
Abstract
Current state-of-the-art de novo long read genome assemblers follow the Overlap-Layout-Consensus paradigm. While read-to-read overlap – its most costly step – was improved in modern long read genome assemblers, these tools still often require excessive RAM when assembling a typical human dataset. Our work departs from this paradigm, foregoing all-vs-all sequence alignments in favor of a dynamic data structure implemented in GoldRush, a de novo long read genome assembly algorithm with linear time complexity. We tested GoldRush on Oxford Nanopore Technologies long sequencing read datasets with different base error profiles sourced from three human cell lines, rice, and tomato. Here, we show that GoldRush achieves assembly scaffold NGA50 lengths of 18.3-22.2, 0.3 and 2.6 Mbp, for the genomes of human, rice, and tomato, respectively, and assembles each genome within a day, using at most 54.5 GB of random-access memory, demonstrating the scalability of our genome assembly paradigm and its implementation.
Similar content being viewed by others


Introduction
Short-read genome assembly methods typically struggle to resolve sequence repeats and often fail to generate assemblies that reach chromosome-scale1. Both prokaryotic and eukaryotic genomes can contain a large proportion of repeats2, with the human genome estimated to be 66–69% repetitive3. Thus, it is imperative that these repetitive regions be sufficiently resolved for a successful de novo genome assembly. Innovations in bioinformatics have emerged to address this challenge, leveraging long-range evidence afforded by various data types, including linked reads4,5, Hi-C6, and long sequencing reads7,8.
Long-read sequencing technology has become increasingly prevalent in recent years. Sequencing throughput, affordability, and the long-read lengths are some of the key reasons9. The long-read lengths, ranging from kilobases to megabases, enable better resolution of structural variants10 and long repeats11. Long reads also enable correct and accurate identification of tandem repeat expansions12.
Oxford Nanopore Technologies (ONT), Plc. (Oxford, UK) and Pacific Biosciences (PacBio), Inc. (Menlo Park, USA) are currently the two preeminent providers of commercial long-read sequencing technology. PacBio generally produces long reads with lower base errors (8–13% and <1% for Continuous Long Reads (CLR) and HiFi, respectively), but with shorter read lengths (typically averaging 30–60 kbp and 10–25 kbp for CLR and HiFi, respectively)13,14 compared to that of ONT (typically 10–100+ kbp)15. Yet, high error rates (1–13%) in ONT reads remain a challenging obstacle to de novo genome assembly15,16,17. Unlike the de novo genome assembly strategies designed for short reads, both long-read sequencing technologies— and especially ONT, require algorithms and data structures that can accommodate mismatches and indels in the sequencing data.
Most long-read genome assemblers follow the Overlap-Layout-Consensus paradigm (OLC), a quadratic run time algorithm in its naïve implementation. OLC consists of three steps. The first step, overlap, typically generates an overlap graph by computing the pairwise alignment of all reads. As datasets often contain tens of millions of reads, finding and storing the detected overlaps is the most computationally- and memory-intensive step in the OLC paradigm, and has been the target of recent innovative algorithms18,19,20. In the second step, layout, the generated read overlap graph is traversed to produce contigs, or contiguous sequences, that reconstruct the underlying genome. The last step, consensus, uses read alignments to infer the most likely nucleotide bases across contigs, and corrects the sequences accordingly21,22.
In recent years, a number of OLC-based de novo long-read genome assemblers have been developed that leverage the long-range evidence provided by the technology. These tools include Flye18, Redbean19, and Shasta20. Each tool brings a different innovation to the table, with implementations of the OLC paradigm aiming to reduce the computational cost and address the high error rates of long reads. For instance, Flye clusters the long reads that are likely to originate from the same genomic locus in a preprocessing step to reduce the number of pairwise comparisons18. Redbean segments each read into 256 bp tiling subsequences, reducing the dynamic programming matrix to a size of 65536 ( = 256 × 256), thus speeding up the pairwise alignment process19. On the other hand, to address the high error rate of long reads, Shasta compresses all homopolymers in the reads using run-length encoding, thereby removing all homopolymer expansion errors, one of the more common error types in the ONT data, and improving the accuracy of alignments in the overlap step of OLC20. While these optimizations have reduced the time it takes to assemble the long sequencing reads and ultimately improve upon the quality of the generated genome assemblies, these tools still have a large memory footprint, requiring upwards of several hundred gigabytes of RAM for assembling a typical 50X human genome dataset.
Long sequencing reads have also enabled the haplotype phasing of haploid genome assemblies to better understand and characterize the genomic diversity of diploid and polyploid organisms23. Existing OLC long-read genome assemblers, such as Shasta24, and haplotype-aware genome assemblers, such as phasebook25 and Verkko26, have been extended or developed to leverage long reads to produce diploid assemblies.
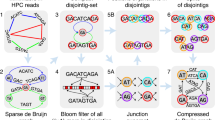
In this work, we present GoldRush, a memory-efficient long-read haploid de novo genome assembler that employs a novel long-read assembly algorithm, which runs in linear time in the number of reads. GoldRush is implemented as a modular pipeline with four main steps: GoldPath, GoldPolish, Tigmint-long7,27, and GoldChain7,28. GoldPath first iterates through the long reads and generates a “golden path”, selected sequences with a ~ 1X representation of the genome of interest. Because the output of GoldPath is a set of raw sequences (termed “goldtigs”), base errors in the golden path are resolved using GoldPolish, a long-read adaptation of the ntEdit+Sealer polishing protocol29. Next, misassemblies (due to chimeric reads) are corrected using Tigmint-long7,27. Finally, the corrected golden path is scaffolded using GoldChain to produce the output genome assembly7,28 (Fig. 1a and Supplementary Fig. 1). The golden path serves as the base assembly for the subsequent steps in GoldRush. Briefly, GoldPath iterates through the reads, querying each read against a dynamic and probabilistic multi-index Bloom filter30,31 (miBf) data structure in turn, and inserts selected sequence or skips over the read depending on the results of the query to generate multiple “silver paths”, which are <1X representations of the target genome (Fig. 1b). The silver paths are then used as input for GoldPath to generate the golden path. Iteration over the long sequencing reads, as opposed to an all-vs-all alignment of reads, allows GoldRush to achieve a linear time complexity in the number of reads. We show that GoldRush produces contiguous and correct genome assemblies with a low memory footprint, and does so without read-to-read alignments, marking an important paradigm shift in the genome assembly of long sequencing reads.

a Raw long reads are first processed by GoldPath to generate the golden path, a ~1X representation of the genome. The golden path is then polished by GoldPolish and corrected for structural errors with Tigmint-long. Finally, GoldChain scaffolds the polished and corrected golden path to generate the final genome assembly. b GoldPath uses the input long sequencing reads or silver path sequences to initialize a miBf data structure. GoldPath then loops over the sequences, and queries each sequence against the miBf. If the sequence is found in the miBf, GoldPath skips it and resumes its iterations. Conversely, if the sequence is not found in the miBf, it is inserted into the miBf and added to the silver/golden path. When GoldPath is constructing a silver path, and if the silver path has not reached the threshold number of bases, GoldPath will continue recruiting bases from the input reads. If the threshold number of bases is reached, GoldPath will check if more silver paths need to be generated. If more silver paths are needed, GoldPath will create them using the same algorithm and parameters, otherwise, it will terminate. Five (by default) silver paths, each representing ~0.9X (by default) coverage of the target genome, are combined to generate a low-coverage subsample input for GoldPath to build the golden path. When creating the golden path, GoldPath will continue iterating over the sequences from the silver paths until all sequences are exhausted.
Results
We assembled the genomes of three different human cell lines (NA24385, HG01243, and HG02055), Oryza sativa (rice), and Solanum lycopersicum (tomato) using GoldRush, Flye, Redbean, and Shasta, and compared the resulting genome assemblies using a variety of length contiguity metrics, genome assembly accuracy markers, including those reported by QUAST32, BUSCO33, the asmgene utility in minimap234, and Merqury35, and their resource usage (see Methods).
Assembly performance
For the genome assemblies of all three human cell lines, GoldRush achieved NG50 lengths between 25.3 and 32.6 Mbp, comparable to both Shasta (29.7–39.6 Mbp) and Flye (26.6–38.8 Mbp), and typically three times more contiguous than the Redbean genome assemblies (8.0–10.9 Mbp) (Supplementary Tables 1–3). Two of the three human GoldRush genome assemblies (NA24385 and HG01243) also had the fewest extensive misassemblies (940 and 1057) among the tools tested; ~2–3 times fewer than Shasta (1682 and 3240), and ~5–7 times fewer than Redbean (4918 and 7052). Despite the relatively low number of structural misassemblies, the NGA50 length for each human GoldRush assembly is around 20 Mbp, indicating that some misassemblies are found in the larger scaffolds and breaking the alignment blocks (Fig. 2a and Supplementary Tables 1–3). In addition to assembling highly contiguous human genomes, GoldRush is also robust in assembling plant genomes, reaching 0.3 and 2.6 Mbp NGA50 lengths for O. sativa and S. lycopersicum, respectively (Fig. 2c and Supplementary Tables 4,5). The O. sativa Shasta genome assembly, on the other hand, had scaffold NG50 and NGA50 lengths of 124,700 bp and 104,593 bp, respectively, only ~4.2 and ~3.6 times longer than the raw reads used as input (N50 = 29,349 bp) for genome assembly, respectively (Supplementary Tables 4,6).

Human individuals NA24385, HG01243, and HG02055 are represented as circles, triangles, and squares, respectively, in a and b. O. sativa and S. lycopersicum are represented as circles and triangles, respectively, in c and d. GoldRush, Flye, Redbean, and Shasta are colored yellow, purple, red, and blue, respectively. The genome assemblies were assessed using QUAST for their contiguity and correctness. Extensive misassemblies and NGA50 length, as determined by QUAST, are shown on the horizontal and vertical axes, respectively, in a and c. Wall clock run time (h) and peak memory (GB) usage of the genome assembly processes were recorded using the unix time command and shown on the horizontal and vertical axes, respectively, in b and d.
GoldRush and Shasta assembled each of the three human genomes in less than a day, executing in 16.6–20.8 h and 4.1–5.0 h, respectively (Fig. 2b and Supplementary Tables 7–9). Both Flye and Redbean required at least 33.7 h to assemble each of the three human genomes, with Redbean assembling two of the datasets (HG01243 and HG02055) in ~68 h, for each (Fig. 2b and Supplementary Tables 7–9). GoldRush is also competitive in assembling the smaller plant genomes, requiring 1.6 and 7.4 h to assemble O. sativa and S. lycopersicum, respectively (Fig. 2d and Supplementary Tables 10,11). GoldRush used, at most, 54.5 GB of RAM to assemble the three human genomes (Fig. 2b and Supplementary Tables 7–9). In comparison, using the same data, Flye and Redbean used between 329.3–502.4 GB (six- to eight-fold more than GoldRush), and Shasta utilized 884.8–1009.2 GB (up to 20-fold more than GoldRush). Similarly, GoldRush required the least amount of RAM to assemble the O. sativa and S. lycopersicum datasets, using at most 45.3 GB (Fig. 2d and Supplementary Tables 10, 11).
GoldPolish base error correction
GoldPolish decreased the number of mismatches or indels per 100 kbp of the NA24385 golden path by ~6.5-fold (1463.7 to 228.5 and 1327.2 to 197.2, respectively) (Supplementary Table 12). This improvement in mismatches and indels translated into a recovery of 12,272 (89.1%) complete BUSCOs, fewer than the 12,920 (93.8%) and 12,988 (94.3%) complete BUSCOs in the Shasta and Flye NA24385 genome assemblies, respectively, but more than the 12,193 (88.5%) complete BUSCOs reconstructed in the Redbean NA24385 genome assembly (Supplementary Table 13). Of the 2461 duplicated genes observed in T2T-CHM1336, a complete telomere-to-telomere reference-grade human genome assembly, 845 were found in the GoldRush assembly in multiple copies, fewer than the 1717 and 1725 found in the Shasta and Flye genome assemblies, respectively, but greater than the 680 found in the Redbean genome assembly (Supplementary Fig. 2 and Supplementary Table 14).
When polishing the genome assemblies of the more erroneous human long-read datasets, HG01243 (estimated 9% error rate) and HG02055 (estimated 11% error rate), GoldPolish reduced the number of mismatches and indels per 100 kbp by 60.8 and 67.4% (to 1372.3 and 980.1) for the former, and by 53.6 and 60.5% (to 1981.7 and 1354.8) for the latter, respectively (Supplementary Tables 6, 15,16). In comparison, the Flye, Redbean, and Shasta assemblies reported 148.7 and 110.6, 324.7 and 354.5, and 195.6 and 100.7 number of mismatches and indels per 100 kbp for HG01243, respectively, and 155.5 and 126.0, 346.1 and 379.1, and 210.2 and 103.9 number of mismatches and indels per 100 kbp for HG02055, respectively (Supplementary Tables 2, 3). The results of running BUSCO on the HG01243 and HG02055 genome assemblies can be found in Supplementary Tables 17,18.
When substituting GoldPolish with Racon37 for polishing the same golden path, the polishing step of the GoldRush pipeline incurred a greater computational cost, requiring over an order of magnitude more memory (602.3 vs 11.0 GB RAM) and taking 19.3% longer (9.9 vs 8.3 h) to complete, but resulted in a more base-accurate genome assembly (157.0 mismatches per 100 kbp and 106.4 indels per 100 kbp) (Fig. 3 and Supplementary Tables 19–21). The improvements in the base accuracy of the resulting NA24385 genome assembly also translated into a higher recovery of complete BUSCOs (12,752, 92.5% complete) (Supplementary Table 22). Polishing the NA24385 golden path with GoldPolish and Racon yielded QV (base quality value) statistics of 28.7 and 31.1, respectively, as assessed with Merqury (Supplementary Table 23).

The average peak memory (GB) for each stage of the assembly of the three human genomes is shown, with the data points (open circles) indicating the peak memory of each assembly and the error bars indicating the standard deviation. Racon can optionally be used for long-read base polishing within GoldRush, if the onboard memory of the computer system is not limited.
To characterize the polishing performance of GoldPolish and Racon in repetitive genomic regions, we compared the resulting QV statistics of the polished assemblies, specifically looking at the statistics in non-repetitive and repetitive regions. GoldPolish polishing yielded QV statistics of 27.7 and 30.0 for non-repetitive and repetitive regions, respectively (Supplementary Table 24). Racon polishing led to QVs of 30.3 and 32.1 for non-repetitive and repetitive regions, respectively (Supplementary Table 25).
Discussion
The GoldRush algorithm is straightforward: collect unique fragments representing the genome to generate a golden path, polish the fragments, correct them for structural misassemblies, and join the polished and corrected fragments together. As GoldRush is built upon this fundamental concept of the golden path, it represents a paradigm shift in the genome assembly of erroneous long reads, no longer requiring the time- and memory-intensive process of all-vs-all sequence alignments. Instead, the golden path, or a ~ 1X read fragment representation of the underlying genome, is constructed by iterating over the read set, and querying a progressive miBf database representing the golden path. We have shown that our genome assembly paradigm yields human genome assemblies that are comparable in contiguity to what can be obtained using different implementations of the OLC algorithm, yet with an order of magnitude smaller memory footprint.
The GoldRush algorithm was designed with no single long-read sequencing technology in mind, making it versatile and platform agnostic. The algorithm is robust to base errors, capable of assembling long-read datasets with estimated error rates ranging from 4 to 20%, and achieving NG50 and NGA50 lengths up to 32.6 and 22.2 Mbp, respectively, for the human data tested (Supplementary Tables 1–6). GoldRush accomplishes this by mitigating the impact of base errors at various stages. For instance, GoldPath uses spaced seeds38 to enable more sensitive detection of erroneous sequences originating from the same genomic locus. In addition, GoldPath only selects sequences with an average Phred39,40 quality ≥15 (by default) for the silver and golden paths. The requirement ensures that the baseline assembly is composed of the higher-quality reads from the set, but may also prevent GoldPath from recruiting sequences derived from genomic loci with reads of insufficient quality. These regions, however, could be rescued downstream if GoldChain scaffolds over the region, as the gap-filling feature can recover the missing sequence. The average Phred quality threshold parameter could also be set to a lower value for more erroneous read datasets. For polishing the golden path sequences, GoldPolish accommodates the higher error rate of long reads by utilizing targeted Bloom filters30 populated with localized k-mers originating exclusively from mapped reads. This approach of using a targeted Bloom filter per goldtig enables the use of smaller k-mer sizes than those used for the original short-read focused ntEdit+Sealer protocol, thus increasing the polishing sensitivity and mitigating errors that would otherwise arise from the use of off-target k-mers. Further, the GoldRush algorithm can reconstruct duplicated genomic regions in the resulting genome assembly. In our tests, we observed that GoldRush reconstructed 34.3% of duplicated human genes in multiple copies, though this figure was higher in Flye and Shasta assemblies (Supplementary Fig. 2 and Supplementary Table 14). This is also recapitulated in the Merqury spectra-cn plots;35 the Flye and Shasta spectrum histograms more closely resemble those of the reference-grade quality NA24385 maternal and paternal genome assemblies41 in comparison to the GoldRush and Redbean spectrum histograms (Supplementary Fig. 3). Since GoldRush uses a completely different paradigm to assemble genomes, it also has different strengths when compared to OLC-based genome assemblers. For instance, in the O. sativa dataset, the N50 length of the reads is 29,349 bp, and a Shasta genome assembly of the data results in an assembly NGA50 length of 104,593 bp—roughly 3.6 times longer. In contrast, the NGA50 length of the GoldRush genome assembly is tenfold greater (307.9 kbp) than the read N50 length (Fig. 2c and Supplementary Tables 4,6). However, when considering which tool performs best on all datasets, none consistently outperforms the other in all the metrics we measured (i.e., run time, memory usage, genome contiguity, genome completeness, and genome correctness).
Yet, GoldRush is consistently more memory-efficient in comparison to other tools in all the datasets tested. This provides the opportunity to assemble long-read data from human-sized or larger genomes to those who do not have access to server-class systems, especially as retail computers with 64 GB RAM or more become more accessible. The memory efficiency of GoldRush is mainly due to the use of the miBf data structure in the GoldPath stage. However, the published version of the miBf data structure31 was intended to serve as a static database, where the user first inserted all the items of interest, and subsequently used the database solely for query operations. For the purpose of GoldPath, we needed a memory-efficient data structure that could also be dynamic, with interleaved insert and query operations. To adjust the miBf for GoldPath, we disabled its ability to rescue information lost to hash collisions, a step that requires all the elements to be inserted, and renders the miBf static afterward. We compensated for this loss of information by using longer tile lengths, t. With longer tiles, there are more queries per given tile, and the increased number of queries would offset the loss of expected hits due to hash collisions.
GoldRush also assembles all the human datasets within a day and, together with Shasta, is faster than all other OLC genome assemblers tested herein. Shasta accomplishes this with heuristics based on MinHash markers to quickly identify potential read-to-read overlaps20. On the other hand, GoldRush achieves this speed with the use of a genome assembly algorithm that has linear time complexity in the number of reads (Supplementary Note 1). Breaking down the time GoldRush spends for completing each stage, we observe that GoldRush devotes more time polishing the golden path with GoldPolish (Supplementary Tables 20, 26, 27), which is already heavily optimized. GoldPolish runs a background Bloom filter building process which continually produces Bloom filters from the mapped reads for the launched pipelines, minimizing waiting time between ending and starting a new polishing run (Supplementary Fig. 4). The GoldPath and GoldChain stages were also optimized to reduce the overall run time of GoldRush. GoldPath utilizes multiple independent silver paths to effectively generate a low-coverage subsample of the original dataset, such that the subset still covers the entire target genome. With enough silver paths, any sections of the genome that are missing in a given silver path should be recovered in the others. This enables GoldPath to generate a golden path without having to process the entire dataset, speeding up the golden path generation considerably.
Further, we observed that running additional rounds of ntLink in the GoldChain stage led to substantial improvements in the contiguity of the final assembly. However, re-mapping the long reads for each round was costly. To remedy this, we implemented a mapping liftover step, allowing ntLink to run multiple rounds of scaffolding without re-mapping the long reads, thus reducing the run time in each subsequent round (Supplementary Table 28). We also implemented two additional features, overlap detection, and gap-filling, in ntLink. While these were introduced specifically for the GoldChain stage of GoldRush, they are also applicable to the general use of ntLink. In earlier versions of ntLink, overlapping sequences were still concatenated end-to-end, which could result in local insertion misassemblies at the contig joints. With the overlap detection and overlap resolution feature, gap estimates from the earlier stages of ntLink are used to identify putative overlaps between adjacent goldtigs (indicated by negative gap estimates), and guide trimming of the overlapping sections. Overlapping goldtigs are expected in the golden path, as reads are evaluated on a tile-by-tile basis in GoldPath.
Polishing the resulting human assemblies with GoldPolish reduces the base error rates by 53.6–85.1%. GoldPolish is built on ntEdit+Sealer29, a k-mer-based base correction algorithm, and requires 2k–1 consecutive high-quality k-mers to correct base mismatches and indels. This requirement is geometrically harder to meet for datasets with higher error rates. In comparison, each of the genome assemblies generated by the Flye and Shasta algorithms has higher base-pair accuracy (Supplementary Tables 1–3, 6). In the Merqury spectra-cn plots, we observe that GoldRush has a greater number of unique k-mers not found in the high-quality short reads compared to competing tools, which is at least partially attributed to gap-filling with uncorrected read sequences (Supplementary Fig. 3).
With the recent release of the ONT Q20+ chemistry and its reported base accuracy of 99%, as well as the continual improvements in basecallers42 and k-mer-based genome analysis solutions43, we expect GoldRush to capitalize on the improvements in these frontiers, and reduce the time spent correcting base errors. Indeed, for the human GoldRush tests, GoldPolish achieved its highest base quality with the fastest run time for the NA24385 dataset, which is estimated to be the least erroneous in our tests (4%) (Supplementary Tables 6, 12, 15, 16, 20, 26, 27). For this dataset, GoldPolish displayed a polishing performance similar to that of Racon37 (Supplementary Tables 12, 21, 23). Racon is computationally more expensive to run, due in part to the input reads and alignments being loaded into memory. In our tests, both GoldPolish and Racon correct repetitive regions better than non-repetitive regions (Supplementary Tables 24, 25).
Lastly, GoldRush is modular. Each step within GoldRush can be substituted for another tool that performs the equivalent function, such as substituting GoldPolish for Racon, allowing GoldRush to easily benefit from any future advances in the field. GoldRush also makes no assumptions about the quality of the input long reads, standing only to gain from future computing and sequencing improvements in the long-read sequencing domain.
We have demonstrated that our memory-efficient and modular long-read genome assembly pipeline, GoldRush, assembles long ONT reads into draft genomes with high contiguity, notably chromosome 18 telomere-to-telomere (HG02055) and several other chromosome arms (Supplementary Fig. 5). We also show that the genome assembly contiguities are comparable to what is achieved with current state-of-the-art tools, but GoldRush uses a fraction of the RAM, lowering the barrier to entry to human long-read genome assembly. With its modular design, memory efficiency, and robust performance in assembling large and complex genomes, we expect our assembly paradigm, and its first implementation, GoldRush, to both be beneficial to the scientific community and expand the reach of long sequencing reads.
Methods
GoldPath
To create the golden path, GoldPath first builds silver paths. A silver path is similar to the golden path, except it is an ~rX (default 0.9X) representation of the genome. The silver paths are combined to generate a low-coverage subsample of the reads. This subsample is then used as input to GoldPath to generate the golden path.
GoldPath builds the silver paths by using a modified miBf 31, a resource-efficient probabilistic data structure, to associate spaced seed (a pattern with care and don’t care positions) derived k-mers with the locus of the genome they are derived from. The miBf is composed of three data structures: a Bloom filter30, a rank array, and an ID array. The Bloom filter is first initialized with h (default 3) sets of spaced seed38 derived k-mers from the input read set using ntHash44,45. Only reads that are at least m bp (default 20,000 bp) long and have an average Phred39,40 quality score higher than P (default 15, representing 97% base accuracy or more, on average) are inserted into the Bloom filter. A rank array is then created to associate each set bit in the Bloom filter with a position in the empty ID array, sized based on the number of set bits in the Bloom filter.
The read set is then iteratively queried against the miBf data structure to determine if a read should be inserted into the miBf and, thus, the silver path. Once a read is processed and determined to contribute new base coverage to the silver path, the read is inserted into the miBf. The read is first split into tiles of length t bp (default 1000), and b (default 10) consecutive tiles are binned into one block. The spaced seed-derived k-mers from one block are hashed and a unique ID associated with the block, synonymous with a genomic locus, is inserted into the position of the miBf ID array that corresponds to the spaced seed-derived k-mer (Supplementary Fig. 6). By binning tiles into blocks, GoldPath clusters together the spaced seed-derived k-mers from a genomic region of length b × t. Each read that is inserted into the miBf is also saved to the silver path.
Like insertion, the querying process first splits the read into tiles of length t. The k-mers in these tiles are hashed using the same spaced seed patterns and queried against the miBf, then the associated ID hits within each tile are tallied in an ID-to-counts table. From these hits, a preliminary best ID hit is associated with each tile. The tiles are considered assigned (found) if the ID with the most hits exceeds a threshold x (default 10), or unassigned (not found) otherwise (Supplementary Fig. 7). Next, we improve the accuracy of the tile’s preliminary best hits by using information from the best hits of neighboring tiles along with the target tile’s own ID-to-counts table (Supplementary Fig. 8 and Supplementary Method 1).
Once GoldPath determines the final assignment of all the tiles in the read, it evaluates the read with three possible outcomes: skip, insert, or trim and insert. If all the tiles in the read are already assigned, the read is skipped since it does not contribute any new base information to the silver path. If all the tiles in the read are unassigned, the read is inserted into the miBf data structure and the silver path in its entirety. Finally, if the read has a mixture of assigned and unassigned tiles, the read is trimmed such that only one assigned tile on either side of the longest stretch of unassigned tiles is retained (Supplementary Fig. 9), and the trimmed read is inserted into both the miBf and silver path.
When the silver path contains or exceeds the threshold number of bases (default genome size × r), the current silver path is finalized, and a new silver path is initialized. The silver path creation continues until the input reads are exhausted or the number of completed silver paths reaches M (default 5). The golden path is then generated using the same algorithm and parameters used for the silver paths, except that the input is now the concatenated sequence files from the silver paths and the golden path is only complete when GoldPath has finished iterating through all of the silver path sequences. Sequences in the golden path construe the contigs of the initial genome assembly to be refined in the later steps of the pipeline, and are henceforth termed “goldtigs”.
GoldPolish
After the golden path is built, the goldtigs are polished to correct mismatches and indels. The polishing protocol, GoldPolish, closely follows the ntEdit+Sealer protocol29, which has been shown to perform well using short sequencing reads. Unlike the currently published paradigm, which stores all the short-read k-mers in a single Bloom filter30 used for correction, GoldPolish uses a targeted approach where each goldtig has a dedicated Bloom filter, each containing a hash representation of k-mers derived from long-read subsets.
To accomplish this, GoldPolish maps the long reads to goldtigs using, by default, minimap234. GoldPolish is also capable of using mappings from other tools, such as ntLink7. For each goldtig, the set of mapped reads are k-merized using a range of different k-mer lengths in order to benefit from a trade-off between specificity (longer k-mers) and sensitivity (shorter k-mers). These k-mers are then inserted into an array of Bloom filters, one Bloom filter for each k-mer size per goldtig. From this, we have a set of k-mers targeted to individual goldtigs that we can use with ntEdit46 and Sealer47 for polishing (Supplementary Fig. 10).
GoldPolish then polishes the goldtigs using the full ntEdit+Sealer pipeline on each individual goldtig using their dedicated Bloom filters. GoldPolish launches multiple ntEdit+Sealer pipelines in parallel to amortize the overhead introduced with each polishing run.
GoldChain
After polishing and correcting the goldtigs, an updated version of the long-read genome scaffolder ntLink7 is used to assemble the goldtigs. To utilize the long-read evidence in building longer sequences, the full long-read set is mapped to the goldtigs using a lightweight minimizer-based approach. Briefly, minimizer sketches are generated for the goldtigs as well as each read for a given k-mer size k and window size w7. The goldtig minimizer sketches are indexed, and for each minimizer in the sketch of a given long-read, this index is queried to find hits between the long read and the goldtigs. Long-read mappings that span multiple goldtigs provide scaffolding evidence. This long-read evidence is stored as a scaffold graph, where the nodes are goldtigs, and the directed edges between the nodes represent evidence that the goldtigs should be joined. This scaffold graph is traversed using abyss-scaffold48, a heuristic-based scaffold layout algorithm, to output the final, contiguated genome assembly.
Three important features have been added to ntLink (v1.3.0+) to adapt the functionality for the de novo long-read genome assembly problem in GoldRush: overlap detection, gap-filling, and scaffolding rounds based on the liftover of sequence mappings.
For the sequence pairs with putative overlaps, minimizer sketches are generated with a lower k and w than the initial ntLink pairing stage to increase the sensitivity of overlap detection (parameters small_k, small_w, defaults 15 and 10, respectively). These sketches are filtered to retain minimizers that fall in the estimated overlapping region with a multiplicity of one in each sequence, ensuring that only non-repetitive minimizers in the sequences are retained. These minimizers are then used to create an undirected minimizer graph, similar to methods employed by the reference-guided scaffolder ntJoin49. In this graph, the minimizers are nodes, and edges between the minimizers indicate that the minimizers are adjacent in at least one of the ordered sequence minimizer sketches, with the edge weights indicating the number of sequences that have that minimizer adjacency. This minimizer graph is filtered to retain edges with a weight of 2, which removes branches and results in a graph consisting of linear path components. Each linear path is a minimizer-based mapping between the putatively overlapping sequence ends. The middle minimizer from the longest mapping is chosen to anchor the sequences to one another, and the coordinates of this minimizer guide the trimming of the detected overlapping regions on the incident sequences. Finally, after this trimming, the sequences are concatenated (Supplementary Fig. 11).
The second major feature added to ntLink uses the mapped long reads to fill gaps between the scaffolded goldtigs. The verbose option in the initial ntLink pairing stage was updated to output the complete long-read mapping information, including the mapped goldtigs and the minimizers (including position and strand on the goldtig and read). For each sequence join induced by ntLink, the verbose mapping information is parsed to identify each read that supports that join, and the associated mapped minimizers (“pass 1 minimizers”). The read with the highest average number of mapped pass 1 minimizers is chosen and subsequently used to fill the scaffold gap. As finding anchoring minimizers as close to the sequence ends as possible is preferred, each chosen read is re-mapped to the flanking sequences using a lower k and w for increased sensitivity (“pass 2 minimizers”). If the mapping is unambiguous, the anchoring pass 2 minimizers closest to the sequence ends are used as cut points for the flanking sequences and the read sequence filling the gap. Otherwise, the pass 1 minimizers are used to determine the gap-filling coordinates (Supplementary Fig. 12). Gap-filling is turned on in GoldRush by default and is run when the target “gap_fill” is specified to the ntLink command.
Finally, liftover-based rounds were integrated into the ntLink code base. We added a step to liftover the mapped minimizer coordinates in the verbose mapping file (described above) from the initial goldtigs to the sequences post-scaffolding. This new mapping file is then input to the ntLink pairing stage in the subsequent ntLink round, which uses the input mapping coordinates instead of re-mapping the reads to infer the scaffold graph. The remaining steps in the ntLink pipeline then proceed as previously described. To invoke these liftover-based rounds, we provided a Makefile “ntLink_rounds”, which runs a specified number of rounds of ntLink (parameter rounds, default 5), lifting over the mapping coordinates between each iteration.
Implementation
The GoldRush pipeline is driven by a Makefile. GoldPath and GoldPolish are coded in C++, and GoldChain is coded in Python. All components of GoldRush utilize the btllib common code library50. The tool can be installed from GitHub or using the conda package manager. Instructions on how to run the GoldRush pipeline are provided on the GitHub page (https://github.com/bcgsc/goldrush). Many of the GoldRush parameters are supplied with default values and can be configured. Only the genome size of the target species and the long reads in a single, uncompressed, multi-FASTQ file are required as input.
Evaluation
To evaluate the performance of GoldRush (v1.0.0), we assembled five genomes from ONT long-read data for three human cell lines (NA24385, HG01243, and HG02055), O. sativa, and S. lycopersicum (Supplementary Table 6). We optimized the parameters of GoldRush for each dataset (Supplementary Figs. 13–17 and Supplementary Table 29). In a separate trial, we also polished the golden paths with Racon37 (v1.5.0) instead of GoldPolish. To assess the polishing performance of GoldPolish and Racon in repetitive and non-repetitive regions of the genome, we first masked the repeats in the assemblies using RepeatMasker51 (v4.1.2) (-e ncbi -species human). Then, to generate an assembly where the non-repetitive regions are masked, we used the complement of the masked repetitive regions. These masked assemblies were then used to compare the polishing performance of GoldPolish and Racon in the repetitive and non-repetitive genomic regions. To compare the performance of GoldRush to current state-of-the-art long-read genome assemblers, we assembled all five datasets with Flye, Redbean, and Shasta. We ran both Flye (v2.9) and Redbean (v2.5) using their default parameters, and Shasta (v0.10.0) using the Nanopore-Plants-Apr2021.conf configuration file for O. sativa and Nanopore-May2022.conf for the other datasets.
All assemblies were analysed using QUAST32 (v5.0.2) (--fast --large --scaffold-gap-max-size 100000 --min-identity 80 --split-scaffold), and the corresponding reference genome (Supplementary Table 30). To assess the contiguity and correctness of the assemblies, we report the NG50 and NGA50 length metrics, and the number of extensive misassemblies (as defined by QUAST). The NG50 length statistic describes that 50% of the genome size is in sequences of NG50 length or longer. The NGA50 length statistic is similar to the NG50 length, but uses alignment blocks instead of sequence lengths for the calculation. To assess the base qualities of the various assemblies, we report the number of mismatches or indels per 100 kbp from QUAST, spectra-cn plots, and QV from Merqury35 (v1.3.0) — the latter a proxy for the log-scaled probability of error for the consensus base calls — using short reads and reference-grade genome assemblies as a comparison (Supplementary Table 31). We also ran BUSCO33 (v5.3.2) using the primates_odb10 lineage to assess the completeness of the human assemblies in the gene space. Finally, to measure the presence of duplicated genes found in the assemblies, we used the asmgene utility in minimap234 (v2.24) (min coverage = 0.99 and min identity = [0.90, 0.99]) using all cDNA sequences annotated in the GRCh38 human reference from Ensembl52 (release 87) and the T2T-CHM1336 (v1.1) genome assembly as the “ground truth” for which genes are considered duplicated. All benchmarking tests were performed on a server-class system with 144 Intel(R) Xeon(R) Gold 6254 CPU @ 3.1 GHz with 2.9 TB RAM.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The GoldRush, Flye, Redbean, and Shasta genome assemblies generated in this study have been deposited in Zenodo at https://doi.org/10.5281/zenodo.788468153. The GoldRush genome assemblies generated for the parameter sweep experiments in Supplementary Figs. 13–17 are available upon request. The accession codes or location of sequencing data used for assembling the draft genomes are listed in Supplementary Table 6. The accession codes of the reference genomes and the short-read dataset used to benchmark GoldRush and comparators’ genome assemblies are provided in Supplementary Tables 30–33.
Code availability
GoldRush (v1.0.0) has been deposited in Zenodo at https://doi.org/10.5281/zenodo.788429154. GoldRush is available at https://github.com/bcgsc/goldrush and released under the GPL-3 license.
References
Treangen, T. J. & Salzberg, S. L. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet. 13, 36–46 (2012).
Haubold, B. & Wiehe, T. How repetitive are genomes? BMC Bioinform. 7, 541 (2006).
de Koning, A. P. J., Gu, W., Castoe, T. A., Batzer, M. A. & Pollock, D. D. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 7, e1002384 (2011).
Afshinfard, A. et al. Physlr: next-generation physical maps. DNA 2, 116–130 (2022).
Coombe, L. et al. ARKS: chromosome-scale scaffolding of human genome drafts with linked read kmers. BMC Bioinform. 19, 234 (2018).
Putnam, N. H. et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 26, 342–350 (2016).
Coombe, L. et al. LongStitch: high-quality genome assembly correction and scaffolding using long reads. BMC Bioinform. 22, 534 (2021).
Qin, M. et al. LRScaf: improving draft genomes using long noisy reads. BMC Genomics 20, 955 (2019).
Adewale, B. Will long-read sequencing technologies replace short-read sequencing technologies in the next 10 years? Afr. J. Lab Med. 9, 1340 (2020).
Sakamoto, Y., Zaha, S., Suzuki, Y., Seki, M. & Suzuki, A. Application of long-read sequencing to the detection of structural variants in human cancer genomes. Comput. Struct. Biotechnol. J. 19, 4207–4216 (2021).
Bongartz, P. Resolving repeat families with long reads. BMC Bioinform. 20, 232 (2019).
Chiu, R., Rajan-Babu, I.-S., Friedman, J. M. & Birol, I. Straglr: discovering and genotyping tandem repeat expansions using whole genome long-read sequences. Genome Biol. 22, 224 (2021).
Liu, Y. et al. Comparison of structural variants detected by PacBio-CLR and ONT sequencing in pear. BMC Genomics 23, 830 (2022).
Hon, T. et al. Highly accurate long-read HiFi sequencing data for five complex genomes. Sci. Data 7, 399 (2020).
Dohm, J. C., Peters, P., Stralis-Pavese, N. & Himmelbauer, H. Benchmarking of long-read correction methods. NAR Genom. Bioinform. 2, lqaa037 (2020).
Liu, L., Yang, Y., Deng, Y. & Zhang, T. Nanopore long-read-only metagenomics enables complete and high-quality genome reconstruction from mock and complex metagenomes. Microbiome 10, 209 (2022).
Luo, J. et al. Systematic benchmarking of nanopore Q20+ kit in SARS-CoV-2 whole genome sequencing. Front. Microbiol. 13, 973367 (2022).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019).
Ruan, J. & Li, H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17, 155–158 (2020).
Shafin, K. et al. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat. Biotechnol. 38, 1044–1053 (2020).
Wajid, B. & Serpedin, E. Review of general algorithmic features for genome assemblers for next generation sequencers. Genom. Proteom. Bioinform. 10, 58–73 (2012).
Li, Z. et al. Comparison of the two major classes of assembly algorithms: overlap–layout–consensus and de-bruijn-graph. Brief. Funct. Genomics 11, 25–37 (2012).
Schrinner, S. D. et al. Haplotype threading: accurate polyploid phasing from long reads. Genome Biol. 21, 252 (2020).
Lorig-Roach, R. et al. Phased nanopore assembly with Shasta and modular graph phasing with GFAse. Preprint at bioRxiv https://doi.org/10.1101/2023.02.21.529152 (2023)
Luo, X., Kang, X. & Schönhuth, A. phasebook: haplotype-aware de novo assembly of diploid genomes from long reads. Genome Biol. 22, 299 (2021).
Rautiainen, M. et al. Telomere-to-telomere assembly of diploid chromosomes with Verkko. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-01662-6 (2023)
Jackman, S. D. et al. Tigmint: correcting assembly errors using linked reads from large molecules. BMC Bioinforma. 19, 393 (2018).
Coombe, L., Warren, R. L., Wong, J., Nikolic, V. & Birol, I. ntLink: A toolkit for de novo genome assembly scaffolding and mapping using long reads. Curr. Protoc. 3, e733 (2023).
Li, J. X., Coombe, L., Wong, J., Birol, I. & Warren, R. L. ntEdit+Sealer: efficient targeted error resolution and automated finishing of long-read genome assemblies. Curr. Protoc. 2, e442 (2022).
Bloom, B. H. Space/time trade-offs in hash coding with allowable errors. Commun. ACM 13, 422–426 (1970).
Chu, J. et al. Mismatch-tolerant, alignment-free sequence classification using multiple spaced seeds and multiindex Bloom filters. Proc. Natl Acad. Sci. USA 117, 16961–16968 (2020).
Mikheenko, A., Prjibelski, A., Saveliev, V., Antipov, D. & Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 34, i142–i150 (2018).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Nurk, S. et al. The complete sequence of a human genome. Science 376, 44–53 (2022).
Vaser, R., Sović, I., Nagarajan, N. & Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746 (2017).
Ma, B., Tromp, J. & Li, M. PatternHunter: faster and more sensitive homology search. Bioinformatics 18, 440–445 (2002).
Ewing, B. & Green, P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8, 186–194 (1998).
Ewing, B., Hillier, L., Wendl, M. C. & Green, P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8, 175–185 (1998).
Jarvis, E. D. et al. Semi-automated assembly of high-quality diploid human reference genomes. Nature 611, 519–531 (2022).
Wick, R. R., Judd, L. M. & Holt, K. E. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 20, 129 (2019).
Sahlin, K. Effective sequence similarity detection with strobemers. Genome Res. 31, 2080–2094 (2021).
Mohamadi, H., Chu, J., Vandervalk, B. P. & Birol, I. ntHash: recursive nucleotide hashing. Bioinformatics 32, 3492–3494 (2016).
Kazemi, P. et al. ntHash2: recursive spaced seed hashing for nucleotide sequences. Bioinformatics https://doi.org/10.1093/bioinformatics/btac564 (2022).
Warren, R. L. et al. ntEdit: scalable genome sequence polishing. Bioinformatics 35, 4430–4432 (2019).
Paulino, D. et al. Sealer: a scalable gap-closing application for finishing draft genomes. BMC Bioinform. 16, 230 (2015).
Jackman, S. D. et al. ABySS 2.0: resource-efficient assembly of large genomes using a Bloom filter. Genome Res. 27, 768–777 (2017).
Coombe, L., Nikolić, V., Chu, J., Birol, I. & Warren, R. L. ntJoin: fast and lightweight assembly-guided scaffolding using minimizer graphs. Bioinformatics 36, 3885–3887 (2020).
Nikolić, V. et al. btllib: a C++ library with Python interface for efficient genomic sequence processing. J. Open Source Softw. 7, 4720 (2022).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker open-4.0. http://www.repeatmasker.org (2013–2015).
Cunningham, F. et al. Ensembl 2022. Nucleic Acids Res. 50, D988–D995 (2022).
Wong, J. et al. Linear time complexity de novo long read genome assembly with GoldRush. Zenodo https://doi.org/10.5281/ZENODO.7884681 (2023).
Wong, J. et al. Linear time complexity de novo long read genome assembly with GoldRush. Zenodo https://doi.org/10.5281/ZENODO.7884291 (2022).
Acknowledgements
This study is supported by the Canadian Institutes of Health Research (CIHR) [PJT-183608, I.B.]; and the National Institutes of Health [2R01HG007182-04A1, I.B.]. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or other funding organizations. The funding organizations did not have a role in the design of the study, the collection, analysis and interpretation of the data, or in writing the manuscript.
Author information
Authors and Affiliations
Contributions
I.B. and R.L.W. conceived the study. J.W., L.C., and V.N. implemented the algorithms. J.W., L.C., E.Z., and P.S. analysed the data. K.M.N. provided input on the design of the algorithm. J.W., L.C., and V.N. created figures and tables with input from co-authors. J.W., L.C., R.L.W., and I.B. wrote the manuscript. R.L.W. and I.B. supervised the research. All authors commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Benjamin Istace and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wong, J., Coombe, L., Nikolić, V. et al. Linear time complexity de novo long read genome assembly with GoldRush. Nat Commun 14, 2906 (2023). https://doi.org/10.1038/s41467-023-38716-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-38716-x
This article is cited by
-
Sexual dimorphism in the tardigrade Paramacrobiotus metropolitanus transcriptome
Zoological Letters (2024)
-
Application of Nanopore Sequencing in the Diagnosis and Treatment of Pulmonary Infections
Molecular Diagnosis & Therapy (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
