted conformation. h Resting state shown as cartoon viewed from the cytosol with wedge loop shown as blue spheres. i The wedge loop occupies a cavity between ARM1, ARM2 and the CLD in the resting state. Ordered residues within the wedge loop are depicted as sticks. Cryo-EM density for the wedge loop is shown as a blue isosurface. j Representative z-score normalized Cal-520-AM fluorescence trace recorded from cells expressing the dARM2 mutant in an IP3R-null background following stimulation by carbachol. N = 3 biologically independent samples. k Aligned first peak of every oscillatory trace (thin lines) normalized to 1. Bold lines represent mean and dashed lines represent 95% confidence interval. l Distribution of peak counts for all oscillatory traces. Individual points represent mean and error bars represent S.E.M. Source data are provided as a Source Data file.
The observed continuum from a symmetric ARM2 extended resting state to a symmetric ARM2 retracted preactivated state suggests that this process is reversible despite the presence of saturating IP3. Consistent with the process being reversible, more particles adopt the resting state than do the ARM2 retracted preactivated and preactivated+Ca2+ states (Fig. 2). Potentially contributing to the favorability of the ARM2 extended state is a loop between Pro897 and Glu958 of the CLD, which we call the wedge loop. In the resting state, a portion of the wedge loop, including Thr926-Ala943, inserts into a cavity surrounded by the CLD, ARM1, ARM2 and ARM3 and adopts an ordered conformation (Fig. 5h–i). Compared to the resting state, ARM2 retraction in the preactivated, preactivated+Ca2+, activated and inhibited states is accompanied by a contraction of this cavity. Modeling the resting state conformation of Thr926-Ala943 into the ARM2 retracted states, where we observed no density for the wedge loop, reveals several steric clashes that would likely disfavor binding of the wedge loop (Supplementary Fig. 15 and Supplementary Table 6).
To assess the relationship between ARM2 retraction and wedge loop binding, we recalculated the ARM2 extended portion of the 3DVA trajectory for the resting-to-preactivated transitions with finer sampling. By aligning the maps based on the strength of the density for the wedge loop, we found that the flexibility of ARM2, as assessed by the local quality of the density, is inversely correlated with the strength of the wedge loop density, indicating that the presence of the wedge loop stabilizes ARM2 in the extended conformation (Supplementary Fig. 15). Moreover, this alignment reveals how the wedge loop dissociates from its binding site in a stepwise fashion. First to dissociate are the residues surrounding Arg931, followed by the N- and C-terminal ends of the loop. Phe936 is the last residue to become disordered, indicating that Phe936 is critical for the interaction.
Flanking Phe936 is the conserved residue Ser934, which can be phosphorylated by protein kinase A (Fig. 5i)60,61,62. Mutation of the residue homologous to Ser934 in hIP3R2 to alanine abrogates the ability of protein kinase A to sensitize hIP3R2 to low-level stimulation by carbachol63. Modeling in a phosphorylated serine at position 934 places the phosphate group in close proximity to Ser937, potentially destabilizing the conformation of the wedge loop and weakening the critical interactions formed by Phe936, suggesting that phosphorylation of Ser934 may influence channel activity by destabilizing the resting state. The residues on and around the wedge loop described here are conserved among the three human IP3R isoforms, suggesting that the wedge loop may serve as a conserved regulatory motif that can influence the equilibrium between ARM2 extension and retraction and thus alter the affinity of the JD site for Ca2+ in all IP3Rs (Supplementary Figs. 10 and 15).
To explore the role of the ARM2-mediated conformational changes in channel activation, we deleted the ARM2 domain (dARM2 mutant; Ala1101-Trp1586) and assessed the effects of its loss on Ca2+ oscillations (Fig. 5j–l). Compared to cells expressing wild-type hIP3R3, carbachol stimulated Ca2+ oscillations were observed less frequently (nWT = 74; ndARM2 = 14) in cells expressing the dARM2 mutant despite both being expressed in a similar fraction of cells (Fig. 5l). Also diminished was the frequency of the Ca2+ spikes. The inter-spike interval was on average 4.7 times longer in cells expressing the dARM2 mutant than in cells expressing hIP3R3. Although the Ca2+ spikes were infrequent, the mean slope of the rising phase of the few responding cells was similar to that of cells expressing wild-type hIP3R3, suggesting that the dARM2 mutant is functional. Thus, while ARM2 is not required for activation or inhibition, its loss appears to reduce the likelihood of exceeding the threshold required for Ca2+ wave propagation50,64. Together, our structural and functional analyses reveal that IP3 binding favors adoption of the preactivated state, which displays a higher apparent affinity for Ca2+, despite the presence of several channel intrinsic features that favor the resting state.
Activation of hIP3R3 by IP3, Ca2+ and ATP
Comparing the closed, Ca2+-bound preactivated+Ca2+ state with the open, Ca2+-bound activated state allows us to observe the conformational changes that enable the pore to open. Although both states share a common ligand-binding profile, large conformational changes can be observed extending from the JD Ca2+ binding site through the JD to the TMD (Fig. 3). The JD, which connects the ligand-binding sites in the cytosolic domain to the TMD, is composed of two interwoven, discontinuous segments of the polypeptide that connect to both the N- and C-terminal ends of the TMD. Together, these connections enable the JD to alter the conformation of both domains of the TMD: the central pore and the peripheral S1-S4 domain (Fig. 6f, h–i). In the closed states, the four JDs assemble into a tetrameric ring structure. In the activated state, the contraction of the ARM3-JD interface induces a ~13˚ clockwise rigid-body rotation of the JDs that disrupts the inter-JD interactions (Fig. 6g).

a Superposition of the pore of the preactivated+Ca2+ (gray) and activated states (red), aligned by the luminal halves of S5 and S6, pore helix and selectivity filter. Front and rear protomers removed for clarity. Blue arrows highlight movement of S6, S4-S5 linker, and gating residues Phe2513 and Ile2517. Arrows show where the pore connects to S1-S4 domain and JD. b–c HOLE diagram showing solvent-accessible surface area of conduction pathway in preactivated+Ca2+ (b) and activated states (c). d–e Top view of constriction in preactivated+Ca2+ (d) and activated (e) states. f Comparison of TMD and JD of a single protomer of preactivated+Ca2+ (gray) and activated (colored) states aligned as in a. Blue arrows highlight the movements of the JD, S1-S4 bundle, S6, and the S4-S5 linker. Bending and rotation of S6 occurs at Gly2514 enabling Phe2513 and Ile2517 to repack behind the pore. g Comparison of JD ring of preactivated+Ca2+ (gray) and activated (colored) states viewed from the cytosol and aligned as in a. Arrows depict the movements that result in JD ring disruption during activation. h The JD (shown here in the activated state) is composed of two fragments JD-A (blue) and JD-B (purple). It is positioned between ARM3 and the TMD, and contributes to the JD Ca2+, ATP, and Zn2+ binding sites. i Inset highlights the ATP and Zn2+ binding sites at the interface between JD-A and JD-B.
The first segment of the JD, which we call JD-A (Glu2111-Met2191), is connected to the peripheral S1-S4 domain. The pore domain and the domain-swapped S1-S4 domain are connected by a short amphipathic S4-S5 linker helix (Fig. 6f). In the closed states, the S4-S5 linkers form a belt around the S6 helices that stabilize the closed pore. In the activated state, the rotation of the JD tilts S1-S4 towards the luminal side of the membrane, relaxing the belt around S6 (Fig. 6a–f and Supplementary Movie 11).
The second segment of the JD, which we call JD-B (Cys2538-Met2608), is directly linked to the cytosolic end of the pore-lining S6 helix. In the activated state, rotation of the JD pulls the S6 helices away from the center of the pore, stabilizing a 13˚ bend and ~30˚ rotation about the helical axis of S6 with Gly2514 being the pivot for both. Together, the tilt and rotation of S6 reposition Phe2513 and Ile2517, which seal the pore in the closed states, out of the ion conduction pathway to create an open pore with a minimum radius of 4 Å (Fig. 6a–e, Supplementary Fig. 16 and Supplementary Movies 9 and 10). In addition to changing the dimensions of the pore, the tilt and rotation of S6 reorient the side chains of Arg2524, Asp2518 and Asp2522, switching the pore from an electropositive to electronegative environment that would be favorable for cation conductance (Supplementary Fig. 16).
Our structural analyses indicate that both segments of the JD contribute to pore opening, with the rotation of JD-A relaxing the belt around the S6 helices and JD-B pulling the S6 helices apart. At the interface between these segments is an ATP (Fig. 6h–i and Supplementary Fig. 13). The adenine moiety of ATP is nestled in a hydrophobic pocket between the two segments lined by Phe2156 from JD-A and Phe2539 and Ile2559 from JD-B (Fig. 6i). The phosphate groups similarly bridge the two segments of the JD with the α-phosphate coordinated by Lys2152 of JD-A and the β-phosphate coordinated by Lys2560 from JD-B. In cells, where ADP and ATP are abundant and the binding site should be predominantly occupied, ADP and ATP likely serve as molecular glue to hold the two discontinuous segments of the JD together. In the absence of ADP or ATP, Ca2+ binding may yield uncoupled movements of the two segments that would be a barrier to opening the pore, consistent with the prevailing model for ATP potentiation through sensitizing the channel to Ca2+ activation without affecting maximal open probability or high-Ca2+ inhibition19,21. Supporting the critical role of a rigid JD domain in channel activation, even a single cysteine-to-serine mutation at the JD Zn2+ binding site results in a complete loss of function without diminishing protein expression or IP3 affinity65.
In addition to the fully-open activated state, our analysis identified several minor classes with partially expanded pores that may represent snapshots of the rearrangements that occur during pore opening (Fig. 7d–e). While the local resolution near the pore of these reconstructions preclude atomic model building, comparing sections of the density maps can inform about how the pore and JDs move during gating. Two of these minor classes belong to the ensemble of preactivated TMD transitions. In one of the classes, two opposing pore-lining S6 helices tilt away from the axis of the pore, while the other two S6 helices are unchanged, establishing a ~C2 pore configuration (Fig. 7d). The S4-S5 linkers also adopt a ~C2 configuration with the S4-S5 linkers of the protomers with displaced S6 helices shifted away from the S6 helix of the neighboring protomer. This uncoupled S4-S5 linker conformation is consistent with our model that the S4-S5 linker belt must be relaxed for the pore to open and appears to be stabilized by an interaction with S1’ of the adjacent protomer (inset in Fig. 7d). In the ~C2 transition, the adjacent S1” tilts towards the pore allowing S1’ to insert underneath the S4-S5 linker of the adjacent protomer, potentially stabilizing this intermediate state. To examine the role of the IP3R-specific S1’ and S1” helices in channel gating, we monitored cytosolic Ca2+ responses following carbachol stimulation in cells transduced with hIP3R3 lacking S1’ and S1” (dS1’-S1”) (Fig. 7g–i). Compared to cells expressing wild-type hIP3R3, cells expressing dS1’-S1” had an increased inter-spike interval and decreased number of oscillatory events per cell, suggesting that S1’ and S1” are required for faithful channel function. Intriguingly, while the analogous linkage between S1 and S2 is a poorly ordered acidic loop in the distantly-related RyRs66, two helices preceding the S1-S4 domain occupy a position similar to S1’-S1” in IP3Rs43. Future studies will be necessary to uncover the role of this unique insertion between S1 and S2 in IP3Rs.

a–f Cryo-EM density maps of the resting (a), preactivated (b), preactivated+Ca2+ (c), ~C2 preactivated TMD transition (d), ~C4 preactivated TMD transition (e), activated (f), and inhibited (g) states, low-pass filtered to 4 Å (overall) or 7 Å (slices). Row 1: Overall cryo-EM density viewed from the side. Row 2: density slice looking from the cytosol at the height of the JD ring with magenta arrows highlighting movements of the JDs. Row 3: density slice looking from the cytosol at the height of the gate with magenta arrows highlighting movements of the S6 helices. Row 4: Side view of a single S1-S4 domain. g Representative z-score normalized Cal-520-AM fluorescence trace recorded from cells expressing the dS1’-S1” mutant in an IP3R-null background following stimulation by carbachol. N = 3 biologically independent samples. h Aligned first peak of every oscillatory trace (thin lines) normalized to 1. Bold line represents mean and dashed lines represent 95% confidence interval. i Distribution of peak counts for all oscillatory traces. Individual points represent mean and error bars represent S.E.M. Source data are provided as a Source Data file.
In the ~C4 subclass, the S4-S5 linkers and the S6 helices of all four protomers are outwardly displaced, creating a partially dilated pore (Fig. 7e). However, compared to the activated state (Fig. 7f), the dilation appears to be incomplete as the cytosolic ends of S6 remain closer together. Comparing the JD in the preactivated+Ca2+ and activated states reveals that the JD also adopts an intermediate conformation. Whereas the JDs are both separated and rotated in the activated state, the JDs in the ~C4 transition are only separated.
Interpolating the ~C2 and ~C4 preactivated TMD transitions into a trajectory that begins with the resting state and ends with the activated state suggests a progression of JD rearrangements that facilitate gating in the pore (Fig. 7a–f). First, retraction of ARM2 in the preactivated state results in a clockwise rotation of the JDs which is further magnified by Ca2+ binding in the preactivated+Ca2+ state. Once Ca2+ is bound, the channel can sample the ~C2 transition, where two opposing JDs shift outwards, disrupting the JD ring and loosening the S4-S5 linker belt leading to an outward movement of two of the four S6 helices. Then, the remaining two JDs are displaced away from the pore axis, resulting in a partial dilation of the pore. Finally, in the activated state, the JDs rotate about the helical axis of S6 to stabilize a fully-open pore where the hydrophobic gating residues Phe2513 and Ile2517 are repacked away from the permeation pathway. Notably, we do not observe any conformational changes in the pore helix or selectivity filter between the high-resolution closed and open states, indicating that the positions of Phe2513 and Ile2517 determine the gating state of the pore.
Subclasses with ~C2 and ~C4 distortions of the pore are also present in the ensemble of resting TMD transitions. In contrast to the preactivated TMD transitions, the JD ring remains intact in these subclasses, suggesting that the pore conformation is not strictly coupled to that of the JD ring (Supplementary Fig. 8). The structural association between the JD ring and TMD in IP3Rs is thus weaker than the associations described between the pore and the cytosolic gating domains of other 6TM cation channels such as the BK channel (Slo1)67.
Mechanisms of high Ca2+ inhibition
Compared to the states with Ca2+ bound solely at the JD site, Ca2+ binding at the CD site in the inhibited state is accompanied by large conformational changes throughout the CD (Fig. 1b–f). The most prominent change is the disruption of the BTF ring, which results in the CDs of the four protomers moving away from one another and towards the membrane. Despite employing the same classification approaches that resulted in identification of several other low-abundance intermediates, we did not observe any transition states between BTF ring intact and BTF ring disrupted states, suggesting that loss of a single interprotomer interaction may be sufficient to disrupt the BTF ring in a highly-cooperative fashion. Due to the presence of a second Ca2+ ion bound at the CD site, and because we previously demonstrated that BTF ring disruption insulates IP3-mediated conformational changes from the channel gate29, we hypothesized that this BTF ring-disrupted conformation is the high-Ca2+ inhibited state of the channel. Consistent with BTF ring disruption being a key aspect of inhibition, mutations at the interface between BTF1 and BTF2 of the neighboring protomer diminish or eliminate carbochol-induced Ca2+ oscillations in cells (Fig. 8d–f and Supplementary Fig. 14). Cells expressing a Trp168Ala/Lys169Ala double mutant displayed no detectable increase in cytoplasmic Ca2+ following carbachol stimulation, while only a single event could be observed in cells expressing a Lys169Ala mutant. These results corroborate mutagenesis experiments that predate structures of a full-length IP3R that yielded a graded effect on IP3-induced Ca2+ release from microsomes, with single mutations at the BTF1-BTF2 interface diminishing release compared to wild-type channels, and two or more mutations resulting in no detectable Ca2+ release68.

a–c Cryo-EM density maps depicting the trajectory of a single protomer’s CD from resting to activated to CD Ca2+-bound inhibited state. a In the resting state, the CD Ca2+ binding site does not exist because ARM2 is extended away from the CLD and ARM1. b ARM2 retraction creates the CD Ca2+ binding site in the preactivated, preactivated+Ca2+, and activated states, but no Ca2+ is yet bound. An interaction between ARM2 and BTF2 of the adjacent protomer restricts the movement of ARM2 and must be released to enable Ca2+ binding. c BTF ring disruption allows ARM2 to move further towards the CLD and bind the CD Ca2+ ion. d Representative z-score normalized Cal-520-AM fluorescence trace recorded from cells expressing the K169A mutant in an IP3R-null background following stimulation by carbachol. N = 3 biologically independent samples. e Aligned first peak of every oscillatory trace (thin lines) normalized to 1. Bold line represents mean and dashed lines represent 95% confidence interval. f Distribution of peak counts for all oscillatory traces. Individual points represent mean and error bars represent S.E.M. Source data are provided as a Source Data file.
Coordination of a Ca2+ in the CD site of the inhibited state is achieved by the N-terminal portion of the CLD and ARM2 rotating towards one another by a total of 3 Å compared to their positions in the activated state (Fig. 8b–c). Through ARM1, the rotation of the CLD pulls BTF1 and BTF2 outwards, away from the BTF domains of the neighboring protomers, while the rotation of ARM2 breaks its interaction with BTF1 of the neighboring protomer. From these observations, Ca2+ binding to the CD site stabilizes the BTF ring disrupted conformation. However, our data cannot discern if Ca2+ binding at the CD site is achieved through an induced fit mechanism or through conformational selection.
Inhibited state particles can form higher-order assemblies
Classification identified two distinct populations of inhibited particles with disrupted BTF rings – individual tetramers and higher-order assemblies of two or more tetramers. The contacts between adjacent tetramers in the assemblies are mediated by two reciprocal interaction surfaces that together create a pseudo two-fold symmetry axis between the two tetramers. Within each of the tetramers, one protomer forms what we call the plus (+) interface through its CLD, one promoter forms what we call the minus (−) interface through its ARM2 domain while the remaining two protomers do not participate in the interaction (Fig. 9a–c). The + interface of one tetramer interacts with the - interface of an adjacent tetramer to create an interaction surface of 2034 Å2. Much of this interface is established by four otherwise disordered linkers that adopt ordered conformations exclusively at the inter-tetramer interface (Supplementary Fig. 11). Loop 1, which is comprised of Ala1556-Asp1587 that connect ARM2 to the CLD, and loop 2, which is comprised of Pro1003-Met1023 in the CLD, contribute to the - side of the interaction (Fig. 9c). Loop 3 (Phe1036-Met1044) and loop 4 (Ser679-Glu690), both of which are in the CLD, contribute to the opposing + side of the interaction.

a, b Top (a) and side (b) views of the octameric assembled inhibited state. c Atomic model showing the four interacting loops at the interface as boxed in a. For clarity, ordered loops are only shown for the tetramer at the right. d Surface representation of a modeling of hypothetical higher-order interactions for the resting, preactivated and activated states shows that steric restrictions imposed by the intact BTF ring allow only a single interaction to form between adjacent tetramers. e BTF ring disruption relieves this restriction and allows higher-order interactions to occur in a reciprocal fashion between adjacent tetramers. Arrows highlight the movements of two CDs that together establish the second inter-tetramer interface.
Higher-order assemblies were notably absent from the other states. By examining potential inter-tetramer interfaces, we found that the ability to form both the + and - interactions was unique to the inhibited state (Fig. 9d–e). The conformational restrictions imposed by an intact BTF ring in the preactivated, preactivated+Ca2+, and activated states permit each tetramer to form only a single interaction with an adjacent tetramer while the extended position of ARM2 precludes either interaction from occurring in the resting states. Although there is a substantial entropic cost to these linkers adopting stable conformations, their extensive interactions suggest that the enthalpic gains from their ordering result in an overall reduction of free energy. In the inhibited state the increased flexibility of the CD following BTF ring disruption may offset this entropic penalty.
Outside of the interfaces and BTF1, which was too poorly-ordered to model, the tetramers in the assemblies are similar to the isolated inhibited state with an all-atom RMSD of 1.2 Å. The largest differences are in the flexible cytosolic domains, which rotate slightly compared to their positions in the isolated inhibited state to enable both the + and - interfaces to interact with the neighboring tetramer. Despite the global structural similarity, the Ca2+-dependence of the assembled inhibited state differs from that of the isolated inhibited state with the assembled inhibited state reaching a plateau at 100 nM Ca2+ while the isolated inhibited state increases in abundance to 10 µM (Fig. 2). The alternative Ca2+-dependence of the assembled inhibited state suggests that it may be functionally distinct, perhaps serving as an alternative mechanism for channel inhibition, which may rationalize how Ca2+ oscillations, which require both activation and high-Ca2+ feedback inhibition48,49,50,51, can be detected in cells expressing the CD site mutant (Fig. 4e–g). Consistent with the assembled inhibited state potentially being functionally distinct from the isolated inhibited state, we did not observe densities that could be attributed to Ca2+ ions in the CD sites although we did observe densities corresponding to ATP, IP3 and the JD Ca2+ ion.
Flexibility of the C-terminal domain is driven by sampling acidic patches on the BTF ring
The CTD forms a four helix coiled-coil that extends through the center of the CD, connecting the JD to the BTF ring in its intact conformations (Supplementary Fig. 18 and Supplementary Table 9). While functional analyses of the CTD have provided conflicting results65,69,70, its central position led to the proposal that it may serve as an allosteric link between the IP3-binding sites in the CD and the pore30. In hIP3R3, the CTD is poorly resolved due to its flexibility. Focused refinement and 3DVA revealed that a portion of the CTD of hIP3R3 alternatively interacts with eight negatively charged patches on the inside of the BTF ring (Supplementary Fig. 18). While the limited resolution precludes building a model for the CTD, a conserved region of positively charged residues from Arg2654 to Arg2659 is the most likely candidate to bind to the negative patches on the BTF ring. The CTD adopts two conformations which are most apparent in the activated state, interacting with four of the eight patches in either ~C2 or ~C4 configurations (Supplementary Fig. 18 and Supplementary Movies 1-8), a noteworthy coincidence given the ~C2 and ~C4 TMD transition states. We investigated the essentiality of the CTD by truncating the channel at Leu2629 and monitoring the effects on IP3R-mediated Ca2+ oscillations. We found that while the CTD deletion (dCTD) mutant produced Ca2+ oscillations with a rising-phase slope that is comparable to wild-type channels, the mean inter-spike interval of 12.2 seconds is significantly shorter (Supplementary Fig. 14). Therefore, while the CTD is not essential for channel activity, CTD deletion does alter Ca2+ dynamics in cells.
Discussion
Here we defined the conformational landscape that underlies the biphasic Ca2+-dependence of IP3Rs and gives rise to IP3R-dependent Ca2+ oscillations in cells. Ordering the states based on their Ca2+ dependence frames a model for the ligand-dependent activation and inhibition of IP3Rs (Fig. 10). IP3 generated in response to extracellular stimuli can bind to the ligand-free channel without altering its global conformation, yielding the low-energy resting state. Once bound to the resting state, IP3 enables the progression through the resting-to-preactivated transitions to the higher energy preactivated state, which appears to have a greater affinity for Ca2+. The increased affinity of the preactivated state for Ca2+ would promote binding of the ~100 nM basal cytosolic Ca2+ at the JD site, unlocking the JD ring and favoring the transition through the ensemble of high-energy intermediate states along the trajectory to the fully-open activated state. Upon opening, IP3Rs release Ca2+ in the cytosol where it can bind to the low-affinity CD site and stabilize the inhibited state to terminate Ca2+ release. With IP3Rs closed, SERCA would be able to pump Ca2+ back into the ER and restore basal Ca2+ concentrations. As Ca2+ is sequestered back into the ER, Ca2+ can dissociate from the low-affinity CD site. When the BTF ring reforms, subsequent Ca2+ release events can then be initiated if IP3 remains abundant, resulting in regenerative Ca2+ oscillations.

Schematic representations depicting the mechanisms of Ca2+- and IP3-dependent activation and Ca2+-dependent inhibition of IP3Rs. Row 1: Side views of the major states with front and rear protomers removed for clarity. Row 2: Cytosolic views of BTF-ring (left) and JD-ring (right). Magenta arrows highlight movements compared to previous state. Row 3: Occupancy of Ca2+-binding sites. Row 4: Correspondence between conformational state and cytosolic Ca2+ during Ca2+ oscillations.
Thus, the conformational landscape of hIP3R3 is comprised of multiple structurally distinct closed states and seemingly only one open state. Notably, ligand binding is not sufficient to determine conformational state, as distinct states exhibit identical ligand-binding profiles. For example, the preactivated+Ca2+ and activated states both bind IP3, ATP, and Ca2+ at the JD site, yet the pore is closed in the preactivated+Ca2+ state and open in the activated state (Fig. 1). Similarly, the resting and preactivated states, as well as the intermediate resting-to-preactivated transitions, all bind IP3 and ATP, but not Ca2+ (Fig. 1). Thus, the free energy gains associated with ligand binding are insufficient to drive the ligand-induced conformational changes, such as priming and gating, to completion. Rather, ligand binding biases the conformational equilibrium to increase the favorability of the high-energy states along the trajectory to activation.
While the trajectory to activation is populated with numerous high-energy states, the resting and inhibited states serve as the lowest energy states in the low and high Ca2+ conditions, respectively. As both of these states are closed, the resting and inhibited states contribute significantly towards establishing the biphasic Ca2+ dependence of IP3Rs. This energetic landscape presents a highly-tunable system where post-translational modifications, protein-protein interactions, cellular metabolites, and other forms of regulation may shift the balance of states to modulate activation or alter the frequency and amplitude of Ca2+ waves without disturbing the principal biphasic Ca2+ dependence of the channel19,21,55,71,72,73. Consistent with this model, we identified several mutations that change the conformational landscape resulting in altered IP3R-dependent Ca2+ oscillation dynamics without abolishing activation or inhibition of the channel (Figs. 4e–g, 5j–l, 7g–i and Supplementary Fig. 12). We also identified a second set of mutations that abolished Ca2+ oscillations, likely by removing one or more critical states from the conformational network (Figs. 4h–j, 8d–f and Supplementary Fig. 12). Thus, our structural landscape provides a framework for understanding how diverse stimuli modulate Ca2+ dynamics in cells24,55,74.
Electrophysiological analyses have demonstrated that inhibition of IP3R3 is highly cooperative, while activation is not19,75. These observations are consistent with the existence of multiple asymmetric states along the trajectory to activation and the complete absence of states along the trajectory to inhibition. The absence of any channels with partially disrupted BTF rings likely arises from the strain that accompanies Ca2+ binding. Once even a single interface in the BTF ring is disrupted, the strain throughout the channel may cause the other interfaces to be pulled apart, resulting in the inhibited state. While the CD adopts several asymmetric states in the resting-to-preactivated transitions, all of the observed states display at least two-fold pseudosymmetry in the TMD. The lack of asymmetric TMD conformations may arise from the domain-swapped arrangement of the S1-S4 domain with respect to the pore, which assures cross-protomer communication. Similar ~C2 states have been observed for TRP channels, which share the domain-swapped 6TM fold76,77,78.
Structural titrations have been previously performed using two ligand-gated ion channels, Slo2.2 and GIRK47,79. Intermediate states were noticeably absent from these analyses, which led to the conclusion that the gating processes were highly cooperative47,79. As electrophysiological analyses of Slo2.2 and GIRK also demonstrate this cooperativity, the correspondence between the structural and functional titrations of Slo2.2, GIRK, and IP3R ion channels indicates that structural titrations can provide mechanistic insights into the processes that underlie protein function.
It has long been appreciated that IP3Rs in cells can function in clusters of multiple channels that display a high degree of synchronicity80. These clusters have been shown to arise in an IP3- and Ca2+-dependent fashion81,82. While our structural interpretation is largely consistent with the functional and cellular evidence, it is unclear if the higher-order assemblies of inhibited channels that we observe correspond to the clusters that have been observed in cells. As the Ca2+-dependency of the inhibited particles in higher-order assemblies differs from those of isolated inhibited state particles, it will be interesting to investigate if the formation of higher-order assemblies contributes to Ca2+-dependent inhibition in cells.
Altogether, our analyses show how structural titrations, the process of determining structures in the presence of varying concentrations of regulatory ligands and co-factors, can reveal how stimuli bias the conformational landscape to modulate protein function.
Methods
hIP3R3 expression
All constructs were N-terminally tagged with 10xHis followed by EGFP (Ca2+ imaging) or mVenus (cryo-EM)83 followed by a human rhinovirus 3 C protease84 cut-site and then human type 3 IP3R. Plasmids were transformed into DH10Bac cells to generate bacmids29. 100–200 µg of purified bacmid were incubated with 400 µg of 25,000 MW polyethyleneimine (PEI; Polysciences Cat# 23966) in 1.4 mL water at 55 °C for 30 minutes to sterilize, then added to 50 mL of Sf9 cells at 1×106 cells/mL grown in suspension at 27–30 °C. The Sf9 TNM-FH (Grace’s modified) media was supplemented with 1% penicillin/streptomycin, 0.1% Pluronic F-68 nonionic surfactant (Gibco Cat# 24040), and 4-8% fetal bovine serum to stabilize the virus. Virus titer was amplified to P3 and separated from cell debris by centrifugation. P3 virus was used to infect mammalian HEK293S GnTI- (ATCC CRL-3022) cells at a density of 3×106 cells/mL at a ratio of 50 mL virus for 800 mL cells and simultaneously stimulated with 4.5 mM valproic acid (VPA; Sigma Cat# P4543). Pellets were harvested from cells by centrifugation at 48–72 hours after infection and snap frozen.
hIP3R3 purification
All surfaces, vessels, and transfer plastics were washed extensively with reverse osmosis water prior to use to minimize contaminating Ca2+. Membrane proteins were solubilized from 2.4 L of pelleted HEK293S GnTI- cells expressing wild-type hIP3R3 for 2 hours by rotation in 2% lauryl maltose neopentyl glycol (LMNG; Anatrace Cat# NG310), 150 mM sodium chloride (NaCl), 20 mM HEPES pH 7.5, 1 mM phenylmethylsulfonyl fluoride (PMSF), 2.5 µg/mL aprotinin (Sigma Cat# A1153), 2.5 µg/mL leupeptin (Alfa Aesar Cat# J61188), 10 µg/mL pepstatin A (GoldBio Cat# P-020-25), 0.5 mM 4-benzenesulfonyl fluoride hydrochloride (AEBSF; EMD Millipore Cat# 101500), and a few flakes of lyophilized deoxyribonuclease (DNAse; Worthington Biochemical Cat# LS002139). The resulting cell lysate was centrifuged at 75,000x g for 40 minutes. The supernatant was incubated with sepharose-coupled GFP nanobody affinity purification beads for 4 hours with gentle agitation85. The protein-GFP-nanobody-bead mixture was isolated in a column and washed with 50 mL of gel filtration buffer containing 150 mM NaCl, 50 mM Tris-HCl pH 8.0, 0.02% LMNG, and 2 mM dithiothreitol (DTT). The protein was eluted from the affinity column by cleavage with genetically modified human rhinovirus 3 C protease overnight. Size exclusion chromatography was performed with a Superose 6 Increase column and the resulting protein peak was pooled and concentrated to 20 mg/mL in a 1 mL, 100 kDa MWCO concentrator (Cytiva VivaSpin Cat# 28932258).
Structural titration sample preparation
Cryo-EM sample blotting paper contributes a significant quantity of contaminating Ca2+ to protein preparations. We opted to produce our own low-Ca2+ blotting paper by treating standard blotting paper (Ted Pella Standard VitroBot Blotting Paper Cat# 47000-100) with an extensive washing protocol. Over several days and multiple buffer exchanges, we treated with approximately 6 L of 100 µM EGTA in reverse osmosis (RO) water, then 6 L of RO water with Ca2+ chelating beads (BIO-RAD Chelex 100 Resin Cat#142-1253), and finally 6 L of RO water alone. The treated paper was then stacked between extensively washed glass plates and subjected to vacuum for 24 hours to remove moisture and resume a flat shape. The treated filter paper is predicted to contain substantially less than 1 mM contaminating Ca2+ (predicted starting condition of blotting paper29) and 100 µM residual EGTA (first wash condition).
To further control our sample Ca2+ concentrations, we engineered a 5X ligand and Ca2+ chelator cocktail. By combining 2 mM each of EDTA (Kd 30 nM), EGTA (Kd 127 nM), BAPTA (Kd 153 nM), HEDTA (Kd 4.8 µM) with 1 mM of ATP (Kd 183 µM), we calculate that our buffer ensures a semi-log-linear relationship between free and total Ca2+ from 1 nM to 300 µM86. The least well-controlled range for free Ca2+ was between 1 nM and 10 nM requiring addition of 864 µM total Ca2+, and the largest was between 10 µM and 100 µM, requiring addition of 2.0 mM total Ca2+. Thus, our total contaminating Ca2+ must be greater than 864 µM to generate a maximum 1-log-fold error in our target free Ca2+ across the entire titratable range, ensuring that we maintain the semi-log-linear relationship between free and total Ca2+ despite contaminating Ca2+. To minimize the impact of widely varying kinetic properties of the chelators, we premixed the 5X solution containing 10 mM of each chelator, 5 mM ATP, 1 mM IP3, and 2.5 mM fluorinated fos-choline-8 (Anatrace Cat# F300F), a detergent that does not interact with hydrocarbons, to protect the protein from the air-water interface. Sensitivity analysis using MaxChelator (https://somapp.ucdmc.ucdavis.edu/pharmacology/bers/maxchelator/webmaxc/webmaxcE.htm) revealed that inaccurate pH was the largest contributor to deviations from the predicted free Ca2+, and thus we carefully adjusted all solutions to pH 8, and added an additional 50 mM Tris pH 8.0 to the ligand and Ca2+ chelator cocktail. CaCl2 and MgCl2 were added in varying quantities to generate the desired free Ca2+ concentration and a constant 3 mM free Mg2+ concentration. During grid preparation, 3.2 µL of purified protein was added to the grid and incubated for 30 seconds, after which we added 0.8 µL of the ligand and Ca2+ chelator cocktail directly to the droplet on the grid, immediately blotted with our low-Ca2+ blotting paper for 2 seconds, then plunge-frozen using a ThermoFisher Vitrobot Mark IV. Since the Ca2+ and chelators are premixed, the free Ca2+ is at equilibrium in the master mix, and pipetting error when adding to the protein on the grid will have no effect on free Ca2+. The only deviations due to pipetting error would be [IP3] and [ATP], both of which are above saturating concentrations and so we assume those to be inconsequential for this analysis. Due to the non-equilibrium nature of grid preparation (e.g. evaporation, temperature changes) we acknowledge that while the semi-log-linear relationship established across the titration range is very precise, there is likely inaccuracy in our nominal free Ca2+ concentrations. The final grid conditions have varying free Ca2+, but constant 200 µM IP3, 1 mM ATP, 3 mM free Mg2+, 1.6 mM dithiothreitol (DTT), 2 mM EDTA, 2 mM EGTA, 2 mM BAPTA, 2 mM HEDTA, 50 mM Tris pH 8.0, 120 mM NaCl, 500 µM fluorinated fos-choline-8, and 159 µM LMNG.
Fura-2 calibration for Ca2+ chelator plus ligand cocktail
Ca2+ calibration curves were generated using Fura-2 (Thermo Scientific Chemicals; J63686.MCR) and Invitrogen Calcium Calibration Buffer Kit (#C3008MP). Briefly, we prepared 50 µL samples for multiple specific free Ca2+ concentrations by mixing buffer A (50 µM Fura-2, 10 mM EGTA, 100 mM KCl, 30 mM MOPS, pH 7.2, 3 mM MgCl2) and buffer B (50 uM Fura-2, 10 mM Ca-EGTA, 100 mM KCl, 30 mM MOPS, pH 7.2, 3 mM MgCl2) in the manner prescribed by the manufacturer. Fura-2 fluorescence emission at 510 nm was collected for an excitation scan from 280 to 450 nm in a clear bottom 96-well plate (Greiner bio-one; #655076) using Molecular Devices SpectraMax M5e microplate reader at room temperature. A calibration curve for Ca2+ was generated by calculating the fluorescence emission ratios at 340 and 380 nm (Supplementary Fig. 1). Following this, 10 µL of the 5X ligand and Ca2+ chelator cocktail (described earlier) with a nominal free Ca2+ concentration of 100-104 nM was added to 40 µL of 62.5 µm Fura-2 (ThermoFisher; #F-1200) diluted in gel filtration buffer (150 mM NaCl, 50 mM Tris-HCl pH 8.0, 0.02% LMNG, and 2 mM dithiothreitol (DTT)) to achieve a final concentration of 50 µM Fura-2 and 1X Ca2+ chelator cocktail. Samples were excited at 340 nm and 380 nm and fluorescence emissions were collected at 510 nm using a Molecular Devices SpectraMax M5e microplate reader at room temperature. GraphPad Prism 9 was used to estimate free Ca2+ concentrations by interpolating the fluorescent emission ratios at 340 nm and 380 nm excitations (Supplementary Fig. 1). Data presented are from three technical replicates.
Cryo-EM data collection, analysis and model building
Images were collected at 0.826 Å/px magnification on an FEI Krios with Gatan K3 detector at 15 e-/pix/sec with 3 sec exposure (0.05 sec/frame) for a total dose of 66 e-/Å2 in automated fashion using SerialEM87,88. Five datasets were collected during the same session for each Ca2+ concentration on a series of grids that were prepared sequentially resulting in 637 movies at 1 nM, 2150 movies at 10 nM, 6126 movies at 100 nM, 1372 movies at 1 µM, and 3136 movies at 10 µM. A sixth dataset of 4312 movies collected at nominal 100 nM free Ca2+ from a grid prepared later in the sequence was collected as a technical replicate to assess experimental error (Supplementary Fig. 18).
All movies were combined and processed starting in CryoSparc Live v3.3.1 for motion correction, CTF estimation, and bias-free autopicking at a rate of 380 picks/micrograph with a gaussian blob of dimensions between 166 and 240 Å, corresponding to the smallest and largest diameter of the known conformational states of IP3Rs. Thus, all of the following classification decisions were made in aggregate and without any a priori knowledge of the dataset from which particle subsets were derived. The over-picked particle stack was extracted in a 512 box and subjected to iterative CryoSparc v3.3.1 Heterogeneous Refinement89 without imposing symmetry. Four references corresponding to the resting, activated, inhibited, and a single consensus average of the preactivated +/- Ca2+ states were used as inputs. These references were previously determined from the combined data using traditional single-particle approaches. The remaining eight input classes were pure noise decoy references generated by randomly sampling a very small number of particles via CryoSparc v3.3.1 Ab-Initio without alignment. The decoy references attract false positives, while the four high-resolution references attract true positives. These references were used for all classifications described herein.
After several rounds of “decoy” classification without imposing symmetry, the particle stack went from 7.8 M particles to 1.7 M particles, with 351k, 117k, 145k, and 1045k residing in the classes obtained from the resting, preactivated, activated, and inhibited references respectively. 2D classification of the discarded classes confirmed that no unintentional removal of true positives occurred. At this stage, each stack was independently subjected to an additional iteration of classification to allow fine separation of states whereby the non-self-references attract particles away from the self-identifying class in cases where the particles deviate from the consensus state in subtle ways. This resulted in six classes that are depicted in the second tier of the cryo-EM workflow figure (Supplementary Fig. 2), with classes that refined to worse than 7 Å being discarded as junk or damaged particles.
Each of these six stacks were refined enforcing C4 symmetry to improve signal for reference-based corrections prior to Bayesian Polishing in Relion v3.1.390. At this stage, optical groups were separated and both local and global CTF parameters were optimized in CryoSparc v3.1.1 during Non-Uniform Refinement91 procedures. Due to the very large number of optical groups, it was found that the fourth-order terms of spherical aberration and tetrafoil92,93 were not being fit accurately in some groups, and hence we did not fit these terms. In aggregate the per particle, per micrograph, and per optical group corrections resulted in improvements for the resting-like stack with strong TMD density (231k particles; 3.5 Å to 2.7 Å), resting-like stack with weak TMD density (108k particles; 3.9 Å to 3.3 Å), preactivated-like stack with weak CD density (83k particles; 4.0 Å to 3.6 Å), activated-like stack (65k particles; 3.7 Å to 3.1 Å), preactivated-like stack with weak TMD density (76k particles; 3.8 Å to 3.2 Å), and inhibited-like stack (1045k particles; 3.2 Å to 2.5 Å). These stacks were subjected to one final round of classification revealing the five primary C4 symmetric states called resting (192k particles; 2.8 Å), preactivated (47k particles; 3.7 Å), preactivated+Ca2+ (31k particles; 3.6 Å), activated (56k particles; 3.1 Å), and inhibited (917k particles; 2.5 Å) states and several heterogeneous conformational ensembles. Notably, local conformational changes can still be observed in the five major states by 3DVA at the end of the hierarchical classification due to the large size and overall flexibility of the channel. Our final maps and the corresponding models represent the average of the particles that comprise these states.
We further improved the C4 symmetric states by performing C4 symmetry expansion and local refinement to correct for subtle local asymmetries in the particles. We used a model to precisely delineate masks surrounding modular units that flex and move in unison: (1) a mask containing a single chain from the tetramer (2) the entire cytosolic domain consisting of residues 1-1697 from a single chain (3) BTF1, BTF2, and ARM1 consisting of residues 1-664 from a single chain (4) CLD, ARM3 consisting of residues 665-1100 and 1586-2074 from a single chain (5) ARM2 consisting of residues 1101-1586 from a single chain (6) TMD, JD consisting of residues 2111-2611 from a single chain. The masks generated from these models were dilated by 4 pixels and a cosine soft-edge was applied for 40 pixels, thereby avoiding ringing and mask artifacts that occur when converting hard edges in real-space to reciprocal space. Therefore, this mask retains 100% of the information at ~3 Å away from the model, and 50% of the information at ~25 Å away from the model. CryoSparc v3.3.1 Local Refinement resulted in resolutions ranging from 2.5 Å (TMD/JD) to 3.3 Å (ARM2) for the resting state, 3.6 Å (TMD/JD) to 6.5 Å (ARM2) for the preactivated state, 3.3 Å (BTF1/BTF2/ARM1) to 4.2 Å (ARM2) for the preactivated+Ca2+ state, 2.9 Å (BTF1/BTF2/ARM1) to 3.3 Å (ARM2) for the activated state, and 2.5 Å (TMD/JD) to 3.4 Å (BTF1/BTF2/RM1) for the inhibited state (Supplementary Figs. 3, 5, 6, 7 and 9). In some highly-heterogeneous cases, the local refinements were subjected to a procedure that will be described in the treatment of the conformational ensembles to improve the resolution (e.g. BTF1/BTF2/ARM1 in the inhibited state).
The local refinements were independently subjected to Phenix v1.20.1-4487 Resolve Cryo-EM94 guided only by experimental density (no model) and employing a lenient mask that contains all proteinaceous and detergent micelle density, an approach we have used previously95,96,97. As part of the procedure, the final maps are sharpened using a half-map derived factor. The resulting density modified and sharpened maps were cropped to a single chain and used for iterative model building using coot98, ISOLDE99 and composite map generation using a 20-residue sliding window cross-correlation (Phenix v1.20.1-4487 Combine Focused Maps)100, which we found to produce artifact-free maps when compared to Chimera’s ‘vop maximum’ command101. For the highest resolution composites (resting, activated, and inhibited) the density-modified local refinements were super-sampled prior to composite generation to aid interpretation of ligands, ions, and waters. Inspection of the resulting composite maps showed that they were free of model-based overfitting, for example density for ions, ligands, and lipids remain intact despite being removed from the input model. The final models were refined against the composite map with Phenix v1.20.1-4487 Real-Space Refinement102.
The remaining classes represent highly-heterogeneous conformational ensembles that we interrogated via 3D variability analysis (3DVA)36. We relaxed our assumptions about symmetry by performing C4 symmetry expansion on each class. For the resting-like ensemble with weak CD density, resting-like ensemble with weak TMD density, preactivated-like ensemble with weak CD density, and the preactivated-like ensemble with weak TMD density, we performed 3DVA with a full channel mask and filter resolution between 5 and 8 Å and clustered each of 3 modes independently into 5 groups. Occasionally, one or two clusters would be populated with very few particles, suggesting that a fewer number of clusters was adequate to represent the underlying heterogeneity. We then refined each class (CryoSparc v3.3.1 Local Refinement due to symmetry expansion) and assessed the resulting structures, selecting the mode of variability that contained our features of interest. From the resting-like and preactivated-like stacks with weak TMD density, we obtained the ~C2 and ~C4 TMD transition states presented in Fig. 7d–e and Supplementary Fig. 8. From the resting-like and preactivated-like stacks with weak CD density, we obtained the asymmetric ARM2 sampling states presented in Fig. 5a. For the ARM2 retraction analysis in Fig. 5b–g and wedge loop analysis in Supplementary Fig. 15, we increased the requested number of clusters to 10 and 20 respectively and selected 6 refinements that appeared to be on a shared trajectory for both ARM2 retraction and loop melting for presentation.
To calculate an octameric reconstruction of two adjacent tetramers in a higher-order assembly, the nonsymmetry expanded stack of 1045k inhibited state particles was subjected to CryoSparc v3.3.1 Heterogeneous Refinement seeded with 24 identical references of the inhibited state. Strong density for an adjacent tetramer was present in three of the resulting classes, totaling 246k particles. This classification was used to quantify the particle distributions for clustered versus isolated inhibited states in Fig. 2. The clustered particles were subjected to C4 symmetry expansion and local refinement using a mask encompassing the entire CD of one protomer, which suggested that the interaction was formed between ARM2 of the central protomer and CLD of the adjacent protomer. To separate the protomers that participate in the inter-tetramer interface from those facing away, we next performed 3DVA with two different masks on the symmetry expanded stack of 984k particles. We first generated a mask comprising the CLD of the central tetramer interacting with ARM2 of the adjacent tetramer to identify protomers that form what we call the + half of the interaction, which yielded 85k particles at 3.3 Å. Then, we generated a mask comprising ARM2 of the central tetramer interacting with the CLD of the adjacent tetramer to classify protomers that form what we call the - half of the interaction, which yielded 88k particles at 3.3 Å. These two populations represent two halves of the ~C2 symmetric interface between two assembled tetramers.
In addition to the masks covering the interfaces, we used the same set of local refinements that we used for the five C4 symmetric states to produce a well-resolved composite map of each protomer in the two associated tetramers. For each population, we first produced a consensus tetrameric alignment, and then proceeded to subject each protomer of the tetramer to the six local refinement masks that were used to produce the C4 symmetric inhibited state (entire protomer, TMD/JD, ARM3/CLD, ARM2, BTF1/BTF2/ARM1, and entire CD). Each of these local refinements was then subjected to Phenix Resolve Cryo-EM, and finally to Phenix Combine Focused Maps to produce a tetrameric composite map of each half of the interaction. For the depiction in Fig. 9a–b, we fit these two tetrameric composite maps together and used Chimera ‘vop maximum’ to create a single volume of the entire assembly.
Once all the particles were assigned to a specific state or ensemble in CryoSparc, we determined the number of particles that originated from each of the Ca2+ concentrations via unique identifiers in the micrograph names in order to calculate the relative abundance of each state at each condition. Because we collected images from two grids prepared at 100 nM Ca2+, we combined the particles and report the weighted average in Fig. 2. We compare the two data sets in Supplementary Fig. 17.
For the depictions of the composite maps in Fig. 1, four copies of the single-chain composite were fit to the consensus C4 refinement and combined using the Chimera ‘vop maximum’ command101. For the depictions of ARM2 density in Fig. 5, unsharpened local CD refinements were shown. For depictions of the overall density or slices at the JD ring, gate, and S1’-S1” in Fig. 6 and Supplementary Fig. 8, the unsharpened consensus refinement maps were low-pass filtered to 4 Å (overall) or 7 Å (zoomed) using ‘relion_image_handler’103,104. For the depictions of the wedge loop density in Supplementary Fig. 15, the resting state composite map was used. For the depictions of the wedge loop density in Supplementary Fig. 15 a B-factor derived from the Guinier plot was used to sharpen the CD local refinements for presentation. All figures depicting models were generated in PyMol (Schrodinger, LLC. 2010. The PyMOL Molecular Graphics System, Version 2.5.3), and all figures depicting density alone were generated in ChimeraX105,106. For the electrostatics calculations in Supplementary Figs. 16 and 18, the Adaptive Poisson-Boltzmann Solver (APBS) algorithm107 was utilized via PyMol plug-in.
High performance computing
The MSK High Performance Computing (HPC) resource provides a GPU cluster built for computing large volume data over a range of applications from drug discovery to deep learning and image processing. It contains 120 nodes connected by a 100 Gigabit ethernet backbone. The nodes used for this project each contain Intel Xeon Platinum 2.2 GHz CPUs and 1 TB DDR4 RAM. Each node also contains four A100 GPUs interconnected using NVLink. The cluster runs the CentOS operating system and is supported by a 4 PB high-speed GPFS-based parallel filesystem. A 200 TB NVMe-based Weka ultra-fast tier was used as scratch space. The CPU to GPU communication is established over PCIE 4.0. The project used IBM Spectrum LSF as the orchestrator of shared resources and parallelization is further achieved by MPI over the ethernet network. All cryo-EM software excluding CryoSparc was maintained via HMS SBGrid108. Multiple sequence alignments were performed using the MUSCLE109 algorithm in DNASTAR LaserGene MegAlign Pro 17.3.
Cloning
Site-directed mutagenesis and cloning for all hIP3R3 constructs detailed in this study was performed using either two concurrent single-primer reactions (PMID: 19566935) or NEBuilder HiFi DNA Assembly kit (NEB Cat# E2621) in Mach1 T1 cells (ThermoFisher; Cat#C862003). Oligonucleotides (Supplementary Table 12) were designed manually for single-primer reactions, whereas NEBuilder (https://nebuilder.neb.com/) was utilized for designing oligos for mutagenesis based on NEBuilder HiFi DNA Assembly. All oligos were acquired from Integrated DNA Technologies.
Adherent cell culture
HEK293T IP3R-null cells were obtained through Kerafast52 and cultured to a confluency of ~75-80% on 100 ×20 mm tissue culture treated dishes in DMEM supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 mg/ml streptomycin at 37 °C with 5% CO2. For imaging, cells were then split in a 1:4 ratio and plated on poly-D-lysine coated, 35 mm diameter, optical quality glass-bottom culture dishes (World Precision Instruments; # FD35PDL-100) and incubated for ~18-24 hours. At ~60% confluency, cells were transduced with a 200 µl baculovirus followed by incubation at 37 C, 5% CO2 for another 24 hours. All constructs used for Ca2+ imaging in this study were overexpressed in HEK293T IP3R-null cells using the BacMam system110.
Ca2+ imaging and data processing
24 hours after baculovirus transduction, cells were gently washed with imaging buffer [20 mM HEPES supplemented Ca2+, Mg2+ free, Hank’s balanced salt solution (ThermoFisher; #14175103)] followed by incubation for 1 hour at 37˚C and 5% CO2 in 1.8 ml of imaging buffer containing 3 mM Cal-520-AM (AAT Bioquest; #21130) Cal-520-AM-loaded cells were removed from the CO2 incubator and equilibrated at room temperature for 5 minutes prior to IP3 stimulation by the addition of 200 µl of 1 mM carbachol (Alfa Aesar; #L06674-06), a Gαq-coupled M3 muscarinic receptor agonist. Carbachol was added at least 10 mm away from the imaging site and allowed to diffuse to a final concentration of 100 µM. Movies of carbachol-induced Ca2+ release in cells were collected at 20x with LD Plan-Neofluar 20X/0.4 Korr M27 objective, for 10 minutes, at 3×3 binning (912×736 pixels post binning), with an exposure time of 250 ms on a Zeiss Axio observer D1 inverted phase-contrast fluorescence microscope equipped with an Axiocam 506 Mono camera (Zeiss). Cal-520-AM imaging was carried out by exciting the sample at 493 nm and monitoring emission at 515 nm using X-Cite Series 120Q illumination system and Zeiss filter set 38 HE.
Ca2+ imaging movies were processed using ImageJ111, Fiji112 and MathWorks MATLAB 9.12.0.1884302 (R2022a) to extract Cal-520-AM fluorescence traces from individual cells. Movie stacks were background-subtracted with a 200-pixel rolling ball radius in ImageJ. Maximum intensity projection of the stack was used to generate a difference of gaussian image, which was used for edge detection and cell segmentation using MATLAB’s Image Processing Toolbox. Traces were then extracted from segmented cells, smoothed over 41 frames using a Savitzky–Golay filter of polynomial order 2, normalized by Z-score, and baseline adjusted using the linear method of MATLAB’s 1-D data interpolation function with a custom MATLAB script called Baseline Fit113. In the baseline-adjusted traces, the smallest observed Ca2+ oscillation peak value was used to manually threshold and identify other peaks automatically. Detected peaks were then used to calculate inter-spike intervals using MATLAB’s Signal Processing Toolbox. All statistical tests were performed using GraphPad Prism 9. Data reported are from 3 independent biological replicates.
For analysis of peaks from individual replicates, traces with transients/oscillations were baseline adjusted in MATLAB using Baseline Fit113 and normalized between 0 and 1. The first peak of each oscillation/transient was identified and a window of 75 seconds on both sides of the peak was extracted and aligned at the peak position. Mean and 95% confidence intervals were calculated using GraphPad Prism and overlayed on traces from a single biological replicate. A 1 second window on both sides of the mean data point corresponding to half maximal intensity were fit to a straight line and used to calculate the mean rising phase for constructs exhibiting transients/oscillations. Traces with oscillations were sorted based on the maximum number of distinguishable peaks and plotted as a fraction of total oscillatory traces.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The Cryo-EM data and atomic coordinates generated in this study have been deposited in the Electron Microscopy Data Bank and Protein Data Bank under accession codes EMD-41323 and 8TK8 for the Resting state structure, EMD-41347 and 8TKD for the Preactivated state structure, EMD-41348 and 8TKE for the Preactivated+Ca2+ state structure, EMD-41349 and 8TKF for the Activated state structure, EMD-41350 and 8TKG for the Inhibited state structure, EMD-41351 and 8TKH for the Labile resting state 1 structure, EMD-41352 and 8TKI for the Labile resting state 2 structure, EMD-41366 and 8TLA for the Higher-order inhibited state 1 structure and EMD-41365 and 8TL9 for the Higher-order inhibited state 2 structure. Cryo-EM data generated in this study have been deposited in the Electron Microscopy Data Bank under accession codes EMD-41324, EMD-41325 and EMD-41326 for the Resting-to-Preactivated transition states, EMD-41327, EMD-41328, EMD-41329, EMD-41330, EMD-41331 and EMD-41332 for the ARM2 retractions states, EMD-41333, EMD-41334, EMD-41335, EMD-41336, EMD-41337 and EMD-41338 for the Wedge loop progression states, EMD-41339 for the ~C2 Preactivated TMD Transition, EMD-41340 for the ~C4 Preactivated TMD Transition, EMD-41341, EMD-41342 and EMD-41343 for the Activated CTD states, EMD-41344 for the ~C2 Resting TMD Transition, and EMD-41345 for the ~C4 Resting TMD Transition. The atomic coordinates of previously published structures used in this study at available at the Protein Data Bank under accession codes 3JAV, 6MU2, 7LHF and 7LHE for rat Type 1 IP3R, 6DQJ and 6UQK for human Type 3 IP3R in a resting state, 6DQS, 6DQZ, and 6DR0 for human type 3 IP3R resting-to-preactivated transition states, 6DQV, 7T3P, 7T3Q, and 7T3R for human type 3 IP3R in preactivated states, 7T3T for human type 3 IP3R in an activated state, 6DRC, 6DR2, 6DRA, and 7T3U for human type 3 IP3R in inhibited states, 1N4K, 3T8S, and 3UJ0 for fragments containing the IP3-binding domain of rat type 1 IP3R, and 5TAP for caffeine- and ATP-bound rabbit RyR1. Source data are provided with this paper. Plasmids are available upon request. Source data are provided with this paper.
Code availability
Custom MATLAB scripts used for fluorescence-based Ca2+ imaging data analysis are available at https://doi.org/10.5281/zenodo.8411195.
References
Lagos-Cabré, R., Ivanova, A. & Taylor, C. W. Ca2+ Release by IP3 Receptors Is Required to Orient the Mitotic Spindle. Cell Rep. 33, 108483 (2020).
Mikoshiba, K. Role of IP3 receptor in development. Cell Calcium 49, 331–340 (2011).
Ahumada-Castro, U. et al. In the Right Place at the Right Time: Regulation of Cell Metabolism by IP3R-Mediated Inter-Organelle Ca2+ Fluxes. Front. Cell Dev. Biol. 9, 629522 (2021).
Wei, C., Wang, X., Zheng, M. & Cheng, H. Calcium gradients underlying cell migration. Curr. Opin. Cell Biol. 24, 254–261 (2012).
Tsai, F.-C., Kuo, G.-H., Chang, S.-W. & Tsai, P.-J. Ca2+ Signaling in Cytoskeletal Reorganization, Cell Migration, and Cancer Metastasis. BioMed Res. Int. 2015, e409245 (2015).
Joseph, S. K. & Hajnóczky, G. IP3 receptors in cell survival and apoptosis: Ca2+ release and beyond. Apoptosis 12, 951–968 (2007).
Kania, E., Roest, G., Vervliet, T., Parys, J. B. & Bultynck, G. IP3 Receptor-Mediated Calcium Signaling and Its Role in Autophagy in Cancer. Front. Oncol. 7, 140 (2017).
Moy, R. H. et al. Functional genetic screen identifies ITPR3/calcium/RELB axis as a driver of colorectal cancer metastatic liver colonization. Dev. Cell 57, 1146–1159.e7 (2022).
Shibao, K. et al. The type III inositol 1,4,5-trisphosphate receptor is associated with aggressiveness of colorectal carcinoma. Cell Calcium 48, 315–323 (2010).
Egorova, P. A. & Bezprozvanny, I. B. Inositol 1,4,5-trisphosphate receptors and neurodegenerative disorders. FEBS J. 285, 3547–3565 (2018).
Foskett, J. K. Inositol trisphosphate receptor Ca2+ release channels in neurological diseases. Pflüg. Arch. - Eur. J. Physiol. 460, 481–494 (2010).
Garcia, M. I. & Boehning, D. Cardiac inositol 1,4,5-trisphosphate receptors. Biochim. Biophys. Acta BBA - Mol. Cell Res. 1864, 907–914 (2017).
Akimzhanov, A. M. & Boehning, D. IP3R function in cells of the immune system. Wiley Interdiscip. Rev. Membr. Transp. Signal. 1, 329–339 (2012).
Iino, M. Biphasic Ca2+ dependence of inositol 1,4,5-trisphosphate-induced Ca release in smooth muscle cells of the guinea pig taenia caeci. J. Gen. Physiol. 95, 1103–1122 (1990).
Bezprozvanny, L., Watras, J. & Ehrlich, B. E. Bell-shaped calcium-response curves of lns(l,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature 351, 751–754 (1991).
Boehning, D., Mak, D.-O. D., Foskett, J. K. & Joseph, S. K. Molecular Determinants of Ion Permeation and Selectivity in Inositol 1,4,5-Trisphosphate Receptor Ca2+ Channels *. J. Biol. Chem. 276, 13509–13512 (2001).
Mak, D.-O. D., McBride, S. & Foskett, J. K. Inositol 1,4,5-tris-phosphate activation of inositol tris-phosphate receptor Ca2+ channel by ligand tuning of Ca2+ inhibition. Proc. Natl. Acad. Sci. 95, 15821–15825 (1998).
Gin, E., Falcke, M., Wagner, L. E., Yule, D. I. & Sneyd, J. A Kinetic Model of the Inositol Trisphosphate Receptor Based on Single-Channel Data. Biophys. J. 96, 4053–4062 (2009).
Foskett, J. K., White, C., Cheung, K.-H. & Mak, D.-O. D. Inositol Trisphosphate Receptor Ca2+ Release Channels. Physiol. Rev. 87, 593–658 (2007).
Nakamura, Y., and Fukami, K. Regulation and physiological functions of mammalian phospholipase C. J. Biochem. (Tokyo), mvw094. https://doi.org/10.1093/jb/mvw094 (2017).
Betzenhauser, M. J., & Yule, D. I. Chapter 12 - Regulation of Inositol 1,4,5-Trisphosphate Receptors by Phosphorylation and Adenine Nucleotides. In Current Topics in Membranes Structure and Function of Calcium Release Channels. (ed. I. Serysheva.) pp. 273–298 (Academic Press, 2010).
Vais, H. et al. ER-luminal [Ca2+] regulation of InsP3 receptor gating mediated by an ER-luminal peripheral Ca2+-binding protein. eLife 9, e53531 (2020).
Patterson, R. L., Boehning, D. & Snyder, S. H. Inositol 1,4,5-Trisphosphate Receptors as Signal Integrators. Annu. Rev. Biochem. 73, 437–465 (2004).
Hajnóczky, G., Robb-Gaspers, L. D., Seitz, M. B. & Thomas, A. P. Decoding of cytosolic calcium oscillations in the mitochondria. Cell 82, 415–424 (1995).
Li, W., Llopis, J., Whitney, M., Zlokarnik, G. & Tsien, R. Y. Cell-permeant caged InsP3 ester shows that Ca2+ spike frequency can optimize gene expression. Nature 392, 936–941 (1998).
Dolmetsch, R. E., Xu, K. & Lewis, R. S. Calcium oscillations increase the efficiency and specificity of gene expression. Nature 392, 933–936 (1998).
Gu, X. & Spitzer, N. C. Distinct aspects of neuronal differentiation encoded by frequency of spontaneous Ca2+ transients. Nature 375, 784–787 (1995).
Fan, G. et al. Gating machinery of InsP3R channels revealed by electron cryomicroscopy. Nature 527, 336–341 (2015).
Paknejad, N. & Hite, R. K. Structural basis for the regulation of inositol trisphosphate receptors by Ca2+ and IP3. Nat. Struct. Mol. Biol. 25, 660–668 (2018).
Fan, G. et al. Cryo-EM reveals ligand induced allostery underlying InsP3R channel gating. Cell Res. 28, 1158–1170 (2018).
Azumaya, C. M., Linton, E. A., Risener, C. J., Nakagawa, T. & Karakas, E. Cryo-EM structure of human type-3 inositol triphosphate receptor reveals the presence of a self-binding peptide that acts as an antagonist. J. Biol. Chem. 295, 1743–1753 (2020).
Baker, M. R. et al. Cryo-EM structure of type 1 IP3R channel in a lipid bilayer. Commun. Biol. 4, 1–11 (2021).
Schmitz, E. A., Takahashi, H. & Karakas, E. Structural basis for activation and gating of IP3 receptors. Nat. Commun. 13, 1408 (2022).
Fan, G. et al. Conformational motions and ligand-binding underlying gating and regulation in IP3R channel. Nat. Commun. 13, 6942 (2022).
Woll, K. A. & Van Petegem, F. Calcium-release channels: structure and function of IP3 receptors and ryanodine receptors. Physiol. Rev. 102, 209–268 (2022).
Punjani, A. & Fleet, D. J. 3D variability analysis: Resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM. J. Struct. Biol. 213, 107702 (2021).
Deng, Z. et al. Cryo-EM and X-ray structures of TRPV4 reveal insight into ion permeation and gating mechanisms. Nat. Struct. Mol. Biol. 25, 252–260 (2018).
Kaftan, E. J., Ehrlich, B. E. & Watras, J. Inositol 1,4,5-Trisphosphate (InsP3) and Calcium Interact to Increase the Dynamic Range of InsP3 Receptor-dependent Calcium Signaling. J. Gen. Physiol. 110, 529–538 (1997).
Yoshikawa, F. et al. Mutational Analysis of the Ligand Binding Site of the Inositol 1,4,5-Trisphosphate Receptor. J. Biol. Chem. 271, 18277–18284 (1996).
Bosanac, I. et al. Structure of the inositol 1,4,5-trisphosphate receptor binding core in complex with its ligand. Nature 420, 696–700 (2002).
Lin, C.-C., Baek, K. & Lu, Z. Apo and InsP3-bound crystal structures of the ligand-binding domain of an InsP3 receptor. Nat. Struct. Mol. Biol. 18, 1172–1174 (2011).
Seo, M.-D. et al. Structural and functional conservation of key domains in InsP3 and ryanodine receptors. Nature 483, 108–112 (2012).
des Georges, A. et al. Structural Basis for Gating and Activation of RyR1. Cell 167, 145–157.e17 (2016).
Ferris, C. D., Huganir, R. L. & Snyder, S. H. Calcium flux mediated by purified inositol 1,4,5-trisphosphate receptor in reconstituted lipid vesicles is allosterically regulated by adenine nucleotides. Proc. Natl. Acad. Sci. 87, 2147–2151 (1990).
Iino, M. Effects of adenine nucleotides on inositol 1,4,5-trisphosphate-induced calcium release in vascular smooth muscle cells. J. Gen. Physiol. 98, 681–698 (1991).
Maeda, N. et al. Structural and functional characterization of inositol 1,4,5-trisphosphate receptor channel from mouse cerebellum. J. Biol. Chem. 266, 1109–1116 (1991).
Hite, R. K. & MacKinnon, R. Structural Titration of Slo2.2, a Na+-Dependent K+ Channel. Cell 168, 390–399.e11 (2017).
Berridge, M. J. Elementary and global aspects of calcium signalling. J. Exp. Biol. 200, 315–319 (1997).
De Young, G. W. & Keizer, J. A single-pool inositol 1,4,5-trisphosphate-receptor-based model for agonist-stimulated oscillations in Ca2+ concentration. Proc. Natl. Acad. Sci. 89, 9895–9899 (1992).
Marchant, J. S. & Parker, I. Role of elementary Ca2+ puffs in generating repetitive Ca2+ oscillations. EMBO J. 20, 65–76 (2001).
Hajnóczky, G. & Thomas, A. P. Minimal requirements for calcium oscillations driven by the IP3 receptor. EMBO J. 16, 3533–3543 (1997).
Alzayady, K. J. et al. Defining the stoichiometry of inositol 1,4,5-trisphosphate binding required to initiate Ca2+ release. Sci. Signal. 9, ra35–ra35 (2016).
Rooney, T. A., Sass, E. J. & Thomas, A. P. Characterization of Cytosolic Calcium Oscillations Induced by Phenylephrine and Vasopressin in Single Fura-2-loaded Hepatocytes. J. Biol. Chem. 264, 17131–17141 (1989).
Meyer, T. & Stryer, L. Calcium Spiking. Annu. Rev. Biophys. Biophys. Chem. 20, 153–174 (1991).
Thurley, K. et al. Reliable Encoding of Stimulus Intensities Within Random Sequences of Intracellular Ca2+ Spikes. Sci. Signal. 7, ra59 (2014).
Arige, V., et al. Functional Determination of Calcium Binding Sites Required for the Activation of Inositol 1,4,5-trisphosphate receptor (2022).
Pietri, F., Hilly, M. & Mauger, J. P. Calcium mediates the interconversion between two states of the liver inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 265, 17478–17485 (1990).
Marshall, I. C. & Taylor, C. W. Two calcium-binding sites mediate the interconversion of liver inositol 1,4,5-trisphosphate receptors between three conformational states. Biochem. J. 301(Pt 2), 591–598 (1994).
Marchant, J. S. & Taylor, C. W. Cooperative activation of IP3 receptors by sequential binding of IP3 and Ca2+ safeguards against spontaneous activity. Curr. Biol. 7, 510–518 (1997).
Soulsby, M. D. & Wojcikiewicz, R. J. H. The type III inositol 1,4,5-trisphosphate receptor is phosphorylated by cAMP-dependent protein kinase at three sites. Biochem. J. 392, 493–497 (2005).
Soulsby, M. D. & Wojcikiewicz, R. J. H. Calcium mobilization via type III inositol 1,4,5-trisphosphate receptors is not altered by PKA-mediated phosphorylation of serines 916, 934, and 1832. Cell Calcium 42, 261–270 (2007).
Wang, D. et al. MusiteDeep: a deep-learning based webserver for protein post-translational modification site prediction and visualization. Nucleic Acids Res. 48, W140–W146 (2020).
Betzenhauser, M. J., Fike, J. L., Ii, L. E. W. & Yule, D. I. Protein Kinase A Increases Type-2 Inositol 1,4,5-Trisphosphate Receptor Activity by Phosphorylation of Serine 937. J. Biol. Chem. 284, 25116–25125 (2009).
Marchant, J., Callamaras, N. & Parker, I. Initiation of IP3-mediated Ca2+ waves in Xenopus oocytes. EMBO J. 18, 5285–5299 (1999).
Uchida, K., Miyauchi, H., Furuichi, T., Michikawa, T. & Mikoshiba, K. Critical Regions for Activation Gating of the Inositol 1,4,5-Trisphosphate Receptor. J. Biol. Chem. 278, 16551–16560 (2003).
Yan, Z. et al. Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature 517, 50–55 (2015).
Hite, R. K., Tao, X. & MacKinnon, R. Structural basis for gating the high-conductance Ca2+-activated K+ channel. Nature 541, 52–57 (2017).
Yamazaki, H., Chan, J., Ikura, M., Michikawa, T. & Mikoshiba, K. Tyr-167/Trp-168 in Type 1/3 Inositol 1,4,5-Trisphosphate Receptor Mediates Functional Coupling between Ligand Binding and Channel Opening. J. Biol. Chem. 285, 36081–36091 (2010).
Schug, Z. T. & Joseph, S. K. The Role of the S4-S5 Linker and C-terminal Tail in Inositol 1,4,5-Trisphosphate Receptor Function. J. Biol. Chem. 281, 24431–24440 (2006).
Hamada, K., Miyatake, H., Terauchi, A. & Mikoshiba, K. IP3-mediated gating mechanism of the IP3 receptor revealed by mutagenesis and X-ray crystallography. Proc. Natl. Acad. Sci. 114, 4661–4666 (2017).
Zhang, S., Fritz, N., Ibarra, C. & Uhlén, P. Inositol 1,4,5-Trisphosphate Receptor Subtype-Specific Regulation of Calcium Oscillations. Neurochem. Res. 36, 1175–1185 (2011).
Taylor, C. W. Regulation of IP3 receptors by cyclic AMP. Cell Calcium 63, 48–52 (2017).
Sneyd, J. et al. On the dynamical structure of calcium oscillations. Proc. Natl. Acad. Sci. 114, 1456–1461 (2017).
Jacob, R., Merritt, J. E., Hallam, T. J. & Rink, T. J. Repetitive spikes in cytoplasmic calcium evoked by histamine in human endothelial cells. Nature 335, 40–45 (1988).
Mak, D.-O. D., McBride, S. & Foskett, J. K. Regulation by Ca2+ and Inositol 1,4,5-Trisphosphate (Insp3) of Single Recombinant Type 3 Insp3 Receptor Channels: Ca2+ Activation Uniquely Distinguishes Types 1 and 3 Insp3 Receptors. J. Gen. Physiol. 117, 435–446 (2001).
Zubcevic, L. et al. Conformational ensemble of the human TRPV3 ion channel. Nat. Commun. 9, 4773 (2018).
Zubcevic, L., Le, S., Yang, H. & Lee, S.-Y. Conformational plasticity in the selectivity filter of the TRPV2 ion channel. Nat. Struct. Mol. Biol. 25, 405–415 (2018).
Singh, A. K. et al. Structural basis of temperature sensation by the TRP channel TRPV3. Nat. Struct. Mol. Biol. 26, 994–998 (2019).
Niu, Y., Tao, X., Touhara, K. K. & MacKinnon, R. Cryo-EM analysis of PIP2 regulation in mammalian GIRK channels. eLife 9, e60552 (2020).
Rahman, T. Dynamic clustering of IP3 receptors by IP3. Biochem. Soc. Trans. 40, 325–330 (2012).
Tateishi, Y. et al. Cluster Formation of Inositol 1,4,5-Trisphosphate Receptor Requires Its Transition to Open State. J. Biol. Chem. 280, 6816–6822 (2005).
Taufiq-Ur-Rahman, Skupin, A., Falcke, M. & Taylor, C. W. Clustering of InsP3 receptors by InsP3 retunes their regulation by InsP3 and Ca2+. Nature 458, 655–659, https://doi.org/10.1038/nature07763 (2009).
Rana, M. S., Wang, X. & Banerjee, A. An Improved Strategy for Fluorescent Tagging of Membrane Proteins for Overexpression and Purification in Mammalian Cells. Biochemistry 57, 6741–6751 (2018).
Walker, P. A. et al. Efficient and Rapid Affinity Purification of Proteins Using Recombinant Fusion Proteases. Bio/Technology 12, 601–605 (1994).
Kirchhofer, A. et al. Modulation of protein properties in living cells using nanobodies. Nat. Struct. Mol. Biol. 17, 133–138 (2010).
Bers, D. M., Patton, C. W., & Nuccitelli, R. Chapter 1 - A Practical Guide to the Preparation of Ca2+ Buffers. In Methods in Cell Biology Calcium in Living Cells., M. Whitaker, ed. (Academic Press), pp. 1–26. https://doi.org/10.1016/B978-0-12-374841-6.00001-3 (2010).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Mastronarde, D. N. SerialEM: A Program for Automated Tilt Series Acquisition on Tecnai Microscopes Using Prediction of Specimen Position. Microsc. Microanal. 9, 1182–1183 (2003).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Zivanov, J., Nakane, T. & Scheres, S. H. W. A Bayesian approach to beam-induced motion correction in cryo-EM single-particle analysis. IUCrJ 6, 5–17 (2019).
Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
Zivanov, J., Nakane, T. & Scheres, S. H. W. Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1. IUCrJ 7, 253–267 (2020).
Terwilliger, T. C., Ludtke, S. J., Read, R. J., Adams, P. D. & Afonine, P. V. Improvement of cryo-EM maps by density modification. Nat. Methods 17, 923–927 (2020).
Alegre, K. O. et al. Structural basis and mechanism of activation of two different families of G proteins by the same GPCR. Nat. Struct. Mol. Biol. 28, 936–944 (2021).
Su, M. et al. Structures of β1-adrenergic receptor in complex with Gs and ligands of different efficacies. Nat. Commun. 13, 4095 (2022).
Liu, S. et al. Differential activation mechanisms of lipid GPCRs by lysophosphatidic acid and sphingosine 1-phosphate. Nat. Commun. 13, 731 (2022).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Croll, T. I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. Sect. Struct. Biol. 74, 519–530 (2018).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. Sect. Struct. Biol. 75, 861–877 (2019).
Pettersen, E. F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. Sect. Struct. Biol. 74, 531–544 (2018).
Scheres, S. H. W. A Bayesian View on Cryo-EM Structure Determination. J. Mol. Biol. 415, 406–418 (2012).
Scheres, S. H. W. RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Goddard, T. D. et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Pettersen, E. F. et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Jurrus, E. et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 27, 112–128 (2018).
Morin, A. et al. Collaboration gets the most out of software. eLife 2, e01456 (2013).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Kost, T. A., Condreay, J. P. & Jarvis, D. L. Baculovirus as versatile vectors for protein expression in insect and mammalian cells. Nat. Biotechnol. 23, 567–575 (2005).
Abramoff, M. D., Magalhães, P. J. & Ram, S. J. Image processing with ImageJ. Biophotonics Int. 11, 36–42 (2004).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Hrovat, M. Baseline Fit. https://www.mathworks.com/matlabcentral/fileexchange/24916-baseline-fit (2022).
Acknowledgements
We thank Jason de la Cruz at the Memorial Sloan Kettering Cancer Center (MSKCC) Richard Rifkind Center for cryo-EM assistance with data collection and the MSKCC High-Performance Computing (HPC) group, in particular Neeraj Harikrishnan and Jamie Cheong, for assistance with data processing. We thank Ellen Zhong for discussions about conformational heterogeneity in cryo-EM data and Elizabeth Campbell, Seth Darst, Melinda Diver and Stephen B. Long for comments on the manuscript. This work was supported by NIH NCI Cancer Center Support grant P30 CA008748 (R.K.H.), NIGMS R01-GM13230704 (R.K.H.), NCI F31-CA243235 (N.P.), the Searle Scholars Program (R.K.H.) and the Josie Robertson Investigators Program (R.K.H.).
Author information
Authors and Affiliations
Contributions
N.P., V.S. and R.K.H. conceptualized the project and contributed to writing the manuscript. N.P. performed the bulk of cryo-EM analysis. V.S. performed the bulk of optical Ca2+ imaging analysis. N.P., V.S. and R.K.H. assisted each other on all experiments and analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Harsh Bansia and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Paknejad, N., Sapuru, V. & Hite, R.K. Structural titration reveals Ca2+-dependent conformational landscape of the IP3 receptor. Nat Commun 14, 6897 (2023). https://doi.org/10.1038/s41467-023-42707-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-42707-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
