
Abstract
As pioneering Fe3O4 nanozymes, their explicit peroxidase (POD)-like catalytic mechanism remains elusive. Although many studies have proposed surface Fe2+-induced Fenton-like reactions accounting for their POD-like activity, few have focused on the internal atomic changes and their contribution to the catalytic reaction. Here we report that Fe2+ within Fe3O4 can transfer electrons to the surface via the Fe2+-O-Fe3+ chain, regenerating the surface Fe2+ and enabling a sustained POD-like catalytic reaction. This process usually occurs with the outward migration of excess oxidized Fe3+ from the lattice, which is a rate-limiting step. After prolonged catalysis, Fe3O4 nanozymes suffer the phase transformation to γ-Fe2O3 with depletable POD-like activity. This self-depleting characteristic of nanozymes with internal atoms involved in electron transfer and ion migration is well validated on lithium iron phosphate nanoparticles. We reveal a neglected issue concerning the necessity of considering both surface and internal atoms when designing, modulating, and applying nanozymes.
Similar content being viewed by others


Introduction
Since the first discovery of Fe3O4 nanoparticles (NPs) with intrinsic peroxidase (POD)-like activity in 2007, nanomaterial-based artificial enzymes (nanozymes) and their extensive applications have rapidly attracted attention over the past decade1,2,3,4,5,6,7,8,9,10. Recently, research efforts on nanozymes have gradually shifted from application-oriented to mechanism-oriented11,12,13,14,15. For example, single-atom nanozymes centered on different metal species have been synthesized with well-defined structures and coordination environments, which facilitate the identification of catalytic centers and unravel the catalytic mechanisms at the atomic level16,17,18,19. Besides, the high substrate selectivity of nanozymes has been achieved by the bionic principle of natural substrate channeling and stepwise screening or by molecular blotting techniques20,21. Given the intricate structure-activity relationships and restricted characterization techniques, however, it is still challenging to understand the explicit mechanism of most nanozymes3,4.
Despite being a pioneering nanozyme, research on the catalytic mechanism of the POD-like activity of Fe3O4 NPs is still limited10,14,15,22,23,24,25. To date, it is generally accepted that high-reactive hydroxyl radicals (·OH) generated by Fenton-like reactions (Eqs. (1) and (2)) involving the surface Fe2+ under acid conditions contribute to the POD-like activity of Fe3O4 NPs24,25. Although the questioning that the POD-like activity of magnetite is mediated by adventitious metal traces, it has been experimentally verified (including this paper) to exclude the interference of metal traces or the leaching effect of Fe ions in reaction solution10,25,26,27,28. Similar to natural horseradish peroxidase, Fe3O4 nanozymes follow the ping-pong mechanism and Michaelis–Menten kinetics10. In addition, their catalytic performances are influenced by particles size, morphology, lattice structure, doping, surface modification, substrates used, and the catalytic environment exposed, all of which could affect the surface-active sites by altering the surface chemistry10,22,23,29,30,31,32. Other individual studies have investigated the absorption, activation, and desorption processes of substrates (e.g., H2O2 and 3, 3′, 5, 5′-tetramethylbenzidine (TMB)) on the surface of Fe3O4 at the atomic level based on density functional theory and developed some descriptors to predict their POD-like activity14,15.
The above mechanistic studies share a theoretical premise: only the surface-active sites play a decisive role in the enzymatic-like property of nanozymes since catalysis occurs mainly on the particle surface or interface. This view is now widely recognized and works for most types of nanozymes1,2,4,11,32,33. For example, in a recent controversial question regarding how to define nanozyme concentration, Liu et al. argued that considering the whole particle or all atomic units within a particle as an enzyme unit would overestimate and underestimate the catalytic activity of nanozymes, respectively, because it is the surface atoms that are truly the catalytic active sites33. However, in the Fenton-like reactions triggered by Fe3O4 nanozymes, we noticed that the reaction rate constant of Eq. (1) is much higher than that of Eq. (2), which implies that the surface-active Fe2+ is hardly recovered after being oxidized. This irreversible oxidation of surface Fe2+ prompts us to ponder if only the surface atoms of the nanozymes, particularly for metal oxide nanozymes, act in enzymatic-like catalysis, would these active sites be exhausted after long-term catalysis, rendering the nanozymes inactive? Nevertheless, no relevant studies can conclusively answer this crucial question.
Here, we propose a neglected issue regarding the POD-like mechanism of nanozymes by characterizing the chemical composition and catalytic activity of the recycled Fe3O4 NPs participating in cyclic POD-like catalysis. Both surface and interior Fe2+ are found to impart POD-like properties to Fe3O4 nanozymes. Generally, Fe2+ inside the particle could transfer its electron to the surface layer, regenerating the surface Fe2+ and sustaining the catalytic reaction. This process is usually coupled with the outward migration of excess oxidized Fe3+, which is probably a rate-limiting step. As the catalysis continues, Fe3O4 is slowly oxidized into γ-Fe2O3 accompanying the depleted enzyme-like activity, similar to the conventional low-temperature oxidation of magnetite, only with different electron receptors. This self-depleting characteristic of nanozymes with internal atoms involved in electron transfer and ion migration is further demonstrated by a typical model material, lithium iron phosphate (LiFePO4), which contains redox-active metal sites and mobile lithium ions (Li+) encapsulated in a rigid phosphate network. This paper reveals that internal atoms may also contribute to nanozyme-catalyzed reactions even though these reactions occur on the surface of NPs, which is thought-provoking when designing, regulating, and applying nanozymes.
Results and discussion
Synthesis and characterization of IONPs
Near-spherical magnetite nanoparticles (Fe3O4 NPs) with an average diameter of 10.16 ± 0.12 nm (Supplementary Fig. 1a) were synthesized using the chemical coprecipitation method24. Maghemite (γ-Fe2O3) and hematite (α-Fe2O3) NPs were derived by calcining the Fe3O4 NPs powder at 200 and 650 °C for 2 h, respectively (Fig. 1a)34. X-ray diffractometer (XRD) and Raman spectra (Supplementary Fig. 1b, c) show the successful synthesis of these three iron oxide NPs (IONPs). These IONPs were uniformly dispersed in an aqueous solution at pH of 3 by ultrasonication (Supplementary Fig. 1d). Their POD-like activities were assessed using different colorimetric substrates, including TMB, 2, 2’-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS), and o-Phenylenediamine (OPD), in the presence of H2O2. The results show that their catalytic activity followed the order of Fe3O4 NPs >> γ-Fe2O3 NPs > α-Fe2O3 NPs (Supplementary Fig. 2). To better quantify their POD-like activity, we calculated their specific activity (anano) according to the specified method35,36, which were 1.79, 0.45, and 0.03 U·mg−1, respectively (Fig. 1b). As previously reported10,24, the higher catalytic ability of Fe3O4 NPs is attributed to the ·OH arising from the surface Fe2+-initiated Fenton-like reaction (Supplementary Figs. 3 and 4). The negligible anano of α-Fe2O3 NPs compared with γ-Fe2O3 NPs is ascribed to the structural effect of the crystal phases37. Briefly, γ-Fe2O3 possess cation vacancies at its octahedral positions and most of these vacancies are located on the particle surface, which can favor the adsorption of the substrate H2O2, resulting in a relatively higher POD-like activity (Supplementary Fig. 5). However, these vacancies do not exist on the surface of α-Fe2O3 due to the change of crystal structure caused by the higher calcination temperature37.

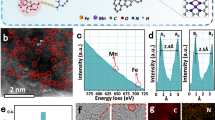
a Illustration of the synthesis process of IONPs. b The specific activity (anano) of these three IONPs with TMB as colorimetric substrates. c Diagram of the cyclic catalysis assay. d Kinetic study of anano values of Fe3O4 NPs with the days of cyclic catalytic reaction. Error bars represent standard deviation from three independent measurements. e Comparison of Fe L2,3 spectra of Fe3O4 NPs before and after 5 days of cyclic POD-like reactions. f The fitted Fe2p XPS spectra of Fe3O4 NPs recycled after catalysis on days 0, 1, 3, and 5. g The Fe L-edge NEXAFS spectra of Fe3O4 NPs and recycled Fe3O4 NPs after 5 days of catalysis in comparison with the reference spectra of FeSO4 and Fe2O3. h Raman spectra of Fe3O4 NPs recycled after catalysis on days 0, 1, 3, and 5. i TEM, HRTEM images, and SAED pattern of Fe3O4 NPs and recycled Fe3O4 NPs after 5 days of catalysis. Images were collected at least three times for each type of NPs.
Cyclic POD-like catalysis of Fe3O4 NPs
To investigate whether the surface Fe2+ of Fe3O4 NPs is depleted after participating in prolonged catalysis, we continuously increased the amount of substrate TMB under sufficient H2O2 with as-synthesized three IONPs as continuous catalysts, and monitored the absorbance changes of TMB oxidation products at 650 nm within 12 h. From Supplementary Fig. 6d–g, even though the TMB was increased from 0.087 to 0.52 mM, the Fe3O4 NPs were still able to continuously and rapidly engage in the catalytic reaction for a long duration (≥12 h) without showing signs of depletion. We speculated two reasons: (1) the amount of substrate is still too low to completely consume the surface-active Fe2+ and (2) the Fe2+ within Fe3O4 NPs provides the impetus for continuous catalysis.
Cyclic POD-like catalytic assays (Fig. 1c) were carried out as validation, which could provide sufficient substrates for Fe3O4 NPs to keep exerting their POD-like capacity. We evaluated the anano of the recycled Fe3O4 NPs within 5 days. The results show that the catalytic ability of Fe3O4 NPs decreased to a level comparable to that of γ-Fe2O3 NPs after five days of cyclic catalysis, while the changes of γ-Fe2O3 NPs were negligible (Fig. 1d and Supplementary Fig. 7). The impact of leached Fe ions in acidic medium on the catalytic activity of IONPs has been excluded (Supplementary Fig. 8 and Supplementary Table 1). It pushed us to wonder how the surface-active Fe2+ of Fe3O4 NPs alone could sustain the TMB oxidation for up to 100 h. Conceivably, if only the surface-active sites are responsible for the enzyme-like performance, nanozymes will deactivate when the surface-active sites are exhausted (Supplementary Fig. 9).
To reveal the potential reasons for the sustained catalytic capacity of Fe3O4 NPs, we characterized the physicochemical properties of the recycled Fe3O4 NPs using different methodologies. The electron energy-loss spectra are a useful tool for revealing the chemical and oxidation state information of iron oxide at high spatial resolution38. In general, the peaks of the transition metal L-edge shift toward higher energy loss with an increasing oxidation state. For iron oxide species, the area ratios of Fe L3/L2 also increase with increasing Fe valence39. As shown in Fig. 1e, both of the Fe3O4 NPs before and after 5 days of cyclic POD-like reactions showed two peaks related to the Fe L3 and L2, with an energy gap of about 13 eV between the two white lines. However, approximately 1.4 eV chemical shift toward high energy loss could be observed from Fe3O4 NPs to recycled Fe3O4 NPs. In addition, the Fe L3/L2 area ratios also increased from 4.7 to 6.1, which indicates the increase in the Fe oxidation state of Fe3O4 NPs after 5 days of POD-like catalysis40. A similar finding was obtained from the XPS analysis of the recycled Fe3O4 NPs on days 0, 1, 3, and 5 of the cyclic catalysis. The X-ray penetration depth of the analyzed sample ranges from 2 to 10 nm for the XPS technique. Since the average diameter of as-synthesized Fe3O4 NPs is around 10 nm, the Fe valence state obtained from the Fe2p fitting analysis can be approximated as the oxidation state of individual Fe3O4 NPs. As shown in Fig. 1f, the Fe2+ in Fe3O4 NPs decreased from the original 30.9% to 0% with the extension of cyclic catalytic days, suggesting that the interior Fe2+ may also be oxidized to Fe3+ in the successive POD-like reactions.
The phase transformation of Fe3O4 NPs caused by internal Fe2+ oxidation was demonstrated by the near-edge X-ray absorption fine structure (NEXAFS) spectroscopy. Figure 1g shows the Fe L-edge NEXAFS spectra of the control Fe3O4 NPs and the recycled Fe3O4 NPs after 5 days of catalysis, in comparison with two reference spectra of FeSO4 and Fe2O3. The increased splitting L3 peak near 708 eV for the recycled Fe3O4 NPs coincides with reference Fe2O341,42,43. Besides, in the Raman spectra of the recycled Fe3O4 NPs, the feature of the A1g mode band gradually shifted from 660 to 700 cm−1 (Fig. 1h), corresponding to a transition from magnetite to maghemite44,45. In addition, transmission electron microscopy (TEM) images (Fig. 1i) and XRD pattern (Supplementary Fig. 10) show that the influence of this transformation on the particle morphology, size, and lattice structure is negligible.
Based on the above characterization results, we conclude that not only the surface Fe2+ but also the interior Fe2+ of the Fe3O4 nanozymes were gradually oxidized by prolonging the reaction time. Simultaneously, the catalytic activity of the recovered NPs gradually decreases with the increase of their oxidation state. Therefore, we suggest that the involvement of Fe2+ inside the particles is responsible for the prolonged catalytic capacity of Fe3O4 nanozymes. Specifically, as shown in Supplementary Fig. 11, when the surface Fe2+ is oxidized to Fe3+ by the Fenton-like reaction, the adjacent Fe2+ inside the particle will continuously transfer its electron outward via the Fe2+-O-Fe3+ chain in the lattice to maintain the catalytic activity of the surface Fe atoms. However, this replenishment of electrons is not infinite. When all the interior Fe2+ are oxidized to Fe3+, the Fe3O4 phase is transformed to γ-Fe2O3 without electrons being transferred to the surface, resulting in the reduction of catalytic activity or even inactivation.
Aeration oxidation kinetics of Fe3O4 NPs
We assume that the oxidation of Fe3O4 nanozymes induced by POD-like catalysis is comparable to the traditional low-temperature (<200 °C) air oxidation of magnetite since the crystal structure of both remains unchanged during the oxidation process46. Both magnetite and maghemite contain 32 O atoms per unit cell. The difference is that the former contains 24 Fe atoms (16 Fe3+ and 8 Fe2+), while the latter has only 21.33 Fe atoms (all Fe3+). Namely, once 8 Fe2+ in magnetite are oxidized to 8 Fe3+ releasing 8 electrons, a charge imbalance will occur (Eq. (3)). To maintain electroneutrality, 2.67 Fe3+ have to migrate to the crystal surface, leaving the cation vacancies (Eq. (4))46. The outward moving Fe3+ will coordinate with the surface absorbed O2- that is ionized by the electrons generated by the oxidation of Fe2+ to Fe3+, and form a film of the solid solution of γ-Fe2O3 in Fe3O434,47,48,49. Therefore, the phase transformation of Fe3O4 to γ-Fe2O3 is a single-phase topological reaction accompanied by the separate migration of electrons and excess Fe3+ 34,47,49.
Lattice defects have been reported to facilitate the outward migration of excess iron ions, thereby accelerating the oxidation process of magnetite48. As verification, we compared the aeration oxidation kinetics of Fe3O4 NPs synthesized by two methods with different degrees of lattice defects. One was prepared by the chemical coprecipitation method as described above (Fig. 1a), which is considered to possess more lattice defects (named cc-Fe3O4 NPs). The other was prepared by the thermal decomposition method (Supplementary Fig. 12) with a relatively complete lattice structure (named TD-Fe3O4 NPs)50. Both Fe3O4 NPs have a similar average particle size (~10 nm) with N(CH3)4+ as a surface stabilizer. Their aqueous solutions were stirred under the same aeration rate (with air) for 12 h at 120 °C. For a better comparison, the oxidation system of cc-Fe3O4 NPs (total 170 mL, 3.6 mg Fe/mL) was much larger than that of TD-Fe3O4 NPs (total 30 mL, 0.45 mg Fe/mL). This implies that individual TD-Fe3O4 could gain more oxygen than cc-Fe3O4 to keep it oxidized. From Fig. 2a, b, both Fe3O4 NPs exhibited electronic transitions in the visible and NIR region due to intervalence charge transfer between Fe2+ and Fe3+ 51, which decreased gradually with oxidation time. At the end of aeration oxidation, little absorption beyond 700 nm was observed, indicating a phase transformation from Fe3O4 NPs to γ-Fe2O3 NPs51. Besides, the color of both suspensions gradually changed from dark-brown to reddish-brown. Notably, despite the less oxygen exposure for individual cc-Fe3O4 NP, its NIR absorption decreased faster than that of TD-Fe3O4 NP, especially during the initial oxidation phase (within 1 h). These results confirm that more lattice defects favor the oxidation reaction of Fe3O4 NPs due to the faster electron and ion transfer.

Variation of UV-Vis-NIR absorption of a cc-Fe3O4 NPs and b TD-Fe3O4 NPs with aeration oxidation time. Insets are photos of the suspensions corresponding to oxidation times at 0, 0.5, 1, 3, 5, 8, 10, and 12 h. All spectra and photos were obtained at the same Fe concentration. c Changes in anano of the oxidized cc-Fe3O4 NPs and TD-Fe3O4 NPs during the aeration oxidation. Error bars represent standard deviation from three independent measurements.
Analogous to aerated oxidation, the rapid electron and ion migration are also considered to facilitate the POD-like catalysis of Fe3O4 NPs, with the only difference being that the electron receptor changed from O2 in aerated oxidation reaction to H2O2 in POD-like reaction. To prove this, the POD-like activity of cc-Fe3O4 NPs and TD-Fe3O4 NPs as well as their variation with aerated oxidation time were investigated. As seen in Supplementary Fig. 13, the POD-like activity of cc-Fe3O4 NPs was higher (2.8 folds) than that of TD-Fe3O4 NPs, despite TD-Fe3O4 NPs having a smaller hydrodynamic diameter and negative surface potential contributing to a strong affinity with TMB. Aeration oxidation kinetic studies show that the POD-like activity of both Fe3O4 NPs decreased with oxidation time (Fig. 2c), along with slight fluctuations in hydrodynamic size and surface potential (Supplementary Fig. 14). However, the decline rate of cc-Fe3O4 NPs was faster than TD-Fe3O4 NPs, particularly in the initial oxidation stage. This phenomenon is consistent with the changes in NIR spectra shown in Fig. 2a, b. These results further confirm that the more lattice defects of Fe3O4 NPs, the easier the migration of excess Fe ions, and thus the higher the POD-like activity. It also means that Fe3O4 NPs with more defect sites are easier to be depleted when involved in a POD-like reaction due to their excellent catalytic capability.
Catalytic mechanism of the POD-like for Fe3O4 NPs
Taken together, the catalytic mechanism of the POD-like activity for Fe3O4 NPs can be summarized as follows (Fig. 3): (1) fenton-like reaction on the surface. Firstly, H2O2 adsorbed on the surface of particles accepts electrons from the surface Fe2+, and then dissociates into highly active ·OH to oxidize the substrates. The surface Fe2+ is oxidized to Fe3+. (2) Internal electrons transfer. Then, the adjacent Fe2+ inside the surface transfers its electron to the surface Fe3+ via the Fe2+-O-Fe3+ chain, retrieving the surface Fe2+ and providing the dynamics for the sustained catalytic reaction. (3) Excess Fe ions outward migration. With the in situ oxidation of internal Fe2+, to maintain electroneutrality, the excess Fe3+ in the lattice has to migrate outward to the surface, leaving cation vacancies. (4) Chemical composition change. With the continuous POD-like catalytic reaction, Fe3O4 NPs are oxidized from the surface to the interior and finally transformed into γ-Fe2O3 NPs. This enzymatic-like reaction-triggered oxidation process of Fe3O4 NPs is thought to be analogous to the conventional low-temperature air oxidation of magnetite, in which iron ion migration is probably a rate-limiting step.

The catalytic POD-like reaction of Fe3O4 NPs occurs along with internal electron transfer and excess Fe ions migration. After prolonged catalysis, Fe3O4 NPs suffer the phase transformation to γ-Fe2O3 NPs with depletable POD-like activity.
As is known, magnetite has an inverse spinel structure, with Fe3+ occupying tetrahedral (A) sites and equal amounts of Fe2+ and Fe3+ occupying octahedral (B) sites, written as (Fe3+)A[Fe2+ Fe3+]BO4. The rapid electron hopping between Fe2+ and Fe3+ on the B-sites, creating an intermediate valence state of Fe2.5+, contributes to the conductivity of magnetite at room temperature, exhibiting a half-metallic nature52. This electron-hopping process has been reported to be limited to available Fe2+–Fe3+ pairs and thus highly depends on the degree of non-stoichiometry of magnetite53. Oxidizing Fe3O4 to non-stoichiometry magnetite (Fe3-δO4) or to γ-Fe2O3, the Fe2+ in B-sites can be replaced by Fe3+ and vacancies, which can be written as (Fe3+)A[Fe2-6δ2.5+]B[Fe5δ3+□δ]BO4 (□ indicates vacancy; δ indicates vacancy parameter, 0 < δ ≤ 1/3). Thus, the number of available Fe2+–Fe3+ pairs decreases while isolated Fe3+ increases. Besides, the formation of cation vacancies due to the surface migration of excess Fe3+ can also disrupt the fast electron-hopping between Fe ions in B-sites. According to the local charge compensation model54, each vacancy is electrically equivalent to an extra −5/2 charge at one B-site, which has to be neutralized by the excess positive charge at the adjacent B-sites. Thus, each vacancy traps 5 Fe3+ and no longer involves in the conduction process53. In general, this disturbed electron-hopping process caused by the reduction of Fe2+–Fe3+ pairs and the formation of cation vacancies is thought to impair the electron transfer to the surface when Fe3O4 nanozymes participate in the sustained POD-like reaction, leading to their depletable catalytic activity.
LiFePO4 NPs as an ideal verification model
To test the above mechanism, we found an ideal model material, LiFePO4, which is commonly applied as cathode material for lithium-ion batteries (LIBs)55,56,57,58. LiFePO4 undergoes redox reactions along with the lithium insertion/extraction during the charge-discharge process (Eqs. (5) and (6)) without changing its ordered-olivine structure (Fig. 4a)55. We speculate that the charging process of LiFePO4 resembles the oxidation process of Fe3O4, both of which involve the oxidation of Fe2+ and the migration of internal ions, which motivated us to focus on whether LiFePO4 NPs also have the POD-like catalytic ability.

a The crystal structure of LiFePO4 and FePO4 viewed along the a, b, c-axis. The olivine structure is maintained during Li-ions insertion and extraction. b The SEM image of as-synthesized LiFePO4 NPs. The image was collected at least three times. Inset is a photo of LiFePO4 NPs aqueous solution. c The POD-like activity of LiFePO4 NPs (6.25 μg Fe/mL) with TMB (1.7 mM) as colorimetric substrates under the presence of H2O2 (0.8 M) in 0.2 M acetate buffer (pH = 3.6). d ESR spectra of spin adducts DMPO/·OH produced by LiFePO4 NPs (10 μg/mL) in the presence or absence of H2O2 (0.165 M) in 0.2 M acetate buffer (pH = 3.6). e Comparison of the anano of as-synthesized LiFePO4 NPs and cc-Fe3O4 NPs. Error bars represent standard deviation from three independent measurements. f Diagram of the POD-like catalytic reaction process of LiFePO4 NPs and Fe3O4 NPs.
Rod-like LiFePO4 NPs with an average length of 321.9 nm and width of 172.2 nm (Fig. 4b) were successfully synthesized using the solvothermal method59 and characterized via various methodologies (Supplementary Fig. 15 and Supplementary Tables 2 and 3). As expected, the POD-like activity of LiFePO4 NPs was demonstrated with different chromogenic substrates including TMB, ABTS, and OPD (Fig. 4c and Supplementary Fig. 16). Also, they follow pH, temperature as well as NPs concentration dependence, and the Michaelis–Menten kinetics (Supplementary Figs. 17 and 18). The optimal pH is about 4.0. The ·OH was shown to be generated in a time- and concentration-dependent manner (Fig. 4d and Supplementary Fig. 19), which is similar to Fe3O4 NPs. We then compared the POD-like activity of LiFePO4 NPs and cc-Fe3O4 NPs using two oppositely charged substrates (TMB and ABTS) at pH 3.6. The results consistently show that LiFePO4 NPs had higher catalytic ability than cc-Fe3O4 NPs (Supplementary Fig. 20), and the anano of LiFePO4 NPs was approximately four times than that of cc-Fe3O4 NPs, despite their larger particle size (Fig. 4e). These results imply that LiFePO4 NPs may share a similar POD-like catalytic mechanism with Fe3O4 NPs, differing in that the rapid Li+ migration in the lattice of LiFePO4 NPs confers them a superior POD-like catalytic activity (Fig. 4f).
Phase transformation of LiFePO4 NPs
The recycled LiFePO4 NPs from three cycles of POD-like catalysis were proven to suffer a phase transformation to FePO4 via multiple characterization techniques. Specifically, the XPS Fe2p peaks of the recycled LiFePO4 NPs were shifted toward the higher binding energy (Fig. 5a), indicating the oxidation of Fe2+ within the NPs. In the XRD pattern (Fig. 5b), the residual LiFePO4 phase (marked with о in the yellow pattern) in the recycled NPs was negligible, proving that almost all LiFePO4 were delithiated and oxidized into FePO4 (marked with +) after cyclic POD-like catalysis. This result was further confirmed by ICP analysis that the Li element content in recycled NPs was almost 0 (Table 1). This phase transformation, as expected, severely impaired the POD-like activity of the recycled LiFePO4 NPs (Fig. 5c), in agreement with the self-depleting characteristic of the Fe3O4 NPs described above.

a The Fe2p XPS spectra, b XRD patterns, c POD-like activities of the control LiFePO4 NPs and the recycled LiFePO4 NPs from three cycles of POD-like reaction. d The CV curves of LiFePO4 NPs and recycled LiFePO4 NPs in lithium acetate buffer solution, compared with that of the Fe3O4 NPs and recycled Fe3O4 NPs. Scanning rate is 2 mV/s. e Comparison the POD-like activity of NaLiFePO4 NPs (0.4 μg Fe/mL) with different Na-doping levels. f Comparison the POD-like activity of commercial LiFePO4, Fe3(PO4)2, and FePO4 (1.25 μg Fe/mL) using TMB (1.7 mM) as substrates in the presence of H2O2 (0.8 M) at pH 3.6. Error bars represent standard deviation from three independent measurements.
As a LIBs cathode material, the reversible lithiation and delithiation characteristic of LiFePO4 contribute to its superior electrochemical performance. We measured the cyclic voltammograms (CV) of the as-prepared LiFePO4 NPs before and after participating in the cyclic POD-like reactions under different scan rates in the electrolyte containing Li+ or Na+. As shown in Supplementary Fig. 21, the increase in redox peak currents (Ip) was proportional to the square root of scan rate (v1/2), implying a diffusion-controlled process of Li+ or Na+ extraction and insertion60. Noticeably, the Ip of the recycled LiFePO4 NPs (i.e., FePO4) was obviously reduced compared to LiFePO4 NPs (Fig. 5d), especially in the electrolyte containing Na+ (Supplementary Fig. 21e–h), indicating that the presence of mobile Li+ contributes significantly to the redox peak current. As a comparison with LiFePO4 NPs, the CV curves of Fe3O4 NPs were also measured under the same conditions. As shown in Supplementary Fig. 22, the Ip also exhibits a linear relation with the v1/2. However, unlike LiFePO4 NPs, the Ip of the recycled Fe3O4 NPs (i.e., γ-Fe2O3) only showed a marginal decrease compared to their counterparts before participating in the catalytic reaction, both of which were found to be similar to the Ip of recycled LiFePO4 NPs under the same scanning rate (Supplementary Fig. 22 and Fig. 5d). This is probably explained by the lack of freely diffusing ions in the lattice of iron oxide and FePO4, which weakens the electron transfer process in their redox reactions. By contrast, LiFePO4 NPs exhibited the highest Ip, ascribed to the availability of Li ions in their crystals.
Mobile Li-ions as the limiting factor
In the field of sodium (Na)-ion batteries, the charge transfer resistances and lattice volume change upon Na+ migration are larger for NaFePO4 electrodes, compared with their Li equivalents due to the larger ionic radius of Na (1.02 Å) than Li (0.76 Å)61. Inspired by this, We partially replaced Li with Na in the lattice of LiFePO4 NPs to explore the potential effect of Na-doping on their POD-like activity. Concretely, three NaLiFePO4 NPs with similar physicochemical properties but different Na-doping amounts were successfully synthesized (Supplementary Fig. 23 and Supplementary Table 4). We then compared their POD-like activities under the same reaction conditions and found that the more Na doping, the lower the POD-like activity (Fig. 5e), indicating that the large Na+ radius hinders the free migration of Na+ and Li+ in the crystal, thereby impairing the electron transfer rate. We attempted to use K-doped LiFePO4 NPs as further proof, however, the large ionic radius of K (1.38 Å) makes it difficult to embed into the electrode materials (Supplementary Fig. 24 and Supplementary Table 4), which is a common issue in K-ion batteries62.
To further prove the decisive role of mobile Li+, we measured the POD-like activity of commercially available LiFePO4, Fe3(PO4)2, and FePO4 materials with similar hydrodynamic dimensions and surface negative potentials (Supplementary Fig. 25). The results show that their POD-like activity follows LiFePO4 >> Fe3(PO4)2 > FePO4 (Fig. 5f), directly confirming that the presence of Fe2+ alone in Fe3(PO4)2 cannot ensure the superior catalytic performance, but the transportable Li+ contributes to the outstanding POD-like activity of LiFePO4.
In summary, the catalytic mechanism of the POD-like activity of Fe3O4 nanozymes is elucidated by characterizing the chemical composition and catalytic activity of the Fe3O4 NPs recycled from the long-term POD-like catalysis. These studies indicate that not only the surface Fe2+, but also the internal Fe2+ contribute to the POD-like activity of Fe3O4 nanozymes. The Fe2+ inside the particle can transfer electrons to the surface, regenerating the surface Fe2+ that is constantly involved in the sustained catalytic reaction. This process is usually accompanied by the outward migration of excess oxidized Fe3+ from the interior of the crystal, which is considered as a rate-limiting step. Analogous to the low-temperature oxidation of magnetite, Fe3O4 NPs participated in the POD-like reaction are eventually oxidized to γ-Fe2O3 NPs with a reduced POD-like capacity. Furthermore, this mechanism is well-validated on LiFePO4 NPs. This work reveals the depletable property of Fe3O4 nanozymes differing from natural enzymes and highlights the potential contribution of internal metal atoms in nanozymes-catalyzed reactions. Meanwhile, these findings provide a theoretical basis for the mechanistic study and rational design of other related nanozymes.
Methods
Chemicals
Ferrous sulfate heptahydrate (FeSO4·7H2O), iron (III) chloride hexahydrate (FeCl3·6H2O), dimethyl sulfoxide, TMB (≥99%), ABTS (98%), OPD (98%), ferric oxide (Fe2O3), iron acetylacetonate [Fe(acac)3], oleic acid (OA), dibenzyl ether, lithium hydroxide (LiOH·H2O), ferric phosphate (FePO4), rhodamine B, terephthalic acid (TA), sodium carbonate (Na2CO3), and sodium acetate (CH3COONa) were purchased from Aladdin (Shanghai, China). Tetramethylammonium hydroxide (TMAOH) and 5,5-dimethyl-1-pyrroline N-oxide (DMPO) were purchased from Sigma Aldrich. Hydrochloric acid (HCl, 36.0–38.0%), nitric acid (HNO3), phosphoric acid (H3PO4, 85% wt), acetic acid (CH3COOH), hydrogen peroxide (H2O2, 30%), ethyl alcohol, ethylene glycol (EG), and potassium hydroxide (KOH) were purchased from Sinopharm Chemical Reagent Co., Ltd. Ferrous phosphate [Fe3(PO4)2] was purchased from Shanghai Maclin Biochemical Technology Co., Ltd. Lithium iron phosphate (LiFePO4) was purchased from Shanghai Xushuo Biological Technology Co., Ltd. All chemicals were used as received without further purification. Deionized water was used throughout the experiments.
Characterization
The particle size and morphology were observed by scanning electron microscopy (SEM, Ultra Plus, Carl Zeiss, Germany). TEM, selective area electron diffraction (SAED), and high-resolution TEM (HRTEM) were taken using transmission electron microscopy (TEM, JEOL JEM-2100F, Japan). The phase and crystalline structure were explored by XRD (Brucker D8 Advance, Germany) and the DIFFRAC plus XRD Commander software (v2.6.1), using Cu Kα radiation. The element composition, chemical structures, and properties of NPs were analyzed by SEM energy dispersive spectroscopy (EDS), ICP-OES coupled with ICP Expert II software (Agilent ICPOES730), Fourier transform infrared spectroscopy coupled with OMNIC software v9.6 (FTIR, Nicolet IS 10, Thermo, USA), Raman spectrometer coupled with WiRE™ software v2.0 (Renishaw Invia, UK) and X-ray photoelectron spectroscopy coupled with Avantage software v5.967 (XPS, Thermo ESCALAB 250Xi, USA). The chemical speciation of Fe in IONPs was determined by NEXAFS at the beamline of 08U1A in the Shanghai Synchrotron Radiation Facility, Shanghai, China. The NEXAFS spectra of the references including FeSO4 and Fe2O3 were also measured. Hydrodynamic diameters and Zeta potential of particles were measured by the dynamic light scattering (DLS, Nano-ZS90, Malvern, England) and Malvern Zetasizer software (v7.12). The UV-Vis-NIR absorption spectra were measured using UV3600 (Shimadzu, Japan) or Microplate Reader (Infinite M200, Tecan, Switzerland). CV was measured using Electrochemical Workstation (CHI760e) and CHI-760E electrochemical software (v20.04). The production of ·OH was detected by the electron spin resonance spectrometer (ESR, Bruker EMXplus, Germany) and Bruker Xenon software (v1.2) at ambient temperature.
Synthesis of IONPs by chemical coprecipitation method
The naked magnetite NPs were synthesized by a modified chemical coprecipitation method, namely, cc-Fe3O4 NPs24. In brief, 2.7025 g of FeCl3·6H2O and 1.39 g FeSO4·7H2O were added to a 100 mL three-necked flask with 10 mL of 2 M HCl reaction solvent. The mixture was stirred at 400 rpm for 10 min under a nitrogen gas (N2) atmosphere. Then, 62.5 mL of 12.5% TMAOH was quickly poured into the above flask and stirred at 700 rpm for 1 h at room temperature. N2 was introduced throughout the experiment to remove O2 from the reaction system. The reaction solution was transferred to a beaker and washed with deoxygenated water three times by magnetic decantation to remove the residual reagents. Part of obtained precipitate was dispersed into deionized water with a pH of 3 to obtain the cc-Fe3O4 NPs suspensions, which were stored at 4 °C for subsequent experiments.
The remaining cc-Fe3O4 NPs precipitates were dried into black Fe3O4 NPs powder under vacuum, and then calcined at 200 and 650 °C for 2 h to obtain γ-Fe2O3 NPs and α-Fe2O3 NPs powder, respectively34. Finally, these three powders were dissolved in a pH of 3 aqueous solutions by ultrasound and stored at 4 °C for subsequent experiments.
Synthesis of Fe3O4 NPs by thermal decomposition method
Firstly, the OA-coated Fe3O4 NPs were synthesized according to the thermal decomposition method (denoted as TD-Fe3O4@OA NPs)50. The chloroform solvent was removed from 4 mL of TD-Fe3O4@OA NPs (1.627 mg Fe/mL) by rotary evaporation. Then, the OA layer on the particle surface was replaced with 15 mL of 0.5% TMAOH under ultrasonic stirring63. TMAOH is a phase transfer small molecule agent which can form an electrostatic double layer on the particle surface and stabilize the particles in the aqueous phase. Next, the reaction system was transferred to 60 mL of the separatory funnel, and 45 mL of chloroform was added. The mixture was thoroughly mixed. After standing for 15 min, the underlying oil phase liquid was discarded. This extraction procedure was repeated three times to fully remove the free OA that was replaced from the particle surface by TMAOH. Finally, the obtained Fe3O4 NPs aqueous solution (denoted as TD-Fe3O4 NPs) was filtered through a 0.22-μm filter and stored at 4 °C for subsequent experiments.
Synthesis of LiFePO4 NPs and Na- or K-doped LiFePO4 NPs
LiFePO4 NPs were successfully synthesized by glycol-based solvothermal method59. In a typical route, 0.27 mL H3PO4 (85% wt) was dropped into 9 mL of EG containing 0.365 g of LiOH·H2O. The mixture was stirred thoroughly until the neutralization reaction was complete, showing a milk-white suspension. Then 9 mL of EG dissolved 0.890 g FeSO4·7H2O was added into the suspension under vigorous stirring. The mixtures slowly turned grayish-green. After stirring for 30 min, the reaction solution was transferred into a sealed hydrothermal reactor, and heated at 180 °C for 10 h. At the end of the reaction, the final products were cooled down to room temperature and washed with deionized water three times.
For Na or K-doped LiFePO4 NPs, the Li element content (8.69 mmol) of 1%, 5%, and 10% in the above method was replaced with Na element in Na2CO3 or K element in KOH. The rest of the synthesis steps remained unchanged. Briefly, for Na-doped LiFePO4 NPs, 0.365 g of LiOH·H2O was replaced with 4.6 mg of Na2CO3 and 0.361 g of LiOH·H2O (denoted as NaLiFePO4−1), 23.0 mg of Na2CO3 and 0.346 g of LiOH·H2O (denoted as NaLiFePO4−5), and 46.1 mg of Na2CO3 and 0.328 g of LiOH·H2O (denoted as NaLiFePO4−10), respectively. For K-doped LiFePO4 NPs, 0.365 g of LiOH·H2O was replaced with 4.9 mg of KOH and 0.361 g of LiOH·H2O (denoted as KLiFePO4−1), 24.3 mg of KOH and 0.346 g of LiOH·H2O (denoted as KLiFePO4−5), and 48.7 mg of KOH and 0.328 g of LiOH·H2O (denoted as KLiFePO4−10), respectively.
The POD-like activity of NPs
The POD-like activity of NPs was measured by different colorimetric substrates, including TMB, ABTS, and OPD, in the presence of H2O2 under different reaction conditions. The absorbance of the colored oxidation products at the corresponding wavelength (TMBox: 650 nm; ABTSox: 415 nm; OPDox: 492 nm) was monitored by a microplate reader and Tecan i-control software (v1.6.19.2). The effects of pH (2.5–11), temperature (10–65 °C), and LiFePO4 NPs concentration (0–6.25 μg Fe/mL) on POD-like activity were also studied.
Measurement of the specific activity (a nano) of NPs
The POD-like specific activity of NPs synthesized in this work was determined according to the modified method specified in the national standard of China (GB/T 37966-2019)35. The following provided the general procedures: (a) monitor the temperature inside the quartz cuvette and incubate all reagents and samples to 25 °C; (b) add 2.000 mL of 0.2 M acetate buffer solution (pH = 3.6) to a reaction container; (c) add 0.100 mL of different concentrations of NPs and 0.100 mL of 10 mg/mL of TMB in order, mix completely and incubate for 60 s at 25 °C; (d) add 0.200 mL of 30% H2O2 and mix completely; (e) immediately transfer the appropriate amount of reaction solution to the cuvette and monitor the changes of absorbance at 650 nm within the specified times using UV-Vis spectroscopy and UV Probe software (v2.42). The initial change rate of absorbance (min−1) is obtained from the slope of the early phase of time course; (f) measurement of reagent blank rate 1 and reagent blank rate 2: 30% H2O2 and NPs are respectively replaced by deionized water. The measurement procedure is as described in Step a–e above; (g) after deducting the reagent blank rate 1 and 2, the POD-like catalytic activity units (bnano) of NPs can be calculated according to Eq. (7):
where bnano is the POD-like catalytic activity units of NPs (U); V is the total volume of reaction solution (μL); ∆A/∆t is the slope of the initial liner portion of absorbance changing over time after correcting with reagent blank rate 1 and 2 (min−1); ε is the molar absorption coefficient of TMB derivative (39,000 mol−1·L·cm−1); l is the optical path of the cuvette (cm).
The specific activity of NPs (anano) can be calculated according to Eq. (8):
where anano is the specific activity of NPs (U·mg−1); mFe is the total Fe element mass contained in added NPs (mg); \(\frac{\triangle {b}_{{{{{{\rm{nano}}}}}}}}{\triangle {{{{{{\rm{m}}}}}}}_{{{{{{\rm{Fe}}}}}}}}\) is the slope of the curve plotting the bnano against different masses of NPs (mFe).
ESR measurement
The production of ·OH was detected by adding 10 μg Fe/mL of cc-Fe3O4 NPs or LiFePO4 NPs and 30 mM of DMPO into 300 μL of acetate buffer (pH = 3.6) with or without 0.5% H2O2. The signal of the spin adduct (DMPO/·OH) was recorded at ambient temperature at the 1st, 5th, and 10th min after all reagents were mixed. The experimental parameters were as follows: 1 G modulation amplitude, 100 kHz modulation frequency, 6.325 mW microwave power, 9.829 GHz resonance frequency.
·OH detection using terephthalic acid as a fluorescent probe
TA is highly selective and can capture ·OH generated in situ, generating 2-hydroxyterephthalic acid (TAOH) with unique fluorescence around 534 nm, which can be detected by fluorescence spectrophotometer and FluorEssence software (v3.8). In this work, 0.2 M H2O2 was added to 0.2 M acetate buffer (pH = 3.6) containing nanozymes (different materials and concentrations) and 0.5 mM TA. After mixing, the fluorescence signal was detected on a fluorescence spectrophotometer at specific times in the range of 350–600 nm with an excitation wavelength of 315 nm.
Cyclic POD-like catalysis of NPs
For IONPs, 0.1 g of Fe3O4 NPs or γ-Fe2O3 NPs powder was added to a 400 mL of acetate buffer (pH 3.6) containing 20 mL TMB (10 mg/mL) and 40 mL of 30% H2O2. This catalytic reaction system lasted for one day (20 h) at 25 °C. After catalysis, Fe3O4 NPs or γ-Fe2O3 NPs in the reaction solution were recycled by magnetic separation, and then ultrasonic washed with deionized water several times. The recycled NPs were re-added into a new reaction system mentioned above to catalyze a new round of POD-like reactions. Totally, 5 rounds (or days) of catalysis were carried out, and each round lasted for 20 h at 25 °C. The Fe3O4 NPs recycled on days 0, 1, 3, and 5 were taken for further characterization.
For LiFePO4 NPs, 1 mL of LiFePO4 NPs (8.4 mg Fe/mL) was added to a 20 mL of acetate buffer (pH 3.6) containing 1 mL TMB (10 mg/mL) and 2 mL of 30% H2O2. This catalytic reaction system lasted for 1 h at 25 °C. After catalysis, LiFePO4 NPs in the reaction solution were recycled by centrifugation, and then ultrasonic washed with deionized water several times. The recycled LiFePO4 NPs were re-added into a new reaction system mentioned above to catalyze a new round of POD-like reactions. Totally, 3 rounds of catalysis were carried out, and each round lasted for 1 h at 25 °C. The LiFePO4 NPs recycled from the 0 and 3 rounds were taken for further characterization.
Aerated oxidation of Fe3O4 NPs
A total of 170 mL of the as-synthesized cc-Fe3O4 NPs suspensions (3.57 mg Fe/mL) was transferred into a 250 mL three-necked flask and heated from room temperature to 120 °C. Air was pumped into the suspension at a constant rate (9 L/min) for 12 h under stirring (350 rpm). Condensation reflux was kept throughout the aeration oxidation. Samples oxidized at 0, 0.5, 1, 3, 5, 8 10, and 12 h were used for subsequent characterization.
As for TD-Fe3O4 NPs, the only difference was that 30 mL of TD-Fe3O4 NPs (0.45 mg Fe/mL) were transferred into a 100 mL three-necked flask. Other steps were consistent with the aeration oxidation of cc-Fe3O4 NPs.
Electrochemical performance of NPs
The CV was used to evaluate the electrochemical performance of NPs in a three-electrode cell35. A total of 10 μL of NPs (100 μg Fe/mL) was mixed with 90 μL of 0.5% Nafion solution, and then dropped 30 μL of the above mixture into the polished glassy carbon electrode, which was dried and used as the working electrode. Mercury/mercurous sulfate (Hg/Hg2SO4) was used as the reference electrode and platinum wire as the counter electrode. The electrolyte solution was 0.2 M lithium acetate or sodium acetate buffer (pH = 3.6). The CV was recorded at different voltage scan rates (ν).
Statistical and reproducibility
All experiments were repeated three times independently, and the data were presented as the mean values ± standard deviations (SD). Error bars shown in this paper represent the SD derived from three repeated measurements. UV probe Software v2.42, Tecan i-control Software v1.6.19.2, Malvern Zetasizer Software v7.12, FluorEssence Software v3.8, Bruker Xenon Software v1.2, CHI-760E electrochemical Software v20.04, WiRE™ 2.0 Software, Thermo Avantage Software v5.967, ICP Expert II Software, OMNIC Software v9.6, and DIFFRAC plus XRD Commander Software v2.6.1 were used for data collection. GraphPad Prism 8 and Origin 2018 were used for data processing and statistical analysis.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available within the article and its Supplementary Information files. Data are available from the corresponding author upon request. Source Data are provided with this paper.
References
Wei, H. et al. Nanozymes: a clear definition with fuzzy edges. Nano Today 40, 101269 (2021).
Liu, Q., Zhang, A., Wang, R., Zhang, Q. & Cui, D. A review on metal- and metal oxide-based nanozymes: properties, mechanisms, and applications. Nanomicro Lett. 13, 1–53 (2021).
Zhang, R., Yan, X. & Fan, K. Nanozymes inspired by natural enzymes. Acc. Mater. Res. 2, 534–547 (2021).
Tang, G., He, J., Liu, J., Yan, X. & Fan, K. Nanozyme for tumor therapy: surface modification matters. Exploration 1, 75–89 (2021).
Wu, Y. et al. Defect engineering in nanozymes. Mater. Today 52, 327–347 (2021).
Kang, T., Kim, Y. G., Kim, D. & Hyeon, T. Inorganic nanoparticles with enzyme-mimetic activities for biomedical applications. Coord. Chem. Rev. 403, 213092 (2020).
Wang, Z., Zhang, R., Yan, X. & Fan, K. Structure and activity of nanozymes: inspirations for de novo design of nanozymes. Mater. Today 41, 81–119 (2020).
Xiang, H., Feng, W. & Chen, Y. Single-atom catalysts in catalytic biomedicine. Adv. Mater. 32, 1905994 (2020).
Liang, M. & Yan, X. Nanozymes: from new concepts, mechanisms, and standards to applications. Acc. Chem. Res. 52, 2190–2200 (2019).
Gao, L. et al. Intrinsic peroxidase-like activity of ferromagnetic nanoparticles. Nat. Nanotechnol. 2, 577–583 (2007).
Cheng, L. et al. Unveiling the actual catalytic sites in nanozyme-catalyzed oxidation of o-Phenylenediamine. Small 17, 2104083 (2021).
Chen, J. et al. Glucose-oxidase like catalytic mechanism of noble metal nanozymes. Nat. Commun. 12, 3375 (2021).
Zhang, R. et al. Unveiling the active sites on ferrihydrite with apparent catalase-like activity for potentiating radiotherapy. Nano Today 41, 101317 (2021).
Shen, X., Wang, Z., Gao, X. & Zhao, Y. Density functional theory-based method to predict the activities of nanomaterials as peroxidase mimics. ACS Catal. 10, 12657–12665 (2020).
Guo, S. & Guo, L. Unraveling the multi-enzyme-like activities of iron oxide nanozyme via a first-principles microkinetic study. J. Phys. Chem. C. 123, 30318–30334 (2019).
Xu, B. et al. A bioinspired five-coordinated single-atom iron nanozyme for tumor catalytic therapy. Adv. Mater. 34, 2107088 (2022).
Jiang, B. & Liang, M. Advances in single-atom nanozymes research. Chin. J. Chem. 39, 174–180 (2021).
Chen, Q. et al. Size-controllable Fe-N/C single-atom nanozyme with exceptional oxidase-like activity for sensitive detection of alkaline phosphatase. Sens. Actuators, B. 305, 127511 (2020).
Xiang, H., Feng, W. & Chen, Y. Single-atom catalysts in catalytic biomedicine. Adv. Mater. 32, 1905994 (2020).
Zhou, Q. et al. Cascaded nanozyme system with high reaction selectivity by substrate screening and channeling in a microfluidic device. Angew. Chem. 134, e202112453 (2022).
Zhang, Z., Zhang, X., Liu, B. & Liu, J. Molecular imprinting on inorganic nanozymes for hundred-fold enzyme specificity. J. Am. Chem. Soc. 139, 5412–5419 (2017).
Raineri, M. et al. Effects of biological buffer solutions on the peroxidase-like catalytic activity of Fe3O4 nanoparticles. Nanoscale 11, 18393–18406 (2019).
Moreno Maldonado, A. C. et al. Free-radical formation by the peroxidase-like catalytic activity of MFe2O4 (M = Fe, Ni, and Mn) nanoparticles. J. Phys. Chem. C. 123, 20617–20627 (2019).
Chen, Z. et al. Dual enzyme-like activities of iron oxide nanoparticles and their implication for diminishing cytotoxicity. ACS Nano 6, 4001–4012 (2012).
Wang, N. et al. Sono-assisted preparation of highly-efficient peroxidase-like Fe3O4 magnetic nanoparticles for catalytic removal of organic pollutants with H2O2. Ultrason. Sonochem. 17, 526–533 (2010).
Nguyen, M., Robert, A. & Meunier, B. “Nanozymes” without catalytic activity should be renamed. hal-03290323 (2021).
Wei, H. & Wang, E. Fe3O4 magnetic nanoparticles as peroxidase mimetics and their applications in H2O2 and glucose detection. Anal. Chem. 80, 2250–2254 (2008).
Gumpelmayer, M. et al. Magnetite Fe3O4 has no intrinsic peroxidase activity, and is probably not involved in alzheimer’s oxidative stress. Angew. Chem. Int. Ed. 57, 14758–14763 (2018).
Dong, H., Fan, Y., Zhang, W., Gu, N. & Zhang, Y. Catalytic mechanisms of nanozymes and their applications in biomedicine. Bioconjugate Chem. 30, 1273–1296 (2019).
Wu, H. et al. Enhanced tumor synergistic therapy by injectable magnetic hydrogel mediated generation of hyperthermia and highly toxic reactive oxygen species. ACS Nano 13, 14013–14023 (2019).
Fan, K. et al. Optimization of Fe3O4 nanozyme activity via single amino acid modification mimicking an enzyme active site. Chem. Commun. 53, 424–427 (2017).
Voinov, M. A., Pagán, J. O. S., Morrison, E., Smirnova, T. I. & Smirnov, A. I. Surface-mediated production of hydroxyl radicals as a mechanism of iron oxide nanoparticle biotoxicity. J. Am. Chem. Soc. 133, 35–41 (2011).
Zandieh, M. & Liu, J. Nanozyme catalytic turnover and self-limited reactions. ACS Nano 15, 15645–15655 (2021).
Colombo, U., Gazzarrini, F., Lanzavecchia, G. & Sironi, G. Magnetite oxidation: a proposed mechanism. Science 147, 1033 (1965).
GB/T 37966-2019, Nanotechnologies—Measurement Method for Peroxidase-like Activity of Iron Oxide Nanoparticles, National Technical Committee on Nanotecnology of Standardization Administration (Beijing, China, 2019).
Jiang, B. et al. Standardized assays for determining the catalytic activity and kinetics of peroxidase-like nanozymes. Nat. Protoc. 13, 1506–1520 (2018).
Masud, M. K. et al. Nanoarchitectured peroxidase-mimetic nanozymes: mesoporous nanocrystalline α- or γ-iron oxide? J. Mater. Chem. B. 7, 5412–5422 (2019).
Song, H. & Zink, J. I. EELS study of differential diffusion of Fe and Co in magnetized silica nanocomposites. J. Phys. Chem. C. 120, 25578–25587 (2016).
Pattammattel, A. et al. Iron speciation in particulate matter (PM2.5) from urban Los Angeles using spectro-microscopy methods. Atmos. Environ. 245, 117988 (2021).
Colliex, C., Manoubi, T. & Ortiz, C. Electron-energy-loss-spectroscopy near-edge fine structures in the iron-oxygen system. Phys. Rev. B. 44, 11402–11411 (1991).
Sun, S. et al. Monodisperse MFe2O4 (M = Fe, Co, Mn) nanoparticles. J. Am. Chem. Soc. 126, 273–279 (2004).
Park, J. et al. One-nanometer-scale size-controlled synthesis of monodisperse magnetic iron oxide nanoparticles. Angew. Chem. 117, 2932–2937 (2005).
Park, J. et al. Ultra-large-scale syntheses of monodisperse nanocrystals. Nat. Mater. 3, 891–895 (2004).
Zhang, L. et al. Facile synthesis of iron oxides/reduced graphene oxide composites: application for electromagnetic wave absorption at high temperature. Sci. Rep. 5, 9298 (2015).
Gao, J. et al. One-step solvothermal synthesis of highly water-soluble, negatively charged superparamagnetic Fe3O4 colloidal nanocrystal clusters. Nanoscale 5, 7026 (2013).
Sun, Y., Ma, M., Zhang, Y. & Gu, N. Synthesis of nanometer-size maghemite particles from magnetite. Colloids Surf., A. 245, 15–19 (2004).
Sidhu, P. S., Gilkes, R. J. & Posner, A. M. Mechanism of the low temperature oxidation of synthetic magnetites. J. Inorg. Nucl. Chem. 39, 1953–1958 (1977).
Colombo, U., Fagherazzi, G., Gazzarrini, F., Lanzavecchia, G. & Sironi, G. Mechanism in the first stage of oxidation of magnetites. Nature 202, 175–176 (1964).
Feitknecht, W. & Gallagher, K. J. Mechanisms for the oxidation of Fe3O4. Nature 228, 548–549 (1970).
Kong, F. et al. Fe3O4@Pt nanozymes combining with CXCR4 antagonists to synergistically treat acute myeloid leukemia. Nano Today 37, 101106 (2021).
Tang, J., Myers, M., Bosnick, K. A. & Brus, L. E. Magnetite Fe3O4 nanocrystals: spectroscopic observation of aqueous oxidation kinetics. J. Phys. Chem. B. 107, 7501–7506 (2003).
Redl, F. X. et al. Magnetic, electronic, and structural characterization of nonstoichiometric iron oxides at the nanoscale. J. Am. Chem. Soc. 126, 14583–14599 (2004).
Voogt, F. C., Hibma, T., Zhang, G. L., Hoefman, M. & Niesen, L. Growth and characterization of nonstoichiometric magnetite Fe3-δO4 thin films. Surf. Sci. s. 331–333, 1508–1514 (1995).
Coey, J. M. D., Morrish, A. H. & Sawatzky, G. A. A Mössbauer study of conduction in magnetite. J. Phys. Colloques 32, C1–C271 (1971).
Yuan, L. et al. Development and challenges of LiFePO4 cathode material for lithium-ion batteries. Energy Environ. Sci. 4, 269–284 (2011).
Wang, J. & Sun, X. Understanding and recent development of carbon coating on LiFePO4 cathode materials for lithium-ion batteries. Energ. Environ. Sci. 5, 5163–5185 (2012).
Wang, J. & Sun, X. Olivine LiFePO4: the remaining challenges for future energy storage. Energ. Environ. Sci. 8, 111–1138 (2015).
Bai, P., Cogswell, D. A. & Bazant, M. Z. Suppression of phase separation in LiFePO4 nanoparticles during battery discharge. Nano Lett. 11, 4890–4896 (2011).
Wang, L. et al. Crystal orientation tuning of LiFePO4 nanoplates for high rate lithium battery cathode materials. Nano Lett. 12, 5632–5636 (2012).
Tang, K., Yu, X., Sun, J., Li, H. & Huang, X. Kinetic analysis on LiFePO4 thin films by CV, GITT, and EIS. Electrochim. Acta 56, 4869–4875 (2011).
Zhu, Y., Xu, Y., Liu, Y., Luo, C. & Wang, C. Comparison of electrochemical performances of olivine NaFePO4 in sodium-ion batteries and olivine LiFePO4 in lithium-ion batteries. Nanoscale 5, 780–787 (2013).
Wang, C. et al. Structural engineering of sulfur-doped carbon encapsulated bismuth sulfide core-shell structure for enhanced potassium storage performance. Nano Res. 14, 3545–3551 (2021).
Mehrmohammadi, M. et al. In vivo pulsed magneto-motive ultrasound imaging using high-performance magnetoactive contrast nanoagents. Nanoscale 5, 11179 (2013).
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2017YFA0205502), National Natural Science Foundation of China (82072067 and 61821002), and the Fundamental Research Funds for the Central Universities.
Author information
Authors and Affiliations
Contributions
Y.Z. conceived and supervised the project; H.D. and Y.Z. designed the studies; H.D. performed the main experiments and wrote the manuscript; W.D. helped with the data analysis. J.D. performed the Raman experiments; R.C. performed the electron microscopy experiments; F.K. provided the TD-Fe3O4 nanoparticles; W.C., M.M., and N.G. helped with article revisions. All the authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dong, H., Du, W., Dong, J. et al. Depletable peroxidase-like activity of Fe3O4 nanozymes accompanied with separate migration of electrons and iron ions. Nat Commun 13, 5365 (2022). https://doi.org/10.1038/s41467-022-33098-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-33098-y
This article is cited by
-
Evaluation of the performance of Fe3O4/MnO2@doxorubicin hybrid nanozymes on multicellular structure and their therapeutic management to limit the growth of human breast cancer cells
Cancer Nanotechnology (2024)
-
Designing nanozymes for in vivo applications
Nature Reviews Bioengineering (2024)
-
Biomimetic synergistic effect of redox site and Lewis acid for construction of efficient artificial enzyme
Nature Communications (2024)
-
Metal-ligand dual-site single-atom nanozyme mimicking urate oxidase with high substrates specificity
Nature Communications (2024)
-
Elucidating the catalytic mechanism of Prussian blue nanozymes with self-increasing catalytic activity
Nature Communications (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
