
Abstract
JAK2V617F mutation is associated with an increased risk for athero-thrombotic cardiovascular disease, but its role in aortic disease development and complications remains unknown. In a cohort of patients with myeloproliferative neoplasm, JAK2V617F mutation was identified as an independent risk factor for dilation of both the ascending and descending thoracic aorta. Using single-cell RNA-seq, complementary genetically-modified mouse models, as well as pharmacological approaches, we found that JAK2V617F mutation was associated with a pathogenic pro-inflammatory phenotype of perivascular tissue-resident macrophages, which promoted deleterious aortic wall remodeling at early stages, and dissecting aneurysm through the recruitment of circulating monocytes at later stages. Finally, genetic manipulation of tissue-resident macrophages, or treatment with a Jak2 inhibitor, ruxolitinib, mitigated aortic wall inflammation and reduced aortic dilation and rupture. Overall, JAK2V617F mutation drives vascular resident macrophages toward a pathogenic phenotype and promotes dissecting aortic aneurysm.
Similar content being viewed by others


Introduction
Aortic aneurysm (AA) is a frequent vascular disease affecting approximately 5% of elderly men in Western countries1 whose incidence is still increasing despite better control of risk factors like smoking and blood pressure. Surgery and endovascular procedure are the only treatments for individuals with aortic aneurysm to limit the risk of aortic rupture and dissection, but the interventions are costly and associated with high morbidity and mortality2. Currently, no drug treatments have been approved for use in aortic aneurysm, which underlines the need for a better understanding of mechanisms underlying the disease in order to implement novel management procedures and therapeutic strategies.
The mechanisms of aortic aneurysm formation are complex, including elastin degradation, collagen remodeling, smooth muscle cell apoptosis and local activation of immune cells such as macrophages3. Several experimental studies have suggested that during the first stages of AA formation, circulating monocytes interact with activated endothelial cells (ECs) and are recruited into the aortic wall, Ly6Chigh monocytes dominating the acute phase of injury while non-classical Ly6Clow monocytes being prevalent thereafter4. Recruitment into the vascular wall depends, at least in part, on GM-CSF5, chemokines/chemokine receptors6, and adhesion molecules. Monocytes locally differentiate into macrophages and, after activation, promote aneurysm formation and complications (dissection and/or rupture) through the release of inflammatory cytokines (including TNF-α, IL-1β) and matrix metalloproteinases (MMPs). Another subset of macrophages, tissue-resident macrophages, which originate from the yolk sac and fetal liver during development7,8, has been recently identified as essential for the maintenance of arterial wall homeostasis through the regulation of smooth muscle cell activity and collagen production9. Tissue-resident macrophages express the lymphatic vessel endothelial hyaluronan receptor-1 (LYVE-1), but their role in aortic aneurysm formation and progression remains unknown.
JAK2V617F is a gain-of-function mutation responsible for enhanced hypersensitivity to growth factors and cytokines10. JAK2V617F mutation promotes the proliferation, the survival and the maturation of hematopoietic progenitors leading to myeloproliferative neoplasms such as polycythemia vera, essential thrombocythemia, and primary myelofibrosis10,11. The presence of JAK2V617F mutation is associated with an increased risk of cardiovascular diseases12. Experimental studies have shown that JAK2V617F mutation in bone marrow cells, mimicking human myeloproliferative neoplasms, accelerates atherosclerosis in hypercholesterolemic mice due to impaired red blood cell efferocytosis and inflammasome activation13, and aggravates post-ischemic cardiac remodeling14. However, the role of JAK2V617F mutation in aortic aneurysm development and complications has never been investigated.
Here, we showed that JAK2V617F mutation is associated with aortic dilation in patients. Moreover, using several complementary genetic and pharmacological approaches in murine models, we found that JAK2V617F mutation drives adventitial tissue-resident macrophages toward a pathogenic inflammatory phenotype causing aortic aneurysm and dissection.
Results
Global JAK2V617F mutation promotes aortopathy in humans, and aortic aneurysm and dissection in mice
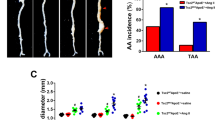
Several epidemiological studies have shown that JAK2V617F gain-of-function mutation is associated with increased risk for athero-thrombotic cardiovascular disease, but to our knowledge nothing has been reported regarding the possible association between this mutation and aortic disease. To address this issue, we first screened JAK2V617F + patients recruited at Saint-Antoine Hospital (N = 382, Paris, France), and identified those who had a recent Body CT-scan (N = 157/382). Mean diameter was measured in different segments of the aorta (ascending, descending thoracic and abdominal) and compared to age- and gender-matched controls by a blinded independent reader (Supplementary Fig. 1). General characteristics of the cohort are detailed in Supplementary Table 1. Interestingly, after multiple adjustment on age, gender, hypertension, diabetes and smocking, we found that the JAK2V617F mutation was associated with significant dilation of both ascending and descending thoracic aorta, but not abdominal aorta (Fig. 1A, B).

A Adjusted risk factor for aorta dilation in a cohort of JAK2V167F + patients (n = 157) and age-gender-matched controls (N = 157), scale bar 2 cm. B CT-scan 3D-reconstruction of JAK2V167F + patient (right) and control (left) showing ascending and descending thoracic aorta. C Paw pictures of 7-week-old Jak2WTcontrol and Jak2V617F HC-EC mice showing palmar erythema in mutant mice, scale bar 1 cm. D Quantitative analysis and representative photomicrographs of spleen size in 7-week-old control Jak2WT(N = 11) and Jak2V617F HC-EC mice (N = 11) (scale bar 1 cm). E red blood cell, platelet, and myeloid cell count of 7-week-old control Jak2WT(N = 5) and Jak2V617F HC-EC mice (N = 9). F Representative photomicrographs and quantitative analysis of the mean aortic diameter in 7-week-old control Jak2WT(N = 11) and Jak2V617F HC-EC mice (N = 7) in the thoracic (ascending and descending) and the abdominal aorta (scale bar 1 mm). G Quantification of the number of elastin layers in the aortic wall by orcein staining in 7-week-old animals (N = 10–12/group), scale bar 50 μm. H Quantification of the collagen content (Cola1 immunostaining) in the aortic wall in 7-week-old animals (N = 9/group), scale bar 50 μm. I Fluorescent Molecular Tomography quantification of matrix metalloproteinase (MMP-2, −3, −9 and −13) activity in aorta of 10-week-old animals (N = 4/group), scale bar 1 mm. J Survival curve of control Jak2WT (N = 15) and Jak2V617F HC-EC mice (N = 25). K Representative photomicrographs of descending thoracic aorta after Orcein staining showing dissecting aortic aneurysm in Jak2V617F HC-EC mouse (representative of 10 aortas/group), scale bar 100 µm. Data are presented as mean values ± SD. [Two-tailed Mann-Whitney test, *P < 0.05, **P < 0.01, ***P < 0.001]. Difference in survival was evaluated using log-rank test. CT computed tomography, HC-EC hematopoietic cells-endothelial cells, MMP matrix metalloprotease. Source data are provided as a Source Data file.
In order to understand the cellular and molecular mechanisms responsible for aortopathy in JAK2V617F+ patients, we used genetically-modified mouse models. First, we studied Jak2V617FWT/Flex VE-Cadherin-Cre+/- mice, herein referred to as Jak2V617F HC-EC mice, in which Cre-mediated recombination occurs in ECs and hematopoietic cells (HCs) due to the expression of VE-cadherin during early embryonic life in a precursor of both ECs and HCs15,16 (Supplementary Fig. 2A). Jak2V617F HC-EC mice developed myeloproliferative neoplasm, as attested by palmar erythema (Fig. 1C), splenomegaly (87 ± 27 vs 560 ± 81 mg; P < 0.0001) (Fig. 1D), and higher red blood cell, platelet and leukocyte counts than littermate controls (Fig. 1E & Supplementary Fig. 2B). At 7 weeks, we performed histological analysis and we found aortic dilation in Jak2V617F HC-EC mice affecting descending thoracic (0.71 ± 0.07 vs 0.80 ± 0.08 mm, P < 0.05) and abdominal (0.74 ± 0.03 vs 0.85 ± 0.08 mm, P < 0.01) aortic regions (Fig. 1F). Notably, both systolic and diastolic blood pressure were significantly lower in Jak2V617F HC-EC mice than in littermate controls, which rules out the possibility that aortic dilation resulted from hypertension (Supplementary Fig. 2C).
At 7 weeks of age, hematopoietic and endothelial JAK2V617F gain-of-function mutation was associated with enhanced elastin degradation, as assessed by the significantly lower number of elastin layers in the abdominal aorta of Jak2V617F HC-EC mice compared with the aorta of control Jak2WT HC-EC mice (3.90 ± 0.12 vs 3.70 ± 0.17 layers, P < 0.05) (Fig. 1G), and reduced content in collagen type 1a (Fig. 1H & Supplementary Fig. 2D), but no difference in α-SMA + smooth muscle cell content (Supplementary Fig. 2E). The decrease in extracellular matrix protein content in Jak2V617F HC-EC mice was accompanied with an increase in global MMP activity in the aortic wall, quantified using ex vivo fluorescence molecular tomography imaging (Fig. 1I) and qPCR (Supplementary Fig. 2F). Interestingly, we observed specifically in Jak2V617F HC-EC mice spontaneous early death (Fig. 1J & Supplementary Fig. 2G), reaching 70% at 20 weeks of age; which was due to aortic dissecting aneurysm (Fig. 1K) and thoracic and/or abdominal hemorrhage. Polyglobulia correction using phenylhydrazine17 in Jak2V617F HC-EC mice did not impact either on aorta dilation or rupture incidence (Supplementary Fig. 3).
JAK2V617F mutation specifically expressed in vascular cells did not induce dissecting aortopathy
To assess the implication of JAK2V617F mutation in vascular cells, we used 2 complementary approaches. First, we generated chimeric mice expressing JAK2V617F mutation only in non-hematopoietic cells by transplanting C57BL/6 bone marrow (BM) cells into lethally irradiated Jak2V617F HC-EC mice and C57BL/6 controls (Supplementary Fig. 4A)16. BM cell transplantation after irradiation allows hematopoietic reconstitution, including perivascular LYVE-1+ tissue-resident macrophages (Supplementary Fig. 5). JAK2V617F mutation only in vascular cells did not induce myeloproliferative neoplasm (Supplementary Fig. 4B) and did not induce elastin degradation (Supplementary Fig. 4D) or aortic dilation (Supplementary Fig. 4C). Moreover, no death was recorded during the 3-month follow-up in the 2 groups of chimeric mice (Supplementary Fig. 4E).
In order to investigate the role of JAK2V617F mutation specifically in endothelial cells in the onset of spontaneous dissecting aortopathy, we next generated Jak2V617FWT/Flex VE-cadherin-Cre-ERT2 (herein referred to as Jak2V617F EC) mice. We crossed Jak2V617FWT/Flex mice with inducible VE-cadherin-Cre-ERT2 mice expressing the Cre recombinase after tamoxifen injection only in endothelial cells (Supplementary Fig. 6A). The absence of hematopoietic recombination in this model, as well as adequate endothelial recombination has been previously validated16,18. As expected, mice did not develop myeloproliferative neoplasm (Supplementary Fig. 6B). Aortic wall structure was preserved in mice expressing JAK2V617F mutation in endothelial cells (Supplementary Fig. 6D). In Jak2V617F EC mice, we observed neither aortic dilation (Supplementary Fig. 6C) nor premature death (Supplementary Fig. 6E). Altogether, these experiments indicate that JAK2V617F mutation in vascular cells was not responsible for dissecting aortopathy.
JAK2V617F mutation specifically expressed in myeloid cells induces dissecting aortopathy
To assess the implication of JAK2V617F mutation in hematopoietic cells, we generated chimeric mice expressing JAK2V617F mutation only in hematopoietic cells by transplanting BM cells from Jak2V617F HC-EC mice into lethally irradiated C57BL/6 mice. Irradiated C57BL/6 mice transplanted with C57BL/6 BM cells were used as controls (Fig. 2A). Hematopoietic expression of JAK2V617F induced a myeloproliferative neoplasm (Fig. 2B), elastin degradation (Fig. 2C) and spontaneous aortic dilation at 1 (Supplementary Fig. 7A) and 3 months (Fig. 2D) after BMT. In addition, irradiated C57BL/6 mice were transplanted with 100% Jak2WT BM cells or a mixture of 80% Jak2WT/20% Jak2V617F HC-EC BM cells. One month after BMT, we observed aorta dilation in chimeric mice receiving 20% Jak2V617F BM cells, despite no major changes in blood cell subsets (Supplementary Fig. 7B, C).

A Experimental protocol of WT or Jak2V617F HC-EC bone marrow cell transplantation in lethally irradiated C57BL/6 mice (7/Group). B Red blood cell, white blood cell and platelet count of 20-week-old chimeric mice (N = 7/group). C Quantification of the number of elastin layers in the aortic wall by Orcein staining at sacrifice (N = 7/group), scale bar 50 μm. D Representative photomicrographs and quantitative analysis of the mean aortic diameter in WT or Jak2V617F HC-EC chimeric mice (N = 7/group) in the thoracic (ascending and descending) and the abdominal aorta, scale bar 0.5 mm. E Red blood cell, white blood cell and platelet count of 7-week-old Jak2WT and Jak2V617F Myel mice (N = 7/group). F Quantification of the collagen content (Cola1 immunostaining) in the aortic wall in 7-week-old Jak2WT(N = 6) and Jak2V617F Myel animals (N = 8), scale bar 50 μm. G Quantification of Cola1 and Cola3 mRNA expression in aorta of 7-week-old Jak2WT (N = 7) and Jak2V617F Myel mice (N = 8). H Fluorescent Molecular Tomography quantification of matrix metalloproteinase (MMP-2, −3, −9, and −13) activity in aorta of 9-week old Jak2WT and Jak2V617F Myel mice (N = 4/group), scale bar 1 mm. I, quantification of Mmp2, Mmp3, Mmp9, Mmp12, Mmp13 mRNA expression in aorta of 7-week-old Jak2WT (N = 5) and Jak2V617F Myel mice (N = 7). J Representative photomicrographs and quantitative analysis of the mean aortic diameter in 7-week-old Jak2WT (N = 7) and Jak2V617F Myel (N = 8) mice in the thoracic (ascending and descending) and the abdominal aorta, scale bar 1 mm. K Survival curve of Jak2WT (N = 11) and Jak2V617F Myel mice (N = 14). Data are presented as mean values ± SD. [Two-tailed Mann-Whitney test, *P < 0.05, **P < 0.01, ***P < 0.001]. Difference in survival was evaluated using log-rank test. HC-EC, hematopoietic cells-endothelial cells; MMP, matrix metalloprotease. Source data are provided as a Source Data file.
Next, to evaluate the implication of JAK2V617F in myeloid cells, we crossed Jak2V617FWT/Flex mice with LysMCre ± mice expressing the Cre recombinase under Lysozyme M promoter, mainly expressed in myeloid cells (Supplementary Fig. 7D). Jak2V617FWT/Flex LysMCre+/−, referred to as Jak2V617F Myel, developed myeloproliferative neoplasm, as attested by palmar erythema (Supplementary Fig. 7E), splenomegaly (Supplementary Fig. 7F), and higher number of circulating red blood cells, neutrophils and monocytes compared to littermate controls. However, platelet count was not different between groups (Fig. 2E). Histological analysis of aorta from 7-week-old Jak2V617F Myel mice phenocopied the vascular phenotype of Jak2V617F HC-EC mice. Indeed, JAK2V617F gain-of-function mutation under LysM promoter led to elastin degradation, as assessed by the significantly lower number of elastin layers in the abdominal aorta of Jak2V617F Myel mice compared with the aorta of control Jak2WT Myel mice (4.5 ± 0.24 vs 4.2 ± 0.09 layers, P < 0.05) (Supplementary Fig. 7G), which was associated with a reduction in collagen type 1a content (Fig. 2F, G) but no difference in α-SMA + smooth muscle cells content (Supplementary Fig. 7H). Global MMP activity in the aortic wall of Jak2V617F Myel mice was significantly increased (Fig. 2H, I). At 7 weeks of age, we observed dilation in ascending thoracic (0.90 ± 0.06 vs 1.12 ± 0.05 mm, P < 0.05), descending thoracic (0.66 ± 0.12 vs 0.86 ± 0.10 mm, P < 0.05) and abdominal (0.72 ± 0.12 vs 0.89 ± 0.11 mm, P < 0.05) aorta in Jak2V617F Myel mice, compared to Jak2WT Myel mice (Fig. 2J). Finally, we observed that all Jak2V617F Myel mice (100%) spontaneously died before 15 weeks of age. Autopsy was systematically performed and confirmed that deaths were due to dissecting aortic aneurysm, mainly associated with thoracic and/or abdominal hemorrhage (Fig. 2K). Taken together, these findings demonstrate that the presence of the JAK2V617F mutation in myeloid cells was responsible for the development of dissecting aortic aneurysm.
In order to understand the mechanisms underlying the lethal JAK2V617F-dependent aortic disease, we performed RNA sequencing of the aorta of 7-week-old Jak2V617F HC-EC and control Jak2WT mice. The whole transcriptomic profile of the aorta was markedly altered by JAK2V617F global mutation, with upregulation of the expression of 137 genes and downregulation of the expression of 70 genes (Fig. 3A). Gene Ontology analysis showed that inflammatory pathways were upregulated in Jak2V617F HC-EC aorta, including those associated with myeloid cell differentiation, homeostasis and development (Fig. 3B). Based on this finding, we investigated the consequences of JAK2V617F mutation on the local inflammatory response. First, we analyzed the immune cell composition in aorta of 7-week-old Jak2V617F HC-EC mice and controls by flow cytometry, after enzymatic digestion. We did not find any significant differences in monocyte and neutrophil counts (Fig. 3C & Supplementary Fig. 8A), but we observed a 3-fold increase in CD45+CD11b+Ly6G-F4/80highCD68high macrophages in the aorta from Jak2V617F HC-EC mice (P < 0.05) (Fig. 3D). This result was confirmed by immunostaining in Jak2V617F HC-EC (Fig. 3E) and Jak2V617F Myel mice (Supplementary Fig. 8B). The significant increase in Il1β, Il6 and Tnfα mRNA expression in aorta of Jak2V617F HC-EC mice supports a vascular pro-inflammatory immune response in mutant mice (Fig. 3F). Notably, most of vascular CD68+ macrophages in mutant mice with aortic dilation expressed LYVE-1 (Fig. 3G) and a significant number expressed Ki-67, a proliferation marker (Supplementary Fig. 8C). However, at more advanced stage, most of CD68+ macrophages in Jak2V617F HC-EC mice with severe aortic aneurysm did not colocalize with LYVE-1+ staining (Fig. 3G). All these observations suggest that macrophage accumulation in mutant mice at early stages unlikely resulted from the differentiation of infiltrating blood monocytes, but most likely from an expansion of tissue-resident macrophages. Transcriptomic analysis confirmed that genes related to the phenotype of tissue-resident macrophages (F13a1, Lyve1, Pf4, and Gas6)19 were enriched in the aorta of Jak2V617F HC-EC mice (Fig. 3H).

A Difference in gene expression in the aorta of 7-week-old Jak2WT (N = 4) and Jak2V617F HC-EC mice (N = 3). B Top Gene Ontology terms mostly enriched among genes upregulated in the aorta of Jak2V617F HC-EC mice when compared to Jak2WT mice. C Flow cytometry quantification of CD45 + CD11clowCMHIIlowCD11b + Ly6G-F4/80lowCD64lowLy6C + monocytes and CD45 + CD11clowCMHIIlowCD11b + Ly6G+ neutrophils in the aorta of 7-week-old Jak2WT (N = 10) and Jak2V617F HC-EC animals (N = 8). D Flow quantification and representative dot plot of CD45 + CD11clowCMHIIlowCD11b + Ly6G-F4/80highCD64high macrophage count in the aorta of 7-week-old Jak2WT (N = 10) and Jak2V617F HC-EC animals (N = 8). E Quantification and representative photomicrographs of CD68 positive macrophages in the aorta of 7-week-old mice (N = 11 in control group and N = 8 in Jak2V617F HC-EC group), scale bar 50 µm. F Quantification of Il6, Tnfα, Il1β, and Il10 mRNA expression by RT-qPCR in the aorta of 7-week-old Jak2WT(N = 8) and Jak2V617F HC-EC animals (N = 9). G Immunofluorescence staining in the aortic wall of Jak2WT and Jak2V617F HC-EC mice, DAPI (Blue), CD68 (Red), LYVE-1 (Green), scale bar 50 μm. resident macrophages were defined as CD68 + LYVE-1+ cells (N = 5/group). H GSEA for genes related to a gene set signature for tissue resident macrophage19 in the aorta of 7-week-old Jak2WT and Jak2V617F HC-EC animals (N = 4/group). Data are presented as mean values ± SD. [Two-tailed Mann-Whitney test, *P < 0.05, **P < 0.01, ***P < 0.001]. HC-EC, hematopoietic cells-endothelial cells; Il, interleukin; Tnf, tumor necrosis factor; GSEA, gene set enrichment analysis. Source data are provided as a Source Data file.
Jak2V617F Myel aortas harbor pro-inflammatory Lyve1 + resident macrophages
To better characterize aortic macrophage populations in Jak2V617F Myel mice, we performed cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq)20 of total CD45+ cells from Jak2WT (n = 5) and Jak2V617F Myel (n = 5) aortas (Fig. 4A). At sacrifice, among Jak2V617F Myel mice, macroscopically observable aortopathy was seen in all mice, 2 mice displayed aortic aneurysm and 3 mice exhibited aortic dilation. Based on the expression of canonical cell surface markers and lineage-specific transcripts, immune cell subsets were identified and annotated (Fig. 4B, Supplementary Fig. 9A, B). Cells corresponding to monocytes/macrophages were extracted and reclustered separately to better resolve discrete subpopulations (Fig. 4A–C). We recovered several populations of LYVE-1+ tissue-resident macrophages (Lyve1, Mrc1; surface CD163, TIMD4, CD169, Fig. 4D) and recruited monocytes/macrophages (Ccr2; surface CD14, Fig. 4D). Differential gene expression analysis revealed three distinct populations of LYVE-1+ tissue-resident macrophages: Res-MGL1hi (cluster A, enriched in Clec10a encoding MGL1), Res-Txniphi (cluster B: Txnip, Trf, Stab1) and Res-S100hi (cluster C enriched in S100-protein encoding genes such as S100a4, S100a6 or S100a10) (Fig. 4C–E). Ccr2+ inflammatory macrophages (cluster D) were enriched for e.g. Spp1, Tnip3 or Il1b (Fig. 4C–E). Populations of type I IFN response macrophages (IFNIC, cluster E:21 Isg15, Ifit), monocytes (cluster F: Plac8, Ly6c2, Chil3, surface Ly6C), and macrophages with high expression of early immediate-early genes22 (cluster G, IEG + : Egr1, Zfp36) were also detected (Fig. 4C–E).

A Experimental design, B UMAP visualization of scRNA-seq profiles of CD45 + cells extracted from Jak2WT and Jak2V617F Myel aortas. C Single-cells annotated as macrophages on the UMAP displayed in B were in silico extracted, reanalyzed with dimensional reduction (UMAP) and clustered identifying 7 major populations (Annotated A to G). D Expression intensity of transcripts (top) and cell surface markers (bottom) characteristic of tissue resident macrophages (Lyve1, Mrc1, TIMD4, CD169) and recruited monocytes/macrophages (Ccr2, CD14) projected on the macrophage UMAP plot. E Expression of selected marker transcripts in the 7 macrophage populations. F Macrophage UMAP plot split according to experimental condition (color code as in B). G Proportions of macrophage populations among total aortic macrophages. H Proportions of tissue resident macrophage subsets among total aortic tissue resident macrophages. I Normalized expression of the indicated transcripts in tissue resident macrophage subpopulations. J Average expression of the indicated cell surface markers (measured by CITE-seq) in tissue resident macrophage subpopulations. UMAP, Uniform Manifold Approximation and Projection for Dimension Reduction, sc single cell, CITE-seq Cellular Indexing of Transcriptomes and Epitopes by Sequencing.
Distribution of macrophage subpopulations was dependent on genotype and disease severity. Pro-inflammatory Ccr2+ macrophages were particularly abundant in aortic aneurysm (59.6% of macrophages), while they were relatively rare in Jak2WT and Jak2V617F Myel with limited aortic dilation (Fig. 4F, G), consistent with our observation of infiltration of Lyve1- macrophages in the media of mutant mice with severe aortic aneurysm (Fig. 3G). The distribution of the different subsets of tissue-resident macrophages differed according to the genotype. Res-MGL1hi were preferentially found in Jak2V617F Myel aortas regardless of aortic disease severity (61.4% and 56.0% of tissue-resident macrophage in Jak2V617F Myel aortas with aortic aneurysm and dilation, respectively), and Res-Txniphi were more abundant in Jak2WT aortas (60.2% of tissue-resident macrophages), whereas Res-S100hi macrophages were evenly distributed across genotypes (Fig. 4G). Moreover, all resident macrophage populations shared expression of markers previously reported as associated with aortic resident macrophages (Lyve1, Folr2, Cbr2, F13a1, Gas6) (Fig. 4D and Supplementary Fig. 9)7,19,23, but Res-MGL1hi macrophages, particularly abundant in Jak2V617F Myel aortas, showed higher expression of pro-inflammatory transcripts (Nlrp3, Tnf, Tnfsf9, Ccl2, Cxcl1, Cxcl2, Cxcl10) (Fig. 4I). Interestingly, Lyve1 expression was significantly increased in Res-MGL1hi compared to Res-Txniphi macrophages, in agreement with our finding of higher expression levels of Lyve1 observed in bulk tissue transcriptome analysis of Jak2V617F HC-EC aortas (Fig. 3H). Differential expression of cell surface markers measured by CITE-seq indicated that besides MGL1, Res-MGL1hi macrophages exhibited higher surface levels of pro-inflammatory proteins such as CD95 (Fas), CD178 (Fas-Ligand), CD40 or JAML (Fig. 4J). All these data confirmed a pro-inflammatory signature in the aorta of Jak2V617F Myel mice but pathways related to angiogenesis and lymphoangiogenesis were not impaired in mutant aortas (Supplementary Fig. 10A, B). Altogether, these results indicate that JAK2V617F mutation in myeloid cells not only increased the number of aortic resident Lyve-1+ macrophages, but also polarized them towards a pro-inflammatory pathogenic phenotype.
The JAK2V617F mutation in vascular tissue-resident macrophages induces dissecting aortic aneurysm
We then sought to generate a genetic model in which the JAK2V617F mutation would be preferentially induced in the tissue-resident macrophages. Our selection of an adequate Cre-driver was guided by data obtained from scRNA-seq analysis. Previous scRNA-seq studies have identified Platelet factor 4 (Pf4), highly expressed in megakaryocytes, as a marker of aortic resident macrophages19,24, which we confirmed using an integrated dataset combining several independent single-cell studies of healthy and diseased mouse aortas25 (Fig. 5A). Likewise, in our newly generated data, Pf4 was highly expressed in LYVE-1+ resident macrophage subsets, but at much lower levels in CCR2+ inflammatory macrophages and monocytes (Fig. 5B). In scRNA-seq data from mouse blood CD11b+ cells26, Pf4 expression was absent from monocytes, but readily detected in contaminating platelets (Supplementary Fig. 11A). In line with our scRNA-seq analysis, previous studies using fluorescent reporter have demonstrated efficient Pf4-Cre-induced recombination in CD206+LYVE-1+ perivascular macrophages24, but limited recombination in circulating monocytes27. Based on these findings, we crossed Jak2V617F WT/Flex mice with Pf4 Cre+/- mice expressing the Cre recombinase under Pf4 promoter, to generate transgenic mice with a preferential recombination of JAK2V617F mutation in tissue-resident macrophages (Fig. 5C). Jak2V617FFlexPf4Cre+/- mice, referred to as Jak2V617F Pf4, developed aortic dilation and aneurysm before red blood cell and white blood cell increased in the blood. Indeed, at 7 weeks of age, red blood cell and white blood cell counts in the blood were not different between Jak2V617F Pf4 and Jak2WT controls (Fig. 5D), but the aorta was significantly dilated in Jak2V617F Pf4 mice (Fig. 5E, F), associated with reduced peri-adventitial collagen 1a content (Fig. 5G, H), increased MMP signature (Fig. 5I) and marked accumulation of LYVE-1+ macrophages (Fig. 5J). The significant increase in Il6 and Tnfα mRNA expression in aorta of Jak2V617F Pf4 mice supports a vascular pro-inflammatory immune response in mutant mice (Fig. 5K). During the follow-up, 89% (34/38) of Jak2V617F Pf4 mice spontaneously died of dissecting aortic aneurysm, whereas only one death was observed in the control group (1/28, 4%) (P < 0.0001) (Fig. 5L).

A Left, cells corresponding to monocytes/macrophages/dendritic cells were extracted from murine healthy and atherosclerotic plaques and separately reanalyzed with dimensional reduction (UMAP)25. Right, expression of Pf4 transcript projected on the vascular monocytes/macrophages/dendritic cells UMAP plot. B Left, cells corresponding to macrophages were extracted from murine aorta and separately reanalyzed with dimensional reduction (UMAP). Right, expression of Pf4 transcript projected on the vascular tissue resident macrophage subsets UMAP plot. C Jak2V617F WT/Flex mice were backcrossed with Pf4 Cre ± mice to generate control Pf4 Cre- Jak2WT and Pf4 Cre + Jak2V617F (Called Jak2V617F Pf4) mice. D Red blood cell, white blood cell and platelet count of 7-week-old Jak2WT and Jak2V617F Pf4 mice (N = 5/group). E, F representative photomicrographs and quantitative analysis of the mean aortic diameter in control 7-week-old Jak2WT (N = 7) and Jak2V617F Pf4 mice (N = 8) in the thoracic (ascending and descending) and the abdominal aorta, scale bar 1 mm. G Quantification of the collagen content (Cola1 immunostaining) in the aortic wall in 7-week-old Jak2WTand Jak2V617F Pf4 animals (N = 5/group), scale bar 50 µm. H Quantification of Cola1 and Cola3 mRNA expression in aorta of 7-week-old Jak2WT (N = 8) and Jak2V617F Pf4 mice (N = 9). I Quantification of Mmp2, Mmp3, Mmp9, Mmp12, Mmp13 mRNA expression in aorta of 7-week-old Jak2WT (N = 8) and Jak2V617F Pf4 mice (N = 9). J Quantification of LYVE-1 + CD68 + tissue resident macrophages in the aorta of 7-week-old mice (Immunostaining, N = 5/group), scale bar 50 µm. K Quantification of Il6, Il1β and Tnfα mRNA expression in aorta of Jak2WT (N = 8) and Jak2V617F Pf4 mice (N = 9). L Survival curve (N > 30/group). Data are presented as mean values ± SD. [Two-tailed Mann-Whitney test, *P < 0.05, **P < 0.01]. Difference in survival was evaluated using log-rank test. UMAP Uniform Manifold Approximation and Projection for Dimension Reduction, Pf Platelet factor, MMP matrix metalloprotease, Il interleukin, Tnf tumor necrosis factor. Source data are provided as a Source Data file.
Pharmacological activation of vascular tissue-resident macrophage activity induces dissecting aortic aneurysm in C57BL/6 mice
Finally, we aimed to test a pharmacological approach in immunocompetent animals to activate in vivo vascular resident macrophages. Using an integrated dataset combining several independent single-cell studies of healthy19,23 (Supplementary Fig. 12A) and diseased mouse aortas (Supplementary Fig. 12B), we found that the erythropoietin receptor (EpoR) was highly expressed in aortic resident macrophages. Flow cytometry analysis in different compartments confirmed that vascular resident macrophages highly and selectively expressed EPOR (Supplementary Fig. 12C), but circulating monocytes did not. Based on these observations, we hypothesized that EPO supplementation might activate vascular tissue-resident macrophages. Eight-week-old C57BL/6 mice were treated intraperitoneally with EPO or PBS, and histological analysis of the aorta were performed after 4 weeks of treatment and survival was recorded during 7 weeks of supplementation (Supplementary Fig. 12D). EPO treatment induced palmar erythema and polycythemia, but did not impact blood leukocyte and platelet counts (Supplementary Fig. 12E, F). EPO treatment induced a similar vascular phenotype to that seen in JAK2V617F mutant mice, with elastin degradation (Supplementary Fig. 12G), thinning of adventitial collagen layer (Supplementary Fig. 12H, I), local increase in proliferating resident macrophages (Supplementary Fig. 12J, K) and enhanced vascular pro-inflammatory responses (Supplementary Fig. 12L, M). EPO treatment promoted aortic dilation after 4 weeks and 7 weeks (Supplementary Fig. 12N, O), as well as aortic aneurysm (0 vs 58 %, P < 0.05) and lethal rupture (0 vs 42%, P < 0.05) (Supplementary Fig. 12P).
Pharmacological depletion of vascular tissue-resident macrophage or JAK/STAT pathway blockade attenuates aortic disease severity in JAK2V617F mutant mice
Taken together, our findings provided strong evidence that JAK2V617F gain-of-function mutation in vascular tissue-resident macrophages promoted dissecting aortic aneurysm. We sought to further evaluate the effect of ablating aortic resident macrophages in the vascular wall on disease severity using Ki20227, an inhibitor of macrophage colony stimulating factor 1 (CSF1) receptor tyrosine kinase. In agreement with a previous report by Lim et al.28, we confirmed that Ki20227 treatment (Fig. 6A) markedly depleted aortic macrophages (Fig. 6B, C), but did not affect red blood cells, white blood cells and platelets in blood (Fig. 6D). To assess the role of aortic resident macrophage depletion on aortic disease development, Ki20227 treatment was orally administered in Jak2WT and Jak2V617F Myel mice alternatively (Fig. 6E) to limit long term hematopoietic toxicity (Supplementary Fig. 13A–C). Ki20227 treatment remarkably abolished lethal aorta rupture in Jak2V617F Myel mice (Fig. 6F). Histological analysis performed at 15 weeks in surviving Ki20227-treated Jak2WT and Jak2V617F Myel mice showed that depletion of vascular tissue-resident macrophages (Fig. 6G) abolished the pro-inflammatory signature in the mutant aorta (Fig. 6H) and alleviated both JAKV617F-mediated aortic remodeling (Supplementary Fig. 13D) and dilation (Fig. 6I).

A Experimental protocol of Ki20227 treatment in C57BL/6 male mice. B Immunostaining of CD68 + LYVE-1+ macrophages (Yellow) in the aorta of Ki20227- or PBS-treated mice (N = 5/group), scale bar 50 μm. C Representative dot plot of CD45 + CD11b + Ly6G-Ly6C-CD64 + F4/80+ macrophage count in the aorta of Ki20227- or PBS-treated C57BL/6 mice. D Red blood cell, white blood cell and platelet count of Ki20227- or PBS-treated C57BL/6 mice. E experimental protocol of PBS or Ki20227 treatment in Jak2WT and Jak2V617F Myel mice. Jak2WT (blue, N = 8) and Jak2V617F Myel (Green, N = 8) mice received Ki20227 and a third group of Jak2V617F Myel (Red, N = 12) only received control diet without pharmacological drug. F survival curve of treated mice. G Immunostaining of CD68 + LYVE-1+ macrophages (Yellow) in the aorta of surviving Ki20227-treated Jak2WT (blue) and Jak2V617F Myel (green) mice (N = 8/group), scale bar 50 μm. H Quantification of Il6, Tnfα, Il1β and Ccl2 mRNA expression by RT-qPCR in the aorta of surviving Ki20227-treated Jak2WT (blue) (N = 8) and Jak2V617F Myel (green) (N = 7) mice. I Quantitative analysis of the mean aortic diameter of surviving Ki20227-treated Jak2WT (blue) and Jak2V617F Myel (green) mice (N = 8/group) in the thoracic ascending (Asc.), thoracic descending (Desc.) and the abdominal (Abd.) aorta. J Experimental protocol describing ruxolitinib (60 mg/kg, daily during 5 weeks) or PBS treatment in Jak2V617F HC-EC mice. K quantification of Il6, Tnfα, Il1β and Ccl2 mRNA expression by RT-qPCR in the aorta of Jak2V617F HC-EC mice treated with PBS (Red) (N = 10) or Ruxolitinib (Green) (N = 8). L Quantitative analysis of the mean aortic diameter of PBS (Red, N = 7) or ruxolitinib (Green, N = 8) -treated Jak2V617F HC-EC mice using ultrasonography in the thoracic ascending (Asc.), thoracic descending (Desc.) and the abdominal (Abd.) aorta. M experimental protocol describing Ruxolitinib (60 mg/kg, daily during 10 weeks) or PBS treatment in Jak2V617F Myel mice. N survival curve of treated mice (N = 11 PBS and N = 7 Ruxolitinib). Data are presented as mean values ± SD. [Two-tailed Mann-Whitney test or Kruskal-Wallis H, I, *P < 0.05, **P < 0.01, ***P < 0.001]. Difference in survival was evaluated using log-rank test. HC-EC, hematopoietic cells-endothelial cells; Il, interleukin; Tnf, tumor necrosis factor. Source data are provided as a Source Data file.
Finally, we tested a pharmacological approach with ruxolitinib, a JAK2 inhibitor, currently used to treat JAK2V617F + patients with MPN. Jak2V617F HC-EC and Myel mice were treated with PBS or ruxolitinib (oral gavage) daily during 5 and 10 weeks, respectively (Fig. 6J, M). Ruxolitinib attenuated the pro-inflammatory signature of aortic wall cells with decreased mRNA expression of Il1β, Il6, Tnfα and Ccl2 in the aorta of ruxolitinib-treated Jak2V617F HC-EC mice, compared to PBS-treated Jak2V617F HC-EC mice (Fig. 6K). Ruxolitinib significantly limited thoracic aorta dilation (Fig. 6L) and decreased lethal aortic aneurysm rupture (Fig. 6N).
Discussion
Here, we identified the JAK2V617F gain-of-function mutation as an independent risk factor for abnormal dilation of the ascending and descending thoracic aorta. We showed that JAK2V617F in mice drives vascular tissue-resident macrophages toward a pro-inflammatory phenotype, responsible for aortic dilation and spontaneous dissecting aortic aneurysm.
Until now, little was known about the impact of JAK2V617F mutation on aortic aneurysm disease. Findings from a recently published study in angiotensin II-infused chimeric hypercholesterolemic Apoe−/− mice, which took place while we conducted our study, agree with ours regarding the association between the JAK2V617F mutation and aortic aneurysm29. Yet, their experimental model was not clinically relevant and did not recapitulate the human disease. Moreover, this study did not provide any mechanistic insights into how JAK2V617F mutation can promote aortopathy and, in particular, made no mention of the central role of resident macrophages in JAK2V617F-induced severe aortopathy. In our study, using several complementary approaches, we showed that mice expressing human JAK2V617F gain-of-function mutation developed spontaneously aortic dilation at early stages and dissecting aortic aneurysm later on, in the absence of any challenge, and we identified the critical impact of JAK2V617F mutation on functions of vascular resident macrophages.
Histological analysis showed increased macrophage content in the aorta of young mutant mice. Early macrophage infiltration in the aortic wall is a hallmark of aortic wall development both in mice and in humans30,31. Macrophages largely participate in aortic wall degradation and remodeling through the production of chemokines, cytokines and proteases. In animal models using angiotensin II to promote aneurysm, macrophages detected in the aneurysmal aorta were shown to mostly derive from circulating monocytes4,32, whereas in Jak2V617F mutant mice, aortic macrophages unlikely derived from blood monocytes at early stages. At 7 weeks of age, Jak2V617F mutant mice developed aortic dilation with no sign of infiltrating neutrophils or monocytes in the vascular wall, but with high numbers of proliferating macrophages expressing LYVE-1, a vascular tissue-resident marker. Global and single cell transcriptomic analysis confirmed that aortic macrophages in Jak2V617F mutant mice exhibited a tissue-resident signature. Vascular tissue-resident Lyve-1+ macrophages have been shown to display protective functions against deleterious aorta remodeling in immunocompetent C57BL/6 mice28. In our study, we found that tissue-resident Res-MGL1hi macrophages, abundant in Jak2V617F Myel aortas, expressed high levels of chemokine transcripts such as Ccl2, Cxcl1, Cxcl2, Cxcl10, as well as high levels of Nlrp3 transcripts, suggesting that JAK2V617F mutation induced a shift in macrophage profile toward a pathogenic pro-inflammatory phenotype. Increased Il1b, Il6 and Tnfα mRNA expression, as well as increased MMP activity in the aorta of 8-week-old Jak2V617F mutant mice, likely resulted from higher content of activated macrophages in the aorta. At more advanced stages, in Jak2V617F mutant mice with large aortic aneurysm, another dominant subset of macrophages expressing Ccr2, but not Lyve1, has been identified in the aortic wall. These Ccr2+ Lyve-1- macrophages most likely derived from recruited blood monocytes in response to local production of chemokines, mainly Ccl2. Ccr2 is highly expressed on classical circulating monocytes and regulate their trafficking. Previous works have reported that in response to Angiotensin II infusion Ccr2-deficient mice are characterized by decreased infiltrating monocytes in the aortic wall, decreased local inflammation and finally less severe aortic disease33,34.
The JAK2V617F mutation has been shown to occur in the majority of patients with polycythemia vera35. The mutation also induces a polycythemia in different mouse models, due to either the proliferation and survival of erythroid progenitors carrying the mutation or local erythropoiesis in the bone marrow resulting from the release of growth factors by mutant myeloid progenitors36. Indirect stimulation of erythropoiesis most likely accounted for polycythemia in Jak2V617F Myel mice since the Jak2 mutation was not detected in erythroid progenitors. Polycythemia by itself might promote arterial dilation and decrease blood pressure through the activation of the endothelial NO synthase37. However, we can rule out the possibility that the Jak2-associated polycythemia was responsible for aortic dilation in mutant mice since aortopathy was observed in Jak2V617F Pf4 mice before polycythemia developed. In addition, disease severity was not affected by pharmacological correction of polyglobulia.
We also found that high doses of EPO induced dissecting aortic aneurysm in immunocompetent C57BL/6 mice, supporting the critical role of the JAK2 pathway in aortic disease, but we did not specifically investigated which cell type was activated in response to EPO supplementation. Our results are in line with a recent study showing that EPO repetitive injection promoted aortic aneurysm in Apoe−/− mice38. The authors found higher pro-inflammatory signature in the aorta of EPO-treated mice and suggested that endothelial cell activation might be involved in EPO-induced abdominal aortic aneurysm. However, in our study performed in normocholesterolemic mice, JAK2V617F gain-of-function mutation in vascular cells, and specifically in endothelial cells, did not promote aortic disease.
The relevance of our results in experimental murine models to the human situation are supported by the present finding of aortic dilation in patients with JAK2V617F mutation. In addition, several case reports also described arterial aneurysms in patients with JAK2+ myeloproliferative neoplasm39,40. More recently, in a genome-wide association study (GWAS) using the UK Biobank, an intronic variant located within the gene encoding JAK2 tyrosine kinase has been linked to aortic aneurysm41. Conversely to what we observed in Jak2V617F mutant mice, dissecting aortic aneurysm is not common in JAK2V617F+ patients. This discrepancy could be explained by the fact that JAK2V617F mutation in human is a somatic mutation occurring at late stages in adult whereas it is a germline mutation in mice who developed severe hematological disease at early stages of life. In addition, this discrepancy between human and mice data might be accounted for by the vascular protection by specific drugs, such as ruxolitinib, that are currently used to treat JAK2V617F+ myeloproliferative diseases. Interestingly, in our study, we showed that ruxolitinib attenuated pro-inflammatory signature in the aortic wall and protected against deleterious aortic remodeling and lethal rupture.
The protection induced by Ki20227 treatment supports the role of resident vascular macrophages in the pathophysiology of JAK2V617F-mediated aortopathy. However, Ki20227 could have off-target effects, such as angiogenesis inhibition42. However, in our mutant models, we did not find any evidence that JAK2V617F mutation impaired angiogenesis in the aortic wall.
Methods
Human samples analysis
We screened in a cohort of JAK2V617F + patients treated in Saint-Antoine Hospital (Assistance Publique-Hôpitaux de Paris), those who had a thoraco-abdominal CT-scan during the last 5 years. Controls were gender- and age-matched patients hospitalized in non-hematology department (Saint-Antoine Hospital) and who had a thoraco-abdominal CT-scan during the same period. Cardiovascular risk factors were recorded and used for multivariate adjustment. Aorta measurements were performed by two radiologists worked on the original axial data sets and used electronic calipers to measure the largest diameter of aorta at three points: (1) the mid ascending aorta at the level of the right pulmonary artery, (2) the mid descending aorta at the level of the aortic valve and (3) the maximal infrarenal aortic diameter. In order to evaluate the effect of the JAK2V617F mutation on the aorta diameter, we perfomed both univariate and multivariate logistic regression. Classical risk factors of aortic aneurysm such as age, arterial hypertension, diabetes, smoking status and gender was included in the multivariate logistic regression. The aorta was considered as dilated if diameter was higher than the average of the whole included patients.
Written informed consent was not required, in compliance with French law on retrospective studies of anonymized data (loi Jardé, MR004). All local medico-administrative data bases are declared to the general register of data processing of the Assistance publique-Hopitaux de Paris (Bureau de la protection des données, France no. 20220106150715).
Murine models
Experiments were conducted according to the guidelines formulated by the European Community for experimental animal use (L358-86/609EEC) and approved by the Ethical Committee of INSERM CEEA34 (MESR26963) and the French Ministry of Agriculture (agreement A75-15-32). Mouse breeding occurred in our animal facility in accordance with local recommendations. Animals were provided with food and water ad libitum. All the animals were maintained under identical standard conditions (housing, regular care and normal chow). Mice were maintained in isolated ventilated cages under specific pathogenfree conditions. Before euthanasia by cervical dislocation, animals were anesthetized with isoflurane (3% in oxygen).
Control mice were matched with littermates of the appropriate, age, sex, and genetic background to account for any variation in data. All mice were on a C57BL/6 background. Jak2V617F HC-EC mice were obtained by crossing VE-cadherin-Cre15 (Jackson Laboratory) transgenic mice with Jak2V617F Flex/WT mice43. Jak2V617F HC-EC mice have been generated by PE Rautou’s group16. Jak2V617F EC mice were obtained by crossing VE-Cadherin-cre-ERT2 transgenic mice (Provided by R.H. Adams, Max Planck Institute for Molecular Biomedicine, Münster, Germany)44 with Jak2V617F Flex/WT mice. Jak2V617F Myel mice were obtained by crossing LysM-Cre transgenic mice (Jackson Laboratory) with Jak2V617F Flex/WT mice. Jak2V617F Pf4 mice were obtained by crossing Pf4-Cre transgenic mice45,46 with Jak2V617F Flex/WT. In all experiments, male and female mice were used. For induction of Cre recombinase expression in Jak2V617F Flex/WT; VE-Cadherin-creERT2 mice, mice were injected intraperitoneally with tamoxifen (Sigma, T5648), 1 mg/mouse/day for 5 consecutive days over 2 consecutive weeks (10 mg in total per mouse) between the age of 5 to 7 weeks. The Flex (Flip-Excision) strategy allows expression of a mutated gene in adulthood, in a temporal and tissue-specific manner47.
Bone marrow transplantation
Six to 8-week-old C57BL/6 J and Jak2V617F HC-EC mice were subjected to medullar aplasia following sublethal total body irradiation (9.5 grays). At day 1, irradiated mice were repopulated intravenously with bone marrow cells (10 millions) isolated from femurs and tibias of age matched Jak2V617F HC-EC and C57BL/6 J CD45.1 or CD45.2 control mice. Mice were followed-up during 1–3 months with repetitive blood cells analysis and aortic diameter measurements by echography.
Blood cell count analysis
Blood was collected through the mandibular vein in a tube pre-coated with EDTA after transient anesthesia (Isoflurane). Blood counts analyses were performed using a Hemavet 950FS analyser (Drew scientific).
Fluorescence molecular tomography (FMT) imaging
Twenty-four hours before sacrifice, Jak2V617F HC-EC and Jak2V617F Myel animals were anesthetized with isoflurane and received 150 μL intravenously of a fluorescent imaging probe MMPsense 680 (NEV 10126, PerkinElmer). The probe is optically silent in its unactivated state and becomes highly fluorescent following activation by MMPs including MMP-2, −3, −9, and −13. Images were acquired using a fluorescence molecular imaging system (FMT 2500TM, VisEnMedical).
In vivo treatment
Erythropoietin (Eprex, Janssen) (EPO) was administered for 5 and 7 consecutive weeks by intraperitoneal injection to 8-week-old C57BL/6 males (5000 UI/kg, 3 times per week). EPO was prepared in PBS. Control mice received the same volume of vehicle (PBS). Ruxolitinib (Jakavi, Novartis) was administered for 5 consecutive weeks (30 mg/kg, 2 times per day) by oral gavage to Jak2V617F HC-EC. Ruxolitinib was prepared from 10 mg commercial tablets in PEG300/5% dextrose mixed at a 1:3 ratio. Control mice were administered the same volume of vehicle (PEG300/5% dextrose). Five-week-old C57BL/6 mice received daily oral administration of Ki202279 by food for 2 weeks. 5-week-old Jak2V617F Myel mice were alternatively fed with Ki20227 and chow diet for 10 weeks. Phenylhydrazine (PHZ) was administered every 3 days by intra peritoneal injection to 6-week-old Jak2V617F HC-EC mice (25 mg/kg body weight). PHZ was prepared in PBS and control mice received the same volume of vehicle.
Histological analysis
The aorta was flushed with cold PBS through the left ventricle and removed from the root to the iliac bifurcation. Abdominal aorta was used for histologic studies. The suprarenal region of the abdominal aorta, subjected to aneurysmal development, was harvested and embedded in both cryostat and paraffin and 7-μm thick sections were used for histology and immunohistochemistry analysis. Sections were stained with Orcein (MMFrance) for detection of elastin layers. The mean number of elastin layers was quantified by a researcher blinded to the experimental protocol (4 measurements/section, 8 sections/mouse). Sections were stained for macrophages using a rat anti– mouse CD68 primary antibody (MCA1957, clone FA-11, Bio Rad) revealed with an anti–rat Cy5 secondary antibody (712-175-153, Jackson ImmunoResearch). The percentage of cellular area positive for CD68 staining (macrophages) was quantified using Histolab software (Microvision) and the CD68 + area/total aortic wall area (media + adventitia) ratio was calculated. For resident macrophages, sections were also stained with a rabbit anti-mouse LYVE-1 (ab33682, polyclonal, Abcam) revealed with an anti–rabbit Cy3 secondary antibody (711-165-152, Jackson ImmunoResearch). Total macrophages (CD68 + ) and resident macrophages (CD68 + LYVE-1 + ) were manually counted (8 sections per mouse for each experiment). Proliferative macrophages were stained with a rabbit anti-mouse Ki67 (RM-9106-S, clone SP6, Epredia) after fixation and permeabilization (Triton 0.01%) and revealed with an anti–rabbit Cy3 secondary antibody (711-165-152, Jackson ImmunoResearch). Total macrophages (CD68 + ) and proliferative macrophages (CD68 + Ki67 + ) were manually counted. The media was stained with a rabbit anti-mouse Alpha Smooth muscle actin (ab5694, polyclonal, abcam) after fixation and permeabilization (Triton 0.01%) and revealed with an anti–rabbit Cy3 secondary antibody (711-165-152, Jackson ImmunoResearch). The quantification was obtained by calculating the aSMA positive area and the total media area using Histolab software (Microvision) and the aSMA+ area/total media ratio was calculated. The adventital collagen was stained with a rabbit anti-mouse Collagen Type I (234167, polyclonal, Merck Millipore) revealed with an anti–rabbit Cy5 secondary antibody (711-175-152, Jackson ImmunoResearch). Total advential collagen positive area was calculated using ImageJ (NIH) software.
In each experiment, 8 sections per mouse were quantified. Histology quantifications were done by a researcher blinded to the experimental protocol. Epifluorescence analysis, brightfield and polarized light imaging were done using Leica DM6000B microscope.
Flow cytometry
FACS analysis was performed on total aortas. The day of sacrifice, animals were perfused with cold PBS and whole aortas were rapidly cleaned and harvested under a dissection microscope. Aortas were minced and incubated 30 min at 37 °C in an enzymatic digestion cocktail (Collagenase I (450 U/ml, Sigma Aldrich), Collagenase XI (125 U/ml, Sigma Aldrich), Hyaluronidase (60 U/ml, Sigma Aldrich), DNAse (60 U/ml, Sigma Aldrich) and Elastase (0.372 U/ml, Sigma Aldrich). Then a mechanic digestion was performed to pass the cells through a 100 µm filter before the staining. Leukocyte suspension was obtained from aorta and extracellular antigens were stained with fluorescent labeled anti-mouse antibodies for 30 min at 4 °C. Flow cytometric acquisitions were done on a BD LSRFortessa (BD Biosciences) and data were analyzed using FlowJo Software (TreeStar, Inc.). Forward scatter (FSC) and side scatter (SSC) were used to gate live cells, red blood cells, debris, cell aggregates and doublets being excluded. Lymphocytes T were identified as CD45 + /CD11b-/CD3 + ; Lymphocytes B as CD45 + /CD11b-/B220 + ; Neutrophils as CD45 + /CD11b + /Ly6G + ; Monocytes as CD45 + /CD11b + /Ly6G-/Ly6C + ; Macrophages as CD45 + /CD11b + /Ly6G-/F4/80high/CD64high; Resident macrophages as CD45 + /CD11b + /Ly6G-/F4/80high/CD64high/Lyve-1 + ; Dendritic cells were CD45 + /CD11chigh/CMHIIhigh.
The following primary conjugated antibodies were used for staining in the aorta: anti-CD45 (Alexa Fluor 700, clone 30-F11, BD Biosciences), anti-CD11b (BV605, clone M1/70, BD Biosciences), anti-Ly6C (APC-Cy7, clone AL-21, BD Biosciences), anti-Ly6G (BUV395, clone 1A8, BD Biosciences), anti-CD64 (BV421, clone X54-5/7.1 Biolegend), anti-F4/80 (PE, clone CI:A3-1, Abcam), anti-CD11c (PerCP-Cy5.5, clone HL3, BD Biosciences), anti-MHC-II (BV510, clone M5/114.115.2, BD Biosciences), anti-CD3 (PE-Cy7, clone 145-2C11, BD Biosciences), anti-CD45R (B220) (BUV496, clone RA3-6B2, BD Biosciences), anti-LYVE1 (clone ALY7, Life Technologies), anti-EPO-R (PE, polyclonal, Clinisciences), anti-CD45.1 (BV605, clone A20, Biolegend), anti-CD45.2 (BV510, clone 104, Biolegend).
The following primary conjugated antibodies were used for the staining in the peritoneal macrophages and in the bone marrow: anti-CD45 (Alexa Fluor 700, clone 30-F11, BD Biosciences), anti-CD11b (BV605, clone M1/70, BD Biosciences), anti-Ly6C (APC-Cy7, clone AL-21, BD Biosciences), anti-Ly6G (BUV395, clone 1A8, BD Biosciences), anti-CD64 (BV421, clone X54-5/7.1 Biolegend), anti-F4/80 (FITC, clone BM8-1, Tonbo), anti-LYVE-1 (clone ALY7, Life Technologies), anti-EPO-R (PE, polyclonal, Clinisciences).
Determination of JAK2V617F allele burden on bone marrow hematopoietic cells
Femur and tibia from control Jak2WT and Jak2V617F HC-EC mice were crushed and filtered in PBS + EDTA 2 mM+FBS 2%. Mature erythroid cells were then lysed in an ammonium buffer. For progenitor cells analysis, an enrichment in CD117 + (Kit + ) cells was performed by magnetic cell sorting using anti-CD117 microbeads according to manufacturer’s instruction (Miltenyi Biotec). Cells were then labeled 30 min at 4 °C with the following antibodies:
for progenitors cells: Biotin-Lin (mix of anti-CD4, CD5, CD8a, Mac-1, B220, Ter119, Gr-1 antibodies, all from BD Bioscience); Kit APC (Biolegend # 105812); Sca 1 BV421 (Biolegend #108128); CD41 APC-Cy7 (Biolegend #133928); CD34 FITC (Biolegend #553733); CD16/32 BV510 (Biolegend #101333) for precursors and mature cells: CD41 APC-Cy7 (Biolegend #133928); Ter119 PE-Cy7 (Biolegend #116222); Gr1 FITC (Biolegend #108406); CD3 PE (Biolegend #100206); CD19 PB (Biolegend #115523). After washing, Steptavidine PerCP-Cy5.5 was added during 15 minutes at 4 °C and cells were then sorted on a FACS Aria LSR (Becton Dickcinson).
The following populations were isolated: LSK (Lin- Kit+ Sca + ); Megakaryocytes-Erythroid Progenitors (MEP) (Lin- Kit+ Sca- CD CD34-CD16/32-41-); Granulocyte-Monocyte Progenitors (GMP): Lin- Kit+ Sca- CD41- CD34 + CD16/32 + ; Megakaryocytic progenitors (MkP, Lin- Kit+ Sca- CD41 + ); T Lymphocytes (CD3 + ); B Lymphocytes (CD19 + ); Granulocytic precursors (CD19-CD3-Gr1 + ); Erythroid precursors (CD19-CD3-Ter119 + ).
Cells were frozen at −80 °C and DNA extracted using Nucleospin Tissue extraction kit (Macherey Nagel). The JAK2V617F allele burden was determined using a droplet digital PCR technic (Biorad QX200 system). The following primer and probes were used:
mJAK2-FwdCGAAGCAGCAAGCATGATGAG
mJAK2-RevGAGAGTAAGTAAAGCCAcCTGCT
mJAK2-Probe WTHEX-ATTATGGTGTCTGTGTCTGTGGAG-BHQ
Fwd mJAK2 ddPCRAAGCCCAGGTGATGGATGAGG
Rev mJAK2 ddPCRGTTACACGAGTCACCCATAATC
Probe mJAK2V617F ddPCRFAM-TACGAAGTTATTGGATTTCCTGCG-BHQ
The amplification program was as follows: 95 °C during 10′, before 40 cycles at 94 °C during 30′ and 57 °C during 1′, and a final step at 98 °C during 10′. Results were analyzed with Quantasoft software and expressed as the proportion of mutated allele among the number of copies of total JAK2 detected.
Bulk RNA sequencing & analysis
After RNA extraction, RNA concentrations were obtained using nanodrop or a fluorometric Qubit RNA assay (Life Technologies, Grand Island, New York, USA). The quality of the RNA (RNA integrity number) was determined on the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) as per the manufacturer’s instructions.
To construct the libraries, 100 ng of high quality total RNA sample (RIN > 8) was processed using NEBNext Ultra II RNA Library Prep Ki (New England BioLabs) according to manufacturer instructions. Briefly, after purification of poly-A containing mRNA molecules, mRNA molecules are fragmented and reverse- transcribed using random primers. Replacement of dTTP by dUTP during the second strand synthesis will permit to achieve the strand specificity. Addition of a single A base to the cDNA is followed by ligation of Illumina adapters. Libraries were quantified by qPCR using the KAPA Library Quantification Kit for Illumina Libraries (KapaBiosystems, Wilmington, MA) and library profiles were assessed using the DNA High Sensitivity LabChip kit on an Agilent Bioanalyzer. Libraries were sequenced on an Illumina Nextseq 500 instrument using 75 base-lengths read V2 chemistry in a paired-end mode. After sequencing, a primary analysis based on AOZAN software (ENS, Paris) was applied to demultiplex and control the quality of the raw data (based of FastQC modules/version 0.11.5). Obtained fastq files were then aligned using STAR algorithm (version 2.5.2b) and quality control of the alignment realized with Picard tools (version 2.8.1). Reads were then counted using Featurecount (version Rsubread 1.24.1) and the statistical analyses on the read counts were performed with the DESeq2 package version 1.14.1 to determine the proportion of differentially expressed genes between two conditions.
Raw fastq files were mapped to mouse genome GRCm38 release 96 with STAR 2.7.1a (Spliced Transcripts Alignment To a ref. 48. Normalization and differential expression analysis was performed with DESeq2 1.32.0 package (https://doi.org/10.1186/s13059-014-0550-8). Adjusted p-values were calculated using Benjamini and Hochberg’s approach, and genes with adjusted p-value <0.05 were considered as differentially expressed and selected for functional enrichment using clusterProfiler package v4.0.5 (https://doi.org/10.1093/bioinformatics/btq064).
CITE-seq
Jak2WT (n = 5) and Jak2V617F Myel (n = 5) mice under isoflurane anesthesia received 2 µg of anti-CD45.2-APC (clone 104, BioLegend) i.v. 5 min before sacrifice to label circulating immune cells19 and were killed by cervical dislocation. Upon collection of the aorta using a stereomicroscope, aneurysm development was macroscopically detected in 2 Jak2V617F Myel mice. Aortas were collected and enzymatically digested in RPMI medium containing 450 U/ml collagenase I (Sigma-Aldrich C0130), 125 U/ml collagenase XI (Sigma-Aldrich C7657), 60 U/ml Hyaluronidase (Sigma-Aldrich H3506) and 60U/ml DNAse1 (Roche #11284932001) for 45 min at 37 °C with agitation, washed in PBS + 1% FCS and passed through a 70 µm cell strainer. After washing in PBS + 1%FCS, aortic cells were resuspended in erythrocyte lysis buffer (ACK buffer) for 5 min and washed again with PBS + 1% FCS. Cells were plated in round bottom 96 well plates, and incubated for 10 min on ice with anti-CD16/32 (BioLegend, Clone 93, 10 µg/ml in PBS + 1% FCS) to block unspecific binding of antibodies to Fc receptors. Cells were subsequently labeled in a final volume of 60 µl for 25 minutes at 4 °C with a mix containing CITE-seq20 antibodies (TotalSeq-A, BioLegend, see table below), 1:1000 Fixable Viability Dye e780 (ThermoFisher Scientific 65-0865-14), 2 µg/ml anti-CD45.2 BV421 (Jak2WT samples) or 2 µg/ml anti-CD45.2 Alexa488 (Jak2V617F samples). Each unique sample was labeled with a specific Hashtag antibody for cell hashing49 (Hashtag 1 to 5: Jak2WT aortas, Hashtag 7 and 9: Jak2V617F Myel with aneurysm; Hashtag 6, 8, and 10: Jak2V617F Myel without aneurysm; final concentration 1:100, BioLegend TotalSeq-A).
TotalSeq-A antibody against: | BioLegend Cat# | Dilution |
Ly6G | 127655 | 500 |
CD11b | 101265 | 500 |
CD62L | 104451 | 500 |
IA_IE | 107653 | 500 |
CD54 (ICAM1) | 116127 | 500 |
Ly6C | 128047 | 800 |
CD115 | 135533 | 500 |
CXCR4 | 146520 | 500 |
Msr1 | 154703 | 500 |
CD64 | 139325 | 500 |
FCeRIa | 134333 | 500 |
CCR3 | 144523 | 500 |
CD49d | 103623 | 500 |
CD80 | 104745 | 500 |
CD117 | 105843 | 500 |
Sca1 | 108147 | 500 |
CD11c | 117355 | 500 |
TIM4 | 130011 | 500 |
CX3CR1 | 149041 | 500 |
XCR1 | 148227 | 500 |
F4/80 | 123153 | 500 |
CD86 | 105047 | 500 |
CD135 | 135316 | 500 |
CD103 | 121437 | 500 |
CD169 | 142425 | 500 |
CD8a | 100773 | 500 |
SiglecH | 129615 | 500 |
CD19 | 115559 | 500 |
CD3 | 100251 | 500 |
CD63 | 143915 | 500 |
CD9 | 124819 | 500 |
CD163 | 155303 | 500 |
NK1.1 | 108755 | 500 |
CD127 | 135045 | 500 |
CD68 | 137031 | 500 |
Sirpa | 144033 | 500 |
ITGB7 | 321227 | 500 |
CD4 | 100569 | 500 |
CD26 | 137811 | 500 |
MGL2 | 146817 | 500 |
TCRgd | 118137 | 500 |
CCR2 | 150625 | 500 |
CD44 | 103045 | 500 |
CD21/35 | 123427 | 500 |
CD43 | 143211 | 500 |
Armenian Hamster IgG Isotype Ctrl | 400973 | 500 |
Rat IgG1, κ Isotype Ctrl | 400459 | 500 |
Rat IgG2a, κ Isotype Ctrl | 400571 | 500 |
Rat IgG2b, κ Isotype Ctrl | 400673 | 500 |
CD47 | 127535 | 500 |
SiglecF | 155513 | 500 |
CD137 | 106111 | 500 |
CD36 | 102621 | 500 |
CCR5 | 107019 | 500 |
CD278 | 117409 | 500 |
PIR-A_B | 144105 | 500 |
CD5 | 100637 | 500 |
CD304 | 145215 | 500 |
CD40 | 124633 | 500 |
CD14 | 123333 | 500 |
CD95 | 152614 | 500 |
CD300c_d | 148005 | 500 |
IL1RL1 | 145317 | 500 |
TCRbeta | 109247 | 500 |
Mac2 | 125421 | 500 |
CD137L | 107109 | 500 |
CD178 | 106613 | 500 |
CD55 | 131809 | 500 |
CD226 | 128823 | 500 |
TIGIT | 142115 | 500 |
CD39 | 143813 | 500 |
JAML | 128507 | 500 |
CXCR5 | 145535 | 500 |
MGL1 | 145611 | 500 |
CD24 | 101841 | 500 |
After labeling, cells were washed twice in PBS + 1% FCS and pooled. Viable leukocytes were sorted with exclusion of contaminating circulating leukocytes (CD45.2-APC+) using a BD FACS Aria III with a 100 µm nozzle. The amount of sorted cells was adjusted so that cells from Jak2WT (CD45.2-BV421+) and from Jak2V617F aortas (CD45.2-Alexa488+) each represented ~50% of the total sorted cells. Cells were sorted in PBS supplemented with 1% PBS, and washed twice post-sort in PBS + 0.04% UltraPure BSA (ThermoFisher AM2616). Cells were counted in Trypan blue to assess viability (>80%). A total of 20,000 cells were loaded at a concentration of 500 cells/µl in the 10× Genomics Chromium (Single Cell 3′ v3 reagents, 10× Genomics) and scRNA-seq, ADT and HTO libraries prepared and sequenced as described in26.
Single-cell RNA-seq data analysis
A total of 10× Genomics data, including HTO and ADT libraries, was demultiplexed using Cell Ranger software (Pipeline Version 6.0.2). Mouse mm10 reference genome was used for the alignment and counting steps. The Feature-Barcode matrix obtained from Cell Ranger was further analyzed in Seurat v3.1.150. The “HTODemux” function was used to identify sample of origin of single cells, exclude multiplets (i.e. cells positive for more than one hashtag signal) and cells with no detectable hashtag signal. Cells with more than 10% mitochondrial transcripts were excluded from further analysis. Data were normalized using the “NormalizeData” function, and 2000 variable features identified with the “FindVariableFeatures” function using the “vst” selection method. Data were scaled using the “ScaleData” function and principal component (PC) analysis performed. Clustering was performed using the “FindNeighbors” and “FindClusters” functions with 20 PCs and a resolution parameter of 0.3. Uniform Manifold Approximation and Projection (UMAP) dimensional reduction was performed using 20 PCs. Immune cell populations were identified based on CITE-seq signal for established cell surface markers (see Results). Cells corresponding to monocytes/macrophages were subset in a new Seurat object and reclustered. This refined analysis revealed clusters of monocyte/macrophages with very low RNA content that were considered as low quality/dead cells and removed from further analysis. The curated monocyte/macrophage dataset was analyzed with clustering (resolution 0.8) and dimensional reduction as described above. 2 clusters of Ccr2+ macrophages with a clearly overlapping pro-inflammatory gene expression profile were manually pooled into the “Ccr2+ Inflammatory” macrophage cluster. Enriched transcripts and cell surface markers in monocyte/macrophage populations were determined using the “FindAllMarkers” function. Data visualizations were generated using built-in functions in Seurat (e.g. “FeaturePlot”, “DotPlot”) and dittoSeq (https://github.com/dtm2451/dittoSeq).
Quantitative real-time polymerase chain reaction
Thoracic aortas were harvested after flushing with cold PBS and were frozen at −80 °C. Total RNA was extracted from frozen mouse thoracic aorta samples with Qiagen columns (RNeasy MiniSpin Columns) using a polytron (T25 basic, IKA, Labortechnik). The phase containing RNAs was then recuperate and washed with molecular biology water. RNA quality control and concentration were performed using Nanodrop 2000 (Thermofisher scientific). and then stored at −80 °C. Reverse transcription was done following manufacturer instruction (kit QuantiTect Reverse Transcription (Qiagen)).
Real-time fluorescence monitoring was performed with the Applied Biosystems, Step One Plus Real-Time PCR System with Power SYBR Green PCR Master Mix (Eurogentec). qPCR was performed in triplicate for each sample. GAPDH cycle threshold was used to normalize gene expression: (F: 50 -CGTCCCGTAGACAAAATGGTGAA-30; R: 50 - GCC GTGAGTGGAGTCATACTGGAACA-30). Relative expression was calculated using the 2-delta-delta CT method followed by geometric average, as recommended.
The following primer sequences were used:
Il1β: Forward 5′-GAAGAGCCCATCCTCTGTGA-3′; Reverse 5′-GGGTGTGCCGTCTTTCATTA-3′; Tnfα: Forward 5′-GATGGGGGGCTTCCAGAACT-3′, Reverse: 5′-CGTGGGCTACAGGCTTGTCAC-3′, Il6: Forward 5′-TGACAACCACGGCCTTCCCTA-3′, Reverse 5′-TCAGAATTGCCATTGCACAACTCTT-3′, Il10: Forward 5′-CTCCTAGAGCTGCGGACTGCCTTCA-3′, Reverse 5′-CTGGGGCATCACTTCTACCAGGTAAAA-3′, Ccl2: forward 5′-CCCCACTCACCTGCTGCTA-3′, reverse 5′-TACGGGTCAACTTCACATTCAAA-3′
Blood pressure
Non-invasive systolic and diastolic blood pressure monitoring using tail cuff was done using Visitech BP-2000. Mice were trained for blood pressure 2 days prior to taking readings. Blood pressures were measured at 7-week-old. In each animal, the system automatically performed 4 measurements first, which were not recorded, then 10 consecutive measurements that were recorded.
Ultrasound study
We used Vevo 2100 ultrasound imager (VisualSonics) equipped with MS550D 22–55 MHz transducer. During imaging, each mouse was anesthetized with 2% isoflurane administered through an adapted nose cone. The mouse was then placed in a supine position on a heated platform to maintain its body temperature at approximately 37 °C. The hair over the areas being imaged was removed with a depilatory cream. Degassed ultrasound gel was used as a coupling medium. The ultrasound device was placed on the thorax and then on the abdomen of the mouse. Sagittal and transversal images were acquired.
Statistical analysis
Values are expressed as mean ± SEM. Differences between values were examined using the nonparametric two-tailed Mann-Whitney, Kruskal-Wallis tests when appropriate and were considered significant at P < 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
CITE-seq and single-cell RNA-sequencing data generated for this report has been deposited in Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE214880). Bulk RNA sequencing data generated for this report has been deposited in Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE215455).
All other data generated or analysed during this study are included in this published article (and its supplementary information files). Source data are provided with this paper.
Change history
23 December 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41467-022-35645-z
References
Nordon, I. M., Hinchliffe, R. J., Loftus, I. M. & Thompson, M. M. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat. Rev. Cardiol. 8, 92–102 (2011).
Mehta, C. K. et al. Management of acute aortic syndromes from initial presentation to definitive treatment. Am. J. Emerg. Med. 51, 108–113 (2022).
Davis, F. M., Rateri, D. L. & Daugherty, A. Abdominal aortic aneurysm: novel mechanisms and therapies. Curr. Opin. Cardiol. 30, 566–573 (2015).
Mellak, S. et al. Angiotensin II mobilizes spleen monocytes to promote the development of abdominal aortic aneurysm in Apoe−/− mice. Arterioscler Thromb. Vasc. Biol. 35, 378–388 (2015).
Ye, P. et al. GM-CSF contributes to aortic aneurysms resulting from SMAD3 deficiency. J. Clin. Invest 123, 2317–2331 (2013).
Iida, Y. et al. Peptide inhibitor of CXCL4-CCL5 heterodimer formation, MKEY, inhibits experimental aortic aneurysm initiation and progression. Arterioscler Thromb. Vasc. Biol. 33, 718–726 (2013).
Ensan, S. et al. Self-renewing resident arterial macrophages arise from embryonic CX3CR1(+) precursors and circulating monocytes immediately after birth. Nat. Immunol. 17, 159–168 (2016).
Swirski, F. K., Robbins, C. S. & Nahrendorf, M. Development and function of arterial and cardiac macrophages. Trends Immunol. 37, 32–40 (2016).
Lim, H. Y. et al. Hyaluronan receptor LYVE-1-expressing macrophages maintain arterial tone through hyaluronan-mediated regulation of smooth muscle cell collagen. Immunity 49, 1191 (2018).
Vainchenker, W. & Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 129, 667–679 (2017).
James, C. et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 434, 1144–1148 (2005).
Marnell, C. S., Bick, A. & Natarajan, P. Clonal hematopoiesis of indeterminate potential (CHIP): Linking somatic mutations, hematopoiesis, chronic inflammation and cardiovascular disease. J. Mol. Cell Cardiol. 161, 98–105 (2021).
Wang, W. et al. Macrophage inflammation, erythrophagocytosis, and accelerated atherosclerosis in Jak2 (V617F) Mice. Circ. Res. 123, e35–e47 (2018).
Sano, S. et al. JAK2 (V617F)-mediated clonal hematopoiesis accelerates pathological remodeling in murine heart failure. JACC Basic Transl. Sci. 4, 684–697 (2019).
Oberlin, E., El Hafny, B., Petit-Cocault, L. & Souyri, M. Definitive human and mouse hematopoiesis originates from the embryonic endothelium: a new class of HSCs based on VE-cadherin expression. Int J. Dev. Biol. 54, 1165–1173 (2010).
Poisson, J. et al. Erythrocyte-derived microvesicles induce arterial spasms in JAK2V617F myeloproliferative neoplasm. J. Clin. Invest. 130, 2630–2643 (2020).
Zhao, B. et al. Loss of pleckstrin-2 reverts lethality and vascular occlusions in JAK2V617F-positive myeloproliferative neoplasms. J. Clin. Invest. 128, 125–140 (2018).
Kilani, B. et al. Comparison of endothelial promoter efficiency and specificity in mice reveals a subset of Pdgfb-positive hematopoietic cells. J. Thromb. Haemost. 17, 827–840 (2019).
Cochain, C. et al. Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ. Res. 122, 1661–1674 (2018).
Stoeckius, M. et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017).
Zernecke, A. et al. Integrated scRNA-seq analysis identifies conserved transcriptomic features of mononuclear phagocytes in mouse and human atherosclerosis. bioRxiv, 2020.2012.2009.417535 (2020).
Van Hove, H. et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat. Neurosci. 22, 1021–1035 (2019).
Zernecke, A. et al. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ. Res. 127, 402–426 (2020).
McKinsey, G.L. et al. A new genetic strategy for targeting microglia in development and disease. Elife 9 (2020).
Zernecke A. et al. Integrated scRNA-seq analysis identifies conserved transcriptomic features of mononuclear phagocytes in mouse and human atherosclerosis. BioRxiv (2020).
Vafadarnejad, E. et al. Dynamics of cardiac neutrophil diversity in murine myocardial infarction. Circ. Res 127, e232–e249 (2020).
Abram, C. L., Roberge, G. L., Hu, Y. & Lowell, C. A. Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J. Immunol. Methods 408, 89–100 (2014).
Lim, H. Y. et al. Hyaluronan receptor LYVE-1-expressing macrophages maintain arterial tone through hyaluronan-mediated regulation of smooth muscle cell collagen. Immunity 49, 326–341 e327 (2018).
Yokokawa, T. et al. Crucial role of hematopoietic JAK2 V617F in the development of aortic aneurysms. Haematologica 106, 1910–1922 (2021).
Saraff, K., Babamusta, F., Cassis, L. A. & Daugherty, A. Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II-infused, apolipoprotein E-deficient mice. Arterioscler Thromb. Vasc. Biol. 23, 1621–1626 (2003).
Raffort, J. et al. Monocytes and macrophages in abdominal aortic aneurysm. Nat. Rev. Cardiol. 14, 457–471 (2017).
Vandestienne, M. et al. TREM-1 orchestrates angiotensin II-induced monocyte trafficking and promotes experimental abdominal aortic aneurysm. J. Clin. Invest. 131 (2021).
Daugherty, A. et al. Angiotensin II infusion promotes ascending aortic aneurysms: attenuation by CCR2 deficiency in apoE-/- mice. Clin. Sci. (Lond.) 118, 681–689 (2010).
de Waard, V. et al. Systemic MCP1/CCR2 blockade and leukocyte specific MCP1/CCR2 inhibition affect aortic aneurysm formation differently. Atherosclerosis 211, 84–89 (2010).
Ishii, T., Bruno, E., Hoffman, R. & Xu, M. Involvement of various hematopoietic-cell lineages by the JAK2V617F mutation in polycythemia vera. Blood 108, 3128–3134 (2006).
Ramos, P. et al. Macrophages support pathological erythropoiesis in polycythemia vera and beta-thalassemia. Nat. Med. 19, 437–445 (2013).
Ruschitzka, F. T. et al. Nitric oxide prevents cardiovascular disease and determines survival in polyglobulic mice overexpressing erythropoietin. Proc. Natl Acad. Sci. USA 97, 11609–11613 (2000).
Zhang, M. et al. Erythropoietin promotes abdominal aortic aneurysms in mice through angiogenesis and inflammatory infiltration. Sci. Transl. Med. 13 (2021).
Ogawa, S., Yoshino, M. & Hara, T. Extracranial carotid artery aneurysm with myeloproliferative neoplastic cell invasion. J. Vasc. Surg. Cases Innov. Tech. 6, 243–246 (2020).
Law, N.L., Villada, F.A. & Kruse, M.J. Rupture of splenic artery aneurysm in a man with polycythemia vera and acquired von Willebrand syndrome. BMJ Case Rep. 14(2021).
Ashvetiya, T. et al. Identification of novel genetic susceptibility loci for thoracic and abdominal aortic aneurysms via genome-wide association study using the UK Biobank Cohort. PLoS ONE 16, e0247287 (2021).
Kubota, Y. et al. M-CSF inhibition selectively targets pathological angiogenesis and lymphangiogenesis. J. Exp. Med. 206, 1089–1102 (2009).
Hasan, S. et al. JAK2V617F expression in mice amplifies early hematopoietic cells and gives them a competitive advantage that is hampered by IFNalpha. Blood 122, 1464–1477 (2013).
Wang, Y. et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465, 483–486 (2010).
Tiedt, R., Schomber, T., Hao-Shen, H. & Skoda, R. C. Pf4-Cre transgenic mice allow the generation of lineage-restricted gene knockouts for studying megakaryocyte and platelet function in vivo. Blood 109, 1503–1506 (2007).
Mansier, O. et al. Description of a knock-in mouse model of JAK2V617F MPN emerging from a minority of mutated hematopoietic stem cells. Blood 134, 2383–2387 (2019).
Schnutgen, F. & Ghyselinck, N. B. Adopting the good reFLEXes when generating conditional alterations in the mouse genome. Transgenic Res. 16, 405–413 (2007).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Stoeckius, M. et al. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 19, 224 (2018).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019).
Acknowledgements
This work was supported by Inserm (Z.M., S.T., A.T., H.A.O.), Fondation Lefoulon-Delalande (RAR) and the British Heart Foundation (Z.M.). We would like to thank Panagiota Arampatzi and the Core Unit Systems Medicine of the University Würzburg for assistance with single-cell RNA-seq analysis.
Author information
Authors and Affiliations
Contributions
R.A.R. and M.V. conducted experiments, acquired data, analyzed data, performed statistical analysis and wrote the manuscript. J.R.L., T.M., J.C., J.P., L.L., B.E., C.K., J.V. conducted experiments, acquired data, analyzed data. P.H., F.F., L.A. and M.L. participate in human study. O.M. and C.J. provided transgenic mice. M.D. analysis transcriptomic profile in the aorta of Jak2V617F HC-EC mice. A.Z., G.R., A.E.S., and C.C. performed scRNA seq experiments and analysis. A.T., Z.M., S.T., P.E.R. drafted of the manuscript. H.A.O. designed the experiments, analyzed data, performed statistical analysis, drafted of the manuscript and obtained funding for the project. M.D. and R.B. performed statistical analysis, All authors discussed and critically revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests
Peer review
Peer review information
Nature Communications thanks Guo-Ping Shi and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Al-Rifai, R., Vandestienne, M., Lavillegrand, JR. et al. JAK2V617F mutation drives vascular resident macrophages toward a pathogenic phenotype and promotes dissecting aortic aneurysm. Nat Commun 13, 6592 (2022). https://doi.org/10.1038/s41467-022-34469-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-34469-1
This article is cited by
-
Genetic dissection of the impact of lncRNA AI662270 during the development of atherosclerosis
Journal of Translational Medicine (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
