
Abstract
Ruthenium (Ru) is the one of the most promising catalysts for polyolefin hydrogenolysis. Its performance varies widely with the support, but the reasons remain unknown. Here, we introduce a simple synthetic strategy (using ammonia as a modulator) to tune metal-support interactions and apply it to Ru deposited on titania (TiO2). We demonstrate that combining deuterium nuclear magnetic resonance spectroscopy with temperature variation and density functional theory can reveal the complex nature, binding strength, and H amount. H2 activation occurs heterolytically, leading to a hydride on Ru, an H+ on the nearest oxygen, and a partially positively charged Ru. This leads to partial reduction of TiO2 and high coverages of H for spillover, showcasing a threefold increase in hydrogenolysis rates. This result points to the key role of the surface hydrogen coverage in improving hydrogenolysis catalyst performance.
Similar content being viewed by others



Introduction
Plastic waste represents a significant threat to the environment due to its leakage into the oceans and soil. Plastic consumption increased drastically during the recent pandemic1, triggering a soaring expansion of incineration, causing extensive CO2 emissions2, and landfilling of consumer packaging materials, foams, films, and personal protection equipment3. Polypropylene (PP) is a large-volume polymer frequently used in packaging, fabrics, and textiles, including face masks. Mechanical recycling fails to deal with mixed PP waste streams since composites usually contain pigments, dyes, antioxidants, and plasticizers, leading to an inferior product. PureCycle4 has demonstrated that solvent extraction can provide consumer-grade PP, but solvent use increases cost and complexity. A possible approach to extend the life cycle is the catalytic conversion of PP waste under solvent-free conditions.
Hydrogenolysis is a low-energy valorization route of PP and polyethylene (PE)5, producing liquid products, including lubricant base oil6. Ru and Pt nanoparticles on carbon or oxide supports have been the hydrogenolysis catalysts of choice7,8,9,10,11. Ru, in particular, is more active than Pt but forms copious amounts of methane, and reaction times are long. Its performance varies widely with the support, but the reasons remain unclear7,10,11,12,13,14. For example, over Ru/ZrO2, the strong PE binding leads to the over-cracking of the reaction intermediates to methane15. On Ru/WOx/ZrO2, on the other hand, hydrogen spillover from Ru stores hydrogen in reducible surface polytungstate domains, thus effectively removing reaction intermediates and suppressing methane formation. Recent work on Ru deposited on redox-active CeO2 support showed 83% liquid yield. Controlling hydrogen availability on the catalyst and polyolefin binding may effectively control the chemistry, but ways to achieve this are lacking.
Here, we introduce a systematic approach to tune the reducibility of the TiO2 support and the metal-support interactions (MSI). Formation of TiOx overlayer reported initially in ref. 16 is known to poison the metal surface and inhibit hydrogen chemisorption. For the Ru/TiO2 catalyst, the MSI enhanced by the lattice matching between RuOx and rutile TiO2 leads to higher catalytic activity in CO2 hydrogenation17. We perform extensive characterization and correlate the electronic properties and hydrogen storage to catalytic performance in PP hydrogenolysis. We show that extensive reduction of TiO2 via hydrogen spillover from Ru modifies the hydrogen storage capacity of the Ru nanoparticles. This boosts the hydrogenolysis activity and reduces the liquid molecular weight and the reaction time.
Results
Ru/TiO2 catalyst characterization
We changed the pH in the Ru deposition step, using aqueous NH3, to modify the catalyst (catalysts are labeled as Ru/TiO2-x, where x indicates the synthesis pH) while ensuring similar Ru loadings (~3 wt%), surface areas (100 m2/g) (Table 1), and pore volume. According to XPS quantitative analysis, all samples have similar surface Ru/Ti atomic ratios consistent with the same Ru loadings. XRD patterns (Supplementary Fig. 1) show relatively broad reflections of the TiO2 anatase support with no sign of crystalline Ru. Ex situ UV-Vis spectra reveal comparable bandgap energies, typical of pure anatase TiO2 (Table 1). All samples contain predominantly 1.4–1.6 nm Ru nanoparticles evenly distributed on TiO2 (STEM images in Fig. 1a, b and particle size distribution in Supplementary Fig. 2). TiO2 consists of elongated cylindrical, highly crystalline particles of 35–40 nm length and ~10 nm diameter. In situ Ru K-edge XANES spectra (Supplementary Fig. 3) after pre-reduction with H2 at 250 °C indicate metallic Ru0 and EXAFS spectral analysis (Table 1, Fig. 1c, Supplementary Fig. 3, Supplementary Table 1) show a coordination number of ~8.5, consistent with the STEM images.

a, b STEM images of Ru/TiO2-1 and Ru/TiO2-8, respectively; c EXAFS spectra of two samples and Ru foil standard; d FTIR of adsorbed CO at −296 °C at different CO pressures; e Zoom in of the 2240–2150 cm−1 region.
TGA results (Supplementary Fig. 4) show that ~10 mmol//\({{{{{{\rm{g}}}}}}}_{{{{{{{\rm{TiO}}}}}}}_{2}}\) of NH3 is retained after impregnation, an order of magnitude higher than Ti4+ on the surface18. NH3 desorbs in H2/He at ~250 °C, indicating strong interaction with the catalyst. NH3 can reduce and modify the TiO2 surface after high-temperature treatment19,20. Based on DRIFTS spectra (Supplementary Fig. 5) of Ru/TiO2-8 directly after impregnation and drying and before the reduction in H2, ammonia is adsorbed in several modes. Two possible mechanisms are proposed to explain the NH3 role during sample pre-reduction (Supplementary Figs. 6–8). XPS analysis shows no signs of N-doping of TiO2 after reduction.
To investigate the modification of the Lewis acidity, we used FTIR of adsorbed CO at −196 °С (Fig. 1d, e). At high CO pressure, Lewis acid site bands appear at 2206 and 2179 cm−1 due to highly electrophilic four-coordinated Ti4+(O4) sites on the particle edges and the less acidic five-coordinated Ti4+(O5) sites on the 101 crystal planes, respectively21. The concentration of Ti4+(O4) sites is similar in both samples. The intensity of both bands decreases with evacuation time, but the 2206 cm−1 band is reduced more slowly. Ru/TiO2-8 retains some CO on both sites even after prolonged evacuation, whereas Ru/TiO2-1 only marginally on the stronger Ti4+(O4) sites (Fig. 1e), indicating stronger binding and Lewis acidity on the former (Supplementary Fig. 9). DRIFTS demonstrates a smaller density of surface Ti-OH groups on the Ru/TiO2-8 and Ru/TiO2-12 than on Ru/TiO2-1 (Supplementary Fig. 10). Partial dehydroxylation enhances the Lewis acidity of Ti4+ sites due to the redistribution of the electron density on the surface22.
In addition to Ti4+–CO adducts, FTIR spectra show that Ru particles get partially oxidized by CO forming multiple Run+(CO)m (m = 1–4) carbonyls with ν(CO) vibration at 2136, 2106, 2084, and 2055 cm−1 (Supplementary Fig. 11)23. Due to the low-temperature CO dissociation, clusters of Run+ and Ru0 species form. The stronger interaction of TiO2 and Ru and the TiOx peripheral layer shifts the broad ν(CO) band to lower wavenumbers24 (from 2077 to 2059 cm−1, Fig. 1d), epitomizing a proximal MSI in the Ru/TiO2-8 catalyst.
Hydrogen binding on Ru/TiO2
H2 temperature-programmed desorption (TPD) (Supplementary Fig. 12) indicates a similar amount of strongly chemisorbed hydrogen on the Ru (low-temperature peak at 160 °C)25 and TiO2 (high-temperature shoulder at 250–300 °C)26. Temperature-programmed reduction (TPR) in H2 flow shows a spike in hydrogen adsorption at 80–90 °C, due to binding on Ru, and a broad peak starting at ca. 250 °C, due to partial TiO2 reduction (Supplementary Fig. 13). Interestingly, Ru/TiO2-8 and Ru/TiO2-12 show more pronounced hydrogen uptakes at 150-200 °C than Ru/TiO2-1. Hydrogen pulse chemisorption at 35 °C (Table 1) shows that Ru/TiO2 samples have different hydrogen uptakes. During an experiment, hydrogen can spillover to the TiO2 support obscuring the real Ru dispersion values. Thus the total H2 uptake inferred from pulse chemisorption depends on the spillover capacity of TiO2, rendering accurate quantification of the exposed Ru surface area impossible with the available methods. Also, the pretreatment temperature before pulse chemisorption (300 °C) is insufficient to remove all chemisorbed hydrogen from the sample. Thus, apparent uptakes may be higher for catalysts with weaker chemisorption.
To decouple Ru from support contributions to the hydrogen binding and activation, we used 2H MAS NMR of chemisorbed D2 (Fig. 2). Chemisorption of 2H on Ru clusters is accompanied by quadrupole interactions with the surrounding electric field gradient (EFG), leading to characteristic sidebands in the NMR spectra. Spectra contours depend on the quadrupole coupling constant (Qcc), a function of the largest EFG component (Vzz), and the asymmetry parameter η, defined as (Vxx − Vyy)/Vzz. One can obtain the EFG parameters, sensitive to the deuteron local surroundings and the binding type27,28.

a Solid-state 2H MAS NMR spectra of Ru/TiO2-8 measured at 4 kHz of spinning. b Fitting of experimental spectra. c Zoom in low-frequencies alongside three components used for fitting, resolved via deconvolution. d–f Deconvoluted signals of weakly bonded D on Ru (d), strongly bonded D on Ru in atop configuration, e, and Ti-OD and Ti←O+D2 groups on TiO2 (f). g DFT-deduced structure of atop bonded D on a Ru cluster. h, i Spectra at 10 kHz MAS for Ru/TiO2−1 (h) and Ru/TiO2−8 (i).
Figure 2a shows a complex line shape after sample reduction followed by saturation with 1 torr of D2. Deconvolution yields several components (Fig. 2b, c; high-resolution data at 10 kHz shown in Supplementary Fig. 14). The first component (Qcc~123–130 kHz/η~0.7–1.0) corresponds to Ti-OD formed by deuterium spillover, confirmed by measuring a D2O-treated pure TiO2 sample (Fig. 2f, Supplementary Fig. 15, Supplementary Table 2). Also, a simple H/D isotope exchange leads to the formation of Ti-OD groups29, and their presence in the spectra is not a fingerprint of spillover.
The latter peak has a symmetric EFG (η~0.1) and low Qcc of ca. 70 kHz, distinctly different from the Ti-OD signal (Fig. 2e, Supplementary Table 3), and consistent with Run-D hydrides27. The second component corresponds to deuterons bonded to Ru in atop conformation (see parameters in Supplementary Tables 3 and 4), confirmed using DFT calculations (Supplementary Fig. 16). Figure 2g shows an optimized structure of a Ru cluster on TiO2 with a D atom, in good agreement with a previous report30, underscoring the preferential formation of atop H instead of (Ru)2-H bridging binding. The EFG parameters depend on the Ru particle size and the support. For an unsupported Ru12 cluster, EFG is more symmetric, unlike the experimental data. Smaller or larger clusters than Ru12 on TiO2 provide a less adequate EFG. Thus, 2H NMR, combined with DFT, provides insights into MSI and particle size effects.
The third component gives a very broad single line with no sidebands (Fig. 2d) at a low resonance frequency (−4 to −4.4 kHz, or −53 to −57.3 ppm). These features are not standard for Run-D hydrides, observed previously for free-standing Ru clusters27. The negative chemical shift is similar to chemisorbed hydrogen on Ru particles on SiO2 and TiO2, inferred from static 1H NMR31,32. The deuteron interaction with the Ru conduction electrons leads to the so-called Knight shift responsible for the −53 ppm peak33. On Ru/SiO2, this peak stems from the overlap of different signals due to strongly and weakly chemisorbed hydrogen31. We exclude deuteron binding to an oxygen vacancy in TiO2 because it would lead to resonances close to 0 kHz at −1.07 ppm34 or −0.66 ppm and −0.78 ppm35. Static 1H NMR studies showed that the exact position and linewidth are highly affected by Na doping of a Ru/TiO2 catalyst36, i.e., this deuteron is sensitive to interactions with the support. In our case, direct interaction via Fermi contact between Ti3+ paramagnetic sites on the partially reduced TiOx support can affect the position and intensity of this peak.
Sample evacuation at 100 °C leads to a significant reduction in the intensity of the −53.2 ppm peak and shifting to −45.8 ppm (lower Knight shift) (Supplementary Figs. 17, 18, Supplementary Table 5). Interestingly, the ratio of Ti-OD and Ru-Datop groups remains constant, indicating comparable stability. Thus, the resonance at −53.2 ppm corresponds to the weakly bonded deuteron, not entirely captured in the TPD. Unlike the Ru-Datop signal, these deuterons are trapped by the Ru’s conduction electrons or some Ti3+ charged center of the support. Based on the thermal stability of the adsorbed deuterium studied by 2H MAS NMR and TPD, all three types of surface deuterons are stable at 25 °C and desorb only upon heating.
The 2H MAS NMR spectra reveal significant differences in the deuterons (Fig. 2h,i, Supplementary Fig. 19). The NH3-treated samples have a much higher content of Ru-bonded deuterium than Ti-OD groups. Higher resolution 10 kHz MAS spectra (Fig. 2h, i) show that Ru-related peaks at 0.18 kHz (2.1 ppm) and ~−4.49 kHz (−58.0 ppm) over Ru/TiO2-8 are more pronounced than on Ru/TiO2-1 The data combined (Supplementary Table 6, 7) reveals that Ru/TiO2-8 has a higher absolute amount of Ru-bonded deuterium. This was further supported by a direct comparison of relative distributions of different deuterons. Thus, NH3-treated supports promote hydrogen binding to Ru clusters.
Dynamics of hydrogen-Ru/TiO2 interactions
Hydrogen can partially reduce the TiO2 support by hydrogen spillover (Fig. 3)29, forming OH groups, and injecting electrons into TiO2, creating Ti3+ sites as a new state in the bandgap (Fig. 3a)37. Thermal excitation could also cause electron delocalization and populate the titania conduction band (CB)38. Specifically, hydrogen spillover creates a Ti3+ shallow trap, 0.1–0.2 eV below the CB edge, and a broadband in the FTIR spectra (Fig. 3c), frequently used to study spillover39,40. Electrons residing in the CB produce a power-law type spectrum distinct from shallow traps.

a Hydrogen spillover schematic over Ru/TiO2. b H2 binding on the metal-support interface. c Theoretical IR spectra for free electrons in the TiO2 conduction band and Ti3+ shallow trapped electrons. d, e Transmission FTIR transient spectra when switching from pure He to H2/He flow at 200 °C on Ru/TiO2−1 and Ru/TiO2−8, respectively. f Steady-state FTIR spectra for both samples at 250 °C and 0.14 bar H2. g, h In situ Raman spectra for Ru/TiO2-1 and Ru/TiO2-8 at 200 °C in He and H2/He flows, respectively (lines show Lorentzian curve fitting). i–k NAP-XPS of Ru/TiO2 samples under high vacuum and 1 mbar H2 pressure at 200 °C in T 2p (i), O 1 s (j), and Ru 3d (k) regions.
On Ru/TiO2-1, hydrogen binding leads primarily to shallow trap electrons (Fig. 3d). Due to the reversible spillover, this partial reduction is sensitive to temperature and hydrogen pressure (Supplementary Figs. 20, 21). A much broader background increase is seen on Ru/TiO2-8 due to the additional partial CB filling (Fig. 3e, Supplementary Fig. 22). The Ti-OH groups give a slight negative peak of ν(OH) at 3665 cm−1, and a new peak for δ(HOH) at 1614 cm−1 emerges due to the recombinative dehydroxylation of vicinal Ti-OH groups into water. Water formation stimulates the population of CB electrons41. FTIR shows that Ru/TiO2-1 is mainly reduced into localized Ti3+ states, whereas NH3-treated samples produce delocalized CB electrons (Fig. 3f). Hydrogen spillover is activated only at sufficiently high H coverage on Ru particles30, i.e., the low H coverage on the Ru/TiO2-1 sample (detected by 2H NMR) does not promote spillover and TiO2 reduction to the same extent.
In situ Raman spectroscopy at 200 °C in He and H2/He flows (Fig. 3g, h) corroborates this result. The Eg(1) vibrational mode of TiO2 anatase (~144 cm−1) is sensitive to the concentration of electrons42. On Ru/TiO2-1, the peak position is unaffected by hydrogen (Fig. 3g). In contrast, on Ru/TiO2-8 it shifts by ~3 cm−1 due to forming CB electrons by hydrogen; shallow traps (Ti3+ states) do not contribute.
The extent of reduction and associated charge transfer were monitored using NAP-XPS. Upon introducing hydrogen at 200 °C to Ru/TiO2-8, the Ti 2p3/2 peak (458.69 eV; Fig. 3i, Supplementary Fig. 23, Supplementary Table 8) and the main lattice oxygen peak in the O 1 s region43 (Fig. 3j) shift to lower binding energies by 0.15 eV due to modest support reduction. No shifts are evident on Ru/TiO2-1, indicating low receptivity toward hydrogen. Since the penetration depth of NAP-XPS corresponds to 2–3 nm, the Ru 3d and Ti 2p signals are collected from the whole Ru nanoparticle volume and 1–2 atomic layers of the underlying TiO2 support44. Thus, changes in the XPS spectra reflect charge distribution and MSI, but are not sensitive to the formation of the narrow TiOx peripheral layer observed in the FTIR of CO experiments (Fig. 1d).
The Ru 3d doublet overlaps with the C 1 s signal caused by carbonaceous deposits on the initial TiO2 (Fig. 3k). Ru/TiO2-1 and Ru/TiO2-8 have slightly different binding energy (BE): 279.70 and 280.12 eV, respectively (Supplementary Table 8). Such values are typical for small Ru particles on TiO244. Upon exposure of Ru/TiO2-1 to hydrogen at 200 °C, the BE of the Ru 3d5/2 peak increases by 0.12 eV; over Ru/TiO2-8, it shifts by 0.61 eV, indicative of Ruδ+ species. H2 activation at the Ru-TiO2 interface leads to negatively charged H− attached to the metal and H+ binding to the nearest oxygen atom (Fig. 3b)45. This heterolytic hydrogen activation leads to a partial positive charge on Ru due to forming negatively charged hydrides (H−) (Fig. 3b). The direct charge transfer from Ru to TiO2 CB leads to a partial reduction of TiO2, as reported in CO2 reduction46. Ruδ+-H− pairs upon H2 adsorption were also reported on Ru/carbon nanotubes47. Ru on NH3-treated supports allows spillover of H2, forming delocalized electrons, reducing the support more extensively, and increasing the positive charge of Ru.
PP hydrogenolysis
PP hydrogenolysis data over the three Ru/TiO2 samples with two polymer/catalyst weight ratios, 20 and 40, is shown in Fig. 4a–c and Supplementary Figs. 22, 23. The liquid yield increases with time and reaches 74% (Ru/TiO2-8) and 65–70% (Ru/TiO2-12) at 6 h, compared to only 63% at much longer times of 20 h (Ru/TiO2-1) reported earlier6. The conversion of the solid residue follows the same trend. The liquid product’s weight-average molecular weight (Mw) (Fig. 4c, Supplementary Tables 9, 10) is lower over Ru/TiO2-8 and Ru/TiO2-12 than Ru/TiO2-1.

a, b Yields of liquids (a) and solid residue (b) vs. time. c Mw of liquid vs. time. d, e Effect of substitution of H2 with D2 on liquid molecular weight distribution over Ru/TiO2-1 (d) and Ru/TiO2-8 (e) catalysts. f Effect of hydrogen pressure on liquid yield over Ru/TiO2-1 (the dotted line extrapolates the data, using a simple A → B → C kinetic scheme). Conditions: 250 °C, 30 bar H2, 2 g PP, 0.05 g catalyst (a, c) or 0.1 g catalyst (b, d–f) for a PP/catalyst ratio of 40 and 20, respectively.
PP conversion follows6 (i) an initial polymer consumption forming a “heavy” liquid; (ii) a decrease of the liquid Mw; and (iii) consumption of the liquid to light gases (~85% methane). When the Mw reaches a critical value, cascade hydrogenolysis to gas starts. Ru/TiO2-8 and Ru/TiO2-12 substantially increase the solid consumption and the liquid C–C bond hydrogenolysis compared to Ru/TiO2-1, leading to lighter products (Fig. 4c, Supplementary Tables 9, 10).
All catalysts show similar methane formation up to ca. 10% at long reaction times due to excessive liquid hydrogenolysis (Supplementary Figs. 24, 25, Supplementary Table 11). To study liquid gasification in more detail, we performed experiments at a higher polymer to catalyst ratio of 20 (Fig. 4b). The liquid stability increases in the order: Ru/TiO2-12 < Ru/TiO2-8 < Ru/TiO2-1 in line with the liquid Mw.
In the PE conversion over Ru/ZrO2 and Ru/WOx/ZrO2, the hydrogen availability on Ru was crucial in controlling the hydrogenolysis selectivity to liquids vs. light gases15. A high intrinsic H coverage favors liquid products. Conversely, a low H coverage promotes a sequential cascade of C–C rupture to methane.
We hypothesize that the performance differences among catalysts stem from the H coverage on Ru. We perform experiments of varying H2 pressure (Fig. 4f). An increased hydrogen pressure leads to higher reaction rates over Ru/TiO2-1; the liquid yield reaches ca. 60% in 6 h (40 bar H2) and 3 h (50 bar H2), much faster than the 30 bar experiment. Still, the liquid to gas decomposition is also accelerated (Supplementary Fig. 26, Supplementary Table 12). This competition leads to a maximum liquid yield. The liquid decomposition on the ammonia-treated Ru/TiO2 samples is less severe, and a maximum is absent at a polymer to catalyst ratio of 20 (Fig. 4a). This maximum is visible at higher catalyst loadings (Fig. 4b, Supplementary Fig. 25) and shifts to shorter reaction times. The increased H coverage at the same H2 pressure drives the improved catalyst performance, consistent with the 2H MAS NMR data of the higher content of both Ru-H species and spillover (Fig. 2). We propose that the different nature of chemisorbed hydrogen may be partially responsible for the variation in the liquid yields over different samples. Thus, a simple increase in surface coverage of hydrogen is insufficient to get a similarly high liquid yield over Ru/TiO2-1 and Ru/TiO2-8.
Alkane hydrogenolysis invokes (Fig. 5) adsorption to the metal, leading to dehydrogenated intermediates, C–C bond breaking of these intermediates, and final hydrogenation and product release. The first step is usually quasi-equilibrated48 due to the lower dehydrogenation barrier than the C–C bond breaking49. Experiments in D2 lead to slower dehydrogenation due to a kinetic isotope effect (KIE) in the dehydrogenation step, while the C–C bond breaking is not strongly affected by the H/D change50.
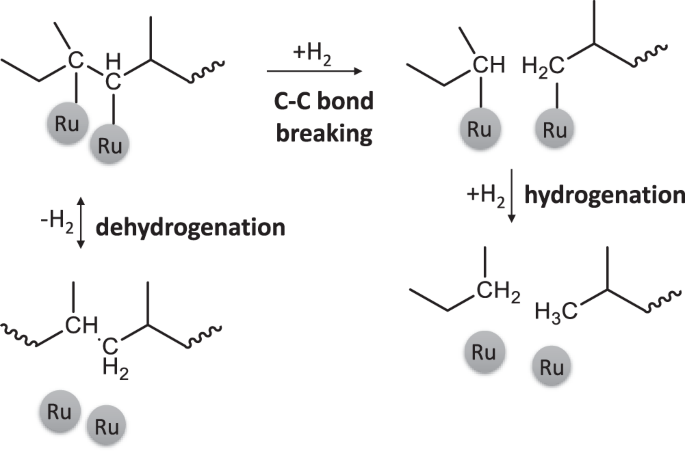
Dehydrogenation happends in both directions, while C–C bond breaking is considered irreversible.
PP hydrogenolysis in D2 is slower6, and the Mw of the liquid is larger, consistent with our data on Ru/TiO2-1 and Ru/TiO2-8 (Fig. 4d, e). A prominent shift in Mw is evident over Ru/TiO2-1 (from 2.67 to 14.12 kDa). Quasi-stationary kinetic analysis with standard transition state theory calculations, based on Fig. 5 (see Supplementary discussion I, Supplementary Fig. 27), shows that increasing the H coverage increases the net hydrogenolysis rate, reduces the KIE, and makes the net reaction rate less sensitive to the H/D exchange. Since Ru/TiO2-8 has a higher H coverage, the reaction rate is less sensitive to deuteration and correlates with the hydrogenolysis data (Fig. 4a). Over the Ru/TiO2-8, a high H coverage pushes further C–C bond breaking, leading to consumption of the initial polymer with no solid residue. On Ru/TiO2-1, the lower H coverage makes dehydrogenation more kinetically relevant and the initial polymer deconstruction slower.
In PE conversion on Ru/ZrO2, higher H coverages accelerate the desorption of reaction intermediates, preventing them from over-cracking, and giving higher liquid selectivity over methane at high H2 pressures. In PP, an increased H coverage boosts all three reaction stages because the PP initial polymer deconstruction is slower than the product desorption. Thus, altering the H coverage primarily alters this reaction step.
Discussion
Ru-based catalysts have demonstrated markedly different performances on various supports, but the reasons have remained elusive. For PE hydrogenolysis over Ru-WOx/ZrO2 catalysts15, a higher H surface coverage shifted the selectivity from terminal to internal C–C bond breaking, reducing methane formation at high hydrogen pressures. Partial reduction of the surface WOx domains provided extra hydrogen storage, increasing H coverage. Hydrogen availability was hypothesized as important, but the generality of this concept and mechanistic insights leading to catalyst improvement have been lacking.
Here, we tuned the catalyst’s electronic properties while holding the Ru particle size and physical characteristics constant by modifying the synthesis using ammonia. Ammonia dehydroxylates the TiO2 surface reducing the density of Ti-OH groups. This, in turn, increases the Lewis acid strength of Ti4+ sites, evidenced by FTIR of CO, creating a more intimate contact of Ru and TiO2 with possible formation of a TiOx peripheral layer, observed in previous reports24. Upon exposure to H2, the untreated (Ru/TiO2-1) sample shows moderate changes. H2 primarily dissociates and binds to Ru in the atop configuration, confirmed by 2H MAS NMR and DFT. Hydrogen spillover from Ru particles to the support leads to T4+ + \(\bar{e}\)→ Ti3+ reduction with subsequent electron trapping in the bandgap states. New Ti-OH groups form simultaneously, consistent with the classical spillover scheme (Fig. 3a).
For the NH3 treated samples (Ru/TiO2-8 and Ru/TiO2-12), spillover reduces TiO2 more extensively and forms delocalized \(\bar{e}\) in the CB in addition to the shallow traps. Ru particles bind more hydrogen, not only covalently (Ru-Hatop) but also through weak interactions involving Ru conduction electrons, revealed by 2H MAS NMR. This electron transfer from Ru to H leads to Ruδ+-H− ion pairs as a new H binding mode on Ru. We speculate that TiO2 CB electrons created by spillover are responsible for activating this new pathway of hydrogen fixation on the Ru/TiO2 periphery.
Due to the higher H coverage on Ru, these catalysts show higher activity in PP hydrogenolysis leading to higher liquid yields and doing so in a shorter time (6 vs. 16 h). Comparison with previously reported PP hydrogenolysis results (Supplementary Table 13) shows that Ru/TiO2-8 provides higher liquid yields at shorter reaction times than Ru/TiO2-1 and Ru/C. Ru/CeO2 shows higher liquid yields (83 vs. 74.1%) at higher catalyst loading and longer reaction times, which make a more detailed comparison hard. A similar acceleration is achieved by increasing the hydrogen pressure. High surface H coverage is manifested in a reduced KIE upon H2/D2 substitution.
Finally, we proposed a simple way to tune metal-support interactions in Ru/TiO2 by adding NH3 as a pH modulator in the Ru deposition step. We demonstrated a boost in hydrogen storage capacity stemming from a pronounced H spillover from Ru to TiO2. 2H MAS NMR proved to be a sensitive, semi-quantitative tool to study hydrogen chemisorption. Coupled with in situ pretreatment and heating, it can provide direct quantitative monitoring of hydrogen species on metal-metal oxide interfaces. Further improvements to the methodology could expand its scope. Raman, FTIR, and NAP-XPS highlight the enhanced spillover is caused by electrons directly filling the conduction bund of TiO2 compared to the standard localized Ti3+ trap sites. This enhances the hydrogen binding capacity for Ru particles due to the relatively weak charge transfer. Technologically, a higher hydrogen coverage increases the liquid yield to ~74% just in 6 h vs. 63% in ~20 h over the conventional catalyst possessing less pronounced MSI. The hydrogen availability drives catalyst improvement and emerges as a general strategy for designing catalysts beyond Ru/TiO2 and PP hydrogenolysis.
Methods
Catalyst preparation
The Ru/TiO2 catalysts were prepared by wetness impregnation using a commercially available anatase TiO2 support (US Research Nanomaterials). Before impregnation, the TiO2 powder was calcined at 450 °C for 6 h in static air. Ru/TiO2-1 was prepared by mixing 2.77 g of Ru precursor solution (ruthenium(III) nitrosyl nitrate solution in dilute nitric acid, Sigma–Aldrich) with 1 g of deionized water and then adding it to TiO2 powder under manual stirring with a glass rod at 70 °C. For Ru/TiO2-8 and Ru/TiO2-12, the pH was adjusted accordingly by adding several drops of aqueous NH3 (25%, Supelco). After impregnation, the catalyst was dried at 100 °C overnight and reduced in H2 (50% in He) flow in a tubular furnace at 300 °C for 2 h (ramp rate 10 °C/min).
Catalyst characterization
The Ru loading was estimated using X-ray fluorescence (XRF) analysis on a Rigaku Supermini 200 WDXRF in a He atmosphere. XRD (X-ray powder diffraction) patterns were obtained on a Bruker D8 diffractometer with 0.05° 2θ step size using Cu Kα radiation (λ 1.54 Å). Weight vs. temperature curves was recorded on a Discovery TGA instrument in a flow of 5%H2/He with 10 °C/min ramp from 30 to 600 °C. N2 sorption isotherms at −196 °C were recorded on a Micromeritics ASAP 2020 instrument. Before measurements, the samples were degassed at 300 °C for 3 h. UV-vis spectra were collected in diffuse-reflectance mode on a spectrometer (JASCO, V-550) with a diffuse-reflectance attachment using an ambient conditions cell with a quartz window and BaSO4 as a standard. The optical bandgap was estimated using the Tauc plot of (ahv)2 vs. hv, where hv is the photon energy in eV, and α is the reflectance. In-house X-ray photoelectron spectra (XPS) were recorded on a Thermo Fisher K-Alpha+ machine with an Al Kα monochromatic source. Before measurements, the samples were reduced in 50% H2/He flow at 300 °C and then deposited on a Cu foil. For binding energy reference, the C 1 s line at 284.6 eV was used. Scanning transmission electron microscopy (STEM) images were acquired on an Aberration Corrected Scanning/Transmission Electron Microscope, JEOL NEOARM TEM/STEM.
Temperature-programmed desorption (TPD) of H2 was recorded on a Micromeritics Autochem II instrument. Approximately 0.2 g of samples were packed in a U-shaped quartz reactor, heated in 10%H2/Ar flow (50 ml/min) to 300 °C with 2 h dwell time. Then samples were cooled to 35 °C in the same 10%H2/Ar flow, then purged isothermally for 30 min with pure Ar. Afterward, samples were heated with 10 °C/min ramping rate to 700 °C in Ar flow with an online thermal conductivity detector (TCD) recording of hydrogen desorption. Temperature-programmed reduction (TPR) with H2 was recorded on the same Autochem II instrument. Samples were pre-reduced in 10%H2/Ar (50 ml/min) flow to 300 °C with 2 h dwell time. Then samples were cooled to 35 °C in the same 10%H2/Ar flow and then heated with 10 °C/min ramping rate to 700 °C with an online TCD recording of hydrogen consumption. Hydrogen pulse chemisorption at 35 °C was measured on the same instrument. Samples were pretreated in 10%H2/Ar flow (50 ml/min) to 300 °C with 2 h dwell time and then for 1 h in pure Ar at the same temperature. Then, the reactor was cooled to 35 °C, and hydrogen was pulsed using 6-port valve with 45 μmol H2 per pulse, calibrated with the empty reactor.
Fourier transform infrared spectroscopy (FTIR) spectra of adsorbed CO at −196 °C were measured on a Nicolet 8700 spectrometer equipped with liquid nitrogen cooling and an MCT detector. The sample was pressed into a self-supported wafer (64 bar/in2) and loaded into a homemade quartz cell connected to the vacuum line. The sample was pretreated at 300 °C for 1 h in a vacuum (<10−4 torr). Then the sample was exposed to 1 torr of H2 gas for 30 min and evacuated for 10 min to reduce Ru. The reduction was repeated four times, followed by a final evacuation for 30 min. Afterward, the sample was cooled to −196 °С and exposed to 1 torr CO to saturate the surface. Then the CO was evacuated till 10−3 torr with spectra measured in ~0.1 torr increments.
Diffuse-reflectance infrared Fourier transform spectroscopy (DRIFT) spectra, which are more sensitive than FTIR to surface OH groups51, were recorded on the same spectrometer with a Praying Mantis attachment and in situ Harrick cell. The samples were reduced in 50%H2/He flow for 2 h at 300 °C followed by flushing with pure He at 300 °C for 30 min. Then the samples were cooled to 35 °C, and DRIFTS spectra were recorded.
FTIR and Raman spectroscopy of hydrogen spillover
Samples were pressed in self-supporting wafers (64 bar/in2) and placed in a homemade transmission IR cell connected to an atmospheric pressure gas flow manifold. Samples were pre-reduced at 300 °C for 2 h in 100 ml flow of 20%H2/He mixture (ramp rate 10 °C/min). Then, the samples were flushed with pure 100 ml/min He flows for 1 h and cooled to treatment temperature (200 or 250 °C). A baseline spectrum was recorded in pure He, and then the gas flow was switched from He to x%H2/He with a hydrogen partial pressure (p(H2)) in the 0.06–0.14 bar range. Spectra were recorded in 10 s increments until complete saturation at a given p(H2) and temperature (~15 mins). Then the baseline spectrum was subtracted to highlight the spillover effect.
Raman spectra were measured on a Horiba LabRAM HR evolution spectrometer using 532 nm green laser (5 mW power), ×50 objective, and 1800 g/mm grating. The detector resolution under these conditions is equal to 0.48 cm−1/pixel. In situ measurements were done using a Harrick Raman cell equipped with a quartz window. Samples were pre-reduced at 300 °C for 2 h in 50 ml flow of 20% H2/He mixture (ramp rate 10°/min). Then, samples were flushed in pure 50 ml/min He flows for 1 h and cooled to treatment temperature (200 or 250 °C). The baseline spectrum was recorded in pure He, and then the gas flow was switched from He to 15%H2/He. Spectra were recorded after 10 min stabilization to ensure completion of spillover. Spectra were baseline corrected and fitted using the Lorentzian function in the Omnic software.
2H MAS NMR spectroscopy of chemisorbed deuterium
The sample was packed in ZrO2 4 mm standard MAS NMR rotor, loaded in a pyrex tube with a valve, and connected to a vacuum line. Then it was heated to 120 °C for 1 h to remove moisture and then to 300 °C for 3 h under 10−4 torr. Then, 1 torr of D2 gas (99.8%, Cambridge Isotope Laboratories, Inc.) was administered through the vacuum line. After reduction for 30 min, the sample was evacuated for 30 min to remove possible traces of water. The treatment with D2 at 300 °C was repeated four times. Then the sample was cooled to room temperature under 1 torr D2 and transferred to a glove box without exposure to air. In nitrogen, the glove box rotor was removed from the tube and sealed. To test the stability of chemisorbed deuterium, in an experiment, the sample was degassed at 100 °C for 30 min at 10−4 torr and only then sealed.
2H MAS NMR spectra were acquired on an 11.7 T Bruker Avance III NMR spectrometer with a 4 mm HX probe at a 2H frequency of 76.77 MHz at magic angle spinning speeds of 4 and 10 kHz. The samples were maintained at room temperature with sample heating due to magic angle spinning taken into account. Data were acquired using a 180°-τ−90° pulse sequence with an interpulse delay of 10 ms to reduce baseline distortions, with a 90° pulse length of 4.25 μs. 16,384–20,480 scans were collected per sample with a pulse delay of 4 s. Spectra were referenced externally to D2O at 4.7 ppm.
Spectra were modeled and fitted using the ssNake software52, and three component refinement of the experimental spectra was performed to estimate the key EFG parameters.
Near ambient pressure X-ray photoelectron spectroscopy (NAP-XPS)
The NAP-XPS spectra were collected under UHV (base pressure of 10−9 mbar) using a SPECS electron spectrometer equipped with PHOIBOS 100 hemispherical energy analyzer and a monochromatic Al Kα X-ray source (1486.7 eV). Spectra were referenced to C1s line at 284.6 eV and fitted using Thermo Avantage software. The samples were pressed on a Cu piece and mounted on a stainless-steel flag-type holder with a K-type thermocouple allowing for in situ temperature readings. After loading the sample in XPS analysis chamber, it was heated to 300 °C and kept at that temperature for 30 min (ramp rate 10°/min). Then 1 mbar hydrogen was dosed for in situ reductions at 300 °C for 40 min. Then hydrogen was evacuated, and the sample was annealed at 300 °C in UHV for 10 min to remove chemisorbed hydrogen. Afterward, the sample was cooled to 200 °C, and initial spectra in UHV were recorded. Then the sample was exposed to 1 mbar hydrogen for 15 min at the same temperature and new spectra were measured. Additional reference spectra were recorded also at 35 °C in UHV.
Reaction tests
Isotactic polypropylene (PP, Mw ~250,000, Mn ~67,000) was purchased from Sigma–Aldrich. An amount of freshly reduced catalyst (0.1 or 0.05 g) was mechanically mixed with 2.0 g of PP using a vortex mixer. The mixture was then transferred into a borosilicate liner of a 50 mL stainless-steel Parr reactor with a 0.7 mL stir bar. The mass ratio of polymer to catalyst was 20 or 40, corresponding to the catalyst loading of 0.1 or 0.05 g. The Parr reactor was sealed and purged six times with pure H2 at 50 bar, charged to 30 bar, and then heated to 250 °C (ramping rate 10 °C/min) using a band heater (Omega Eng.). Stirring was initiated after the temperature reached 160 ± 5 °C to first melt the polymer. The stirring speed was set at 300 rpm; additional experiments showed similar product yields and molecular weight distributions at 400 and 500 rpm but nearly no reactivity without stirring. Reactions were maintained for specified time intervals and then quickly quenched in an ice bath. For H2 pressure variation experiments, the hydrogen pressure was 20, 40, and 50 bar.
Product analysis
After the temperature dropped below 10 °C, the gas from the reactor’s headspace was transferred to a 1 L Tedlar gas sampling bag for analysis. Then the reactor was opened, and the liquid and solid residue were mixed with 20 mL of CH2Cl2, used as a solvent. This slurry was filtered (Whatman, 100 μm), and the solid residue was dried at room temperature overnight with complete evaporation of all CH2Cl2. The solvent was removed from the liquid fraction using a rotary evaporator. The solid and liquid fraction yields were quantified gravimetrically. A GC with an FID detector (Agilent 7890 Series, HP-volamine column) was used for gas analysis. The concentration of hydrocarbons in the gas sample was calculated using a standard C1–C4 calibration mixture. The absolute amount of hydrocarbons in the gas was calculated using the ideal gas law.
An overall balance was calculated according to Eq. (1):
where \({m}_{L},{m}_{s},{m}_{g},{m}_{{{{{{{\mathrm{initial}}}}}}}}\) are the mass of liquid, gas, solid, and initial polymer, respectively. The yield of the i-th group of products was calculated according to Eq. (2):
where mi is the mass of the ith group of products (liquid, gas, or solid).
Liquid products were analyzed using gel permeation chromatography (GPC) using Styragel HR 4, HR 3, and HR 0.5 columns (dimensions 4.6 × 300 mm) connected in tandem using THF as solvent (0.3 ml/min flow rate) and a Waters 2414 refractive index detector (RID). The retention time was calibrated using Polystyrene Standards Kit (Waters, WAT058931).
Density functional theory (DFT) calculations
The TiO2 support was modeled as an anatase structure with the (101) facet exposed. The supported Ru was modeled as a Ru-12 cluster on the TiO2 surface. DFT calculations were performed using the Vienna Ab initio Simulation Package (VASP) package53. The RPBE potential was used in conjunction with the D3 correction54. The energy cutoff was set as 520 eV, and the electron smearing factor was set as 0.1 eV. Electronic structures were convergence until 10−6 eV, and ionic steps were iteratively taken until the force fell below 0.05 eV/A. The Electric Field Gradient (EFG) was calculated using the VASP internal functions. Nuclear quadrupole moments were adapted accordingly55.
Data availability
All data generated or analyszed during this study are included in this published article (and its supplementary information file).
References
Silva, A. L. P. et al. Increased plastic pollution due to COVID-19 pandemic: challenges and recommendations. Chem. Eng. J. 405, 126683 (2021).
Zheng, J. & Suh, S. Strategies to reduce the global carbon footprint of plastics. Nat. Clim. Change 9, 374–378 (2019).
Vanapalli, K. R. et al. Challenges and strategies for effective plastic waste management during and post COVID-19 pandemic. Sci. Total Environ. 750, 141514 (2021).
Moncrief, L. J., Maggie, G., Bryan, B., Eric, N. A. & Elizabeth, H. J. Articles of reclaimed polypropylene compositions. USA patent (2016).
Kots, P. A., Vance, B. C. & Vlachos, D. G. Polyolefin plastic waste hydroconversion to fuels, lubricants, and waxes: a comparative study. React. Chem. Eng. 7, 41–54 (2022).
Kots, P. A. et al. Polypropylene plastic waste conversion to lubricants over Ru/TiO2 catalysts. ACS Catal. 11, 8104–8115 (2021).
Jaydev, S. D., Martín, A. J. & Pérez‐Ramírez, J. Direct conversion of polypropylene into liquid hydrocarbons on carbon‐supported platinum catalysts. ChemSusChem 14, 5179–5185 (2021).
Celik, G. et al. Upcycling single-use polyethylene into high-quality liquid products. ACS Cent. Sci. 5, 1795–1803 (2019).
Zhang, F. et al. Polyethylene upcycling to long-chain alkylaromatics by tandem hydrogenolysis/aromatization. Science 370, 437–441 (2020).
Rorrer, J. E., Beckham, G. T. & Román-Leshkov, Y. Conversion of polyolefin waste to liquid alkanes with Ru-based catalysts under mild conditions. JACS Au, 1, 8–12 (2020).
Nakaji, Y. et al. Low-temperature catalytic upgrading of waste polyolefinic plastics into liquid fuels and waxes. Appl. Cat. B Environ. 285, 119805 (2021).
Rorrer, J. E., Troyano-Valls, C., Beckham, G. T. & Román-Leshkov, Y. Hydrogenolysis of polypropylene and mixed polyolefin plastic waste over Ru/C to produce liquid alkanes. ACS Sustain. Chem. Eng. 9, 11661–11666 (2021).
Jia, C. et al. Deconstruction of high-density polyethylene into liquid hydrocarbon fuels and lubricants by hydrogenolysis over Ru catalyst. Chem. Catal. 1, 437–455 (2021).
Chen, L. et al. Effect of reaction conditions on the hydrogenolysis of polypropylene and polyethylene into gas and liquid alkanes. React. Chem. Eng., 7, 844–854 (2022).
Wang, C. et al. Polyethylene hydrogenolysis at mild conditions over ruthenium on tungstated zirconia. JACS Au 1, 1422–1434 (2021).
Tauster, S., Fung, S. & Garten, R. L. Strong metal-support interactions. Group 8 noble metals supported on titanium dioxide. J. Am. Chem. Soc. 100, 170–175 (1978).
Zhou, J. et al. Interfacial compatibility critically controls Ru/TiO2 metal-support interaction modes in CO2 hydrogenation. Nat. Commun. 13, 1–10 (2022).
Lin, F. et al. Single-facet dominant anatase TiO2 (101) and (001) model catalysts to elucidate the active sites for alkanol dehydration. ACS Catal. 10, 4268–4279 (2020).
Fàbrega, C. et al. Tuning the fermi level and the kinetics of surface states of TiO2 nanorods by means of ammonia treatments. J. Phys. Chem. C. 117, 20517–20524 (2013).
Román, E. L., de Segovia, J., Kurtz, R. L., Stockbauer, R. & Madey, T. E. UPS synchrotron radiation studies of NH3 adsorption on TiO2 (110). Surf. Sci. 273, 40–46 (1992).
Hadjiivanov, K., Lamotte, J. & Lavalley, J.-C. FTIR study of low-temperature CO adsorption on pure and ammonia-precovered TiO2 (anatase). Langmuir 13, 3374–3381 (1997).
Panayotov, D. A. et al. Effect of methanol on the Lewis acidity of rutile TiO2 nanoparticles probed through vibrational spectroscopy of coadsorbed CO. Langmuir 26, 8106–8112 (2010).
Hadjiivanov, K. et al. FTIR study of CO interaction with Ru/TiO2 catalysts. J. Catal. 176, 415–425 (1998).
Zhang, Y. et al. Tuning reactivity of Fischer–Tropsch synthesis by regulating TiOx overlayer over Ru/TiO2 nanocatalysts. Nat. Commun. 11, 1–8 (2020).
Feulner, P. & Menzel, D. The adsorption of hydrogen on ruthenium (001): adsorption states, dipole moments and kinetics of adsorption and desorption. Surf. Sci. 154, 465–488 (1985).
Zhan, Y., Zhou, C., Jin, F., Chen, C. & Jiang, L. Ru/TiO2 catalyst for selective hydrogenation of benzene: Effect of surface hydroxyl groups and spillover hydrogen. Appl. Surf. Sci. 525, 146627 (2020).
Gutmann, T. et al. Hydrido-ruthenium cluster complexes as models for reactive surface hydrogen species of ruthenium nanoparticles. solid-state 2H NMR and quantum chemical calculations. J. Am. Chem. Soc. 132, 11759–11767 (2010).
Walaszek, B. et al. 2H-solid-state-NMR study of hydrogen adsorbed on catalytically active ruthenium coated mesoporous silica materials. Solid State Nucl. Magn. Reson. 35, 164–171 (2009).
Prins, R. Hydrogen spillover. Facts fiction. Chem. Rev. 112, 2714–2738 (2012).
Chen, H.-Y. T., Tosoni, S. & Pacchioni, G. Hydrogen adsorption, dissociation, and spillover on Ru10 clusters supported on anatase TiO2 and tetragonal ZrO2 (101) surfaces. ACS Catal. 5, 5486–5495 (2015).
Engelke, F., Bhatia, S., King, T. S. & Pruski, M. Dynamics of hydrogen at the surface of supported ruthenium. Phys. Rev. B 49, 2730 (1994).
Komaya, T. et al. The influence of metal-support interactions on the accurate determination of Ru dispersion for Ru/TiO2. J. Catal. 149, 142–148 (1994).
Yesinowski, J. P. et al. Distributions of conduction electrons as manifested in MAS NMR of gallium nitride. J. Am. Chem. Soc. 128, 4952–4953 (2006).
Mehta, M. et al. Hydrogen treated anatase TiO2: a new experimental approach and further insights from theory. J. Mater. Chem. A 4, 2670–2681 (2016).
Sinhamahapatra, A., Lee, H.-Y., Shen, S., Mao, S. S. & Yu, J.-S. H-doped TiO2-x prepared with MgH2 for highly efficient solar-driven hydrogen production. Appl. Cat. B Environ. 237, 613–621 (2018).
Komaya, T. et al. Effects of sodium on the structure and Fischer-Tropsch synthesis activity of Ru/TiO2. J. Catal. 152, 350–359 (1995).
Di Valentin, C., Pacchioni, G. & Selloni, A. Reduced and n-type doped TiO2: nature of Ti3+ species. J. Phys. Chem. C. 113, 20543–20552 (2009).
Panayotov, D. et al. Hydrogen spillover on Rh/TiO2: the FTIR study of donated electrons, co-adsorbed CO and H/D exchange. Phys. Chem. Chem. Phys. 17, 20563–20573 (2015).
Mahdavi-Shakib, A., Rich, L. C., Whittaker, T. N. & Chandler, B. D. Hydrogen adsorption at the Au/TiO2 interface: quantitative determination and spectroscopic signature of the reactive interface hydroxyl groups at the active site. ACS Catal. 11, 15194–15202 (2021).
Panayotov, D. A., Burrows, S. P., Yates, J. T. Jr. & Morris, J. R. Mechanistic studies of hydrogen dissociation and spillover on Au/TiO2: IR spectroscopy of coadsorbed CO and H-donated electrons. J. Phys. Chem. C. 115, 22400–22408 (2011).
Panayotov, D. A. & Yates, J. T. Jr. Depletion of conduction band electrons in TiO2 by water chemisorption–IR spectroscopic studies of the independence of Ti–OH frequencies on electron concentration. Chem. Phys. Lett. 410, 11–17 (2005).
Mazzolini, P. et al. Vibrational–electrical properties relationship in donor-doped TiO2 by Raman spectroscopy. J. Phys. Chem. C. 120, 18878–18886 (2016).
Abdel-Mageed, A. M. et al. Steering the selectivity in CO2 reduction on highly active Ru/TiO2 catalysts: Support particle size effects. J. Catal. 401, 160–173 (2021).
Elmasides, C., Kondarides, D., Grünert, W. & Verykios, X. XPS and FTIR study of Ru/Al2O3 and Ru/TiO2 catalysts: reduction characteristics and interaction with a methane–oxygen mixture. J. Phys. Chem. B 103, 5227–5239 (1999).
Whittaker, T. et al. H2 oxidation over supported Au nanoparticle catalysts: evidence for heterolytic H2 activation at the metal–support interface. J. Am. Chem. Soc. 140, 16469–16487 (2018).
Li, X. et al. Controlling CO2 hydrogenation selectivity by metal‐supported electron transfer. Angew. Chem. Int. Ed. 59, 19983–19989 (2020).
Verdinelli, V., Juan, A. & German, E. Ruthenium decorated single walled carbon nanotube for molecular hydrogen storage: A first-principle study. Int. J. Hydrog. Energy 44, 8376–8383 (2019).
Flaherty, D. W. & Iglesia, E. Transition-state enthalpy and entropy effects on reactivity and selectivity in hydrogenolysis of n-alkanes. J. Am. Chem. Soc. 135, 18586–18599 (2013).
Zaera, F. An organometallic guide to the chemistry of hydrocarbon moieties on transition metal surfaces. Chem. Rev. 95, 2651–2693 (1995).
Shi, H., Gutierrez, O. Y., Zheng, A., Haller, G. L. & Lercher, J. A. Mechanistic pathways for methylcyclohexane hydrogenolysis over supported Ir catalysts. J. Phys. Chem. C. 118, 20948–20958 (2014).
Halasz, I., Moden, B., Petushkov, A., Liang, J.-J. & Agarwal, M. Delicate distinction between OH groups on proton-exchanged H-chabazite and H-SAPO-34 molecular sieves. J. Phys. Chem. C. 119, 24046–24055 (2015).
Van Meerten, S., Franssen, W. & Kentgens, A. ssNake: a cross-platform open-source NMR data processing and fitting application. J. Magn. Reson. 301, 56–66 (2019).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B 48, 13115 (1993).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Pyykkö, P. Year-2008 nuclear quadrupole moments. Mol. Phys. 106, 1965–1974 (2008).
Acknowledgements
We thank to Dr. Stavros Caratzoulas for valuable discussions. This work was financially supported by the Center for Plastics Innovation (CPI), an Energy Frontier Research Center funded by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, award number DE-SC0021166. This research used instruments in the Advanced Materials Characterization Lab (AMCL) at the University of Delaware. MAS NMR measurements were made possible by the Delaware COBRE program, supported by a grant from the National Institute of General Medical Sciences—NIGMS (5 P30 GM110758-02) from the National Institutes of Health. STEM was performed at the Singh Center for Nanotechnology at the University of Pennsylvania, supported by the National Science Foundation (NSF) National Nanotechnology Coordinated Infrastructure Program grant NNCI-1542153. Additional support to the Nanoscale Characterization Facility at the Singh Center has been provided by the Laboratory for Research on the Structure of Matter (MRSEC) supported by the National Science Foundation (DMR-1720530). This research used beamline 7-BM (QAS) of the National Synchrotron Light Source II, a U.S. DOE Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. Beamline operations were supported in part by the Synchrotron Catalysis Consortium (U.S. DOE, Office of Basic Energy Sciences, Grant No. DE-SC0012335). This research used resources of the Center for Functional Nanomaterials (CFN), which is a U.S. Department of Energy Office of Science User Facility, at Brookhaven National Laboratory under Contract No. DE-SC0012704.
Author information
Authors and Affiliations
Contributions
P.A.K. conceived the idea, designed the project, and carried out the experiments. T.X. performed DFT calculations and the associated analysis. B.C.V. and C.W. carried out some experiments and contributed to the discussions. C.M.Q. measured and analyzed the MAS NMR spectra. M.D.M. and J.A.B. performed and analyzed the NAP-XPS data. P.K. and E.A.S. took and analyzed the STEM images. N.S.M., L.M., and S.N.E. performed in situ XAS data collection and analysis. D.G.V. supervised the project. P.A.K. and D.G.V. wrote the manuscript, and all authors discussed the results and assisted the manuscript preparation.
Corresponding author
Ethics declarations
Competing interests
D.G.V., P.A.K., B.C.V., and C.W. are authors of a patent application. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Masazumi Tamura and the other, anonymous, reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kots, P.A., Xie, T., Vance, B.C. et al. Electronic modulation of metal-support interactions improves polypropylene hydrogenolysis over ruthenium catalysts. Nat Commun 13, 5186 (2022). https://doi.org/10.1038/s41467-022-32934-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-32934-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
