
Abstract
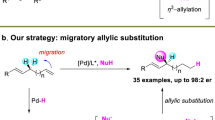
Transition-metal-catalyzed allylic substitution reactions (Tsuji−Trost reactions) proceeding via a π-allyl metal intermediate have been demonstrated as a powerful tool in synthetic chemistry. Herein, we disclose an unprecedented π-allyl metal species migration, walking on the carbon chain involving 1,4-hydride shift as confirmed by deuterium labeling experiments. This migratory allylic arylation can be realized under dual catalysis of nickel and lanthanide triflate, a Lewis acid. Olefin migration has been observed to preferentially occur with the substrate of 1,n-enols (n ≥ 3). The robust nature of the allylic substitution strategy is reflected by a broad scope of substrates with the control of regio- and stereoselectivity. DFT studies suggest that π-allyl metal species migration consists of the sequential β-H elimination and migratory insertion, with diene not being allowed to release from the metal center before producing a new π-allyl nickel species.
Similar content being viewed by others
Introduction
Transition-metal-catalyzed allylic substitution reactions (Tsuji−Trost reactions) have emerged as a powerful tool for the construction of carbon−carbon and carbon−heteroatom bonds with a broad scope of both electrophiles and nucleophiles (Fig. 1a)1. By employing an appropriate ligand, the utilization of a transition metal catalyst, including Pd2,3,4,5, Ir6,7, Rh8,9,10,11,12,13,14, Ru15,16,17,18,19,20, Co21,22,23, Cu24,25,26,27,28,29,30,31,32, Mo33,34,35,36,37,38,39,40,41, W42, and Ni43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60 realizes the control of regio- and stereoselectivity during nucleophilic substitution of the key π-allyl metal intermediate (Int-A), thus to deliver regio- and/or stereomerically pure organic compounds. On the other hand, migratory coupling reactions, combining bond migration with a coupling process, display unique regioselectivity controls, e.g., multiple 1,2-hydride shifts result in the walking of carbon−metal bond over long distances via olefin-metal intermediate (olefin-[M]-Int), leading to remote selectivity in coupling reactions (Fig. 1b)61,62,63,64,65,66,67,68,69,70. Recently, the groups of Kawatsura54 and Stanley57 independently reported nickel-catalyzed arylative substitution of homoallylic carbonates and alcohols respectively, and 1,3-hydride shift was involved during olefin migrations. In contrast, migratory allylic substitution, in which the walking of π-allyl metal species proceeds over the carbon chain via multiple 1,4-hydride shifts, are still unexplored (Fig. 1c). During one migration cycle, sequential β-H elimination and migratory insertion occurs with diene-metal complex (diene-[M]-Int) as the key intermediate71,72.

a Classic allylic substitution reactions. b Migratory coupling reaction via olefin intermediate (well developed). c Migratory allylic substitution reaction via π-allyl metal migration (unexplored). d This work.
Herein, we disclose our recent observations on the migratory dehydroxylative allylic arylation under the catalysis of nickel and lanthanide triflate, a Lewis acid (Fig. 1d).
Results and discussion
Optimization of the reaction conditions
Preliminary attempt began with the substitution reaction of allylic alcohol 1a with PhB(OH)2 (2a) under the catalysis of Ni(cod)2. To our surprise, when electron-rich ligand PCy3 was employed, the reaction does not undergo a direct allylic phenylation pathway to access 3-A or 3-B73. Instead, migratory phenylation product 3 was unexpectedly obtained in 30% yield (Table 1, entry 1), meanwhile the formation of ketone 4 was also detected, generated from 1a via intramolecular transfer hydrogenation. Obviously, migration of allylic species occurs preferentially, and product 3 was consequently produced via coupling reaction with PhB(OH)2 as the last step. Interestingly, base does not favor the Suzuki coupling reaction towards 3 (Table 1, entry 2). Therefore, a series of Lewis acids were investigated as additive to the reaction respectively (Table 1, entries 3–9). To our delight, La(OTf)3 gave the best performance, and the yield was dramatically improved to 68%, with side product 4 less than 5% (Table 1, entry 9). Further ligand screening fails to give better results (Table 1, entries 10–14). It is noteworthy to mention that bidentate ligands completely prevent the formation of 3. The yield of migratory arylation was improved to 75% in the solvent of methyl tert-butyl ether (MTBE) (Table 1, entries 15–19). Increasing the temperature to 110 °C does not give further promotion (Table 1, entry 20). Finally, Ni(cod)2 (10 mol%), PCy3 (20 mol%), La(OTf)3 (20 mol%), and PhB(OH)2 (2.0 equiv) in MTBE at 90 °C for 12 h were defined as the optimal reaction conditions for additional studies.
Scope of the reaction
With the optimized conditions in hand, we started to investigate the scope of this coupling reaction under dual catalysis (Fig. 2). Arylboronic acids irrespectively bearing electron donating groups or electron withdrawing groups all reacted with good yields (3, and 5−10). To our delight, a variety of functional groups, including 2-Me, 2-OMe, 2-F, 3-Me, 3-OMe, 3-F, 3-CF3, 4-Me, 4-tBu, 4-Ph, 4-OMe, 4-F, 4-Cl, 4-CF3, and 4-OCF3 on the benzene ring turned out to be compatible under the standard reaction conditions (11−25). Notably, medicinally prevalent aromatic moieties other than benzene were incorporated smoothly, including 1-naphthyl, 2-naphthyl, 3-indolyl, 2-thiophenyl, and 3-thiophenyl groups (26−30). 1,2-Enol featuring a terminal olefin works as well, affording arylated product 31 in 63% yield correspondingly. Scope of 1,2-enols could also be extended to those bearing long aliphatic chains (32−35). However, migratory phenylation fails to construct a quaternary carbon center in 36. Moreover, 1,2-enol featuring a tertiary alcohol led to tri-substituted alkene via migratory arylation process. The reactions of cyclic 1,2-enols work nicely to afford indene derivative 37, bearing a fused five-membered ring, as well as product owning a fused six- (38) or seven-membered ring (39), and such units have been widely found in various natural products and drug molecules. Finally, it is noteworthy to mention that all the reactions give regio- and stereospecific products via migratory allylic substitutions, exhibiting the synthetic robustness of this dual catalytic protocol.

The reaction was carried out with enaol 1 (0.2 M), ArB(OH)2 (2.0 equiv.), Ni(cod)2 (10 mol%), PCy3 (20 mol%), and La(OTf)3 (20 mol%) in MTBE (1.0 mL) at 90 oC for 12 h.
To learn more about reactivity of the migratory allylic arylation transformation, comparison experiments were carried out under the standard conditions by employing (E)-enol and its stereoisomer (Z)-enol parallelly. The reaction of (E)−1a afforded migratory phenylated product 3 in 72% yield, and almost the same yield (73%) of 3 could be produced starting from its stereoisomer, (Z )−1a (Fig. 3a). Similar results could be obtained with tertiary enol (E)-1b or (Z )-1b as substrates, yielding 59% and 58% of the same product 37 respectively. These outcomes suggest the reaction proceeding via the same intermediate from both stereoisomers, implying the intermediacy of π-allyl metal species. Next, the scope of enols was investigated with OH group and/or olefin moiety at a different position on the carbon chain (Fig. 3b). 1,3-Enol 1c, with a terminal olefin could also be employed to yield 3 in 71%, the same product as the reaction of 1a. 1,4-enol 1d, with olefin distancing one more carbon from OH group gave a decreased yield of 38%. Direct allylic arylation occurs efficiently with allylic alcohol 1e. The reaction of allylic alcohol 1 f, in which two more carbons locate between phenyl and OH group compared with 1e, resulting in the corresponding phenylated product in a decreased yield of 42%, implying consecutive π-allyl metal species migrations before the final coupling with PhB(OH)2. Further studies were carried out by employing 1,n-enols 1g-m (n = 3–10), featuring different distances of olefin unit from OH group. To our delight, these enols are all compatible in the migratory arylation transformations, leading to the migration products in 25–85% yields. These observations make clear that olefin migration preferentially occurs to generate the corresponding allylic alcohol, which further undergoes a substitution reaction with ArB(OH)2 under nickel catalysis. It is worth noting that, in these types of reactions, the preferential olefin migration undergoes via multiple 1,2-hydride shift, and 1,3-hydride shift was also possibly involved in the reaction of 1,3-enols (e.g., 1 g)54. Given the fact that mixtures of olefin or alcohol isomers are abundant industrial feedstocks, and are more easily accessible with less expense than pure isomers, the direct employment of isomers in regio- and stereoconvergent reactions to produce value-added products is of considerable interest. As shown in Fig. 3c, the reaction using a mixture of six regio- and stereoisomers (1:1:1:1:1:1) delivers regio- and stereodefined product 3 in 64% yield. Next, when an alkenylboronic acid was employed in the reaction of (E)−1a, we are glad to obtain the corresponding alkenylated product 3-alkenyl in 24% yield (Fig. 3d). However, cyclopentylboronic acid fails to promote the alkylation reaction of (E)−1a. Finally, asymmetric migratory allylic arylation of enols 1o and 1 h was investigated, and preliminary attempts were conducted to achieve good enantioselective controls with chiral bidentate nitrogen ligands L1 and L2 respectively (91:9 er for 31, 82:18 er for 3), but with low yields in both reactions (for details see page S57-60 in the supplementary discussion section of Supplementary Information).

a Reactivity comparison of (E)- and (Z)-enols. b Reaction of enols with OH group and/or olefin moiety at different positions. c Reaction of a mixture of six regio- and stereoisomers. d Reaction of (E)-1a with an alkenyl or alkyl boronic acid.
Mechanistic studies
To gain a deeper insight of the mechanism of this migratory arylation reaction, control experiments with enol 1n were performed. When the reaction was stopped in 1 h, corresponding product 21 was formed in 18% yield, while diene 40 was not detected via dehydration from 1n (Fig. 4a). Further reaction of diene 40 directly under the standard reaction conditions also failed to yield product 21. When diene 40 was introduced as an additive in the reaction of 1a, migratory arylation product 3 was generated from 1a in 70% yield, while 21 was not detected from diene additive 40. These observations point out that migratory arylation transformations are not proceeding via simple dehydration of enols, and diene cannot access the final products via hydroarylation, indicating that the intermediacy of diene could be ruled out. Moreover, deuterium labeling experiments were carried out to investigate the reaction details during migration (Fig. 4b). When deuterated d2−1a was employed under the standard conditions, 95% deuterium incorporation was observed at 4-position, making clear that a specific 1,4-hydride shift occurs during the migratory process. Finally, no intermolecular H/D exchange was observed from the deuterium cross-over reaction of d2−1a and 1e in a one-pot manner (Fig. 4c), implying the prior formation of the new π-allyl nickel intermediate from diene-Ni-Int before the possible disassociation of diene-ligand from the metal, which differs from many cases involving chain-walking process via olefins57.

a Control experiments. b Deuterium labeling experiments. c Intermolecular cross-over reaction.
To account for the 1,4-hydride shift process observed by the aforementioned deuterium labeling experiments, density functional theory (DFT) calculations were carried out using 1a as model substrate along with the Ni(0)−PCy3 catalytic system (for details of the optimized structures see coordination data sets and free energies in Supplementary Data 1). The free energy profiles of the preferred 1,4-hydride transfer pathways for (Z)-1a and (E)-1a represented by the black line and the blue line, respectively, are shown in Fig. 5, taking the π-allyl nickel species Int1b as the free energy reference. In the case of (E)-1a, isomerization of π-allyl nickel species Int1b affords Int2b, which undergoes β-hydride elimination via Ts1b with an energy barrier of 20.1 kcal.mol−1 (TS1b with respective to int1b) to form the diene-nickel complex Int3b. Hydrometallation of Int4b, generated by the further isomerization of Int3b, needs to overcome an activation barrier of only 5.1 kcal.mol−1 (Ts2b), providing a new and desired π-allyl nickel species Int5. A similar situation can be found in the case of (Z)-1a with slightly higher free energy demanding. These calculations indicate that both stereoisomers of 1a can undergo the 1,4-hydride transfer process to furnish the same π-allyl nickel complex Int5, which is in line with the experimental observation in Fig. 3. Moreover, the dissociation of the diene-nickel complex Int3a to form Int3a-p1 and Int3a-p2 indicated by red line was also considered, and the higher energy barrier rules out the possibility of the formation of diene, consistent with the results experimentally observed in Fig. 4.

Free-energy reaction profiles (kcal mol−1) are presented for the 1,4-hydride transfer process of two stereoisomers of 1a, calculated at the PBE0/combined basis set level at 363 K.
Based on the regiochemical outcome, mechanistic experiments in Fig. 4, and DFT calculation results in Fig. 5, a possible mechanism for the migratory allylic arylation reaction is proposed in Fig. 6. Ligand exchange of Ni(cod)2 with PCy3 generates the active catalyst Ni(PCy3)2, which undergoes the reaction with allylic alcohol 1a via oxidative addition to give π-allyl nickel species Int1. Olefin migration occurs to produce the corresponding allylic alcohols when 1,n-enols (n ≥ 3) are employed, e.g., 1,3-enol 1c. The hydroxyl group performs as a leaving group after being activated by La(OTf)3, a Lewis acid. β-Hydride elimination of Int1 affords diene-nickel complex Int3, which further transforms to a new π-allyl nickel species Int5 via hydrometallation. It is noteworthy to mention that the active NiH species in Int3 is believed to be generated via β-hydride elimination, instead of being formed via oxidative addition of alcohols in many other cases74,75,76,77. Transformation from one π-allyl species (Int1) to the other (Int5) proceeds via an overall 1,4-hydride shift, and disassociation of the diene-ligand in Int3 from the metal seems not possible as confirmed by the deuterium labeling and cross-over studies. Finally, transmetallation of Int5 with ArB(OH)2 produces Int6, which on subsequent reductive elimination leads to the final product 3, releasing the nickel(0) catalyst to close the cycle.

A plausible reaction pathway for migratory dehydroxylative allylic arylation of 1,n-enols.
To sum up, we have developed a migratory dehydroxylative allylic arylation of unactivated 1,n-enols enabled by dual nickel/Lewis acid catalysis with high regio- and stereoselectivity. The reaction proceeds via unprecedented π-allyl nickel species migration involving 1,4-hydride shift as confirmed by deuterium labeling experiments. DFT studies suggest that π-allyl metal species migration consists of the sequential β-H elimination and migratory insertion, with diene not being allowed to release from the metal center before hydrometallation to produce a new π-allyl nickel species. Olefin migration was observed to preferentially occur starting from 1,n-enols (n ≥ 3). The method provides a facile and migratory strategy to construct C(sp2)–C(sp3) bonds with unique selectivity and features a broad substrate scope for both arylboronic acids and 1,n-enols. Further studies on the mechanism, synthetic application, and asymmetric variants are currently ongoing in our laboratory.
Methods
Representative procedure for migratory allylic arylation of 1,n-enols enabled by nickel catalysis
Under a nitrogen atmosphere, a mixture of Ni(cod)2 (5.5 mg, 0.02 mmol), PCy3 (11.2 mg, 0.04 mmol), Lewis acid (0.04 mmol), and ArB(OH)2 (0.4 mmol) was added a solution of corresponding enol (0.2 mmol) in MTBE (1.0 mL). The reaction was sealed and stirred at 90 °C or 100 °C for 12 h. Subsequently, the reaction was cooled down to room temperature and the mixture was evaporated and purified via column chromatography on silica gel (eluent: petroleum ether/ethyl acetate) afforded the desired product.
Data availability
All data generated or analyzed during this study are included in this Article and the Supplementary Information. Details about materials and methods, experimental procedures, mechanistic studies, characterization data, computational details, NMR and HPLC spectra are available in the Supplementary Information. Calculated coordinates are available in the Supplementary Data file. All other data are available from the corresponding author upon request.
References
Trost, B. M. & van Vranken, D. L. Asymmetric transition metal-catalyzed allylic alkylations. Chem. Rev. 96, 395–422 (1996).
Trost, B. M. Pd asymmetric allylic alkylation (AAA). A powerful synthetic tool. Chem. Pharm. Bull. 50, 1–14 (2002).
Trost, B. M. & Crawley, M. L. Asymmetric transition-metal-catalyzed allylic alkylations: applications in total synthesis. Chem. Rev. 103, 2921–2944 (2003).
Milhau, L. & Guiry, P. J. Palladium-catalyzed enantioselective allylic substitution. Top. Organomet. Chem. 38, 95–153 (2011).
Blieck, R., Taillefer, M. & Monnier, F. Metal-catalyzed intermolecular hydrofunctionalization of allenes: easy access to allylic structures via the selective formation of C–N, C–C, and C–O bonds. Chem. Rev. 120, 13545–13598 (2020).
Krautwald, S., Sarlah, D., Schafroth, M. A. & Carreira, E. M. Enantio- and diastereodivergent dual catalysis: α-allylation of branched aldehydes. Science 340, 1065–1068 (2013).
Cheng, Q. et al. Iridium-catalyzed asymmetric allylic substitution reactions. Chem. Rev. 119, 1855–1969 (2019).
Leahy, D. K. & Evans, P. A. Rhodium(I)-catalyzed allylic substitution reactions and their applications to target directed synthesis. In Modern Rhodium-Catalyzed Organic Reactions; Evans, P. A., Ed.; John Wiley & Sons, Inc.: New York, 2005; pp 191–214.
Evans, P. A. & Nelson, J. D. Conservation of absolute configuration in the acyclic rhodium-catalyzed allylic alkylation reaction: evidence for an enyl (σ + π) organorhodium intermediate. J. Am. Chem. Soc. 120, 5581–5582 (1998).
Hayashi, T., Okada, A., Suzuka, T. & Kawatsura, M. High enantioselectivity in rhodium-catalyzed allylic alkylation of 1-substituted 2-propenyl acetates. Org. Lett. 5, 1713–1715 (2003).
Kazmaier, U. & Stolz, D. Regio- and stereoselective rhodium-catalyzed allylic alkylations of chelated enolates. Angew. Chem. Int. Ed. 45, 3072–3075 (2006).
Sidera, M. & Fletcher, S. P. Rhodium-catalysed asymmetric allylic arylation of racemic halides with arylboronic acids. Nat. Chem. 7, 935–939 (2015).
Li, C. & Breit, B. Rhodium-catalyzed dynamic kinetic asymmetric allylation of phenols and 2-hydroxypyridines. Chem. Eur. J. 22, 14655–14663 (2016).
Parveen, S., Li, C., Hassan, A. & Breit, B. Chemo-, regio-, and enantioselective rhodium-catalyzed allylation of pyridazinones with terminal allenes. Org. Lett. 19, 2326–2329 (2017).
Matsushima, Y., Onitsuka, K., Kondo, T., Mitsudo, T. & Takahashi, S. Asymmetric catalysis of planar-chiral cyclopentadienylruthenium complexes in allylic amination and alkylation. J. Am. Chem. Soc. 123, 10405–10406 (2001).
Onitsuka, K., Matsushima, Y. & Takahashi, S. Kinetic resolution of allyl carbonates in asymmetric allylic alkylation catalyzed by planar-chiral cyclopentadienyl-ruthenium complexes. Organometallics 24, 6472–6474 (2005).
Onitsuka, K., Okuda, H. & Sasai, H. Regio- and enantioselective O-allylation of phenol and alcohol catalyzed by a planar-chiral cyclopentadienyl ruthenium complex. Angew. Chem. Int. Ed. 47, 1454–1457 (2008).
Trost, B. M., Rao, M. & Dieskau, A. P. A Chiral Sulfoxide-Ligated Ruthenium Complex for Asymmetric Catalysis: Enantio- and Regioselective Allylic Substitution. J. Am. Chem. Soc. 135, 18697–18704 (2003).
Kawatsura, M. et al. Ruthenium-Catalyzed Regio- and Enantioselective Allylic Amination of Racemic 1-Arylallyl Esters. Org. Lett. 16, 1470–1473 (2014).
Kanbayashi, N. et al. Enantio- and Diastereoselective Asymmetric Allylic Alkylation Catalyzed by a Planar-Chiral Cyclopentadienyl Ruthenium Complex. Chem. Commun. 51, 10895–10898 (2015).
Mizutani, K., Yorimitsu, H. & Oshima, K. Cobalt-Catalyzed Allylic Substitution Reaction of Allylic Ethers with Phenyl and Trimethylsilylmethyl Grignard Reagents. Chem. Lett. 33, 832–833 (2004).
Yasui, H., Mizutani, K., Yorimitsu, H. & Oshima, K. Cobalt- and rhodium-catalyzed cross-coupling reaction of allylic ethers and halides with organometallic reagents. Tetrahedron 62, 1410–1415 (2006).
Han, J.-F., Guo, P., Zhang, X.-G., Liao, J.-B. & Ye, K.-Y. Recent advances in cobalt-catalyzed allylic functionalization. Org. Biomol. Chem. 18, 7740–7750 (2020).
Langlois, J. B. & Alexakis, A. Copper-Catalyzed Enantioselective Allylic Substitution. Top. Organomet. Chem. 38, 235–268 (2011).
Malda, H., van Zijl, A. W., Arnold, L. A. & Feringa, B. L. Enantioselective Copper-Catalyzed Allylic Alkylation with Dialkylzincs Using Phosphoramidite Ligands. Org. Lett. 3, 1169–1171 (2001).
Van Veldhuizen, J. J., Campbell, J. E., Giudici, R. E. & Hoveyda, A. H. A Readily Available Chiral Ag-Based N-Heterocyclic Carbene Complex for Use in Efficient and Highly Enantioselective Ru-Catalyzed Olefin Metathesis and Cu-Catalyzed Allylic Alkylation Reactions. J. Am. Chem. Soc. 127, 6877–6882 (2005).
Yoshikai, N., Zhang, S.-L. & Nakamura, E. Origin of the Regio- and Stereoselectivity of Allylic Substitution of Organocopper Reagents. J. Am. Chem. Soc. 130, 12862–12863 (2008).
Selim, K. B., Matsumoto, Y., Yamada, K. & Tomioka, K. Efficient Chiral N-Heterocyclic Carbene/Copper(I)-Catalyzed Asymmetric Allylic Arylation with Aryl Grignard Reagents. Angew. Chem., Int. Ed. 48, 8733–8735 (2009).
Langlois, J.-B. & Alexakis, A. Copper-Catalyzed Asymmetric Allylic Alkylation of Racemic Cyclic Substrates: Application of Dynamic Kinetic Asymmetric Transformation (DYKAT). Adv. Synth. Catal. 352, 447–457 (2010).
Shi, Y., Jung, B., Torker, S. & Hoveyda, A. H. N-Heterocyclic Carbene–Copper-Catalyzed Group-, Site-, and Enantioselective Allylic Substitution with a Readily Accessible Propargyl(pinacolato)boron Reagent: Utility in Stereoselective Synthesis and Mechanistic Attributes. J. Am. Chem. Soc. 137, 8948–8964 (2015).
You, H., Rideau, E., Sidera, M. & Fletcher, S. P. Non-Stabilized Nucleophiles in Cu-Catalysed Dynamic Kinetic Asymmetric Allylic Alkylation. Nature 517, 351–355 (2015).
Rideau, E., You, H., Sidera, M., Claridge, T. D. W. & Fletcher, S. P. Mechanistic Studies on a Cu-Catalyzed Asymmetric Allylic Alkylation with Cyclic Racemic Starting Materials. J. Am. Chem. Soc. 139, 5614–5624 (2017).
Belda, O. & Moberg, C. Molybdenum-Catalyzed Asymmetric Allylic Alkylations. Acc. Chem. Res. 37, 159–167 (2004).
Trost, B. M. Pd- and Mo-Catalyzed Asymmetric Allylic Alkylation. Org. Process Res. Dev. 16, 185–194 (2012).
Moberg, C. Molybdenum-Catalyzed Asymmetric Allylic Alkylations. Organic Reactions 84, 1–74 (2014).
Trost, B. M. & Hachiya, I. Asymmetric Molybdenum-Catalyzed Alkylations. J. Am. Chem. Soc. 120, 1104–1105 (1998).
Malkov, A. V. et al. Asymmetric Allylic Substitution Catalyzed by C1-Symmetrical Complexes of Molybdenum: Structural Requirements of the Ligand and the Stereochemical Course of the Reaction. Chem. Eur. J. 12, 6910–6929 (2006).
Trost, B. M. & Zhang, Y. Mo-Catalyzed Regio-, Diastereo-, and Enantioselective Allylic Alkylation of 3-Aryloxindoles. J. Am. Chem. Soc. 129, 14548–14549 (2007).
Trost, B. M. & Zhang, Y. Catalytic Double Stereoinduction in Asymmetric Allylic Alkylation of Oxindoles. Chem. -Eur. J. 16, 296–303 (2010).
Trost, B. M. & Zhang, Y. Molybdenum-Catalyzed Asymmetric Allylic Alkylation of 3-Alkyloxindoles: Reaction Development and Applications. Chem. Eur. J. 17, 2916–2922 (2011).
Ozkal, E. & Pericàs, M. A. A Bis(Triazolecarboxamido) Ligand for Enantio- and Regioselective Molybdenum-Catalyzed Asymmetric Allylic Alkylation Reactions. Adv. Synth. Catal. 356, 711–717 (2014).
Lloyd-Jones, G. C. & Pfaltz, A. Chiral Phosphanodihydrooxazoles in Asymmetric Catalysis: Tungsten-Catalyzed Allylic Substitution. Angew. Chem., Int. Ed. Engl. 34, 462–464 (1995).
Chung, K.-G., Miyake, Y. & Uemura, S. Nickel(0)-catalyzed asymmetric cross-coupling reactions of allylic compounds with arylboronic acids. J. Chem. Soc., Perkin Trans. 1, 15–18 (2000).
Jiménez-Aquino, A., Flegeau, E. F., Schneider, U. & Kobayashi, S. Catalytic intermolecular allyl–allyl cross-couplings between alcohols and boronates. Chem. Commun. 47, 9456–9458 (2011).
Kita, Y. et al. Pentacoordinated Carboxylate π-Allyl Nickel Complexes as Key Intermediates for the Ni-Catalyzed Direct Amination of Allylic Alcohols. Chem. Eur. J. 21, 14571–14578 (2015).
Kita, Y., Kavthe, R. D., Oda, H. & Mashima, K. Asymmetric Allylic Alkylation of β-Ketoesters with Allylic Alcohols by a Nickel/Diphosphine Catalyst. Angew. Chem. Int. Ed. 55, 1098–1101 (2016).
Bernhard, Y., Thomson, B., Ferey, V. & Sauthier, M. Nickel-Catalyzed α-Allylation of Aldehydes and Tandem Aldol Condensation/Allylation Reaction with Allylic Alcohols. Angew. Chem. Int. Ed. 56, 7460–7464 (2017).
Chen, Y.-G. et al. Regioselective Ni-Catalyzed Carboxylation of Allylic and Propargylic Alcohols with Carbon Dioxide. Org. Lett. 19, 2969–2972 (2017).
Yang, B. & Wang, Z.-X. Nickel-Catalyzed Cross-Coupling of Allyl Alcohols with Aryl- or Alkenylzinc Reagents. J. Org. Chem. 82, 4542–4549 (2017).
Nazari, S. H. et al. Nickel-Catalyzed Suzuki Cross Couplings with Unprotected Allylic Alcohols Enabled by Bidentate N-Heterocyclic Carbene (NHC)/Phosphine Ligands. ACS Catal 8, 86–89 (2018).
Wang, G., Gan, Y. & Liu, Y. Nickel-Catalyzed Direct Coupling of Allylic Alcohols with Organoboron Reagents. Chin. J. Chem. 36, 916–920 (2018).
Jia, X.-G., Guo, P., Duan, J. & Shu, X.-Z. Dual nickel and Lewis acid catalysis for cross-electrophile coupling: the allylation of aryl halides with allylic alcohols. Chem. Sci. 9, 640–645 (2018).
Wang, Y.-B., Liu, B.-Y., Bu, Q., Dai, B. & Liu, N. In Situ Ring-Closing Strategy for Direct Synthesis of NHeterocyclic Carbene Nickel Complexes and Their Application in Coupling of Allylic Alcohols with Aryl Boronic Acids. Adv. Synth. Catal. 362, 2930–2940 (2020).
Hamaguchi, T., Takahashi, Y., Tsuji, H. & Kawatsura, M. Nickel-Catalyzed Hydroarylation of in Situ Generated 1,3-Dienes with Arylboronic Acids Using a Secondary Homoallyl Carbonate as a Surrogate for the 1,3-Diene and Hydride Source. Org. Lett. 22, 1124–1129 (2020).
Tsuji, H., Takahashi, Y. & Kawatsura, M. Nickel-catalyzed hydroalkylation of 1,3-dienes with malonates using a homoallyl carbonate as the 1,3-diene and hydride source. Tetrahedron Lett 68, 152916 (2021).
Peng, Y. et al. Nickel/Copper-Cocatalyzed Asymmetric Benzylation of Aldimine Esters for the Enantioselective Synthesis of α-Quaternary Amino Acids. Angew. Chem. Int. Ed 61, e202203448 (2022).
Tran, H. N., Nguyen, C. M., Koeritz, M. T., Youmans, D. D. & Stanley, L. M. Nickel-catalyzed arylative substitution of homoallylic alcohols. Chem. Sci. 13, 11607–11613 (2022).
Flaget, A., Zhang, C. & Mazet, C. Ni-Catalyzed Enantioselective Hydrofunctionalizations of 1,3-Dienes. ACS Catal 12, 15638–15647 (2022).
Bhakta, S. & Ghosh, T. Nickel-Catalyzed Hydroarylation reaction: a useful tool in organic synthesis. Org. Chem. Front. 9, 5074–5103 (2022).
Wang, C.-D., Guo, Y.-J., Wang, X.-M., Wang, Z. & Ding, K.-L. Ni-Catalyzed Regioselective Hydroarylation of 1-Aryl-1,3-ButadieneswithAryl Halides. Chem. Eur. J. 27, 15903–1590 (2021).
Hilt, G. Double Bond Isomerisation and Migration New Playgrounds for Transition Metal-Catalysis. ChemCatChem 6, 2484–2485 (2014).
He, Y., Cai, Y. & Zhu, S. Mild and Regioselective Benzylic C−H Functionalization: Ni-Catalyzed Reductive Arylation of Remote and Proximal Olefins. J. Am. Chem. Soc. 139, 1061–1064 (2017). For recent examples of chain-working via olefin migration by nickel catalysis, see.
Vasseur, A., Bruffaerts, J. & Marek, I. Remote Functionalization through Alkene Isomerization. Nat. Chem. 8, 209–219 (2016).
Molloy, J. J., Morack, T. & Gilmour, R. Positional and Geometrical Isomerisation of Alkenes: The Pinnacle of Atom Economy. Angew. Chem. Int. Ed. 58, 13654–13664 (2019).
Liu, Q., Liu, X. & Li, B. Base-Metal-Catalyzed Olefin Isomerization Reactions. Synthesis 51, 1293–1310 (2019).
Chen, F. et al. Remote Migratory Cross-Electrophile Coupling and Olefin Hydroarylation Reactions Enabled by in Situ Generation of NiH. J. Am. Chem. Soc. 139, 13929–13935 (2017).
Kapat, A., Sperger, T., Guven, S. & Schoenebeck, F. E-Olefins through intramolecular radical relocation. Science 363, 391–396 (2019).
Gao, J. et al. Nickel-Catalyzed Migratory Hydrocyanation of Internal Alkenes: Unexpected Diastereomeric-Ligand-Controlled Regiodivergence. Angew. Chem. Int. Ed. 60, 1883–1890 (2021).
Long, J., Xia, S., Wang, T., Cheng, G.-J. & Fang, X. Nickel-Catalyzed Regiodivergent Cyanation of Allylic Alcohols: Scope, Mechanism, and Application to the Synthesis of 1,n-Dinitriles. ACS Catal 11, 13880–13890 (2021).
Ding, Y., Long, J., Sun, F. & Fang, X. Nickel-Catalyzed Isomerization/Allylic Cyanation of Alkenyl Alcohols. Org. Lett. 23, 6073–6078 (2021).
Feng, X. & Du, H. Application of Chiral Olefin Ligands in Asymmetric Catalysis. Chin. J. Org. Chem. 35, 259–272 (2015).
Huang, Y. & Hayashi, T. Chiral Diene Ligands in Asymmetric Catalysis. Chem. Rev. 122, 14346–14404 (2022).
Demel, P., Keller, M. & Breit, B. o-DPPB-Directed Copper-Mediated and -Catalyzed Allylic Substitution with Grignard Reagents. Chem. Eur. J. 12, 6669–6683 (2006).
Xiao, L.-J. et al. Nickel(0)-Catalyzed Hydroarylation of Styrenes and 1,3-Dienes with Organoboron Compounds. Angew. Chem. Int. Ed. 57, 461–464 (2018).
Lv, X.-Y., Fan, C., Xiao, L.-J., Xie, J.-H. & Zhou, Q.-L. Ligand-Enabled Nickel-Catalyzed Enantioselective Hydroarylation of Styrenes and 1,3-Dienes with Arylboronic Acids. CCS Chem 1, 328–334 (2019).
Chen, Y.-G. et al. Nickel-catalyzed Enantio-selective Hydroarylation and Hydroalkenylation of Styrenes. J. Am. Chem. Soc. 141, 3395–3399 (2019).
Marcum, J. S., Taylor, T. R. & Meek, S. J. Enantioselective Synthesis of Functionalized Arenes by Nickel-Catalyzed Site-Selective Hydroarylation of 1,3-Dienes with Aryl Boronates. Angew. Chem. Int. Ed. 59, 14070–14075 (2020).
Acknowledgements
We acknowledge the National Natural Science Foundation of China (Grant No. 22271054, C.Z.), the China Postdoctoral Science Foundation (2022M713667, B.X.), the “1000-Youth Talents Plan”, and Fudan University (start-up grant) for financial support. We also thank Mr. Jun Zhang for his help in revisions, Prof. Huadong Wang and Bing-Tao Guan for insightful and fruitful discussions. This research paper is dedicated to Prof. Li-Xin Dai on the occasion of his 100th birthday.
Author information
Authors and Affiliations
Contributions
C.Z. conceived the project, analyzed the data, and wrote the manuscript. D.Z. performed the most of experiments. B.X. did the DFT calculations. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, D., Xu, B. & Zhu, C. Migratory allylic arylation of 1,n-enols enabled by nickel catalysis. Nat Commun 14, 3308 (2023). https://doi.org/10.1038/s41467-023-38865-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-38865-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
