
Abstract
Electrochemical CO2 reduction (CO2R) is an approach to closing the carbon cycle for chemical synthesis. To date, the field has focused on the electrolysis of ambient pressure CO2. However, industrial CO2 is pressurized—in capture, transport and storage—and is often in dissolved form. Here, we find that pressurization to 50 bar steers CO2R pathways toward formate, something seen across widely-employed CO2R catalysts. By developing operando methods compatible with high pressures, including quantitative operando Raman spectroscopy, we link the high formate selectivity to increased CO2 coverage on the cathode surface. The interplay of theory and experiments validates the mechanism, and guides us to functionalize the surface of a Cu cathode with a proton-resistant layer to further the pressure-mediated selectivity effect. This work illustrates the value of industrial CO2 sources as the starting feedstock for sustainable chemical synthesis.
Similar content being viewed by others
Introduction
Electrochemical CO2 reduction (CO2R) to chemicals offers one form of upgrading/utilizing captured CO21,2,3. CO2R has demonstrated a wide range of products, including carbon monoxide (CO), formate/formic acid, alcohols, and hydrocarbons, at industrially relevant reaction rates4,5,6. When powered using renewable electricity, CO2R can reduce the carbon intensity of the otherwise fossil fuel-based production of these carbon compounds7,8,9,10. To date, most studies have focused on electrolysis of ambient pressure CO211,12. In actuality, many processes involving CO2 work at pressure (PCO2), with CO2 typically in dissolved form (1–110 bar)13. PCO2 is the effluent of industrial processes such as natural gas reforming and ethylene oxide production (3–20 bar)14,15. Depressurizing these CO2 sources to accommodate existing ambient pressure CO2R cells incurs an energy penalty and unnecessarily takes downhill the total energetic value of the reactant.
PCO2 also benefits from high CO2 solubility in aqueous solutions. At ambient pressures, CO2’s low solubility diminishes current densities to the vicinity of few ~10 milliamperes per square centimeter16,17. Dissolving PCO2 in liquid electrolyte delivers much more reactant to the CO2R catalyst: the CO2 concentration increases from 0.03 M under ambient pressure to 1.16 M under 50 bar18. In addition, adopting dissolved PCO2 during CO2R could stabilize the bulk catholyte pH at ~6.2, which is otherwise alkalized amid stoichiometric OH− production19.
In prior studies that sought to lever pressure in aqueous-based CO2R20,21, PCO2 was reduced to CO or formate22. These include examinations of altered CO2R product selectivity on various metal catalysts under high pressure23,24,25,26. A Ni wire electrode that had no CO2R activity under ambient pressure showed 23% formic acid selectivity under 60 bar25. Enhanced formate selectivity was seen on Sn using PCO227,28. Theoretical modeling and control experiments were also conducted to understand CO2R under high pressure29,30,31,32,33. More recently, PCO2 has been found to transform Cu-based catalysts to become formate-selective34.
Although these results have shown the impact of pressure on CO2R, the underlying mechanism of the pressure-dependent CO2R selectivity has yet to be systemically revealed. In particular, the local microenvironment near the CO2R electrode (such as the concentrations of key species, pHs, etc.) under the influence of pressure is critical to the final CO2R pathway, but has been rarely observed. This task is beyond the capability of prevailing operando tools for electrochemistry, such as Raman spectroscopy35,36, because the electrode in a high-pressure aqueous-based CO2R cell is immersed deep inside the liquid electrolyte. The working distance of commercial Raman systems is limited to several millimeters with an excitation power of dozens of milliwatts and the Raman signals of species dissolved in liquids are susceptible to strong background interference37. Consequently, reaction mechanisms and cathode design principles relevant to high-pressure CO2R remain largely unexplored.
Here we examine pressurization in the 1–50 bar range and find that several catalysts, including Cu, Au, Ag, and Sn, become formate selective in aqueous CO2R systems. Quantitative operando Raman spectroscopy, custom-built for high-pressure CO2R cells, and density functional theory (DFT) calculations, taken together indicate higher CO2 coverage and lower proton concentration on the cathode surface under elevated pressure, each favoring the formate formation. Guided by the pressure-dependent reaction mechanism, we devised a proton-resistant Cu/polypyrrole (Cu/PPy) cathode, which was then assembled into a narrow-gap aqueous flow cell for more selective and active CO2 to formate conversion.
Results
Impact of pressure on aqueous-based CO2R
The impact of pressure on CO2R reaction pathways was evaluated on Cu, Au, Ag, and Sn in a two-compartment high-pressure H-cell (Fig. 1a). We used 0.5 M KHCO3 aqueous solution saturated with CO2 under different pressures as the electrolyte, Pt foil as the counter electrode, and Ag/AgCl (saturated KCl) as the reference electrode. The gas headspace in each compartment was minimal, and three optical windows were fixed in the cathode chamber for ensuing operando Raman spectroscopy.

a Schematic of the customized high-pressure H-cell for CO2R performance evaluation and operando Raman spectroscopy. b FEs toward CO2R products and H2, and CO2R partial current densities on the Cu catalyst under different pressures at −1.1 V vs. RHE. c FEs toward formate and CO2R partial current densities on Au, Ag, and Sn catalysts under different pressures at −1.1 V vs. RHE. d FEs toward formate and total current densities on the Cu catalyst in an ambient pressure H-cell (1 bar CO2), an ambient pressure gas-fed flow cell (1 bar CO2), and an H-cell filled with 50 bar CO2/Ar mixture (1 bar CO2 and 49 bar Ar). j denotes the current density. Error bars represent the standard deviation of three independent measurements.
We first prepared a Cu nanoparticle catalyst (Supplementary Fig. 1a). Cyclic voltammetry, x-ray powder diffraction (XRD), and x-ray photoelectron spectroscopy (XPS) indicate metallic Cu(0) as the dominant CO2R active site (Supplementary Figs. 1b–d)34. Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) determine that the catalyst surface is rich in (111)-oriented planes (Supplementary Figs. 1e, f)38,39. In good agreement with the literature3,40, the as-prepared Cu catalyst converts CO2 to diverse C1 and C2 products under ambient pressure. At −1.1 V versus the reversible hydrogen electrode (vs. RHE), the formate Faradaic efficiency (FE) is only 14.2% and the other CO2R products (20.5% of CO, 6.6% of CH4, 15.2% of C2H4, and 8.4% of C2H5OH) account for 50.7% (Fig. 1b). Surprisingly, at the same cathode potential, the Cu catalyst becomes more formate selective when the pressure increases. Under 50 bar, we note a formate FE of 68.1% and a ten-fold increase in the FE ratio of formate to other CO2R products, whereas the collective FE of the rest C1/C2 products drops below 7% (Fig. 1b and Supplementary Fig. 2). The partial current density to CO2R increases from 61.5 mA cm−2 under ambient pressure to 140.2 mA cm−2 under 50 bar (Fig. 1b).
Similar observations hold for other common CO2R catalysts targeting CO or formate. We screened Au, Ag, and Sn nanoparticle catalysts (Supplementary Fig. 3) that have been extensively reported41,42. At −1.1 V vs. RHE, the Au and Ag catalysts convert CO2 to CO (FE > 62%) with modest yields toward formate (FE < 5%) under ambient pressure (Fig. 1c and Supplementary Fig. 4). Strikingly, at the same cathode potential, both catalysts exhibit a notable increase in formate selectivity under elevated pressure. Under 50 bar, a formate selectivity of 63.8% and 81.7% is achieved on Au and Ag, respectively (Fig. 1c and Supplementary Fig. 4). Likewise, the formate-producing Sn catalyst shows higher formate selectivity under higher pressure (Fig. 1c and Supplementary Fig. 4). The universal increase in formate FEs on Cu, Au, Ag, and Sn suggests that high pressure regulates CO2R toward the carbon hydrogenation route (*CO2 → *HCOO → HCOO−) regardless of the nature of the catalyst, while compromising the original oxygen hydrogenation pathway (*CO2 → *COOH → *CO/CO) on Cu, Au, and Ag43. The trend of the CO2R partial current density to increase with higher pressure is maintained across all tested catalysts (Figs. 1b, c), implying a denser population of adsorbed CO2 that is accessible to the active sites.
Mechanistic study
These recurring phenomena motivated us to probe the CO2R reaction mechanism under the influence of high pressure. We first ruled out the contribution of changes in the catalyst intrinsic properties: Scanning electron microscopy (SEM) and XRD reveal negligible differences in morphologies and crystal structures of the Cu, Au, and Ag catalysts before and after tests under pressure (Supplementary Figs. 5 and 6). After extended CO2 electrolysis under 50 bar, the Cu, Au, and Ag catalysts all resume their original CO2R behaviors under ambient pressure (Supplementary Fig. 7).
We then assessed the influence of CO2 availability. In a gas CO2-fed flow cell under ambient pressure (Supplementary Fig. 8a), Cu, Au, and Ag manifest regular CO2R performance with higher current densities compared to ambient pressure H-cell measurements (Fig. 1d and Supplementary Fig. 8b). This implies that the availability of gas-phase CO2 primarily affects CO2R reactivity rather than the reaction pathway. In another experiment, we saturated the electrolyte in the high-pressure H-cell with a mixture of 1 bar CO2 and 49 bar Ar (Supplementary Fig. 8c), so as to control the CO2 solubility the same as that under ambient pressure. The Cu, Au, and Ag catalysts are more formate selective than the ambient pressure scenarios, albeit to a reduced extent compared to the 50 bar CO2 case (Fig. 1d and Supplementary Fig. 8d). This indicates that, while the pressure-dependent CO2R performance can be partially explained by the higher availability of dissolved CO2, there may be other critical contributors. We speculate that the pressure might pose impact on the cathode/electrolyte interface and alter the adsorption energy of the intermediates.
To test this hypothesis, we sought to ascertain the species distributions and pH variations in the vicinity of the cathode surface using operando Raman spectroscopy. We built a Raman system employing an 18 W continuous-wave laser as the excitation source to realize a >300 mm working distance (Supplementary Fig. 9)44. This setup enhanced the signal-to-noise ratio compared to conventional ones: the incident laser beam was highly focused and transmitted along the cathode surface, and the scattered Raman signals were collected, collimated, and screened in the perpendicular direction (Fig. 1a and Supplementary Fig. 9; details in the Supplementary Materials).
With this operando Raman platform, we examined the Cu catalyst in 0.5 M KHCO3 saturated with 50 bar CO245. When the laser beam is positioned at the cathode surface (details in the Supplementary Materials), the acquired blended Raman spectrum at −0.9 V vs. RHE displays features of HCOO−, dissolved CO2, and HCO3− (Fig. 2a and Supplementary Fig. 10). In particular, a strong characteristic peak of HCOO− emerges at 1356 cm−1, arising from the C − O symmetric stretch46, which cannot be observed under the same conditions without applying a potential (Fig. 2a). This HCOO− peak is rarely reported using commercial Raman spectrometers because it is overlaid by a HCO3− peak at 1368 cm−1 (Supplementary Fig. 10). When switching to more negative potentials of −1.0 and −1.1 V vs. RHE, the peak intensity of HCOO− at 1356 cm−1 increases whereas that of the dissolved CO2 at 1280 cm−1 decreases (insets of Fig. 2a). The HCO3− peak intensity at 1019 cm−1 varies negligibly throughout the experiment, as indicated by the unchanged peak profile (Fig. 2a). No CO32− signal is found, primarily due to the saturation of CO2 (Fig. 2a and Supplementary Fig. 10). No other CO2R liquid products are detected, such as methanol and ethanol47, in agreement with the CO2R performance of Cu under high pressure (Fig. 1b). These results confirm that the carbon source of HCOO− originates from dissolved CO2, instead of HCO3− or CO32− in the electrolyte.

a Operando Raman spectra acquired on the Cu surface at open-circuit potential, −0.9, −1.0, and −1.1 V vs. RHE under 50 bar. Insets show the characteristic peaks of dissolved CO2 at 1280 cm−1 (C = O symmetric stretch) and HCOO− at 1356 cm−1 (C − O symmetric stretch). b Operando Raman spectrum acquired on the Cu surface at −1.1 V vs. RHE under 50 bar and its fitting curve. Dashed lines mark band assignments for dissolved CO2, HCO3−, and HCOO−. c Measured concentrations of dissolved CO2 and HCOO− against the distance from the Cu surface at −1.1 V vs. RHE under 50 bar. Error bars represent the standard deviation of thirty consecutive measurements.
We then applied the operando Raman system to quantify the HCOO−, dissolved CO2, and HCO3− concentrations in the vicinity of the cathode by a hybrid fitting and calibration method (Fig. 2b and Supplementary Figs. 11–13; details in the Supplementary Materials). This allows us to map the key species concentrations as a function of distance (x) from the cathode surface ranging from 0 μm (the surface) to 250 μm. At −1.1 V vs. RHE under 50 bar, the HCOO− concentration decreases from 0.032 M at x = 0 μm to 0.016 M at x = 150 μm, whereas the dissolved CO2 concentration increases from 0.59 M to 0.72 M (Fig. 2c). The absolute value of the HCOO− concentration gradient (0.12 mM μm−1) in the region of 0 ≤ x ≤ 150 μm is lower than that of the dissolved CO2 (0.81 mM μm−1), possibly because the dissolved CO2 is adsorbed on the cathode as a reactant, whereas HCOO− is released to the electrolyte as a product. We also used the species concentration profiles and equilibrium constants to depict the pH variations as a function of x11,19, suggesting a local pH of 12.3 on the cathode surface.
These findings motivated us to investigate the CO2R reaction mechanism at high pressure using DFT. In CO2R, oxygen or carbon atoms of CO2 can be protonated to *COOH or *HCOO, respectively—the key intermediates for CO2R in branching to *CO vs. HCOOH pathways (Supplementary Fig. 14)48. In light of the pressure-dependent CO2 solubility and local microenvironment near the cathode surface (Fig. 2c), we sought to explore the impact of CO2 coverage on CO2R on a Cu(111) facet—the dominant facet of the as-prepared Cu catalyst (Supplementary Fig. 1f). As depicted in Fig. 3a, b, the calculated energy diagrams on the periodic Cu(111) surface indicate the potential-determining steps (PDS) for two CO2R pathways—the formation of *COOH for *CO pathway (PDSCO) and the hydrogenation of *HCOO for HCOOH pathway (PDSHCOOH), as seen in previous reports49. The free energies of forming *COOH and *HCOO both increase with the CO2 coverage varying from 1/9 monolayer (ML) to 3/9 ML. However, with higher CO2 coverages, the free energy change (ΔG) of PDSCO increases, while that of PDSHCOOH decreases. Specifically, at a CO2 coverage of 1/9 ML, the ΔG of PDSCO is notably lower than that of PDSHCOOH, indicating that the *CO pathway is dominant. The situation is reversed when the CO2 coverage gradually increases to 3/9 ML, where the ΔG of PDSCO increases to 1.04 eV and that of PDSHCOOH decreases to 0.75 eV—that means, the *CO pathway become more difficult whereas HCOOH production become more energetically favorable (Fig. 3c). The DFT models reveal that the pressure-dependent CO2 coverage plays a crucial role in shifting the CO2R product selectivity towards formate/formic acid. It is important to note that due to the imprecise portrayal of carbon-oxygen double bonds in DFT, we focus on the variation trends of the free energies, instead of their absolute values50. We then studied the effect of CO2 coverage on the side reaction—the hydrogen evolution reaction (HER; Supplementary Fig. 15). The free energy diagram was calculated based on the Langmuir-Hinshelwood (LH)-type mechanism, and the adsorption of *H was identified as the PDS for HER (\({{{{{{\rm{PDS}}}}}}}_{{{{{{{\rm{H}}}}}}}_{2}}\)). As the CO2 coverage increases from 0 to 2/9 ML, the adsorption strength of *H is slightly decreased, but no further decrease is observed at a higher CO2 coverage of 3/9 ML (Fig. 3d). The insignificant ΔG of \({{{{{{\rm{PDS}}}}}}}_{{{{{{{\rm{H}}}}}}}_{2}}\) suggests the weak impact of CO2 coverages on HER, which is consistent with the experimental observation of the slight decrease in H2 selectivity under elevated pressures. (Supplementary Fig. 16). The interplay of electrochemical measurements, quantitative operando Raman studies, and theoretical calculations elucidate how pressure regulates CO2R pathways: Elevating CO2 pressure increases the availability of dissolved CO2, favoring the formate formation and stimulating the CO2R reactivity. Higher CO2R reactivity, with its concomitant faster OH− production, alkalizes the microenvironment near the cathode surface.

a Free energy diagram from CO2 toward *CO. b Free energy diagram from CO2 toward HCOOH. c Free energy changes of PDS in CO2R for *CO or HCOOH production on Cu(111) with various CO2 coverages. d Free energy diagram of HER on Cu(111) with various CO2 coverages.
Theory-guided electrode design
The reaction mechanism and experimental results imply that further elevating pressure beyond 50 bar can increase CO2 coverage and formate productivity (Supplementary Fig. 16). On the other hand, the selectivity of formate under high pressure is predominantly constrained by competing HER, which is weakly influenced by CO2 coverage and pressure (Fig. 3d). Therefore, we sought to retrofit the Cu cathode surface to suppress HER, so that the formate yield under high pressure can be further improved. We turned our attention to polypyrrole (PPy), capable of limiting the diffusion of excess protons to the electrode surface with its electropositive pyrrole-N group51,52. We posited that the controlled assembly of PPy and Cu can lower the local proton concentration near the Cu surface, thus inhibiting the HER (Fig. 4a)19,53 and promoting the formate selectivity. To test this postulation, we used an electrochemical anodization method to grow an ultrathin PPy layer on the surface of the Cu catalyst (Supplementary Fig. 17a; details in the Materials and Methods). Fourier transform infrared (FTIR) spectroscopy confirms the formation of PPy (Supplementary Fig. 17b). TEM and scanning TEM (STEM) images, and the corresponding electron energy loss spectroscopy (EELS) mapping indicate an epitaxial growth of PPy on the Cu surface with a thickness <2 nm (Supplementary Fig. 17c, d). The deconvolved N 1 s and C 1 s peaks in XPS spectra reveal the presence of polaron (C–N+) and bipolaron (C = N+) in PPy (Fig. 4b). STEM and HRTEM images show no noticeable structural change of Cu on the as-prepared Cu/PPy catalyst (Supplementary Figs. 18a, b). Cu 2p XPS spectra indicate that the PPy layer does not substantially alter electronic structure of the Cu surface (Supplementary Fig. 18c), consistent with the theoretical simulation of the charge density difference (Supplementary Fig. 18d).

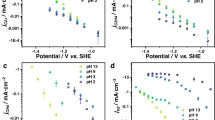
a Schematic of the proton-resistant cathode surface functionalized by the PPy layer. b N 1 s and C 1 s XPS spectra of the Cu/PPy catalyst. FEs and partial current densities toward formate on Cu and Cu/PPy catalysts as a function of c pressure at −1.1 V vs. RHE, and d cathode potential under 50 bar. j denotes the current density.
Using operando Raman spectroscopy, we determine that the local HCOO− concentration on the Cu/PPy surface (0.057 M) is higher than that of Cu (0.032 M) at −1.1 V vs. RHE under 50 bar, validating the function of PPy to promote formate production (Supplementary Fig. 19). This phenomenon translates to other cathode potentials ranging from −0.9 to −1.2 V vs. RHE (Supplementary Fig. 19). We then evaluated the CO2R performance of Cu/PPy in the high-pressure H-cell. The Cu/PPy catalyst exhibits a notable increase in formate selectivity and productivity compared to Cu over a wide range of pressures from 10 to 50 bar (Fig. 4c) at −1.1 V vs. RHE. Under 50 bar, the FE toward formate surpasses 82% at −1.1 V vs. RHE, and the formate partial current density exceeds 200 mA cm−2 at −1.21 V vs. RHE (Fig. 4d). In contrast, the Cu catalyst is limited to FEs <70% and partial current densities <130 mA cm−2 toward formate (Fig. 1b). Bare PPy, on the other hand, shows almost no CO2R activity on its own (Supplementary Fig. 20), something we attribute to its weak adsorption of CO2R intermediates (Supplementary Fig. 18e).
We then integrated the Cu/PPy cathode into a narrow-gap aqueous flow cell (Fig. 5a, Supplementary Fig. 21a, b). The flow cell employed 1 M KHCO3 and 0.5 M K2SO4 saturated with 50 bar CO2 as the catholyte and anolyte, respectively, and RuO2/Ti foam as the anode. The catholyte and anolyte channels were ultraslim ( ~ 0.3 mm) to minimize the ohmic loss (Supplementary Fig. 21c). The narrow-gap aqueous flow cell manifests a maximal formate FE of 84.7% with a full cell voltage of 2.85 V at 200 mA cm−2 (Fig. 5b and Supplementary Fig. 22a). The formate partial current density reaches 310 mA cm−2 with a FE of 77.5% and a cell voltage of 3.85 V at 400 mA cm−2 (Fig. 5b and Supplementary Fig. 22a). The cell voltage and formate FE remain largely stable over the course of a 12 h chronopotentiometric operation at 400 mA cm−2 (Fig. 5c). Formate is confirmed to be the only liquid product (Fig. 5b and Supplementary Fig. 22b).

a Schematic of the high-pressure narrow-gap aqueous flow cell. b FEs and partial current densities at total current densities of 200, 300, 400 and 500 mA cm−2 on Cu/PPy in the high-pressure narrow-gap aqueous flow cell fed by 50 bar PCO2. No other liquid products were detected. j denotes the current density. c Prolonged electrolysis of 50 bar PCO2 to formate on Cu/PPy in the narrow-gap aqueous flow cell at a constant current density of 400 mA cm−2. The full cell voltage and formate FE were well retained over 12 h continuous operation.
Discussion
In summary, we report here on the role of pressure in regulating aqueous-based CO2R pathways that is catalyst independent. We developed operando methods for high-pressure conditions, including a quantitative operando Raman system capable of probing the local microenvironment near the electrode in high-pressure aqueous-based CO2R cells. The work sheds light on cathode design principles and suggests further avenues for commodity chemicals from PCO2.
Methods
Electrodes preparation
Metal electrodes were prepared using galvanostatic electrodeposition in a three-electrode setup using an electrochemical workstation (BioLogic SP-150 Potentiostat). Commercial Cu foam (MTI Corporation, 99.9%, 0.30 mm thick) or carbon paper (Toray 120, Fuel Cell Store, 0.30 mm thick) was used as the working electrode. An Ag/AgCl electrode (CH Instruments, saturated KCl) was used as the reference electrode, and a Pt foil (Tianjin Aida Hengsheng Technology Development Co., Ltd, >99.99%, 1 × 1 cm) was used as the counter electrode. All chemicals and reagents were used as received without further purification.
For the Cu electrode, a piece of Cu foam (1 × 2 cm) was washed by acetone (Sigma-Aldrich, ≥ 99.5%), ethanol (Sigma-Aldrich, ≥99.8%), 0.5 M HCl (Sigma-Aldrich, 37%), and deionized water (Millipore, 18.2 MΩ cm) successively, each for 5 min under sonication. The Cu foam was then electrochemically anodized in 1.0 M KOH (Sigma-Aldrich, ACS reagent) for 20 min at 10 mA cm−2 to obtain Cu(OH)2 nanowire arrays. Finally, the sample was electrochemically reduced in 0.5 M KHCO3 (Macklin Inc., ≥99.9%) for 20 min at −5 mA cm−2 to form the Cu electrode. For Cu/PPy electrode, the aforementioned Cu(OH)2 nanowire arrays were further anodized in 0.1 M pyrrole (Alfa Aesar, > 98.0%) and 0.01 M KOH for 2 min at 5.0 mA cm−2, followed by being reduced in 0.5 M KHCO3 for 20 min at −5 mA cm−2 (Supplementary Fig. 17a).
For the Au electrode, the dendritic Au nanoparticles (Supplementary Fig. 3a) were formed on carbon paper (1 × 2 cm) through galvanostatic electrodeposition for 20 min at −5.0 mA cm−2. The electrolyte solution consisted of (i) 50 mM HAuCl4 (Alfa Aesar, 99.9%) dissolved in 0.1 M H2SO4 (Sigma-Aldrich, 95.0–98.0%) as the Au precursor and (ii) 0.15 mM Pluronic F-127 (Innochem Co., Ltd., average molecular weight ~10,000) as the structure-directing agent.
For the Ag electrode, the dendritic Ag nanoparticles (Supplementary Fig. 3b) were formed on carbon paper using the same protocol as the Au electrode, except for the electrolyte solution. The electrolyte solution was 50 mM AgNO3 (Sigma-Aldrich, ≥99.0%) dissolved in 0.4 M NH3·H2O (Sigma-Aldrich, 28.0–30.0% NH3 basis) and 0.15 mM Pluronic F-127.
For Sn electrode, the hierarchical flake-like Sn nanoparticles (Supplementary Fig. 3c) were formed on carbon paper using the same protocol as the Au and Ag electrodes, except for the electrolyte solution. The electrolyte solution was 50 mM SnCl2 (Innochem Co., Ltd., 99%) dissolved in 0.4 M Na4P2O4 (Sigma-Aldrich, ≥95.0%) and 0.15 mM Pluronic F-127.
Material characterization
Scanning electron microscopy (SEM) images were collected using FEI Quanta 600 FEG ESEM operated at 15 kV. Transmission electron microscopy (TEM) images were acquired using FEI Tecnai G2 Spirit Twin operated at 120 kV. High-resolution TEM (HRTEM), scanning TEM (STEM), and electron energy loss spectroscopy (EELS) were performed by FEI Titan 80-300 equipped with a field emission gun and spherical aberration corrector operated at 300 kV. EELS mapping was collected using a post-column filter in diffraction mode. X-ray photoelectron spectroscopy (XPS) was operated using Kratos Analytical AMICUS/ESCA 3400 equipped with an Mg-anode Kα excitation x-ray source (hν = 1253.6 eV) at 10 kV, 10 mA, and 2×10−6 Pa. The measured binding energies were calibrated based on C 1 s binding energy at 284.8 eV. X-ray powder diffraction (XRD) was carried out using the Bruker D8 Advance with a Cu Kα radiation. Fourier transform infrared (FTIR) spectroscopy was performed using the Thermo Scientific Nicolet 6700 FTIR spectrometer.
High-pressure H-cell
The customized two-compartment high-pressure H-cell (Fig. 1a and Supplementary Fig. 8c) was made of Teflon-lined titanium. The two compartments were separated by a proton exchange membrane (Nafion 117, Fuel Cell Store). Each compartment contained 120 mL of 0.5 M KHCO3 aqueous solution with a gas headspace <15 mL, and was connected to an independent pressure regulator. The working electrode (1 × 0.5 cm) and reference electrode (Ag/AgCl with saturated KCl, Gaoss Union) were placed inside the cathode compartment, and the counter electrode (Pt foil, 1 × 2 cm) was placed inside the anode compartment. A small hole (diameter <0.5 mm) was drilled on the top of the reference electrode to balance its internal and external pressure. Prior to the tests, electrolytes in each compartment were first purged for 5 min using the feed gas (99.995% CO2 or its mixture with 99.999% Ar, Air Liquide), and then saturated with the feed gas by stirring for 30 min to reach equilibrium under the desired pressure (from 1 to 50 bar). The pressure of the two compartments were kept identical. The gas products in the headspace of the cathode compartment were sampled using 10 mL air-tight syringes from an outlet relief valve, and then injected into a gas chromatograph (GC) system.
Ambient-pressure gas-fed flow cell
The customized ambient-pressure gas-fed flow cell comprised three compartments made of poly(methyl methacrylate) (PMMA): a gas compartment with serpentine channel, a catholyte compartment, and an anolyte compartment (Supplementary Fig. 8a). The size of each compartment was 0.5 cm (height) × 0.5 cm (width) × 1 cm (length). The working electrode was housed between the gas and catholyte compartments, with the catalyst layer facing the catholyte. An Ag/AgCl electrode (saturated KCl) and a RuO2/Ti foam (Gaoss Union, 1.5 × 1.5 cm) were used as the reference and counter electrode, respectively. The anolyte and catholyte compartments were separated by a Nafion 117 membrane. 0.5 M KHCO3 electrolytes were supplied to the catholyte and anolyte compartments and recirculated at a constant flow rate of 10 mL min−1 using a peristaltic pump (Longer Pump, BT100-2J). CO2 was delivered to the gas compartment at a constant flow rate of 20 sccm using a mass flow controller (Cole-Parmer, Masterflex Proportional Flowmeter Controller), and the gas effluents were extracted for detection by GC.
High-pressure narrow-gap aqueous flow cell
The high-pressure narrow-gas aqueous flow cell system (Supplementary Fig. 21a) consisted primarily of a narrow-gap aqueous flow cell, two high-pressure high-performance liquid chromatography pumps (HPLC pump; Sanotac SP6010), two backpressure valves (Beijing Xiong Chuan Technology Co. LTD), two Teflon-lined titanium tanks containing 1.0 M KHCO3 catholyte and 0.5 M K2SO4 anolyte, respectively, and a CO2 gas cylinder. The high-pressure narrow-gap aqueous flow cell was assembled by stacking the following components in order: a Cu/PPy cathode sandwiched by two polytetrafluoroethylene (PTFE) gaskets (with a 0.5 × 1 cm window as the reactive area) as the catholyte compartments, a Nafion 117 membrane, and a RuO2/Ti foam anode sandwiched by two aforementioned PTFE gaskets as the anolyte compartments (Fig. 5a and Supplementary Fig. 21b). These components were fixed and sealed by two titanium plates with channels and ports. The catholyte and anolyte were pressurized by high-pressure HPLC pumps and equilibrated by backpressure valves. Prior to each experiment, the air in the catholyte was purged out by bubbling CO2 under atmospheric pressure. Then, the pressures of CO2 and electrolytes were simultaneously and gradually increased by adjusting the gas cylinder valve and backpressure valves. Both cathode and anode compartments of the narrow-gas aqueous flow cell were pressurized to 50 bar and held for 30 min to achieve the equilibrium solubility of CO2 in catholyte. During CO2R, the electrolytes were recirculated at a constant flow rate of 10 mL min−1. The anodic O2 and cathodic CO2/CO2R products were discharged separately through the outlets of each backpressure valves. The CO2R gas products were collected by a syringe and analyzed by GC, while the liquid products were sampled by withdrawing the catholyte solution through the sampling port every hour and analyzed by NMR.
Electrochemical measurements
All electrochemical measurements were conducted using an electrochemical workstation (BioLogic SP-150 Potentiostat) at room temperature. Cathode potentials in three-electrode systems (H-cells and gas-fed flow cell) were recorded with iR compensation, where the cell resistance was determined using a current-interrupt method, and the potential was manually corrected after each measurement. And then converted to the reversible hydrogen electrode (RHE) scale using the following equation:
Full-cell voltages of the high-pressure narrow-gap aqueous flow cell were measured using the chronopotentiometry method. The currents were normalized to the geometric area of the working electrodes.
Product analysis
Gas products were analyzed using a GC (Trace 1310, Thermo Scientific) equipped with Molecular Sieve 5 A and Porapak N columns. Ar (Al Khafrah Industrial Gases, 99.999%) was used as the carrier gas. CO, CH4, C2H4, C2H6, C3H6, and C3H8 were quantified using a flame ionization detector (FID) with a methanizer. H2 was quantified using a thermal conductivity detector (TCD). The volumes of gas products were derived from the output peak areas based on calibration curves.
Liquid products were analyzed by a 1H NMR (Bruker, 600 MHz) using water suppression method. Each liquid sample was prepared by mixing 490 µL of the electrolytes with 110 µL of the internal standards (20 ppm of dimethyl sulfoxide in D2O).
The Faradaic efficiency (FE) of a specific product (p) was calculated using the following equation:
where z denotes the number of the electrons transferred to one p molecule, n represents the total moles of the product, F is the Faradaic constant (F = 96,485 C mol−1), and Q indicates the total number of electrons transferred.
Operando Raman spectroscopy
The schematic of the custom-built operando Raman system is illustrated in Supplementary Fig. 9. The high-pressure H-cell was placed on a mechanical sample stage. The excitation source was a 532 nm continuous-wave narrow-band laser (Coherent, Inc. Verdi G18) with a maximum power of 18 W. The laser was highly focused by a spherical convex lens (focus length f = 500 mm) in the probing region and transmitted along the cathode surface. To locate the cathode surface (x = 0 µm), we first moved the sample stage until the laser beam was cut by the cathode, and then moved the sample stage backward until the laser beam fully appeared. The species concentrations against the distance from the cathode surface were acquired by controlling the sample stage for line scan. The Raman signal was collected by a Nikon micro lens (f = 105 mm, F#2.8) through the perpendicular window of the cathode compartment (Fig. 1a, Supplementary Figs. 8c and 9) and collimated by a digital single-lens reflex (DSLR) prime lens (Samyang, f = 135 mm, F#2.2). The combination of a 532 nm notch filter with a half-wave plate (HWP) and a wire grid polarizer was placed in the collimated beam to filter out the Rayleigh signal and stray light background. The beam was then rotated by 90° and passed through the slit of an astigmatism-free spectrometer (Princeton Instrument Isoplane 320). The design of the spectrometer avoided the bowing effect and allowed the integration along the spatial direction without degrading the spectral resolution. An electron multiplication charge-coupled devices (EMCCD) camera (Princeton Instruments, ProEM:1600 200) imaged the Raman signal with a 0.01 nm per pixel dispersion along the spectral direction. Full binning along the laser propagation direction and 1 s exposure time were applied to ensure a high signal-to-noise ratio54. The spatial resolution along the laser propagation was about 8.6 mm, and the resolution along the vertical direction was around 50 μm.
As depicted in Supplementary Fig. 10, some Raman peaks of the key species (HCO3−, HCOO−, and dissolved CO2) overlapped. We therefore employed a hybrid fitting and calibration method to rigorously convert the areas under the Raman peak to concentrations. First, the Raman spectra were acquired for standard aqueous solutions of KHCO3, HCOOK, and dissolved CO2 with known concentrations (Supplementary Fig. 11). Taking HCOOK as an example, the spectra of its ν2 and v5 bands at a concentration of 0.4 M were fitted with the summation of the corresponding Voigt functions (Supplementary Fig. 12a), which settled the parameters of the center wavelength, Raman line shape, and area ratio of the two bands. Then, we fitted the spectra of 0.025, 0.05, 0.1, 0.2 and 0.3 M HCOOK based on the as-obtained parameters and set the peak area as the free parameter. In this way, the relationship between the area of fitted function and the HCOO− concentration was established (Supplementary Fig. 13a). Following the same procedure, we can quantify the concentrations of other species in the vicinity of the cathode surface.
Theoretical methods
All density functional theory (DFT) calculations were performed by the Vienna ab initio simulation program (VASP)55,56. The core-valence interactions were calculated by the project augmented wave (PAW) method with 450 eV as the cut-off energy57,58. The generalized gradient approximation in the Perdew–Burke–Ernzerhof functional (GGA-PBE) was applied to describe the exchange-correlation correction effect59. The DFT-D3 method was used to consider the dispersion correction of the van der Waals force60. For the geometry optimization, the self-consistent iteration must reach 10−6 eV for the energy convergence and 0.01 eV Å−1 for the force convergence.
The (3 × 3) Cu(111) model consisted of four Cu atomic layers, where the two bottom layers were fixed to mimic the bulk material and the rest of atoms were relaxed. To account for both explicit solvation and field effects, we incorporated one charged water layer onto the Cu(111) surface at the intermediates according to studies of Nørskov et al.61, where the optimal water structure are obtained via using a minima-hopping algorithm49. Here, the periodic structure of one charged water layer, consisting of five water molecules and one hydronium molecule in the (3 × 3) cell, closely resembles the hexagonal ice-like structure using previously in various DFT-based studies of adsorption and proton-coupled electron transfer (PCET) kinetics on Pt(111)62,63, and has been widely used in the CO2R studies64. The vacuum space was set at about 15 Å in z-axis to avoid interactions between the periodic images. To investigate the impact of CO2 coverage, we conducted a comprehensive analysis involving the adsorption of one, two, or three CO2 molecules onto the periodic (3 × 3) Cu(111) surface. Two key reaction pathways of CO2R toward *CO or HCOOH were considered as follows:
CO2 to HCOOH:
CO2 to *CO:
Additionally, the elementary steps of HER were listed as below:
where * represents the active site or the adsorbed intermediate. The proton-electron pair was treated with the computational hydrogen electrode (CHE) model49. The PDS in the three reaction pathways was identified based on the most positive change in free energy. A more positive change in PDS indicated a more difficult reaction pathway. The optimized structures for all reactions were presented in the Supplementary Figs. 14 and 15. The free energy change of each elementary step for the production of *CO, HCOOH(l), or H2(g) was obtained by the correction of Gibbs free energy (G) at room temperature (T = 298.15 K), using the following equation:
where EDFT is the energy changes of DFT calculations, ZPE is the zero-point energy, Cp is the heat capacity, and S is the entropy.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
Code availability
Vienna ab initio simulation package (VASP) for the DFT calculations is available at https://www.vasp.at/.
References
Masel, R. I. et al. An industrial perspective on catalysts for low-temperature CO2 electrolysis. Nat. Nanotechnol. 16, 118–128 (2021).
Bushuyev, O. S. et al. What should we make with CO2 and how can we make it? Joule 2, 825–832 (2018).
Wang, G. et al. Electrocatalysis for CO2 conversion: from fundamentals to value-added products. Chem. Soc. Rev. 50, 4993–5061 (2021).
Garcia de Arquer, F. P. et al. CO2 electrolysis to multicarbon products at activities greater than 1 A cm−2. Science 367, 661–666 (2020).
Gao, D., Arán-Ais, R. M., Jeon, H. S. & Roldan Cuenya, B. Rational catalyst and electrolyte design for CO2 electroreduction towards multicarbon products. Nat. Catal. 2, 198–210 (2019).
Birdja, Y. Y. et al. Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels. Nat. Energy 4, 732–745 (2019).
De Luna, P. et al. What would it take for renewably powered electrosynthesis to displace petrochemical processes? Science 364, 350 (2019).
Chen, C., Khosrowabadi Kotyk, J. F. & Sheehan, S. W. Progress toward commercial application of electrochemical carbon dioxide reduction. Chem 4, 2571–2586 (2018).
Gao, W. et al. Industrial carbon dioxide capture and utilization: state of the art and future challenges. Chem. Soc. Rev. 49, 8584–8686 (2020).
Smith, W. A., Burdyny, T., Vermaas, D. A. & Geerlings, H. Pathways to industrial-scale fuel out of thin air from CO2 electrolysis. Joule 3, 1822–1834 (2019).
Huang, J. E. et al. CO2 electrolysis to multicarbon products in strong acid. Science 372, 1074–1078 (2021).
Chen, X. et al. Electrochemical CO2-to-ethylene conversion on polyamine-incorporated Cu electrodes. Nat. Catal. 4, 20–27 (2020).
Li, J., Guo, J. & Dai, H. Probing dissolved CO2(aq) in aqueous solutions for CO2 electroreduction and storage. Sci. Adv. 8, eabo0399 (2022).
Verma, S., Lu, S. & Kenis, P. J. A. Co-electrolysis of CO2 and glycerol as a pathway to carbon chemicals with improved technoeconomics due to low electricity consumption. Nat. Energy 4, 466–474 (2019).
Snyder, B. F., Layne, M. & Dismukes, D. E. A cash flow model of an integrated industrial CCS-EOR project in a petrochemical corridor: a case study in Louisiana. Int. J. Greenh. Gas Con. 93, 102885 (2020).
Sun, Z., Ma, T., Tao, H., Fan, Q. & Han, B. Fundamentals and challenges of electrochemical CO2 reduction using two-dimensional materials. Chem 3, 560–587 (2017).
Kibria, M. G. et al. Electrochemical CO2 reduction into chemical feedstocks: from mechanistic electrocatalysis models to system design. Adv. Mater. 31, e1807166 (2019).
Duan, Z., Sun, R., Zhu, C. & Chou, I. M. An improved model for the calculation of CO2 solubility in aqueous solutions containing Na+, K+, Ca2+, Mg2+, Cl−, and SO42−. Mar. Chem. 98, 131–139 (2006).
Gu, J. et al. Modulating electric field distribution by alkali cations for CO2 electroreduction in strongly acidic medium. Nat. Catal. 5, 268–276 (2022).
Gabardo, C. M. et al. Combined high alkalinity and pressurization enable efficient CO2 electroreduction to CO. Energy Environ. Sci. 11, 2531–2539 (2018).
Dufek, E. J., Lister, T. E., Stone, S. G. & McIlwain, M. E. Operation of a pressurized system for continuous reduction of CO2. J. Electrochem. Soc. 159, F514–F517 (2012).
Ramdin, M. et al. High pressure electrochemical reduction of CO2 to formic acid/formate: a comparison between bipolar membranes and cation exchange membranes. Ind. Eng. Chem. Res. 58, 1834–1847 (2019).
Hara, K., Tsuneto, A., Kudo, A. & Sakata, T. Electrochemical reduction of CO2 on a Cu electrode under high-pressure: factors that determine the product selectivity. J. Electrochem. Soc. 141, 2097–2103 (1994).
Todoroki, M., Hara, K., Kudo, A. & Sakata, T. Electrochemical reduction of high pressure CO2 at Pb, Hg and In electrodes in an aqueous KHCO3 solution. J. Electroanal. Chem. 394, 199–203 (1995).
Kudo, A., Nakagawa, S., Tsuneto, A. & Sakata, T. Electrochemical reduction of high pressure CO2 on Ni electrodes. J. Electrochem. Soc. 140, 1541–1545 (1993).
Hara, K., Kudo, A. & Sakata, T. Electrochemical reduction of carbon dioxide under high pressure on various electrodes in an aqueous electrolyte. J. Electrochem. Soc. 391, 141–147 (1995).
Scialdone, O. et al. Electrochemical reduction of carbon dioxide to formic acid at a tin cathode in divided and undivided cells: effect of carbon dioxide pressure and other operating parameters. Electrochim. Acta 199, 332–341 (2016).
Proietto, F., Rinicella, R., Galia, A. & Scialdone, O. Electrochemical conversion of CO2 to formic acid using a Sn based cathode: Combined effect of temperature and pressure. J. CO2 Util. 67, 102338 (2023).
Mizuno, T. et al. Effect of temperature on electrochemical reduction of high-pressure CO2 with In, Sn, and Pb electrodes. Energy Sources 17, 503–508 (1995).
Köleli, F. & Balun, D. Reduction of CO2 under high pressure and high temperature on Pb-granule electrodes in a fixed-bed reactor in aqueous medium. Appl. Cataly. A-Gen. 274, 237–242 (2004).
Siahrostami, S., Moshar, A. & Farmand, M. M. High pressure-high temperature CO2 electroreduction on Sn granules in a fixed-bed reactor. Fresen. Environ. Bull. 12, 1202–1206 (2003).
Morrison, A. R. T. et al. Modeling the electrochemical conversion of carbon dioxide to formic acid or formate at elevated pressures. J. Electrochem. Soc. 166, E77–E86 (2019).
Proietto, F., Galia, A. & Scialdone, O. Electrochemical conversion of CO2 to HCOOH at Tin cathode: Development of a theoretical model and comparison with experimental results. ChemElectroChem 6, 162–172 (2019).
Li, J. et al. Electroreduction of CO2 to formate on a copper-based electrocatalyst at high pressures with high energy conversion efficiency. J. Am. Chem. Soc. 142, 7276–7282 (2020).
Handoko, A. D., Wei, F., Jenndy, Yeo, B. S. & Seh, Z. W. Understanding heterogeneous electrocatalytic carbon dioxide reduction through operando techniques. Nat. Catal. 1, 922–934 (2018).
Deng, Y. & Yeo, B. S. Characterization of electrocatalytic water splitting and CO2 reduction reactions using in situ/operando Raman spectroscopy. ACS Catal. 7, 7873–7889 (2017).
Lu, X. et al. In situ observation of the pH gradient near the gas diffusion electrode of CO2 reduction in alkaline electrolyte. J. Am. Chem. Soc. 142, 15438–15444 (2020).
Lei, Q. et al. Investigating the origin of enhanced C2+ selectivity in oxide-/hydroxide-derived copper electrodes during CO2 electroreduction. J. Am. Chem. Soc. 142, 4213–4222 (2020).
Vavra, J., Shen, T. H., Stoian, D., Tileli, V. & Buonsanti, R. Real‐time monitoring reveals dissolution/redeposition mechanism in copper nanocatalysts during the initial stages of the CO2 reduction reaction. Angew. Chem. Int. Ed. 133, 1367–1374 (2020).
Kim, C. et al. Tailored catalyst microenvironments for CO2 electroreduction to multicarbon products on copper using bilayer ionomer coatings. Nat. Energy 6, 1026–1034 (2021).
Qiao, J., Liu, Y., Hong, F. & Zhang, J. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 43, 631–675 (2014).
Han, N., Ding, P., He, L., Li, Y. & Li, Y. Promises of main group metal-based nanostructured materials for electrochemical CO2 reduction to formate. Adv. Energy Mater. 10, 1902338 (2019).
Seifitokaldani, A. et al. Hydronium-induced switching between CO2 electroreduction pathways. J. Am. Chem. Soc. 140, 3833–3837 (2018).
Yang, C., Ezendeeva, D., Yu, T. & Magnotti, G. Temperature dependent Raman spectra of ammonia ranging from 3150 cm(-1) to 3810 cm(-1) for combustion applications. Opt. Express 29, 33234–33244 (2021).
Khan, I. Strategies for improved electrochemical CO2 reduction to value-added products by highly anticipated copper-based nanoarchitectures. Chem. Rec. 22, e202100219 (2022).
Yasaka, Y., Saito, Y. & Kimura, Y. Role of hydrogen-bond interactions in CO2 capture by wet phosphonium formate ionic liquid: a Raman spectroscopic study. Chemphyschem 19, 1674–1682 (2018).
Emin, A., Hushur, A. & Mamtimin, T. Raman study of mixed solutions of methanol and ethanol. AIP Adv. 10, 065330 (2020).
Vasileff, A. et al. Selectivity control for electrochemical CO2 reduction by charge redistribution on the surface of copper alloys. ACS Catal. 9, 9411–9417 (2019).
Peterson, A. A., Abild-Pedersen, F., Studt, F., Rossmeisl, J. & Nørskov, J. K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 3, 1311–1315 (2010).
Yoo, J. S., Christensen, R., Vegge, T., Norskov, J. K. & Studt, F. Theoretical insight into the trends that guide the electrochemical reduction of carbon dioxide to formic acid. ChemSusChem 9, 358–363 (2016).
Kim, D. H. et al. Facile surface modification of anion-exchange membranes for improvement of diffusion dialysis performance. J. Colloid Interface Sci. 416, 19–24 (2014).
Jo, A. et al. Enhanced electrochemical conversion of CO2 to CO at bimetallic Ag-Zn catalysts formed on polypyrrole-coated electrode. J. Catal. 393, 92–99 (2021).
Otero, T. F., Martinez, J. G., Fuchiwaki, M. & Valero, L. Structural electrochemistry from freestanding polypyrrole films: full hydrogen inhibition from aqueous solutions. Adv. Funct. Mater. 24, 1265–1274 (2014).
Yang, C., Tang, H. & Magnotti, G. High-speed 1D Raman analyzer for temperature and major species measurements in a combustion environment. Opt Lett 45, 2817–2820 (2020).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev., B Condens. Matter 47, 558–561 (1993).
Kresse, G. & Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B Condens. Matter 49, 14251–14269 (1994).
Blochl, P. E. Projector augmented-wave method. Phys. Rev. B Condens. Matter 50, 17953–17979 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Montoya, J. H., Shi, C., Chan, K. & Norskov, J. K. Theoretical insights into a CO dimerization mechanism in CO2 electroreduction. J. Phys. Chem. Lett. 6, 2032–2037 (2015).
Gohda, Y., Schnur, S. & Gross, A. Influence of water on elementary reaction steps in electrocatalysis. Faraday Discuss. 140, 233–244 (2009).
Bondarenko, A. S. et al. The Pt(111)/electrolyte interface under oxygen reduction reaction conditions: an electrochemical impedance spectroscopy study. Langmuir 27, 2058–2066 (2011).
Xie, Y. et al. High carbon utilization in CO2 reduction to multi-carbon products in acidic media. Nat. Catal. 5, 564–570 (2022).
Acknowledgements
This work was financially supported by the Baseline Fund (BAS/1/1413-01-01) and Center Competitive Fund (URF/1/1975-16-01) to X.L. from King Abdullah University of Science and Technology (KAUST). E.H.S. and D.S. acknowledge the Natural Sciences and Engineering Research Council (NSERC) of Canada and the Ontario Research Fund: Research Excellence Program. DFT calculations were performed on the Niagara supercomputer at the SciNet HPC Consortium. G.M. thanks the KAUST Research Funding Office (URF/1/3715-01-01 and BAS/1/1388-01-01).
Author information
Authors and Affiliations
Contributions
X.L., E.H.S., D.S., and G.M. supervised the project. X.L. and L.H. conceived the idea. L.H. and G.G. fabricated and characterized the samples and conducted electrochemical experiments. C.Y. and L.H. carried out the operando Raman measurements. X.Y.L. and P.O. performed the theoretical calculations. R.K.M. and Y.X. assisted the electrode assembly in the narrow-gap aqueous flow cell. C.T.Y., Y.H., R.K.M., and K.X. contributed to data analysis and manuscript editing. L.H. and X.L. wrote the manuscript. All authors discussed the results and assisted with the manuscript preparation.
Corresponding authors
Ethics declarations
Competing interests
X.L. has filed provisional patent application no. 63/244,449 regarding the high-pressure electrochemical CO2 reduction system. The authors declare no other competing interests.
Peer review
Peer review information
Nature Communications thanks the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, L., Gao, G., Yang, C. et al. Pressure dependence in aqueous-based electrochemical CO2 reduction. Nat Commun 14, 2958 (2023). https://doi.org/10.1038/s41467-023-38775-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-38775-0
This article is cited by
-
Pressure-induced generation of heterogeneous electrocatalytic metal hydride surfaces for sustainable hydrogen transfer
Nature Communications (2024)
-
Steering electroreduction of carbon dioxide to valuable C3+ products
Science China Materials (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
