
Abstract
Azepinone derivatives are privileged in organic synthesis and pharmaceuticals. Synthetic approaches to these frameworks are limited to complex substrates, strong bases, high power UV light or noble metal catalysis. We herein report a mild synthesis of azepinone derivatives by a photochemical generation of 2-aryloxyaryl nitrene, [2 + 1] annulation, ring expansion/water addition cascade reaction without using any metal catalyst. Among the different nitrene precursors tested, 2-aryloxyaryl azides performed best under blue light irradiation and Brønsted acid catalysis. The reaction scope is broad and the obtained products underwent divergent transformations to afford other related compounds. A computational study suggests a pathway involving a step-wise aziridine formation, followed by a ring-expansion to the seven-membered heterocycle. Finally, water is added in a regio-selective manner, this is accelerated by the added TsOH.
Similar content being viewed by others
Introduction
Azepinones and related seven-membered nitrogen-containing heterocycles are distinguished by their versatility in organic synthesis1 and ubiquity in various natural products as well as pharmaceutically important compounds (Fig. 1)2,3,4,5. Traditionally, these compounds were prepared by a strong base-mediated cyclization of complex diester substrates2. High-power (>300 W) UV-light promoted decompositions of aryl azides in the presence of nucleophiles also delivered azepinone derivatives6,7,8,9. Recently, Rh-catalyzed annulations of benzamides with α,β-unsaturated aldehydes and ketones provided another option for accessing these privileged heterocycles10. Despite these advances, a metal-free method using readily available substrates under mild conditions is still challenging and of high utility for synthesis of pharmaceuticals and natural products and the generation of libraries of derivatives.

a Azepinone-based cyclin-dependent kinase inhibitor. b Azepinone-based antitumor compound. c Azepinone-based antibacterial agent. d Azepinone-based vitamin D receptor.
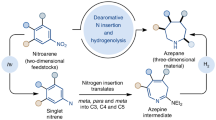
There is a continuing demand for selective and efficient C − N bond formations for challenging organic synthesis. Organic reactions forging C − N bonds through nitrene intermediates have opened up new avenues for the synthesis of N-heterocycles11,12,13. The applicability of these transformations has advanced with the development of new nitrene precursors. In this context, a series of reagents, including azides, 2H-azirines, isoxazole derivatives, aza-pyridium ylides and sulfilimines, were elegantly designed14. Methods for preparing aza-heterocycles based upon transition metal-catalyzed nitrene transfer from these nitrene equivalents to functionalized alkynes have been frequently reported by Liu15,16,17, Ye18,19,20,21, Hashmi22,23, and others14. The direct generation of nitrene intermediates, generally promoted by photoirradiation, high temperatures or transition metal catalysts, allows to form C − N bonds, including C − H amination, C − H amidation and alkene aziridation24,25,26,27. Among these nitrene transfer reactions, those using visible light source under metal-free conditions are comparatively rare and attractive26,28,29. Ortho alkyl-, alkenyl- or aryl-substituted aryl nitrenes have garnered significant attention as such aryl nitrene intermediates can undergo C − H insertions to afford carbazoles or indoles (Fig. 2a)28,30,31,32. To date, the chemistry of 2-aryloxyaryl nitrenes has not yet been explored. Very recently, we disclosed a blue light-promoted synthesis of unprotected carbazoles and indoles by utilizing 2-substituted aryl sulfilimines as nitrene precursors28. Based on our ongoing interest in sulfilimine chemistry22,23,28,33,34,35,36, we envisaged a synthesis of 10H-phenoxazine 4a from 2-phenoxyphenyl sulfilimine 1a by a photochemical intramolecular C − H amination (Fig. 2b). The desired product 4a, however, was not observed. An unexpected seven-membered ring product, 2H-azepinone 5a, was formed instead and its structure was confirmed by single crystal X-ray structure analysis. Here, a mild synthesis of azepinone derivatives by leveraging the reactivity of 2-aryloxyaryl nitrene intermediates is presented.

a Previous work: synthesis of carbazoles and related heterocycles through 2-alkyl, 2-alkenyl or 2-aryl arylnitrene intermediates. b This work: synthesis of azepinone derivatives through 2-aryloxyaryl nitrene intermediates.
Results
Reaction optimization
Optimization commenced with sulfilimine 1a as substrate (Table 1). Blue light performed better than UVA (entries 1–2). The product yield decreased to 14% when the solvent was changed from THF to toluene and further decreased in DCE (entries 3-4). Adding 10 equivalents of water is beneficial for product formation (entry 5). Other nitrene precursors were also tested. While phosphanimine 2a did not react under the standard reaction conditions, the product yield was raised to 65% by using azide substrate 3a (entries 6–7). Adding more H2O afforded decrease in the product yield (entries 8–9). The addition of 50 mol% TsOH significantly improved the reaction (entry 10). Both 5 mol% and 100 mol% TsOH led to less efficient reactions (entries 11–12). Other protonic acids and a Lewis acid, Zn(OTf)2, delivered the desired product in no higher than 69% yield (entries 13–16). A control experiment in the dark demonstrated that light is essential to this transformation (entry 17).
Substrate scope
We then examined the reaction scope and limitations under the optimal reaction conditions (Table 1, entry 10). As shown in Fig. 3, different R1 groups at the azido arene moiety, including chloro, trifluoromethyl, 3-methyl, 4-methyl, 6-methyl as well as methoxy groups, were well tolerated, and the corresponding products 5a-5f were obtained in moderate to good yield. Product 5g was further transformed into acetyl derivative 5g’. Next, R2 substituents were varied to investigate the functional group tolerance further. Azide 3h bearing a methyl group at ortho position (R2 = 2′-methyl) was able to undergo the desired cascade reaction, selectively generating product 5h in 47% yield. When R2 was varied from an alkyl group (ethyl or tert-butyl) to an aryl substituent (phenyl or thiophenyl) at the para positions, the reactions proceeded smoothly and product yield ranged from 40% to 67%. A dihydroindene-based substrate 3m afforded a single isomer, 5m, in good yield. The para-bromo and -chloro substrates 5n-5o were very well tolerated and the obtained products are valuable starting materials for further cross coupling strategies. The meta-chloro substrate 3p delivered two isomers 5p and 5p′ in 91% combined yield with low levels of regioselectivity. 3′,5′-Dichloro and 3′,5′-ditrifluoromethyl groups remained untouched, the products 5q and 5r were isolated in high yield. A complex substrate 3s was also tested, enabling 53% yield of the desired product 5s. Encouraged by the performance of aryloxyaryl azides, we became interested in evaluating heteroaryloxy and alkoxy type substrates under the standard conditions. Although the thiophene and 2-pyridinyl substrates (3t and 3u) remained unreactive, 3-pyridinyl substrate 3 v regioselectively afforded 1,3-diazepinone 5v in 10% yield with 20% conversion. An attempt to synthesize N-alkyl azepinone 5w by engaging alkoxy substrate 3w was unsuccessful.

Reaction conditions: 3 (0.2 mmol), H2O (2.0 mmol), TsOH•H2O (0.1 mmol), THF (2.0 mL, 0.1 M), blue LEDs, room temperature. Isolated yields of 5 are given. Compound 5g’ was obtained by a subsequent transformation: pyridine (1.5 eq.), acetic anhydride (1.2 eq.), CH2Cl2 (1.0 mL, 0.1 M), RT.
Our attention then turned to the synthetic utilizations of the obtained products (Fig. 4). The unprotected phenol products 5 can be protected by different protecting groups, such as methyl, propargyl and trifluoromethanesulfonyl, resulting in products 6, 7, 8, 11 in high yield. The generated triflate group can further be coupled with phenylacetylene or a complex arylboronic acid in excellent yield. Compound 11, containing a triflate and a bromo substituent, was able to efficiently react with 2 equivalents of para-tolylboronic acid under palladium catalysis.

All reactions are performed on a 0.2 mmol scale. Isolated yields are given.
Mechanistic studies
In an effort to elucidate the reaction mechanism, we conducted an isotope labeling experiment by adding D2O instead of H2O (Fig. 5). As a result, 50% D was found in the CH2 group and >95% D was found in the OH group of the product, which suggested that both the proton in OH group and one of the protons in the CH2 group of the product originated from H2O. We further conducted an 18O-labeling experiment by using 18O water (98% 18O). Mass spectrometry does not indicate any two-fold incorporation, 90% 18O were incorporated, but due to the fragmentation pathways, MS did not allow to assign the 18O to either the ketone or the hydroxyl group. Thus the obtained product 5a” was analyzed by 13C NMR (150 MHz). The carbonyl carbon signal at 167.12 ppm could be assigned to the 16O carbonyl by comparison with the unlabeled product 5a, the signal at 167.09 ppm thus belongs to the 18O labeled 5a” (Supplementary Fig. 14). Thus the carbonyl oxygen of product 5a” indeed is derived from the external water nucleophile.

The H/D ratios were determined by 1H NMR spectroscopy with trimethoxybenzene as the internal standard. The incorporation of the 18O atom was detected by MS and a second carbonyl peak in the 13C NMR spectrum.
It is well established that light decomposes aryl azides into open-shell singlet nitrenes37. To confirm this, we utilized TD-DFT calculations at the CPCM/CAM-B3LYP/def2-TZVP//CPCM/CAM-B3LYP/6-31G(d) level of theory to investigate the mechanism of photoactivation (Fig. 6). In the calculations, we used substrate 3a as a representative for the aryl azides. The first excitation was identified at 301 nm with a negligible oscillator strength (f = 0.0003), therefore it was not computationally investigated further. The second excitation at 263 nm has a significant oscillator strength with f = 0.1923. This excitation mainly corresponds to the transition of an electron from the HOMO to the LUMO of substrate 3a. Attempts to optimize this excited state, however, always collapsed to the S1 state, implying that the internal conversion (IC) from S2 to S1 should be extremely fast. The optimization of this excited state results in a structure with a much weakened Na–Nb bond, as supported by the elongation of the corresponding bond from 1.232 Å in 3a to 1.434 Å in 3a*. The resultant 3a* could then serve as a branching point for several pathways, three of which are depicted in Fig. 6. This structure can be involved in breaking the Na–Nb bond via transition structure TSS1 to form open-shell singlet structure AOSS, or it can give triplet structure T1 via MECP S1/T1, or it can relax non-radiatively to structure 3a by crossing MECP S1/S0. According to the results of the calculations, TSS1 has a significantly lower energy than both MECP S1/T1 and MECP S1/S0. This implies that open-shell singlet structure AOSS should be formed significantly faster than other species, which is completely consistent with previous findings, indicating that light-driven decomposition of aryl azides produces directly open-shell singlet nitrenes37.

The relative potential energies are given in kcal/mol.
To understand how the title reaction proceeds, we investigated its mechanistic details with the aid of the density functional theory (DFT) by using the SMD/M06-2X/def2-TZVP//SMD/M06-2X/6-31G(d) calculations in THF (for details, see Supplementary Data 1–2). The unrestricted open-shell singlet AOSS is computed to be 14.6 kcal/mol higher in energy than 3a (Fig. 7a). We have also calculated the restricted closed-shell singlet for structure A and found this structure (ACSS) with a free energy of 26.1 kcal/mol is far less stable than AOSS (Fig. 7a). The intersystem crossing (ISC) to the triplet state gives AT, which is 22.9 kcal/mol more stable than AOSS38. The intramolecular aziridation was then investigated from AOSS and AT. Our DFT calculations demonstrate that both AOSS and AT intermediates are reactive toward the aziridation. The unrestricted open-shell intermediate directly gives Zwitterion intermediate B via transition structure TSAOSS, which is only 5.1 kcal/mol higher in energy than AOSS. AT is connected to BT on the triplet surface by surmounting an activation free energy of 17.9 kcal/mol via transition structure TSAT. The resultant triplet intermediate BT then undergoes a spin flip transition via MECP1 to form more stable Zwitterion intermediate B on the single surface; the MECP1 is found to be only 0.6 kcal/mol above BT. The exergonicity for transformation 3a → AT + N2 is further supported by our additional calculations at the SMD/CCSD(T)/def2-TZVP//SMD/M06-2X/6-31 G(d) level of theory for N2 release from a related system (phenyl azide, Supplementary Fig. 15d); the single-point calculations using CCSD(T) yield an ΔE of +1.3 kcal/mol for the N2 release, suggesting that the reaction is exergonic with ΔG = −11.1 kcal/mol when the entropy contribution from the SMD/M06-2X/6-31 G(d) calculations is taken into account (Supplementary Fig. 15d). The Mulliken spin density analysis for structure AT indicates that the two single electrons are mainly located on the nitrogen atom with a contribution of 1.664 (Fig. 7a). The rest of the electrons, due to the π conjugation effect are distributed on the carbon atoms of the six-membered ring bonded to the N atom with a very negligible contribution from the oxygen atom (0.045, for details, see Supplementary Fig. 16). A similar electron distribution was also obtained for AOSS, with the nitrogen atom contributing the most with a value of 0.529 (Supplementary Fig. 16).

a The calculated energy profile of the aziridation step. b The calculated energy profile of the aziridation step for the ring-closing and first ring-opening steps. c The calculated energy profile of the hydrolysis and second ring-opening steps. Free energies (potential energies) calculated at the SMD/M06-2X/def2-TZVP//SMD/M06-2X/6-31G(d) level of theory in THF are given in kcal/mol. The triplet pathway is shown in green. The pink values are the Mulliken spin density on selective atoms in AT and BT.
Once B has formed, it is involved in a ring-closing process and furnishes C (Fig. 7b). This process is predicted to occur extremely fast with a calculated activation barrier as low as 1.2 kcal/mol. Intermediate B is also prone to participate in an aromatization process to give a phenoxazine. Our DFT calculations indicate that although the aromatization process from B is predicted to be fast with an activation barrier of ~11 kcal/mol (Supplementary Fig. 17), it should take place much slower than the ring-closing transformation B → C, resulting in no phenoxazine being formed during the light-promoted reaction. This is in contrast to the observations in our recent report28 where the nitrene produced from sulfilimines practically only affords carbazoles attributed to favourability of the aromatization process over the ring-closing (Supplementary Fig. 18). We also investigated the direct formation of C from BT (Supplementary Fig. 20) and found that it is 1.8 kcal/mol less favorable than the stepwise pathway following the sequence BT → B → C. Once C has formed, it undergoes ring expansion to give tricyclic intermediate D (Fig. 7b). This step is computed to be exergonic by about 11.9 kcal/mol and occurs with a small activation barrier of 6.0 kcal/mol. Next, product 5a is generated by the hydrolysis of intermediate D followed by a second ring-opening process. The hydrolysis starts with the addition of a proton from water to the Cb atom of D. Similar to the previous studies, a water cluster of three water molecules was employed for this step (Fig. 7c)39,40. The presence of the π-donor atoms on the Ca atom, particularly the N atom, makes the Cb atom in D so basic that it deprotonates water with a moderate activation free energy of 25.8 kcal/mol to give the ion pair E (Supplementary Fig. 19). Subsequently, the in situ generated hydroxide ion is trapped by the carbocation stabilized by the oxygen and nitrogen atoms in E and produces intermediate F. Finally, the breaking of the Ca–Ob bond (the second ring-opening process) via transition structure TSF directly affords final product 5a. This means that when the Ca-Ob bond is completely broken, the proton transfer from Oa to Ob occurs spontaneously. Efforts to identify a transition structure in which the proton transfer occurs prior to the Ca-Ob bond breaking were unsuccessful.
Two important points can emerge from these calculations: (i) the hydrolysis mechanism proposed by the computations explains well the isotope labeling experiments outlined in Fig. 5 and (ii) the rate-determining step is found to be the hydrolysis step with ΔG‡ = 25.8 kcal/mol. This relatively high activation barrier rationalizes why the yield is moderate when only water is used as the additive.
Figure 8 shows the role of TsOH in catalyzing the hydrolysis and second ring-opening steps. The use of TsOH decreases the overall barrier to 19.1 kcal/mol, resulting in the reaction proceeding experimentally with a much higher yield. As shown in Fig. 8, the hydrolysis step is commenced by protonation of the Cb atom of D through transition structure TSD with an activation free energy of 9.5 kcal/mol. The reaction is then followed by the formation of hydrogen-bonded adduct H, which subsequently gives intermediate I through trapping the carbocation by the TsO--stabilized water. Finally, the second ring-opening process from I takes place by overcoming a barrier of 11.9 kcal/mol via TSI. The relatively low barrier calculated for this step confirms that TsOH plays a crucial role in its facilitation. In short, the hydrolysis process is determined to be the rate-determining step for the formation of product 5a. The activation free energy of this step is calculated to be 25.8 kcal/mol (Fig. 7c), whereas TsOH facilitates the hydrolysis, allowing it to proceed with a lower activation free energy of 19.1 kcal/mol (Fig. 8).

Free energies (potential energies) calculated at the SMD/M06-2X/def2-TZVP//SMD/M06-2X/6-31G(d) level of theory in THF are given in kcal/mol.
For the sake of completeness, we also investigated computationally the possibility of formation of the normally produced Bʹ and Cʹ (Fig. 9)6,7,8,9. Although the formation of such intermediates is calculated to be kinetically feasible, those are thermodynamically highly unstable with respect to intermediate D. As a result, once formed, they can involve a reverse process with an activation energy of 16.6 kcal/mol (free energy difference between TSAʹ and Bʹ) to yield the thermodynamically stable intermediate D from which the final product 5a is formed by the above discussed mechanism.

Free energies (potential energies) calculated at the SMD/M06-2X/def2-TZVP//SMD/M06-2X/6-31G(d) level of theory in THF are given in kcal/mol.
In summary, we have reported a green method for the synthesis of azepinone derivatives by using readily available 2-aryloxy aryl azides as aryl nitrene precursors under blue light irradiation. An isotope labeling experiment suggested that H2O is key to the formation of the final products. A detailed computational study rationalized the reaction pathway. The current reaction features a broad scope, mild conditions, and good efficiency, providing an alternative method for the synthesis of these seven-membered aza-heterocycles without using any transition metals. The isolated products are versatile substrates for further transformations.
Methods
Synthesis of substituted compound 5
Under nitrogen atmosphere, to a solution of substrate 3 (0.2 mmol) in 2.0 mL THF were added TsOH·H2O (19 mg, 0.1 mmol, 0.5 eq.) and H2O (36 mg, 2.0 mmol, 10 eq.). The resulting mixture was irradiated at room temperature with 29 W blue LEDs for 48 h. The solvent was removed under reduced pressure and the residue was purified by column chromatography on silica gel to provide the desired product 5.
Data availability
Experimental procedures, mechanistic studies, characterization data, copies of NMR spectra and computational details are available in the Supplementary Information. Crystallographic parameters for compound 5a are available free of charge from the Cambridge Crystallographic Data Centre under deposition number CCDC 2026384 (5a).
References
Dubinina, G. G. & Chain, W. J. Reactions of azepinones with electrophiles. Tetrahedron Lett. 52, 939–942 (2011).
Schultz, C. et al. Paullones, a series of cyclin-dependent kinase inhibitors: synthesis, evaluation of CDK1/cyclin B inhibition, and in vitro antitumor activity. J. Med. Chem. 42, 2909–2919 (1999).
Le Diguarher, T. et al. Synthesis of N,N′-disubstituted 3-aminobenzo[c] and [d]azepin-2-ones as potent and specific farnesyl transferase inhibitors. Bioorg. Med. Chem. Lett. 14, 767–771 (2004).
Huigens, R. W. III et al. A ring-distortion strategy to construct stereochemically complex and structurally diverse compounds from natural products. Nat. Chem. 5, 195–202 (2013).
Carson, M. W. et al. HDX reveals unique fragment ligands for the vitamin D receptor. Bioorg. Med. Chem. Lett. 24, 3459–3463 (2014).
Huisgen, R. Angew. Chem. 67, 756 (1955).
Doering, W., von, E. & Odum, R. A. Ring enlargement in the photolysis of phenyl azide. Tetrahedron 22, 81–93 (1966).
Takeuchi, H. & Koyama, K. Photolysis and thermolysis of phenyl azide in acetic acid. Trapping of 1-azacyclohepta-1,2,4,6-tetraene and nucleophilic aromatic substitution. J. Chem. Soc. Perkin Trans. 1 1269–1273 (1982).
Schrock, A. K. & Schuster, G. B. Photochemistry of phenyl azide: chemical properties of the transient intermediates. J. Am. Chem. Soc. 106, 5228–5234 (1984).
Shi, Z., Grohmann, C. & Glorius, F. Mild rhodium(III)-catalyzed cyclization of amides with α,β-unsaturated aldehydes and ketones to azepinones: application to the synthesis of the homoprotoberberine framework. Angew. Chem. Int. Ed. 52, 5393–5397 (2013).
Plietker, B. & Röske, A. Recent advances in Fe-catalyzed C–H aminations using azides as nitrene precursors. Catal. Sci. Technol. 9, 4188–4197 (2019).
Dequirez, G., Pons, V. & Dauban, P. Nitrene chemistry in organic synthesis: still in its infancy? Angew. Chem. Int. Ed. 51, 7384–7395 (2012).
Soderberg, B. C. G. Synthesis of heterocycles via intramolecular annulation of nitrene intermediates. Curr. Org. Chem. 4, 727–764 (2000).
Ye, L.-W. et al. Nitrene transfer and carbene transfer in gold catalysis. Chem. Rev. 121, 9039–9112 (2021).
Giri, S. S. & Liu, R.-S. Gold-catalyzed [4+3]- and [4+2]-annulations of 3-en-1-ynamides with isoxazoles via novel 6π-electrocyclizations of 3-azahepta trienyl cations. Chem. Sci. 9, 2991–2995 (2018).
Tsai, M.-H., Wang, C.-Y., Raja, A. S. K. & Liu, R.-S. Gold-catalyzed annulations of N-aryl ynamides with benzisoxazoles to construct 6H-indolo[2,3-b]quinoline cores. Chem. Commun. 54, 10866–10869 (2018).
Sahani, R. L. & Liu, R.-S. Development of gold-catalyzed [4+1] and [2+2+1]/[4+2] annulations between propiolate derivatives and isoxazoles. Angew. Chem. Int. Ed. 56, 1026–1030 (2017).
Liu, X. et al. Copper-catalyzed azide–ynamide cyclization to generate α-imino copper carbenes: divergent and enantioselective access to polycyclic N-heterocycles. Angew. Chem. Int. Ed. 59, 17984–17990 (2020).
Zhu, X.-Q. et al. Zinc-catalyzed asymmetric formal [4+3] annulation of isoxazoles with enynol ethers by 6π electrocyclization: stereoselective access to 2H-azepines. Angew. Chem. Int. Ed. 59, 1666–1673 (2020).
Zhou, A.-H. et al. Atom-economic generation of gold carbenes: gold-catalyzed formal [3+2] cycloaddition between ynamides and isoxazoles. Chem. Sci. 6, 1265–1271 (2015).
Shu, C. et al. Generation of α-imino gold carbenes through gold-catalyzed intermolecular reaction of azides with ynamides. J. Am. Chem. Soc. 137, 9567–9570 (2015).
Tian, X., Song, L. & Hashmi, A. S. K. α-Imino gold carbene intermediates from readily accessible sulfilimines: intermolecular access to structural diversity. Chem. Eur. J. 26, 3197–3204 (2020).
Tian, X. et al. Sulfilimines as versatile nitrene transfer reagents: facile access to diverse aza-heterocycles. Angew. Chem. Int. Ed. 58, 3589–3593 (2019).
Kobayashi, Y., Masakado, S. & Takemoto, Y. Photoactivated N-Acyliminoiodinanes applied to amination: an ortho-Methoxymethyl group stabilizes reactive precursors. Angew. Chem. Int. Ed. 57, 693–697 (2018).
Guo, Y., Pei, C., Jana, S. & Koenigs, R. M. Synthesis of trifluoromethylated aziridines via photocatalytic amination reaction. ACS Catal. 11, 337–342 (2021).
Guo, Y., Pei, C., Empel, C., Jana, S. & Koenigs, R. M. Photochemical nitrene transfer reactions of iminoiodinanes with sulfides. ChemPhotoChem 6, e202100293 (2022).
Guo, Y. et al. Intermolecular amination of allenes via 2-fold photocatalytic nitrene transfer reactions. Chem. Catal. 2, 2012–2023 (2022).
Tian, X., Song, L. & Hashmi, A. S. K. Synthesis of carbazoles and related heterocycles from sulfilimines by intramolecular C−H aminations. Angew. Chem. Int. Ed. 59, 12342–12346 (2020).
Guo, Y., Pei, C. & Koenigs, R. M. A combined experimental and theoretical study on the reactivity of nitrenes and nitrene radical anions. Nat. Commun. 13, 86 (2022).
Alt, I. T., Guttroff, C. & Plietker, B. Iron-catalyzed intramolecular aminations of C(sp3)−H bonds in alkylaryl azides. Angew. Chem. Int. Ed. 56, 10582–10586 (2017).
Alt, I. T. & Plietker, B. Iron-catalyzed intramolecular C(sp2)−H amination. Angew. Chem. Int. Ed. 55, 1519–1522 (2016).
Smith, P. A. S. & Brown, B. B. The synthesis of heterocyclic compounds from aryl azides. I. bromo and nitro carbazoles. J. Am. Chem. Soc. 73, 2435–2437 (1951).
Antoni, P. W. et al. Dibenzothiophenesulfilimines: a convenient approach to intermolecular rhodium-catalysed C−H amidation. Chem. Eur. J. 26, 8235–8238 (2020).
Tian, X. et al. N-pyridinyl sulfilimines as a source for α-imino gold carbenes: access to 2-amino-substituted N-fused imidazoles. Org. Lett. 21, 1598–1601 (2019).
Tian, X. et al. Gold(III)-catalyzed formal [3 + 2] annulations of N-acyl sulfilimines with ynamides for the synthesis of 4-aminooxazoles. Org. Lett. 21, 2937–2940 (2019).
Tian, X., Song, L., Rudolph, M., Rominger, F. & Hashmi, A. S. K. Synthesis of 2-aminoindoles through gold-catalyzed C–H annulations of sulfilimines with N-arylynamides. Org. Lett. 21, 4327–4330 (2019).
Smith, P. A. S. In Azides and Nitrenes; Reactivity and Utility. Scriven, E. F. V., Ed.; Academic: New York, 1984; p 95.
Ess, D. H. & Cook, T. C. Unrestricted prescriptions for open-shell singlet diradicals: using economical ab initio and density functional theory to calculate singlet–triplet gaps and bond dissociation curves. J. Phys. Chem. A 116, 4922–4929 (2012).
Krauter, C. M., Hashmi, A. S. K. & Pernpointner, M. A new insight into gold(I)-catalyzed hydration of alkynes: proton transfer. ChemCatChem 2, 1226–1230 (2010).
Farshadfar, K., Chipman, A., Hosseini, M., Yates, B. F. & Ariafard, A. A modified cationic mechanism for PdCl2-catalyzed transformation of a homoallylic alcohol to an allyl ether. Organometallics 38, 2953–2962 (2019).
Acknowledgements
L.S. and X.T. are grateful to the CSC (China Scholarship Council) for a PhD fellowship. We gratefully acknowledge the generous allocation of computing time from the Australian National Computational Infrastructure and University of Tasmania, and the Australian Research Council (grant number DP180100904) for financial support. We thank Petra Krämer for her UV-vis measurements.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
X.T. and A.S.K.H. conceived and designed the project. L.S. performed experiments and analyzed the experimental data. A.A., K.F., and F.S. conducted DFT calculations. F.R. analyzed the X-ray crystal structure. L.S., X.T., K.F., A.A., and A.S.K.H. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Pascale Maldivi, and the other, anonymous, reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Song, L., Tian, X., Farshadfar, K. et al. An unexpected synthesis of azepinone derivatives through a metal-free photochemical cascade reaction. Nat Commun 14, 831 (2023). https://doi.org/10.1038/s41467-023-36190-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-36190-z
This article is cited by
-
Single-atom skeletal editing of 2H-indazoles enabled by difluorocarbene
Science China Chemistry (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
