
Abstract
Imitating the natural photosynthesis to synthesize hydrocarbon fuels represents a viable strategy for solar-to-chemical energy conversion, where utilizing low-energy photons, especially near-infrared photons, has been the ultimate yet challenging aim to further improving conversion efficiency. Plasmonic metals have proven their ability in absorbing low-energy photons, however, it remains an obstacle in effectively coupling this energy into reactant molecules. Here we report the broadband plasmon-induced CO2 reduction reaction with water, which achieves a CH4 production rate of 0.55 mmol g−1 h−1 with 100% selectivity to hydrocarbon products under 400 mW cm−2 full-spectrum light illumination and an apparent quantum efficiency of 0.38% at 800 nm illumination. We find that the enhanced local electric field plays an irreplaceable role in efficient multiphoton absorption and selective energy transfer for such an excellent light-driven catalytic performance. This work paves the way to the technique for low-energy photon utilization.
Similar content being viewed by others
Introduction
Artificial photosynthesis offers an appealing way to store intermittent solar energy by directly converting CO2, H2O and sunlight into hydrocarbon compounds1,2,3,4. Therewith, a myriad of semiconductors and metal nanocatalysts have been studied, demonstrating their feasibility in light-driven CO2 reduction reaction (CO2RR)5,6,7. Nonetheless, the low utilization efficiency of the solar spectrum, especially the low-energy photons, and the sluggish process for energy coupling into the reactant molecules are the two critical obstacles that limit their further expansion toward technoeconomic applications. Moreover, the selective production of high-energy-value hydrocarbons involving multiple electron-proton transfer steps through CO2RR is another great challenge constraining their potential8.
Plasmonic metal nanoparticles exhibit strong interaction with incident light in the form of localized surface plasmon resonance (LSPR)9,10. The absorption wavelength center of such LSPR can be easily tuned from ultraviolet (UV) to near-infrared (NIR) range just by changing the geometry structure (including symmetry, size, etc.) of plasmonic nanomaterials11,12, allowing them to have the wide possibility as light-harvesting units for solar-to-chemical energy conversion13,14,15,16,17,18,19. Recently, it was demonstrated that specific plasmonic nanostructures are able to convert CO2 under laser irradiation with the help of organic additives20,21, suggesting their potential in CO2RR. In addition, the rates of plasmon-induced catalytic reaction exhibit a super-linear power law dependence on the number of incident photons, providing a positive relationship between quantum efficiency and photon flux22. Amidst the fascinating development of such plasmonic materials, it is discovered that the mismatch of quick LSPR relaxation (up to ~100 ps) and slow chemical reduction kinetics (several milliseconds or seconds) in time scale severely retards the conversion efficiency of solar energy23,24,25. To break this bottleneck, versatile co-catalysts are employed to establish an effective bridge for transferring hot carriers from plasmonic materials into reactant molecules13,14,15,16,17,26.
In this respect, the precise design of the co-catalyst toward efficient hot carrier accepting ability and unique CO2 chemisorption capability is highly sought. Here we employ Au rod as a plasmonic light-harvesting unit and copper-palladium (CuPd) alloy shell as a co-catalyst to realize efficient plasmon-induced artificial photosynthesis in the absence of the additional sacrificial agent, achieving nearly 100% CH4 product selectivity under full-spectrum light illumination. As deciphered by in situ near ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) and density functional theory (DFT) calculations, the localized electric field, which leads to the emergence of a new isolated state above the Fermi energy (Ef) and the differentiation of electron transfer within different molecular orbitals, enables such a unique solar-to-chemical energy conversion. By optimizing the reaction system, an impressive CH4 production rate of 0.55 mmol g−1 h−1 is achieved via a gas-solid biphase system, demonstrating a record-high apparent quantum efficiency (AQE) of 0.38% at 800 nm NIR light illumination.
Results
Plasmon-induced CO2 reduction with water
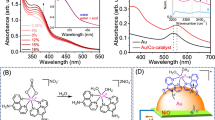
The CuPd co-catalyst can capture CO2 molecules and enhance the CO2 density on the catalyst surface, which allows the reactants to locate within the range of the plasmon-induced local field, increasing the opportunity for their further activation and conversion (see Fig. 1a). Typically, we obtain the Au rod@CuPd core-shell composites via a wet chemical synthesis (Fig. S1). The shell thickness and Pd/Cu ratio of the Au rod@CuPd core-shell composites can be easily controlled by changing the amount of metal precursors (see Tables S1, S2, and Figs. S2–S5 for the list of prepared samples). Taking Au rod@CuPd2 as an example, the transmission electron microscopy (TEM) image (Fig. S3a) shows that the rod structure is preserved after the deposition of the CuPd shell onto the Au surface. The fast Fourier transform patterns and magnified images of the selected area in Fig. S6, a,b, are well-fitted with a pure face-centered cubic crystal structure viewed along the [110] and [100] directions, respectively. The ordered lattice fringes with a spacing of 2.35 and 2.05 Å are attributed to the (111) and (002) planes of Au lattice, respectively, indicating that precursor ions are epitaxially reduced on the surface of Au rods to form a CuPd single-crystal shell. Such a result can be also supported by the X-ray diffraction (XRD) analysis (Fig. S7) and the elemental mapping profiles (Fig. S8). In addition, the Pd and Cu K-edge X-ray absorption near-edge structure (XANES, Fig. S9) and Fourier transformed extended X-ray absorption fine structure (EXAFS, Fig. S10) spectra of the prepared samples show that the total coordination numbers of Pd and Cu are smaller than 12 (see Table S3), further confirming that the Pd and Cu elements are located in the outermost shell of these samples. Then we employ UV-vis extinction spectra to evaluate the light-harvesting ability of the prepared samples. As shown in Fig. S11, two peaks around 510 and 740 nm are the typical characteristics of the transversal and longitudinal LSPR modes of Au nanorod, respectively27,28. It is discovered that, compared with the Cu/Pd molar ratio, the CuPd thickness has a more prominent influence on the optical properties, allowing the remarkable broadening of the absorption range of prepared samples into the NIR region.

a Schematic illustration of the role of CuPd co-catalyst in capturing CO2 molecules. Plasmonic catalysis usually takes place very close to the catalyst surface (i.e., within the range of the plasmon-induced local field). For the pure Au nanorods, the probability of the pure Au nanorods and CO2 molecules to contact through the collision is very low (left), resulting in low CO2 conversion efficiency. For the Au rod@CuPd, CuPd co-catalyst can capture CO2 molecules and enhance the CO2 concentration on the catalyst surface, increasing the opportunity for their further activation and conversion (right). b Average production rates of CH4 over Au rod@CuPd2 with different shell thicknesses. c Time-dependent CH4 production over Au rod@CuPd2 in 24 h. d Calculated AQEs (color dots) over Au rod@CuPd2 under different monochromatic light illumination, in reference to its UV-vis extinction spectrum (black line). The error bars represent the standard deviation of the experiments. The catalytic experiments of (b,c) are all carried out in CO2-saturated water and CO2 atmosphere without any sacrificial agent under 400 mW cm–2 full-spectrum light illumination.
Upon having a comprehensive understanding of the structural information and optical properties of the Au rod@CuPd, we assess their efficacy for CO2RR in a conventional reactor consisting of CO2-saturated water and suspended catalysts (see Fig. S12). After continuous illumination for 3 h, only CH4 can be detected with a maximum production rate of 15.6 μmol g−1 h−1, ca. 39 times that of Au rod, demonstrating the excellent catalytic performance of Au rod@CuPd2 (Fig. 1b). The production rates of CH4 show a volcano-shaped relationship with the CuPd/Au ratios, which can be attributed to the competition of increased surface active sites and aggravated electron-phonon scattering in the thickened CuPd shell16. The linear time-dependent CH4 production shown in Fig. 1c corroborates the excellent durability of Au rod@CuPd2. Moreover, C2H6 and C2H4 can also be detected after the long-term test, suggesting the presence of multiple proton-coupled electron transfer processes during the reaction, which is uncommon in conventional photocatalysis. In the meantime, after normalizing the CH4 production rates by the amount of Cu (orange dots in Fig. S13), the catalytic performances of samples with different Cu/Pd atomic ratios are similar, suggesting the inseparable role of Cu in CO2RR. The wavelength-dependent apparent quantum efficiencies (AQEs), which are in good agreement with the extinction spectrum of Au rod@CuPd2, are up to 0.1% at 800 nm (see Fig. 1d). This result explicitly suggests the unique features of Au rod@CuPd2 in effectively utilizing low-energy photons in the absence of the sacrificial agent (see also Fig. S14). In cyclic tests under 800 nm monochromatic light illumination, the production rates of CH4 are well maintained for 10 runs (each run for 3 h, Fig. S15), verifying the long-term stability of Au rod@CuPd2. Furthermore, hydroxyl radicals (•OH) are detected as the main oxidative product by electron spin resonance spectroscopy (Fig. S16).
The verification of product origin is another key aspect. The conditions for various control experiments and corresponding results are illustrated in Fig. S17 (see also Table S4 for details). Unambiguously, catalyst, light, CO2 and H2O are keys for CO2RR over Au rod@CuPd2 and the CH4 can hardly be produced from the direct photolysis or pyrolysis of the carboneous compounds (e.g., surfactants and reducing agents for catalyst synthesis). We further employ synchrotron radiation vacuum UV photoionization mass spectrometry (SVUV-PIMS) to convincingly identify the origin of the CH4 product. As shown in Fig. S18, a,b, the peaks at m/z = 16 and 17 can be assigned to 12CH4 and 13CH4 when using 12CO2 and 13CO2 as the feed gas, respectively, firmly suggesting that the CO2 feed gas is indeed the carbon source of produced CH4. In addition, we employ offline sum frequency generation vibrational spectroscopy (SFG-VS) to monitor the surface intermediates formed during the photocatalytic reaction. As shown in Fig. S19, a,b, after illumination for 5 min, three SFG-VS signals appear at 2880, 2945, and 2965 cm−1, which can be assigned to the symmetric stretching (νs C–H), Fermi-resonance (νFermi C–H) and asymmetric stretching (νas C–H) modes of the generated methyl groups, respectively29. This observation firmly demonstrates the evolution of CO2 and H2O to methyl groups at the catalytic interface.
Mechanism for plasmon-induced catalytic process
The direct charge transfer mechanism provides a reasonable explanation for the unique selectivity in plasmon-induced catalytic process30,31,32. Nevertheless, in the case of our work, a critical issue is that the energy of a single low-energy photon (~1.55 eV for 800 nm light) is insufficient for reaching the energy barrier of CO2RR without adding sacrificial agent. To gain insight into such a low-energy photon utilization ability for CO2RR, we employ in situ NAP-XPS to examine the plasmon-induced light-matter interaction in the catalytic system. All NAP-XPS spectra have been calibrated with the Au 4 f core level of Au foil, which is mounted beside the sample (see Fig. S20). Under the dark condition, no obvious fluctuation arises in the binding energy of Cu (Fig. 2a, b). Once turning on the light, the binding energy of the Cu peak instantaneously decreases (Stage I), and then is gradually reduced (Stage II) under subsequent light illumination. When the light is turned off, the binding energy of Cu increases immediately (Stage III) but cannot return to the initial level. Based on these observations, we can infer that the changes in Cu involve two conceivable processes, where one is fast and reversible (Stage I and III), and the other is slow and irreversible (Stage II). For Stage I and III, electron energy fluctuation appears when the interaction of incident light with catalyst changes. This phenomenon can be attributed to the accumulation of hot electrons above the Fermi level (Ef), whose lifetime typically lasts only a few hundred femtoseconds24,33. For Stage II, it is accompanied by the gradual disappearance of the CuII satellite peak in Fig. 2a, suggesting that the Cu elements are gradually reduced under light illumination by the hot electrons generated by LSPR relaxation (see also Figs. S21 and S22). Such a chemical state change is relatively sluggish and can be responsible for the slow process. By analyzing the changes of Pd 3d NAP-XPS spectra in Fig. 2c, d, we can confirm that Pd shows similar three-stage behavior to Cu 2p3/2. From the viewpoint of catalysis, the slow photoreduction and the fast hot-electron accumulation above the Ef are both beneficial to improve the chemical potential of the catalyst34,35, which is vital to break its catalytic limitation. In the meantime, we also monitor the C 1 s spectra through in situ characterization. As shown in Fig. S23, two peaks at 284.4 and 293.2 eV attributed to the adsorbed C species (Cad species) and gaseous CO2 (g-CO2), respectively, can be observed. The relative increase in peak area (see Fig. S21) and positive shift of the binding energy of g-CO2 peak under light conditions represent the continuous adsorption of CO2 molecules and their conversion process to hydrocarbons36,37,38.

a and b In situ NAP-XPS spectra of Cu 2p3/2 (a) and contour plot for the amplified top view of the selected box-shaped area (b). c, d In situ NAP-XPS spectra of Pd 3d5/2 (c) and contour plot for the amplified top view of the selected box-shaped area (d). The in situ NAP-XPS data are recorded continuously for 1 h over Au rod@CuPd2 at 0.25 mbar CO2 atmosphere and light illumination is introduced into the catalytic system in the middle 30 min (marked in orange color). Representative peak positions are marked with open squares in (b) and (d). The squares and lines in black and white indicate unilluminated and illuminated conditions, respectively.
To reveal the mechanism how CO2 is activated with the catalyst upon light irradiation, we further investigate the key role of unoccupied adsorbate states, by analyzing the projected density of states (PDOS, Fig. 3a) of our catalytic system consisting of catalyst and CO2 molecule. With CuPd as catalytic sites, the CO2 molecule can form hybrid states with the catalyst upon adsorption. Upon introduction of the electric field to the hybrid states, the calculation indicates that the state peaks of O1 2p and O2 2p shift toward lower energy and form a larger overlap area with the more localized Pd 4dz and Cu 3dz (parallel to the electric field) states, enhancing the stability of the key adsorption configuration39. More importantly, a series of new quasi isolated trap states emerge above the Ef in the presence of the electric field as illustrated in Fig. 3a. Compared to the continuous states near Ef of PdCu metal, these quasi isolated trap states can effectively extend the lifetime of hot electrons and increase the probability of electron re-excitation by another low-energy photon40,41, which makes the utilization of low-energy photons possible. Such a multiphoton absorption behavior can be corroborated by the superlinear law dependence between CH4 production rate and light intensity (see Figs. S24–S27)19,20,22. Taken together, the information gleaned by NAP-XPS and theoretical calculation can put forward the mechanism for plasmon-induced charge transfer in CO2RR as shown in Fig. 3b. Generally, there are three key processes for initiating and enhancing the plasmon-induced CO2RR, including (i) fast excitation for accumulating hot electrons above the Fermi level (Ef), (ii) slow photoreduction process for elevating the chemical potential of the catalytic sites, and (iii) emergence of new quasi isolated trap states for re-exciting the electrons, gaining sufficient energy for triggering CO2RR. During the light-driven CO2RR, the three distinct processes work together to realize the utilization of low-energy NIR light for the reaction.

a The projected density of states (PDOS) of CO2 adsorbed on a CuPd (100) surface in the absence (left) and presence (right) of electric field pointing toward the surface. The main quasi isolated trap states are marked with orange asterisks. b Schematic illustration for the plasmon-induced energy transfer in CO2RR, where the y-axis is energy (E) and the gray shading indicates filled electronic states. Three key processes for initiating and enhancing the plasmon-induced CO2RR include (i) fast excitation and accumulation of electrons above the Fermi level (Ef), (ii) plasmon-induced slow photoreduction of Cu and Pd, and (iii) formation of new isolated trap state (origin shading) by coupling CO2 molecule with metal orbitals and re-excitation of the electrons above the Fermi level, which eventually result in the CO2RR process. c NOCV orbitals relevant to the most important deformation density in CO2 and H2O on a CuPd (100) surface in the absence (middle column) and presence of electric field toward (right column) or away from (left column) the surface. Donor NOCV on the left and acceptor NOCV on the right are displayed in each panel. Isovalue is set to 0.015.
Given the key role of hybrid states in the mechanism, we further look into the hybrid orbitals composed of catalytic sites and CO2 or H2O molecules through DFT calculations. The optimized geometries and adsorption energies of molecules on CuPd (100) surface in different field conditions are illustrated in Fig. S28. Although the electron transfers from CuPd (100) surface to H2O and CO2 molecules exhibit similar trends (see Fig. S29), the CO2 and H2O display different natural orbitals for chemical valence (NOCVs) (Fig. 3c). In detail, when electric field points toward the metal surface, the electrons of CO2 are redistributed from O lone-pair orbital to its antibonding π* orbital, weakening the C–O bond and increasing the bond length. When the electric field points away from the metal surface, the lone-pair electrons transfer to the σ orbital between O and the metal atom. However, the electron transfer in adsorbed H2O molecules only oscillates between the bonding orbital of H2O and the orbital of Pd atoms. Thus the electric field provides scant assistance to the cleavage of the O–H bond. Based on these results, it is confirmed that the local electric field can facilitate the directional electron transfer to CO2 molecules, which contributes to the selective production of hydrocarbons.
Performance enhancement based on catalytic mechanism
Upon demonstrating the opportunity of utilizing low-energy photons in plasmon-induced CO2RR, it remains an open question whether it is possible to further improve the photon utilization for enhanced multiphoton absorption. Given the super-linear relationship between plasmonic catalytic rate and photon flux (i.e., the light intensity, Fig. S26), the inevitable scattering loss of incident light becomes the main limitation of the conventional reaction system (see Fig. 4a). To overcome the limitation, we design a gas-solid reaction system with a spherical structure to realize the re-incidence of scattered photons, which works only for the plasmonic catalytic system rather than conventional photocatalysis. By optimizing the volume of H2O and the partial pressure of CO2, the CH4 production rate of the light-driven CO2RR performed in such a system reaches 0.55 mmol g−1 h−1 (7.5 mmol gCu−1 h−1) under 400 mW cm−2 full-spectrum light illumination (Fig. 4b), ca. 35 times higher than that in the reaction system allowing only a single pass of the incident photon. To verify the catalytic nature, turnover number (TON) is calculated (see Supplementary Text for detail). A TON of 9.6 per exposed Cu atoms is obtained after 3 h of illumination given that no catalyst deactivation has been observed in long-term operation. In the meantime, the control experiments are also performed in the dark at an elevated temperature (see Fig. S30 and Table S5), which matches the local temperature on the surface of catalyst nanoparticles under photoexcitation, to confirm the indispensability of light illumination. This conversion rate, enabled by the reaction system design taking advantage of plasmonic catalysis mechanism, far exceeds the current literature record (Fig. 4c and Table S6). Notably, the AQE in the gas-solid reaction at 800 nm is promoted to 0.38%. Such a superior photoconversion efficiency under low-energy NIR light illumination, which is even comparable with the efficiencies of the representative works in CO2RR with high-energy photons, allows our Au rod@CuPd2 catalyst to hit the record-high CO2RR AQEs under NIR light illumination (Fig. 4d and Table S7).

a Schematic illustration of the gas-solid reaction, where mixed vapor of CO2 and H2O with centrally installed catalyst nanoparticles contained in a spherical reactor is illuminated with full-spectrum light. During the reaction, the scattered photons can be reflected to the catalyst surface, leading to efficient multi-absorption processes. b Average production rates of CH4 through plasmon-induced CO2RR using the gas-solid reaction device over Au rod@CuPd2 under different CO2 pressure and H2O volume. The catalytic experiments are carried out in the mixed vapor of CO2 and H2O without any sacrificial agent under 400 mW cm–2 full-spectrum light illumination. c, d The comparison of production rates (c, see Table S5) and AQEs (d, see Table S6) of Au rod@CuPd2 reported in this work with the records reported in the literature for the last 5 years on artificial photosynthesis, which directly convert CO2, H2O and sunlight into compounds without sacrificial agent.
Discussion
This work emphasizes the irreplaceable role of plasmon-induce local fields in the solar-to-chemical energy transfer process, providing a riveting picture of the potential of plasmon-induced catalysis toward achieving broadband artificial photosynthesis, particularly with the utilization of low-energy photons, and renders new insights for pushing forward the future development of plasmonic catalysis. Such a technique for low-energy photon utilization can potentially be integrated with the existing techniques available for UV and visible light in the future.
Methods
Materials
Potassium(II) tetrachloropalladate (K2PdCl4, 99.99%) was obtained from Beijing HWRK Chem Co., LTD. Sodium borohydride (NaBH4, 98%) was obtained from Sigma Aldrich. 13CO2 isotope was obtained from Sigma Aldrich. Copper(II) chloride dihydrate (CuCl2·2H2O) was obtained from Aladdin. Gold(III) chloride tetrahydrate (HAuCl4·4H2O, 99.95%), ascorbic acid (AA, 99.7%), hexadecyl trimethyl ammonium bromide (CTAB, 99.0%) and hexadecyl trimethyl ammonium chloride (CTAC, 98.0%) were purchased from Sinopharm Chemical Reagent Co., Ltd. The water used in all experiments was deionized. All the chemical reagents were used as received without further purification.
Synthesis of Au rods
High-quality Au rods encircled by {100} and {110} facets were prepared through a modified seed-mediated growth process42. In the preparation of seed solution, an aqueous solution of CTAB (0.2 M, 2.25 mL) was mixed with 2.25 mL of 0.5 mM HAuCl4, and kept stirring in a 20 mL glass vessel. Subsequently, 157.5 μL ice-cold aqueous solution of NaBH4 (16.4 mM) was quickly added. Then the seed solution was kept vigorous stirring, resulting in the formation of a brownish yellow solution. After stirring for 1 min, it was kept at 30 °C for 30 min before further use.
In a typical synthetic process of Au rods, 195 mL CTAB (0.1 M) was kept in a 250 mL conical flask under magnetic stirring at room temperature. Subsequently, 5 mL aqueous solution of HAuCl4 (20 mM) and 5 mL aqueous solution of AgNO3 (4 mM) were injected into the conical flask successively. Then the aqueous solutions of AA (0.1 M) were dropped into the above solution until the color of the solution turned from orange to colorless. After another 300 μL AA (0.1 M) was added, 1 mL prepared seed solution was quickly added into the flask with a pipette. After reaction for 1 h, the products were centrifuged with deionized water for three times at 11,000 rpm for 10 min. The precipitation was dispersed in deionized water and measured by inductively coupled plasma mass spectrometry (ICP-MS) for further use.
Synthesis of Au rod@CuPd2
The co-deposition of Cu and Pd atoms on Au rod was achieved via an epitaxial growth method. In a typical synthetic process for Au rod@CuPd2 sample, 70.8 mL aqueous solution containing 0.1 M hexadecyl trimethyl ammonium chloride (CTAC) was kept in a 100 mL round-bottom flask under magnetic stirring at 50 °C, followed by the addition of a suspension containing 4 mg Au rods. Then the aqueous solutions of K2PdCl4 (1 mL, 10 mM) and CuCl2 (1 mL, 10 mM) were added into the flask. Subsequently, 2.4 mL aqueous solution of ascorbic acid (0.1 M) was added into the flask with a pipette immediately. After reaction for 1 h, the products were centrifuged with deionized water for three times at 9500 rpm for 10 min before being re-dispersed in 4 mL deionized water.
Synthesis of Au rod@CuPdx-y
Other Au rod@CuPdx-y samples were synthesized by following similar procedures except for the use of different amounts of K2PdCl4 and CuCl2. The usage of K2PdCl4 and CuCl2: (1) 1.2 mL K2PdCl4 and 800 μL CuCl2 for Au rod@CuPd2.5, (2) 1.4 mL K2PdCl4 and 600 μL CuCl2 for Au rod@CuPd3.6, (3) 1.6 mL K2PdCl4 and 400 μL CuCl2 for Au rod@CuPd6.1, (4) 1.8 mL K2PdCl4 and 200 μL CuCl2 for Au rod@CuPd8.8, (5) 1.9 mL K2PdCl4 and 100 μL CuCl2 for Au rod@CuPd14.2, (6) 200 μL K2PdCl4 and 200 μL CuCl2 for Au rod@CuPd2-0.1, (7) 500 μL K2PdCl4 and 500 μL CuCl2 for Au rod@CuPd2-0.2, (8) 1.5 mL K2PdCl4 and 1.5 mL CuCl2 for Au rod@CuPd2-0.9, and (9) 2.0 mL K2PdCl4 and 2.0 mL CuCl2 for Au rod@CuPd2-1.2.
In situ NAP-XPS characterization
In situ NAP-XPS was performed at the X-ray Photoelectron Spectroscopy and UV Photoelectron Spectroscopy Endstation (BL24A1) in the National Synchrotron Radiation Center (NSRRC). The apparatus was composed of three chambers, including a sample load-lock chamber, a preparation chamber and an analysis chamber. The analysis chamber was equipped with an SPEC Phoibos 150 energy analyzer. The catalyst was dropcast onto a clear silicon wafer, and dried at room temperature. The samples were then loaded into the load-lock chamber and delivered to preparation chamber for 10 min Ar ion sputtering treatment. High-purity CO2 (99.9999%) was introduced into the analysis chamber with the partial pressure maintained at 0.25 mbar. Afterwards, the samples were delivered to the analysis chamber. NAP-XPS were recorded with 1170 eV photons while a Xe lamp source (PLS-SXE300D, Beijing Perfectlight) was used as a light source. The Cu 2p, Pd 3d and C 1s spectra were recorded continuously for 1 h and light illumination was introduced into the analysis chamber in the middle 30 min. The incident photon energy was calibrated against Au 4f core level measured with an Au foil mounted beside the sample.
Sum frequency generation vibrational spectroscopy characterization
We performed the offline SFG-VS study on Au rod@CuPd2 with a home-built SFG-VS spectrometer. Briefly, an 1 kHz Ti:Sapphire laser amplification system (Spitfire Ace, Spectra-Physics) was employed to produce a ~80 fs pulse with the center wavelength at 800 nm and a pulse energy of 6 mJ. About 3 mJ of this output was fed into an optical parametric amplifier (TOPAS Prime, Light Conversion) to generate the femtosecond broadband infrared (IR) pulse, while another portion of the 800 nm fundamental beam (~1.2 mJ) was guided into a 4f pulse shaper to produce the narrowband upconversion pulse centered around 800 nm with a FWHM of ~7 cm−1. The broadband IR and narrowband upconversion pulses overlapped at the sample surface both spatially and temporally with their incident angles fixed at 45° and 63° against the surface normal, respectively. The sum frequency generation signals were acquired in a reflective geometry with a high-resolution optical spectrometer (SR-500i-A, Andor Inc.) coupled with a CCD detector (Du970P-BVF, Andor Inc.). Before the SFG-VS measurements, the sample films were deposited on the flat side of a CaF2 half cylinder support and washed with water vapor for 20 min to totally remove the hydrocarbon surfactant. The newly prepared films were transferred into CO2-saturated ultrapure water environment enclosed by a Teflon vessel, followed by the exposure to the broadband irradiation (Xe lamp) for 20 min. During the offline SFG-VS experiment, the averaged pulses of IR and upconversion pulses were kept at 3.0 and 9.0 μJ, respectively, for Au rod@CuPd2 at ppp (p-SFG, p-Vis, and p-IR) and ssp (s-SFG, s-Vis, and p-IR) polarization to avoid any surface damage. All SFG signals were averaged for 600 k shots.
Characterization
Transmission electron microscopy (TEM) images were taken on a Hitachi Model H-7700 microscopy at an accelerating voltage of 100 kV. High-resolution transmission electron microscopy images and energy dispersive spectroscopy mapping profiles were recorded on a JEOL ARM-200F field-emission transmission electron microscope at an accelerating voltage of 200 kV. Powder XRD patterns were collected using a Japan Rigaku DMax-γA rotating anode X-ray diffractometer equipped with diffracted beam graphite monochromator (Cu Kα radiation (λ = 1.54178 Å)). The concentrations of various elements in the prepared samples were measured with a Thermo Scientific PlasmaQuad 3 inductively-coupled plasma mass spectrometry (ICP-MS) after dissolving them with a mixture of HCl and HNO3 (3:1, volume ratio). Ultraviolet-visible (UV-Vis) extinction spectra were recorded in the spectral region of 300–1000 nm with an Agilent Technologies Cary 60 spectrometer. The samples were diluted with deionized water to the same concentration before being measured in a quartz cuvette.
Photocatalytic CO2 reduction reaction measurements
The light-driven catalytic CO2RR was performed in a homemade quartz tube. Typically, 1 mg catalyst sample (based on inductively coupled plasma mass spectrometry, ICP-MS measurements) was dispersed in deionized water (18.25 MΩ cm) in the tube to reach a total suspension volume of 4 mL, which was then vacuumed and saturated with high-purity CO2 (99.9999%) for three times and sealed with a glass stopper after filled with 1 atm high-purity CO2 (99.9999%). Subsequently the suspension was vigorously stirred, and illuminated using a Xe lamp source (PLS-SXE300D, Beijing PerfectLight) at a light intensity of 400 mW cm–2 (full-spectrum light). The resultant reaction gas was then analyzed by gas chromatography (GC, 7890 A, Agilent) equipped with a flame ionization detector (FID). The control experiments in dark environment were performed under the same conditions except the exclusion of light with an aluminum foil or the exclusion of carbon dioxide with high-purity Ar (99.9999%).
Photocatalytic CO2 reduction measurements in a gas-solid biphase reactor
The light-driven catalytic CO2RR was performed in a 10 cm diameter spherical reactor equipped with a 1 cm diameter quartz window as the light inlet. The inside of the spherical reactor was uniformly coated with foamed polytetrafluoroethylene, whose reflectance is 0.978, 0.991, 0.989 and 0.988 at 400, 600, 800 and 1000 nm, respectively. Typically, 0.5 mg catalyst sample (based on ICP-MS measurements) was dispersed evenly on a 3 cm diameter quartz wool sheet, which was then gently dried in an inert atmosphere and installed in the center of the integrating sphere reactor facing the light inlet. Subsequently, 0–0.4 mL CO2-saturated water was dropped on the quartz wool sheet. After the reactor was sealed with the quartz window, high-purity CO2 (99.9999%) was introduced to purify the reactor and filled to 0–2 atm. Then a Xe lamp source (PLS-SXE300D, Beijing PerfectLight, 400 mW cm–2, full spectrum) was employed as a light source to irradiate the catalyst from the light inlet. The resultant reaction gas was then analyzed by gas chromatography (GC, 7890 A, Agilent) equipped with a FID. The control experiments in dark environment were performed under the same conditions except the exclusion of light with an aluminum foil or the exclusion of CO2 with high-purity Ar (99.9999%).
Computational details
DFT calculations with periodic boundary conditions (PBC) were carried out using the program ADF-BAND 2019. Perdew-Burke-Ernzerhof exchange-correlation functional with DFT-D3 (BJ) dispersion correction and TZP basis set incorporated with ZORA approach to include the scalar relativistic effects were employed under the spin-unrestricted scheme. A 5 × 5 slab with three atomic layers was used to mimic the Pd (100) surface with Cu doping. During the geometry optimizations, only the top layer was relaxed and the two bottom layers were frozen at the bulk positions. A 3 × 3 × 1 k-point grid was used to sample the first Brillouin zone under the two-dimensional PBC. A \(\varGamma\)-only k-mesh was adopted in the energy decomposition analysis for extended systems (pEDA) with the extension of natural orbitals for chemical valence. Since the strong interaction of photons and plasmonic metal can result in an alternating electric field, which is localized near the metal surface following the law of sines (E = Asint, where E, A and t represent electric field intensity, amplitude and time, respectively). In order to investigate the influence of the electric field induced by LSPR on the adsorbed molecules, the adsorption energy and bond length of these molecules were simulated under specific electric field conditions. Given the monotonicity and periodicity of the sine function, the three most representative conditions were studied. Specifically, when t = 0 or π, t = π/2 and t = 3π/2, and A = 0.04 au, the E values are 0, 0.04 and −0.04 au, respectively. Taken together, the external electric fields with the directions pointing toward/away the substrate were applied in simulations considering the field created from surface plasmon polariton.
Data availability
All data supporting the findings of this study are available in the article and its Supplementary Information. Source data for the following figures are provided with this paper. Fig 1b–d, Fig. 2a–d, Fig. 3a, Fig. 4b–d, Fig. S4a–g, Fig. S5a–g, Fig. S7, Fig. S9a and b, Fig. S10a and b, Fig. S11a and b, Fig. S13, Fig. S14, Fig. S15, Fig. S16a and b, Fig. S18a and b, Fig. S19a and b, Fig. S20, Fig. S21, Fig. S22a and b, Fig. S23a and b, Fig. S24a and b, Fig. S25, Fig. S26, Fig. S27. Source data are provided with this paper.
Change history
22 March 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41467-023-37241-1
References
Smith, P. T., Nichols, E. M., Cao, Z. & Chang, C. J. Hybrid catalysts for artificial photosynthesis: merging approaches from molecular, materials, and biological catalysis. Acc. Chem. Res. 53, 575–587 (2020).
Maeda, K. Metal-complex/semiconductor hybrid photocatalysts and photoelectrodes for CO2 reduction driven by visible light. Adv. Mater. 31, e1808205 (2019).
Zhang, B. & Sun, L. Artificial photosynthesis: opportunities and challenges of molecular catalysts. Chem. Soc. Rev. 48, 2216–2264 (2019).
Chen, S., Qi, Y., Li, C., Domen, K. & Zhang, F. Surface strategies for particulate photocatalysts toward artificial photosynthesis. Joule 2, 2260–2288 (2018).
Li, X., Yu, J., Jaroniec, M. & Chen, X. Cocatalysts for selective photoreduction of CO2 into solar fuels. Chem. Rev. 119, 3962–4179 (2019).
Wan, L. et al. Cu2O nanocubes with mixed oxidation-state facets for (photo)catalytic hydrogenation of carbon dioxide. Nat. Catal. 2, 889–898 (2019).
Wu, X. et al. Photocatalytic CO2 conversion of M0.33WO3 directly from the air with high selectivity: insight into full spectrum-induced reaction mechanism. J. Am. Chem. Soc. 141, 5267–5274 (2019).
Yu, S., Wilson, A. J., Kumari, G., Zhang, X. & Jain, P. K. Opportunities and challenges of solar-energy-driven carbon dioxide to fuel conversion with plasmonic catalysts. ACS Energy Lett. 2, 2058–2070 (2017).
Olson, J. et al. Optical characterization of single plasmonic nanoparticles. Chem. Soc. Rev. 44, 40–57 (2015).
Hartland, G. V. Optical studies of dynamics in noble metal nanostructures. Chem. Rev. 111, 3858–3887 (2011).
Skrabalak, S. E., Au, L., Li, X. & Xia, Y. Facile synthesis of Ag nanocubes and Au nanocages. Nat. Protoc. 2, 2182–2190 (2007).
Cortie, M. B. & McDonagh, A. M. Synthesis and optical properties of hybrid and alloy plasmonic nanoparticles. Chem. Rev. 111, 3713–3735 (2011).
Hu, C. et al. Surface plasmon enabling nitrogen fixation in pure water through a dissociative mechanism under mild conditions. J. Am. Chem. Soc. 141, 7807–7814 (2019).
Zheng, Z., Tachikawa, T. & Majima, T. Plasmon-enhanced formic acid dehydrogenation using anisotropic Pd-Au nanorods studied at the single-particle level. J. Am. Chem. Soc. 137, 948–957 (2015).
Zheng, Z., Tachikawa, T. & Majima, T. Single-particle study of Pt-modified Au nanorods for plasmon-enhanced hydrogen generation in visible to near-infrared region. J. Am. Chem. Soc. 136, 6870–6873 (2014).
Huang, H. et al. Unraveling surface plasmon decay in core-shell nanostructures toward broadband light-driven catalytic organic synthesis. J. Am. Chem. Soc. 138, 6822–6828 (2016).
Guo, J. et al. Boosting hot electrons in hetero-superstructures for plasmon-enhanced catalysis. J. Am. Chem. Soc. 139, 17964–17972 (2017).
Mukherjee, S. et al. Hot electrons do the impossible: plasmon-induced dissociation of H2 on Au. Nano Lett. 13, 240–247 (2013).
Zhang, X. et al. Product selectivity in plasmonic photocatalysis for carbon dioxide hydrogenation. Nat. Commun. 8, 14542 (2017).
Yu, S., Wilson, A. J., Heo, J. & Jain, P. K. Plasmonic control of multi-electron transfer and C–C coupling in visible-light-driven CO2 teduction on Au nanoparticles. Nano Lett. 18, 2189–2194 (2018).
Yu, S. & Jain, P. K. Plasmonic photosynthesis of C1–C3 hydrocarbons from carbon dioxide assisted by an ionic liquid. Nat. Commun. 10, 2022 (2019).
Christopher, P., Xin, H., Marimuthu, A. & Linic, S. Singular characteristics and unique chemical bond activation mechanisms of photocatalytic reactions on plasmonic nanostructures. Nat. Mater. 11, 1044–1050 (2012).
Zhan, C. et al. From plasmon-enhanced molecular spectroscopy to plasmon-mediated chemical reactions. Nat. Rev. Chem. 2, 216–230 (2018).
Zhang, Y. et al. Surface-plasmon-driven hot electron photochemistry. Chem. Rev. 118, 2927–2954 (2018).
Polte, J. et al. Nucleation and growth of gold nanoparticles studied via in situ small angle X-ray scattering at millisecond time resolution. ACS Nano 4, 1076–1082 (2010).
Aslam, U., Chavez, S. & Linic, S. Controlling energy flow in multimetallic nanostructures for plasmonic catalysis. Nat. Nanotech. 12, 1000–1005 (2017).
Link, S. & El-Sayed, M. A. Spectral properties and relaxation dynamics of surface plasmon electronic oscillations in gold and silver nanodots and nanorods. J. Phys. Chem. B 103, 8410–8426 (1999).
Jana, N. R., Gearheart, L. & Murphy, C. J. Wet chemical synthesis of high aspect ratio cylindrical gold nanorods. J. Phys. Chem. B 105, 4065–4067 (2001).
Feng, R. J., Li, X., Zhang, Z., Lu, Z. & Guo, Y. Spectral assignment and orientational analysis in a vibrational sum frequency generation study of DPPC monolayers at the air/water interface. J. Chem. Phys. 145, 244707 (2016).
Boerigter, C., Campana, R., Morabito, M. & Linic, S. Evidence and implications of direct charge excitation as the dominant mechanism in plasmon-mediated photocatalysis. Nat. Commun. 7, 10545 (2016).
Rao, V. G., Aslam, U. & Linic, S. Chemical requirement for extracting energetic charge carriers from plasmonic metal nanoparticles to perform electron-transfer reactions. J. Am. Chem. Soc. 141, 643–647 (2019).
Boerigter, C., Aslam, U. & Linic, S. Mechanism of charge transfer from plasmonic nanostructures to chemically attached materials. ACS Nano 10, 6108–6115 (2016).
Linic, S., Aslam, U., Boerigter, C. & Morabito, M. Photochemical transformations on plasmonic metal nanoparticles. Nat. Mater. 14, 567–576 (2015).
Kim, Y., Smith, J. G. & Jain, P. K. Harvesting multiple electron-hole pairs generated through plasmonic excitation of Au nanoparticles. Nat. Chem. 10, 763–769 (2018).
Kim, Y., Dumett Torres, D. & Jain, P. K. Activation energies of plasmonic catalysts. Nano Lett. 16, 3399–3407 (2016).
Favaro, M. et al. Subsurface oxide plays a critical role in CO2 activation by Cu(111) surfaces to form chemisorbed CO2, the first step in reduction of CO2. Proc. Natl Acad. Sci. USA 114, 6706–6711 (2017).
Kattel, S. et al. CO2 hydrogenation over oxide-supported PtCo catalysts: the role of the oxide support in determining the product selectivity. Angew. Chem. Int. Ed. 55, 7968–7973 (2016).
Deng, X. et al. Surface chemistry of Cu in the presence of CO2 and H2O. Langmuir 24, 9474–9478 (2008).
Wu, H., Zhang, N., Wang, H. & Hong, S. Adsorption of CO2 on Cu2O (111) oxygen-vacancy surface: first-principles study. Chem. Phys. Lett. 568–569, 84–89 (2013).
Wang, X. & Long, R. Thermal-driven dynamic shape change of bimetallic nanoparticles extends hot electron lifetime of Pt/MoS2 catalysts. J. Phys. Chem. Lett. 12, 7173–7179 (2021).
Liang, L. et al. Infrared light-driven CO2 overall splitting at room temperature. Joule 2, 1004–1016 (2018).
Nikoobakht, B. & El-Sayed, M. A. Preparation and growth mechanism of gold nanorods (NRs) using seed-mediated growth method. Chem. Mater. 15, 1957–1962 (2003).
Acknowledgements
We acknowledge financial support from the National Key R&D Program of China (2020YFA0406103), NSFC (22122506, 22075267, 22109152, 22232003, 21725102, 91961106, 21950410514, 22109148), Strategic Priority Research Program of the CAS (XDPB14), Youth Innovation Promotion Association of CAS (2019444), Open Funding Project of National Key Laboratory of Human Factors Engineering (SYFD062010K), and China Postdoctoral Science Foundation (2021M693063). The in situ NAP-XPS experiments were performed at the X-ray Photoelectron Spectroscopy and UV Photoelectron Spectroscopy Endstation (BL24A1) in the National Synchrotron Radiation Center (NSRRC). XAFS measurements were performed at the beamline 1W1B of the Beijing Synchrotron Radiation Facility (BSRF) and beamline BL14W1 of the Shanghai Synchrotron Radiation Facility (SSRF). We thank the support from USTC Center for Micro- and Nanoscale Research and Fabrication.
Author information
Authors and Affiliations
Contributions
R.L. and Y.X. supervised the project. C.H. and J.L. designed the experiments. C.H. carried out the experiments and analyzed the results. X.C. performed DFT simulations. J.J. and He L. helped to synthesize the catalysts. D.W. helped to evaluate the catalyst performance. Y.W.Y., S.C., H.J., C.H.W., and L.S. contributed to in situ near ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) characterization. Hao L. and Z.L. contributed to the off-line sum frequency generation vibrational spectroscopy (SFG-VS) characterization. C.H., J.L., R.L., and Y.X. wrote the paper with input and comments from the other authors. C.H., X.C., and J.L. equally contributed to this work.
Corresponding authors
Ethics declarations
Competing interests
A patent application was authorized by China National Intellectual Property Administration (patentee: University of Science and Technology of China; names of inventors: Y.X., C.H., R.L.; Patent Application No. 202110378508.5; status of application: receipt; specific aspect of manuscript covered in the patent application: A method for catalytic conversion of carbon dioxide driven by infrared light). The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jennifer Strunk and the other anonymous reviewer(s) for their contribution to the peer review of this work. Peer review reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hu, C., Chen, X., Low, J. et al. Near-infrared-featured broadband CO2 reduction with water to hydrocarbons by surface plasmon. Nat Commun 14, 221 (2023). https://doi.org/10.1038/s41467-023-35860-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-35860-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
