
Abstract
TRF1 is an essential component of the telomeric protective complex or shelterin. We previously showed that dysfunctional telomeres in alveolar type II (ATII) cells lead to interstitial lung fibrosis. Here, we study the lung pathologies upon telomere dysfunction in fibroblasts, club and basal cells. TRF1 deficiency in lung fibroblasts, club and basal cells induced telomeric damage, proliferative defects, cell cycle arrest and apoptosis. While Trf1 deletion in fibroblasts does not spontaneously lead to lung pathologies, upon bleomycin challenge exacerbates lung fibrosis. Unlike in females, Trf1 deletion in club and basal cells from male mice resulted in lung inflammation and airway remodeling. Here, we show that depletion of TRF1 in fibroblasts, Club and basal cells does not lead to interstitial lung fibrosis, underscoring ATII cells as the relevant cell type for the origin of interstitial fibrosis. Our findings contribute to a better understanding of proper telomere protection in lung tissue homeostasis.
Similar content being viewed by others


Introduction
Telomeres are heterochromatic structures at the chromosome ends, which are essential for chromosome stability. In mammals, telomeric DNA consists of TTAGGG tandem repeats bound by the so-called shelterin complex, which encompasses TRF1, TRF2, TIN2, POT1, TPP1 and RAP1 protein1,2. Shelterin complex ensures telomere protection by preventing end-to-end chromosome fusions, telomere fragility, and activation of DNA damage response3. With each cell division, telomeres shorten due to the incomplete replication of chromosome ends4,5. Telomere shortening can be compensated through the de novo addition of telomeric repeats by telomerase, a reverse transcriptase composed of a catalytic subunit (TERT) and an RNA component (Terc)6.
Specifically, TRF1 has a relevant role in shelterin complex assembly7,8,9. TRF1 is also important to prevent telomere fusions, telomeric DNA damage and multitelomeric signals, as well as for the replication of telomeric DNA10,11. Moreover, TRF1 was reported to be essential for the induction and maintenance of pluripotency12,13. Conditional deletion of Trf1 in specific cell types has demonstrated and important role in tissue regeneration and homeostasis10,13,14,15.
Idiopathic pulmonary fibrosis (IPF) is a fibrosing interstitial lung disease characterized by the histopathological pattern of usual interstitial pneumonia. IPF affects 3 million people worldwide with a median survival time from diagnosis of 2–4 years16,17,18. Interestingly, between 8% and 15% of familial IPF cases are associated with mutations in telomerase or in telomere-protective proteins19,20,21,22,23,24. Interestingly, sporadic cases of IPF, not associated with telomerase mutations, also show shorter telomeres compared to age-matched controls, with 10% of the patients showing telomeres as short as the telomerase mutation carriers19. Telomerase mutations have also been found in up to 1% of smokers showing chronic obstructive pulmonary disease (COPD), also leading to abnormally short telomeres25.
Lung fibroblasts are mesenchymal cells of the interstitium with a key role in the formation and extension of alveolar septa and in alveolar epithelial proliferation and differentiation. After alveolar or endothelial-cell injury or immune activation and inflammation, fibroblasts can be activated and proliferate and differentiate into myofibroblasts, which further contribute to pulmonary fibrosis26,27. On the other hand, alveolar type II (ATII) cells are localized in the gaseous alveolar surfaces, and their main function is the secretion of surfactant to prevent alveolar collapse. ATII cells have been reported as progenitor cells for ATI cells, having a key role in lung repair after injury28,29,30. Club cells are airway epithelial cells whose main roles are detoxification of xenobiotic and oxidizing molecules, secretion of antimicrobial peptides, and promotion of mucociliary clearance. Club cells are primary progenitor cells, since the play a key role in bronchiolar epithelial repair through their ability to self-renew and differentiate into ciliated and goblet cells31,32,33,34. Basal cells are only present in the mouse trachea, whereas in humans, they extend to the respiratory bronchioles. In a steady state basal cells are quiescent; however, in response to injury, airway basal cells become activated and operate as stem/progenitor cells capable of self-renewal and differentiation into ciliated and secretory cells33,35,36.
Noteworthy, we have shown that induction of telomere dysfunction in ATII cells is sufficient to induce progressive and lethal pulmonary fibrosis in mice15. However, lung pathological consequences of telomere dysfunction in fibroblasts, club and basal cells have not yet been investigated. On this basis, we have conditionally deleted in mice the sheltering component Trf1 in fibroblasts, club and basal cells to examine the pathological consequences of dysfunctional telomeres on the lung. Here, we show that depletion of TRF1 in fibroblasts, Club and basal cells does not lead to interstitial fibrosis, underscoring ATII cells as the relevant cell type for the origin of interstitial lung fibrosis.
Results
Efficient Trf1 deletion in lung fibroblasts upon tamoxifen administration
We induced a short-term telomere dysfunction in fibroblasts by genetically deleting the shelterin component Trf1 in these cells, which have been reported to contribute to pulmonary fibrosis26,27. In particular, to specifically delete Trf1 in lung fibroblasts we generated a Trf1lox/lox KFPLox-LSL-Lox Col1a2-CreERT2 mouse model, in which Cre recombinase expression is controlled by the collagen, type I, alpha 2 (Col1a2) promoter, which is specific for fibroblasts37,38. CreERT2 is conditionally activated by tamoxifen (TMX) administration37. Moreover, a transgene encoding for the katushka fluorescent protein (KFP) that contains a stop cassette flanked by lox sequences was introduced as a reporter to monitor Cre activity15 (Fig. 1a). TMX was administered intraperitoneally (i.p.) for five consecutive days during the first week and once a week until the sacrifice and sample collection on week (W) 7, and during the follow-up of survival until W12 (Fig. 1b). First, we assessed the fluorescence intensity of KFP in Trf1+/+ KFP+/+ Cre+/t, Trf1+/+ KFP+/t Cre+/t (Col1a2-Cre; Trf1+/+) and Trf1lox/lox KFP+/t Cre+/t (Col1a2-Cre; Trf1lox/lox) mice by in vivo fluorescence imaging. As expected, KFP fluorescence was solely detected in KFP+/t mice. Fluorescence intensity was apparently higher in the lungs of Col1a2-Cre; Trf1lox/lox mice compared to Col1a2-Cre; Trf1+/+ (controls) mice after 7 weeks of TMX treatment (Fig. 1c). Accordingly, the amount of KFP-positive cells per 40X high-power field (HPF) evaluated by immunohistochemistry was higher in the lungs of Col1a2-Cre; Trf1lox/lox mice compared to control mice (Fig. 1d, e). We did not find differences in survival between Col1a2-Cre; Trf1+/+ (Trf1+/+, controls) and Col1a2-Cre; Trf1Δ/Δ (Trf1Δ/Δ) mice for as long as 12 weeks of TMX treatment (Fig. 1f). Immunofluorescence staining with antibodies against COL1A2 and TRF1 clearly demonstrated that the majority of COL1A2-positive cells stained negative for TRF1 (93%) in the lungs of Trf1Δ/Δ mice (Fig. 1g, h). Moreover, we performed an immune-telomere-Q-FISH for the quantification of mean telomere spot intensity and average number of telomeres in COL1A2-positive cells, which are readouts of telomere length10. The results did not show any significant difference either in telomere fluorescence or in the numbers of detectable telomeres between wild-type and Trf1Δ/Δ fibroblasts, indicating that Trf1 deletion in lung fibroblasts does not result in telomere length changes (Fig. 1g, i, j).

a Generation of the conditional knockout mouse model in which Trf1 was deleted in fibroblasts using the Cre recombinase driven by the Col1a2 promoter. Trf1lox, KFPLox-LSL-Lox, and Col1a2-CreERT2 alleles are depicted before and after Cre-mediated excision. b Tamoxifen (TMX) treatment, survival rate assessment and sample collection. Eight-to 10-week-old male Trf1+/+ KFP+/t Cre+/t (Col1a2-Cre; Trf1+/+) and Trf1lox/lox KFP+/t Cre+/t (Col1a2-Cre; Trf1lox/lox) mice were i.p. injected with TMX for five consecutive days during the first week and once a week until the sacrifice and sample collection on week (W) 7, and during the follow-up of survival until W12. c Representative images of fluorescence intensity of katushka fluorescent protein (KFP) in Trf1+/+ KFP+/+ Cre+/t, Trf1+/+ KFP+/t Cre+/t and Trf1lox/lox KFP+/t Cre+/t mice. Representative immunostainings for KFP (d), and quantification of KFP positive cells per 40X high-power field (HPF) (e) in lung sections from Trf1+/+ KFP+/t Cre+/t and Trf1lox/lox KFP+/t Cre+/t mice. f Kaplan–Meier survival curves of Col1a2-Cre; Trf1+/+ (Trf1+/+, controls) and Col1a2-Cre; Trf1Δ/Δ (Trf1Δ/Δ) mice upon TMX treatment. g Representative immunofluorescence stainings for COL1A2 (green) and TRF1 (red) (white arrowheads indicate COL1A2+ fibroblasts with deletion of TRF1), and immune-telomere-Q-FISH in COL1A2+ fibroblasts (Cy3Tel probe (red), COL1A2+ cells (green), and nuclei stained with DAPI (blue)) in lung sections from Trf1+/+ and Trf1Δ/Δ mice. Quantification of the proportion of double COL1A2+-TRF1+ fibroblasts (h) and mean telomere spot intensity (i) and average number of telomeres (j) in COL1A2+ cells from Trf1+/+ and Trf1Δ/Δ mice. Data are expressed as mean ± SEM (the number of mice is indicated in each case). **p < 0.01 (Mann–Whitney or unpaired t tests). Animal survival was assessed by the Kaplan–Meier analysis, using the log Rank (Mantel–Cox) test). Source data are provided as a Source Data file.
Trf1 deletion in lung fibroblasts increases telomeric damage, cell cycle arrest and apoptosis, and reduces proliferation of fibroblasts
In order to study the effects of TRF1 deficiency on lung fibroblasts, we performed double immunostainings of COL1A2 with markers of DNA damage (γH2AX), cell cycle arrest (p16 and p21), apoptosis (C3) and proliferation (Ki67), as well as an an immuno-telomere-Q-FISH with the DNA damage marker 53BP1 to identify telomeric induced foci (TIF) in COL1A2 positive cells in lung sections from Trf1Δ/Δ mice and controls (Fig. 2a–g). Trf1Δ/Δ mice exhibited increased numbers of lung fibroblasts with DNA damage, cell cycle arrest, apoptosis and proliferation, as well as increased proportion of COL1A2+ cells with more than 1 TIF (Fig. 2a–g).

a Representative lung immunostainings for COL1A2 (purple) and γH2AX (brown; black arrowheads indicate double COL1A2+-γH2AX+ fibroblasts), COL1A2 (purple) and p16 and p21 (brown; red arrowheads indicate double COL1A2+-p16+ and COL1A2+-p21+ fibroblasts), COL1A2 (purple) and C3 (brown; green arrowheads indicate double COL1A2+-C3+ fibroblasts), and COL1A2 (purple) and Ki67 (brown; orange arrowheads indicate double COL1A2+-Ki67+ fibroblasts), as well as representative lung images of telomeric induced foci (TIF) in COL1A2+ cells (COL1A2 (purple), Cy3Tel probe (red), 53BP1+ cells (green; white arrowheads indicate TIF) and nuclei stained with DAPI (blue)) in lung sections from Trf1+/+ and Trf1Δ/Δ mice. Quantification of COL1A2+-γH2AX+ (b), COL1A2+-p16+ (d), COL1A2+-p21+ (e), COL1A2+-C3+ (f), and COL1A2+-Ki67+ (g) fibroblasts per 40X high-power field (HPF), as well as the proportion (%) of COL1A2+ cells with more than 1 TIF in lung sections from Trf1+/+ and Trf1Δ/Δ mice (c). h Representative images of Trf1+/+ and Trf1Δ/Δ lungs (H&E), BALF cytospin preparations (May-Grünwald Giemsa (MGG)) and Sirius Red staining, and Vimentin immunostainings in lung sections from Trf1+/+ and Trf1Δ/Δ mice. Quantification of total (i) and differential BALF cell counts for neutrophils (j), lymphocytes (k) and macrophages (l), and Lung tissue mRNA expression levels of Tnf (m) Il1b (n), Il6 (o) and Ifn-γ (p) (Th1 inflammation) in Trf1+/+ and Trf1Δ/Δ mice. Quantification of alveolar collagen (Sirius Red) (q) and Vimentin (r) positive areas (%), and lung resistance (LR) (s) and dynamic compliance (Cdyn) (t) evaluated by plethysmography in Trf1+/+ and Trf1Δ/Δ mice. Data are expressed as mean ± SEM (the number of mice is indicated in each case). **p < 0.01 (Mann–Whitney or unpaired t tests). Source data are provided as a Source Data file.
Next, to evaluate the potential lung pathological effects of Trf1 deletion in lung fibroblasts, we assessed cellularity and total protein concentration in bronchoalveolar lavage fluid (BALF), mRNA expression of Th1 inflammation markers, Sirius Red staining (collagen deposition), immunostaining for Vimentin (fibroblast presence), as well as lung function by plethysmography (lung resistance (LR) and dynamic compliance (Cdyn)) in our mouse cohorts (Fig. 2h–t). We found elevated BALF cell counts for neutrophils and lymphocytes in Trf1Δ/Δ as compared to wild-type mice (Fig. 2h–l). Nevertheless, lung mRNA expression of Th1 inflammation markers Tnf, Il1b, Il6 and Ifn-γ (Fig. 2m–p), as well as alveolar Sirius Red and Vimentin-stained areas (Fig. 2q, r) were not found significantly changed between experimental groups, indicating that the short-term deletion of Trf1 specifically in fibroblasts does not result in either inflammation nor fibrosis. In agreement, lung function was not altered as judged by similar lung resistance (LR) and dynamic compliance (Cdyn) in both genotypes (Fig. 2s, t).
Trf1 deletion in lung fibroblasts exacerbates profibrotic pathologies upon bleomycin-induced pulmonary fibrosis
As we did not see significant lung pathologies in Col1a2 mutants, we set to study the effect of TRF1 deficiency in fibroblasts in the context of a bleomycin (BLM)-induced fibrosis model. For this purpose, eight-to 10-week-old male Trf1+/+ KFP+/t Cre+/t (Col1a2-Cre; Trf1+/+) and Trf1lox/lox KFP+/t Cre+/t (Col1a2-Cre; Trf1lox/lox) mice were intraperitoneally (i.p.) administered with TMX for five consecutive days during the first week and then once a week until week (W) 7 to induce the deletion of Trf1 in Col1a2+ fibroblasts (Fig. 3a). Then, at W7, animals were intra-tracheally instilled with either a single dose of 0.8 mg/kg of BLM or saline solution (Fig. 3a). First, we performed an immunofluorescence staining with antibodies against COL1A2 and TRF1 that clearly demonstrated that the majority of COL1A2-positive cells stained negative for TRF1 (88%) in the lungs of Trf1Δ/Δ mice. Upon BLM challenge, only the 33 % of COL1A2-positive cells stained negative for TRF1 at the end-point three weeks post-BLM (Fig. 3b, c).

a Tamoxifen (TMX) was intraperitoneally (i.p.) injected to eight-to 10-week-old male Trf1+/+ KFP+/t Cre+/t (Col1a2-Cre; Trf1+/+) and Trf1lox/lox KFP+/t Cre+/t (Col1a2-Cre; Trf1lox/lox) mice for five consecutive days during the first week and then once a week until week (W) 7. Then, at W7, animals were intra-tracheally instilled with either a single dose of 0.8 mg/kg of bleomycin (BLM) or saline (controls). Sacrifice and sample collection were performed at W10. b Representative immunofluorescence stainings for COL1A2 (green) and TRF1 (red) (white arrowheads indicate COL1A2+ fibroblasts with deletion of TRF1), Sirius Red stainings and immunostainings for Vimentin, Smooth Muscle Actin (SMA) and Ki67 (SMA (purple) and Ki67 (brown); green arrowheads indicate double SMA+-Ki67+ fibroblasts), and E-cadherin in lung sections from control and BLM-challenged Trf1+/+ and Trf1Δ/Δ mice. Quantification of the proportion of double COL1A2+-TRF1+ fibroblasts in COL1A2+ cells (c), and airway collagen (Sirius Red) (d), Vimentin (e), SMA (f) and E-cadherin (h) positive areas (%), and SMA+-Ki67+ fibroblasts per 40X high-power field (HPF) (g) in lung sections from control and BLM-challenged Trf1+/+ and Trf1Δ/Δ mice. Lung tissue mRNA expression levels of Ccl12 (recruitment of fibrocytes) (i), Col1a1 (j), Col1a2 (k), Col3a1 (l), Col4a1 (m), Col5a1 (n) and Col6a1 (o) (collagen markers), and TGFB1 (myofibroblast differentiation) protein levels (p) in lung homogenates from control and BLM-challenged Trf1+/+ and Trf1Δ/Δ mice. Quantification of lung resistance (LR) (q) and dynamic compliance (Cdyn) (r) evaluated by plethysmography in control and BLM-challenged Trf1+/+ and Trf1Δ/Δ mice. Data are expressed as mean ± SEM (the number of mice is indicated in each case). *p < 0.05, **p < 0.001, ***p < 0.001 (Dunn–Sidak test for multiple comparisons). Source data are provided as a Source Data file.
In order to assess collagen deposition we performed Sirius Red stainings in our mouse cohorts. Collagen deposition was found significantly increased after BLM challenge, being this increment more pronounced in Trf1Δ/Δ mice (Fig. 3b, d). As we observed that Trf1 deletion in fibroblasts increased collagen deposition in the lungs of Trf1Δ/Δ mice after BLM treatment, we next studied the expression of several lung profibrotic markers. To this end, we performed immunostainings for Vimentin and Smooth Muscle Actin (SMA) (fibroblast abundance) and E-Cadherin (Epithelial-Mesenchymal Transition, EMT), as well as double immunostainings of SMA with the proliferation marker Ki67 in lung sections of our mouse cohorts (Fig. 3b, e–h). Of note, Vimentin, SMA and E-Cadherin positive areas, as well as the number of SMA proliferating cells were found significantly incremented after BLM challenge, being this increase more pronounced in Trf1Δ/Δ mice (Fig. 3b, e–h). We next assessed lung tissue mRNA expression of Ccl12 (recruitment of fibrocytes), Col1a1, Col1a2, Col3a1, Col4a1, Col5a1 and Col6a1 (collagen markers), as well as an ELISA to quantify TGFB1 protein levels on lung homogenates (Fig. 3i–p). In agreement with increased fibroblast abundance (Fig. 3b, e, f), these markers were found elevated upon BLM treatment, being this increase more pronounced in Trf1Δ/Δ mice (Fig. 3i–p).
To better evaluate the potential lung pathological effects of Trf1 deletion in lung fibroblasts upon BLM treatment, we assessed lung function by plethysmography (Fig. 3q, r). We observed that lung resistance (LR) was increased after BLM challenge in Trf1+/+ and Trf1Δ/Δ mice compared to controls but this increment was higher in Trf1Δ/Δ mice (Fig. 3q). Of note, dynamic compliance (Cdyn) was only found significantly decreased in Trf1Δ/Δ mice after BLM treatment (Fig. 3r).
Trf1 deletion in lung fibroblasts exacerbates bleomycin-induced inflammatory response
To evaluate the effect of Trf1 deficiency in fibroblasts on lung inflammation, we assessed cellularity and total protein concentration in bronchoalveolar lavage fluid (BALF), as well as performed immunostainings for the quantification of neutrophils (MPO), T lymphocytes (CD4 and CD8) and macrophages (F4/80) in lung sections from our mouse cohorts (Fig. 4a–k). Specifically, neutrophil and lymphocyte counts in BALF were found elevated in Trf1Δ/Δ control mice as compared to Trf1+/+ mice. Of note, BLM treatment resulted in increased total and differential cell counts, being this increase significantly higher in Trf1Δ/Δ mice in the case of total cells and neutrophils (Fig. 4a–f). Moreover, BLM challenge increased total protein concentration in BALF, being this increment more pronounced in Trf1Δ/Δ mice (Fig. 4g). Accordingly, Trf1Δ/Δ control lungs exhibit increased presence of MPO+ neutrophils, CD4+ and CD8+ lymphocytes and F4/80+ macrophages compared to Trf1+/+ mice. BLM treatment induced an increase in the presence of these immune cell types along with F4/80+ macrophages that was more pronounced in Trf1Δ/Δ mice (Fig. 4h–k).

a Representative BALF cytospin preparations (May-Grünwald Giemsa (MGG)) and immunostainings for MPO (red arrowheads indicate MPO+ neutrophils), CD4 and CD8 (green and black arrowheads indicate CD4+ and CD8+ T lymphocytes) and F4/80 (orange arrowheads indicate F4/80+ macrophages) in lung sections from control and BLM-challenged Trf1+/+ and Trf1Δ/Δ mice. Quantification of total (b) and differential BALF cell counts for eosinophils (c), neutrophils (d) lymphocytes (e) and macrophages (f) and total protein concentration in BALF (g), of control and BLM-challenged Trf1+/+ and Trf1Δ/Δ mice. Quantification of lung MPO (h), CD4 (i), CD8 (j) and F4/80 (k) positive cells per 40X high-power field (HPF), and lung tissue mRNA expression levels of Tnf (l) Il1b (m), Il6 (n) and Ifn-γ (o) (Th1 inflammation) and Il4 (p), Il10 (q) and Il13 (r) (Th2 inflammation) in control and BLM-challenged Trf1+/+ and Trf1Δ/Δ mice. Data are expressed as mean ± SEM (the number of mice is indicated in each case). *p < 0.05, **p < 0.01, ***p < 0.001 (Dunn–Sidak test for multiple comparisons). Source data are provided as a Source Data file.
We next quantified lung tissue mRNA expression of Th1 (Tnf, Il1b, Il6 and Ifng) and Th2 (Il4, Il10 and Il13) inflammation markers (Fig. 4l–r). BLM challenge increased the expression of Th1 and Th2 inflammation markers, and this increment was more pronounced in Trf1Δ/Δ mice in the case of Il6, Ifng, Il4, Il10 and Il13 (Fig. 4l–r). These results clearly show that deletion of Trf1 in fibroblasts exacerbates the BLM-induced inflammatory response.
Efficient Trf1 deletion in club cells upon tamoxifen administration
Next, we induced telomere dysfunction in club cells by genetically deleting Trf1 in these cells, which constitute a lung stem cell population that plays a key role in bronchiolar epithelial repair through their ability to self-renew and differentiate into ciliated and goblet cells31,32. To delete Trf1 in club cells, we generated the Trf1lox/lox KFPLox-LSL-Lox Scgb1a1-CreERT2 mouse model, in which Cre recombinase expression is controlled by the secretoglobin, family 1A, member 1 (Scgb1a1) promoter, which is specific for club cells (Fig. 5a). CRE-ERT2 is conditionally activated by TMX administration31. In addition, a transgene encoding for the KFP that contains a stop cassette flanked by lox sequences was introduced as a reporter to monitor Cre activity15 (Fig. 5a). TMX was administered i.p. for five consecutive days during the first week and once a week until the end-point. Mice were sacrificed at W22 post-tamoxifen treatment (Fig. 5b). We did not find significant differences in survival between Scgb1a1-Cre; Trf1+/+ (Trf1+/+, controls) and Scgb1a1-Cre; Trf1Δ/Δ (Trf1Δ/Δ) male and female mice (Fig. 5c and Supplementary Fig. 1a). Of note, immunostaining with antibodies against SCGB1A1 and TRF1 clearly demonstrated that the majority of SCGB1A1-positive club cells stained negative for TRF1 (97%) in the lungs of Trf1Δ/Δ male and female mice (Fig. 5d, e and Supplementary Fig. 1b). To measure telomere length specifically in club cells, we performed an immune-telomere-Q-FISH for the quantification of mean telomere spot intensity and average number of telomeres in SCGB1A1-positive cells. No differences neither in mean telomere length nor in the number of telomeric spots were found between male wild-type and Trf1Δ/Δ club cells, indicating that TRF1 depletion does not affect telomere length homeostasis in this cell type (Fig. 5d, f, g).

a Generation of the conditional knockout mouse model in which Trf1 was deleted in club cells using the Cre recombinase driven by the Scgb1a1 promoter. Trf1lox, KFPLox-LSL-Lox, and Scgb1a1-CreERT2 alleles are depicted before and after Cre-mediated excision. b Tamoxifen (TMX) treatment, survival rate assessment and sample collection. Eight-to 10-week-old male Trf1+/+ KFP+/t Cre+/t (Scgb1a1-Cre; Trf1+/+) and Trf1lox/lox KFP+/t Cre+/t (Scgb1a1-Cre; Trf1lox/lox) mice were i.p. injected with TMX for five consecutive days during the first week and once a week until the sacrifice and sample collection on week (W) 22. c Kaplan–Meier survival curves of Scgb1a1-Cre; Trf1+/+ (Trf1+/+, controls) and Scgb1a1-Cre; Trf1Δ/Δ (Trf1Δ/Δ) mice upon TMX treatment. d Representative immunostainings for SCGB1A1 (blue) and TRF1 (brown) (red arrowheads indicate SCGB1A1+ club cells with deletion of TRF1), and immune-telomere-Q-FISH in SCGB1A1+ club cells (Cy3Tel probe (red), SCGB1A1+ cells (green), and nuclei stained with DAPI (blue)) in lung sections from Trf1+/+ and Trf1Δ/Δ mice. Quantification of SCGB1A1+-TRF1+ cells (%) (e), mean telomere spot intensity (f) and average number of telomeres (g) in SCGB1A1+ cells in Trf1+/+ and Trf1Δ/Δ mice. Quantification of total white blood cells (h), neutrophils (i), eosinophils (j), lymphocytes (k) and macrophages (l) in peripheral blood from Trf1+/+ and Trf1Δ/Δ mice. Data are expressed as mean ± SEM (the number of mice is indicated in each case). *p < 0.05, ***p < 0.001 (Mann–Whitney or unpaired t tests). Animal survival was assessed by the Kaplan–Meier analysis, using the log Rank (Mantel–Cox) test). Source data are provided as a Source Data file.
In order to evaluate the effect of Trf1 deficiency in club cells on peripheral blood, we evaluated total and differential white blood cell counts in male and female mice. We only found a slight increment in differential blood cell counts for neutrophils and eosinophils in Trf1Δ/Δ male mice (Fig. 5h–l and Supplementary Fig. 1c–g).
Trf1 deletion in club cells increases telomeric damage, cell cycle arrest and differentiation, and reduces proliferation of club cells
In order to evaluate the effects of Trf1 deficiency on club cells, we assessed the total number of club and basal cells, the numbers of club cells positive for γH2AX, p21, Ki67 and SOX2 as a read out for DNA damage, cell cycle arrest, proliferation and differentiation, respectively, as well as an immuno-telomere-Q-FISH with the DNA damage marker 53BP1 to identify telomeric induced foci (TIF) in SCGB1A1 positive cells in lung sections from Trf1Δ/Δ mice and controls (Fig. 6a–h). First, we performed double immunostainings with the club and basal cell markers SCGB1A1 and p63, and with SCGB1A1 and γH2AX, p21, Ki67 and SOX2 (Fig. 6a–d, f–h) in Trf1+/+ and Trf1Δ/Δ male mice. The results show that Trf1Δ/Δ mice exhibited a reduced number of SCGB1A1 positive club cells and conversely increased presence of p63 positive basal cells per epithelium length in distal airways (Fig. 6a–c). Moreover, Trf1Δ/Δ mice also showed increased number of γH2AX, p21 and SOX2 positive club cells, as well as decreased number of proliferating Ki67 positive club cells per epithelium length (Fig. 6a, d, f–h). Trf1Δ/Δ mice also showed increased proportion of SCGB1A1+ cells with more than 1 TIF (Fig. 6a, e). Lung resistance (LR) and dynamic compliance (Cdyn) were addressed by plethysmography in Trf1Δ/Δ mice and controls. Notably, only Trf1Δ/Δ male mice showed a slight but significant increase in LR (Fig. 6i, j and Supplementary Fig. 1h, i).

a Representative immunostainings for SCGB1A1 (blue) and p63 (brown; green arrowheads indicate p63+ basal cells), SCGB1A1 (blue) and γH2AX (brown; black arrowheads indicate double SCGB1A1+-γH2AX+ club cells), SCGB1A1 (blue) and p21 (brown; red arrowheads indicate double SCGB1A1+-p21+ club cells), SCGB1A1 (purple) and Ki67 (blue; blue arrowheads indicate double SCGB1A1+-Ki67+ club cells), and SCGB1A1 (blue) and SOX2 (brown; orange arrowheads indicate double SCGB1A1+-SOX2+ club cells), as well as representative images of telomeric induced foci (TIF) in SCGB1A1+ cells (SCGB1A1 (purple), Cy3Tel probe (red), 53BP1+ cells (green; white arrowheads indicate TIF) and nuclei stained with DAPI (blue)) in lung sections from Trf1+/+ and Trf1Δ/Δ mice. Quantification of SCGB1A1+ (b), p63+ (c), and double SCGB1A1+-γH2AX+ (d), SCGB1A1+-p21+ (f), SCGB1A1+-Ki67+ (g) and SCGB1A1+-SOX2+ (h) club cells per epithelium length (mm), as well as the proportion (%) of SCGB1A1+ cells with more than 1 TIF (e) in Trf1+/+ and Trf1Δ/Δ mice. Quantification of lung resistance (LR) (i) and dynamic compliance (Cdyn) (j) evaluated by plethysmography in Trf1+/+ and Trf1Δ/Δ mice. Data are expressed as mean ± SEM (the number of mice is indicated in each case). *p < 0.05, **p < 0.01 (Mann–Whitney or unpaired t tests). Source data are provided as a Source Data file.
Trf1 deletion in club cells increases lung inflammation and airway remodeling
To study the pathological consequences of Trf1 deficiency in club cells on the lung, we quantified cellularity and total protein concentration in BALF, as well as performed immunostainings for the quantification of neutrophils (MPO), T lymphocytes (CD4 and CD8) and macrophages (F4/80) in lung sections from Trf1Δ/Δ male mice and controls, as well as lung mRNA expression of Th1 inflammation markers (Fig. 7a–n). We found that total and differential cell counts for neutrophils, lymphocytes and macrophages as well as total protein concentration in BALF, an indicator of vascular permeability were elevated in Trf1Δ/Δ as compared to wild-type control male mice (Fig. 7a–f). Accordingly, we found increased presence of neutrophils, CD4 and CD8 T lymphocytes and macrophages in lung sections from Trf1Δ/Δ male mice, as well as incremented lung mRNA expression of Th1 inflammation markers Tnf, Il1b, Il6 and Ifn-γ (Fig. 7g–n). These differences were not observed in female mice with the exception of a slight increment in the number of neutrophils in BALF (Supplementary Fig. 1j–n).

a Representative BALF cytospin preparations (May-Grünwald Giemsa (MGG)) and immunostainings for MPO (neutrophils), CD4 and CD8 (T lymphocytes) and F4/80 (macrophages) in lung sections from Trf1+/+ and Trf1Δ/Δ mice. Quantification of total (b) and differential BALF cell counts for neutrophils (c), lymphocytes (d) and macrophages (e), and total protein concentration in BALF (f) of Trf1+/+ and Trf1Δ/Δ mice. Quantification of lung MPO (g), CD4 (h), CD8 (i) and F4/80 (j) positive cells per 40X high-power field (HPF), and lung tissue mRNA expression levels of Tnf (k), Il1b (l), Il6 (m) and Ifn-γ (n) (Th1 inflammation) in Trf1+/+ and Trf1Δ/Δ mice. o Representative stainings for Sirius Red (alveolar parenchyma and airways) and immunostainings for Collagen I, Vimentin and SMA (airways) in lung sections from Trf1+/+ and Trf1Δ/Δ mice. Quantification of airway collagen (Sirius Red) (p), airway Collagen I (q) and Vimentin (r) positive areas (%), and airway smooth muscle (SM) thickness (SMA) (µm) (s) in Trf1+/+ and Trf1Δ/Δ mice. Data are expressed as mean ± SEM (the number of mice is indicated in each case). *p < 0.05, **p < 0.01, ***p < 0.001 (Mann–Whitney or unpaired t tests). Source data are provided as a Source Data file.
To further investigate the consequences of Trf1 deficiency in club cells on the lung, we performed a Sirius Red staining (collagen deposition), as well as immunostainings for Collagen I (collagen deposition), Vimentin and SMA (fibroblast abundance) in Trf1Δ/Δ male mice and controls. We did not find changes in alveolar collagen content between both experimental groups. On the other hand, airway collagen (Sirius Red), Collagen I and Vimentin stained areas, as well as airway smooth muscle (SM) thickness were found significantly increased in Trf1Δ/Δ male mice (Fig. 7o–s). Conversely, airway collagen (Sirius Red) and Vimentin-stained areas, as well as SM thickness were not significantly incremented in Trf1Δ/Δ female mice (Supplementary Fig. 1j, o, p, q). These results indicate that telomere dysfunction in club cells results in pathological pattern exclusively in the airways but not in the lung parenchyma of male mice, thus ruling out that telomere dysfunction in club cells is sufficient to induce pulmonary fibrosis in mice. Instead, these pathologies are reminiscent of airway remodeling observed in asthmatic patients, characterized by increased airway smooth muscle mass and sub-epithelial fibrosis39.
Efficient Trf1 deletion in lung basal cells upon tamoxifen administration
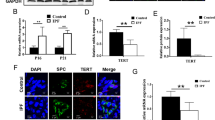
Next, we induced a telomere dysfunction in basal cells by genetically deleting Trf1 in these cells, which in response to injury, become activated and operate as progenitor cells capable of self-renewal and differentiation into ciliated and secretory cells33,35,36. To delete Trf1 in basal cells we generated the Trf1lox/lox KFPLox-LSL-Lox p63-CreERT2 mouse model, in which Cre recombinase expression is controlled by the transformation related protein 63 (p63) promoter, which is specific for basal cells CRE-ERT2 is conditionally activated by TMX administration40. In addition, a transgene encoding for the KFP that contains a stop cassette flanked by lox sequences was introduced as a reporter to monitor Cre activity (Fig. 8a). TMX was administered i.p. for five consecutive days during the first week and once a week until the end-point that was set at week 10 (W10) after the beginning of TMX treatment (Fig. 8b and Supplementary Fig. 2a). Following TMX administration, p63-Cre; Trf1Δ/Δ male and female mice exhibited a decreased survival compared to p63-Cre; Trf1+/+ (control) mice, which was more marked in male mice (Fig. 8C and Supplementary Fig. 2a). Histopathological analysis at death point show that Trf1Δ/Δ mice exhibited abnormal pathologies in the skin, tongue, esophagus and non-glandular stomach, characterized by the presence of hyperkeratosis, dysplastic epithelial hyperplasia, dermal invasion, nuclear atypias and epithelial lamina propria invasion with inflammation (Fig. 8d). Immunostaining with antibodies against p63 and TRF1 clearly demonstrated that the majority of p63-positive cells stained negative for TRF1 (97%) in the lungs of Trf1Δ/Δ male and female mice (Fig. 8e, f and Supplementary Fig. 2b).

a Generation of the conditional knockout mouse model in which Trf1 was deleted in basal cells using the Cre recombinase driven by the p63 promoter. Trf1lox, KFPLox-LSL-Lox, and Scgb1a1-CreERT2 alleles are depicted before and after Cre-mediated excision. b Tamoxifen (TMX) treatment, survival rate assessment and sample collection. Eight-to 10-week-old male Trf1+/+ KFP+/t Cre+/t (p63-Cre; Trf1+/+) and Trf1lox/lox KFP+/t Cre+/t (p63-Cre; Trf1lox/lox) mice were i.p. injected with TMX for five consecutive days during the first week and once a week until the sacrifice and sample collection on week (W) 10. c Kaplan–Meier survival curves of p63-Cre; Trf1+/+ (Trf1+/+, controls) and p63-Cre; Trf1Δ/Δ (Trf1Δ/Δ) mice upon TMX treatment. d Abnormal pathologies observed in Trf1Δ/Δ mice in the skin (hyperkeratosis (H), dysplastic epithelial hyperplasia (DEH) and dermal invasion (DI)), tongue (hyperkeratosis (H), dysplastic epithelial hyperplasia (DEH), nuclear atypias (NA) and epithelial lamina propria invasion with inflammation (LPI + I)), esophagus (hyperkeratosis (H) and nuclear atypias (NA)) and stomach (hyperkeratosis (H), dysplastic epithelial hyperplasia (DEH) and epithelial lamina propria invasion with inflammation (LPI + I)). e Representative immunostainings for p63 (blue) and TRF1 (brown) (red arrowheads indicate p63+ basal cells with deletion of TRF1), and immune-telomere-Q-FISH in p63+ club cells (Cy3Tel probe (red), p63+ cells (green), and nuclei stained with DAPI (blue)) in lung sections from Trf1+/+ and Trf1Δ/Δ mice. Quantification of p63+-TRF1+ cells (%) (f), mean telomere spot intensity (g) and average number of telomeres (h) in SCGB1A1+ cells in Trf1+/+ and Trf1Δ/Δ mice. i–m Quantification of total white blood cells (i), neutrophils (j), eosinophils (k), lymphocytes (l) and macrophages (m) in peripheral blood from Trf1+/+ and Trf1Δ/Δ mice. Data are expressed as mean ± SEM (the number of mice is indicated in each case). *p < 0.05, **p < 0.01, ***p < 0.001 (Mann–Whitney or unpaired t tests). Animal survival was assessed by the Kaplan–Meier analysis, using the log Rank (Mantel–Cox) test). Source data are provided as a Source Data file.
Quantification of mean telomere spot intensity and average number of telomeres in p63-positive cells by immune-telomere-Q-FISH showed no significant changes in the lungs of Trf1Δ/Δ male mice as compared to control mice (Fig. 8e, g, h).
To evaluate the consequences of Trf1 deficiency in basal cells on peripheral blood, we evaluated total and differential white blood cell counts. We only found increased total and differential blood cell counts for neutrophils and lymphocytes in Trf1Δ/Δ male mice (Fig. 8i–m) and for neutrophils and eosinophils in Trf1Δ/Δ female mice (Supplementary Fig. 2c–g).
Trf1 deletion in basal cells increases telomeric damage and cell cycle arrest, and reduces proliferation of lung basal cells
To evaluate the effect of Trf1 deficiency on basal cells, we assessed the total number of basal as well as double positive for p63 and either γH2AX, p21 or Ki67 as a read out for DNA damage, cell cycle arrest and proliferation, respectively, as well as an immuno-telomere-Q-FISH with the DNA damage marker 53BP1 to identify telomeric induced foci (TIF) in p63 positive cells in lung sections from in Trf1Δ/Δ male mice and controls (Fig. 9a–g). The Trf1Δ/Δ mice showed a reduced number of p63 positive basal cells per epithelium length, without significant changes in the number of club cells (Fig. 9a–c). Furthermore, Trf1Δ/Δ mice showed increased number of γH2AX and p21, as well as decreased number of Ki67 positive basal cells per epithelium length (Fig. 9a, d, f, g). Trf1Δ/Δ mice also exhibited increased proportion of p63+ cells with more than 1 TIF (Fig. 9a, e). In addition, lung resistance (LR) and dynamic compliance (Cdyn) were evaluated by plethysmography in Trf1Δ/Δ mice and controls. Of note, Trf1Δ/Δ male mice exhibited a slight increment in LR that was not observed in females, indicating that TRF1 depletion in basal cells in the airways affects male pulmonary function (Fig. 9h, i and Supplementary Fig. 2h, i).

a Representative immunostainings for p63 (blue) and SCGB1A1 (brown; green arrowheads indicate p63+ basal cells), p63 (blue) and γH2AX (brown; black arrowheads indicate double p63+-γH2AX+ basal cells), p63 (blue) and p21 (brown; red arrowheads indicate double p63+-p21+ basal cells), and p63 (blue) and Ki67 (purple; orange arrowheads indicate double p63+-Ki67+ basal cells), as well as representative images of telomeric induced foci (TIF) in p63+ cells (p63 (purple), Cy3Tel probe (red), 53BP1+ cells (green; white arrowheads indicate TIF) and nuclei stained with DAPI (blue)) in lung sections from Trf1+/+ and Trf1Δ/Δ mice. Quantification of p63+ (b), SCGB1A1+ (c) and double p63+-γH2AX+ (d), p63+-p21+ (f) and p63+-Ki67+ (g) basal cells per epithelium length (mm), as well as the proportion (%) of p63+ cells with more than 1 TIF (e) in Trf1+/+ and Trf1Δ/Δ mice. Quantification of lung resistance (LR) (h) and dynamic compliance (Cdyn) (i) evaluated by plethysmography in Trf1+/+ and Trf1Δ/Δ mice. Data are expressed as mean ± SEM (the number of mice is indicated in each case). **p < 0.01 (Mann–Whitney or unpaired t tests). Source data are provided as a Source Data file.
Trf1 deletion in lung basal cells increases lung inflammation and airway remodeling
To gain insight into how Trf1 deficiency in basal cells affect normal lung homeostasis, we quantified cellularity and total protein concentration in BALF, as well as performed immunostainings for the quantification of neutrophils (MPO), T lymphocytes (CD4 and CD8) and macrophages (F4/80) in lung sections from Trf1Δ/Δ male mice and controls. In addition, we assessed lung mRNA expression of Th1 inflammation markers (Fig. 10a–n). We observed that total and differential BALF cell counts for neutrophils and lymphocytes were found elevated in Trf1Δ/Δ mice, with the exception of macrophages which were found significantly reduced (Fig. 10a–e). Trf1Δ/Δ female mice exhibited increased presence of neutrophils and lymphocytes as well as decreased number of macrophages in BALF (Supplementary Fig. 2j–n). Total protein concentration in BALF, an indicator of vascular permeability, was also found incremented in Trf1Δ/Δ male mice (Fig. 10f). In accordance, we found increased presence of neutrophils, CD4 and CD8 T lymphocytes and macrophages in lung sections from Trf1Δ/Δ male mice, and incremented lung mRNA expression of Th1 inflammation markers Tnf, Il1b, Il6 and Ifn-γ (Fig. 10g–n).

a Representative BALF cytospin preparations (May-Grünwald Giemsa (MGG)) and immunostainings for MPO (neutrophils), CD4 and CD8 (T lymphocytes) and F4/80 (macrophages) in lung sections from Trf1+/+ and Trf1Δ/Δ mice. Quantification of total (b) and differential BALF cell counts for neutrophils (c), lymphocytes (d) and macrophages (e), and total protein concentration in BALF (f) of Trf1+/+ and Trf1Δ/Δ mice. Quantification of lung MPO (g), CD4 (h), CD8 (i) and F4/80 (j) positive cells per 40X high-power field (HPF), and lung tissue mRNA expression levels of Tnf (k), Il1b (l), Il6 (m) and Ifn-γ (n) (Th1 inflammation) in Trf1+/+ and Trf1Δ/Δ mice. o Representative stainings for Sirius Red (alveolar parenchyma and airways) and immunostainings for Collagen I, Vimentin and SMA (airways) in lung sections from Trf1+/+ and Trf1Δ/Δ mice. p–s Quantification of airway collagen (Sirius Red), airway Collagen I and Vimentin positive areas (%), and airway smooth muscle (SM) thickness (SMA) (µm) in Trf1+/+ and Trf1Δ/Δ mice. Data are expressed as mean ± SEM (the number of mice is indicated in each case). **p < 0.01 (Mann–Whitney or unpaired t tests). t Pathological consequences of telomere dysfunction in fibroblasts, club and basal cells in the lung. Dysfunctional telomeres in alveolar type II (ATII) cells led to alveolar DNA damage, senescence and apoptosis, as well as to interstitial lung fibrosis15. TRF1 deficiency in Club and basal cells induced telomeric damage and cell cycle arrest, and reduced proliferation of these cell types. TRF1 deletion in fibroblasts increased telomeric damage, cell cycle arrest, apoptosis and proliferation in this cell type. Noteworthy, depletion of TRF1 in fibroblasts, Club and basal cells did not lead to interstitial lung fibrosis. Source data are provided as a Source Data file.
Next, to further study the pathological consequences of Trf1 deficiency in basal cells on the lung, we performed a Sirius Red staining (collagen deposition), as well as immunostainings for Collagen I (collagen deposition), Vimentin and SMA (fibroblast abundance) in Trf1Δ/Δ male mice and controls. We did not find changes in alveolar collagen content between experimental groups. However, in the airways there was a clear increase in collagen (Sirius Red), Collagen I and Vimentin-stained areas, as well as smooth muscle (SM) thickness in Trf1Δ/Δ male mice (Fig. 10o–s). Of note, airway collagen (Sirius Red) and Vimentin-stained areas, as well as SM thickness were not significantly incremented in Trf1Δ/Δ female mice (Supplementary Fig. 2j, o, p, q). Again, we did not find any signs of lung parenchymal fibrosis, thus suggesting that telomere dysfunction in basal cells is not sufficient to lead to pulmonary fibrosis in mice.
Additionally, we studied the lung pathological consequences of Trf1 deletion in basal cells when deleted from early embryonic development onwards. For this purpose, we studied the Trf1Δ/Δ p53−/− K5-Cre mouse model10. The Trf1Δ/Δ p53−/− K5-Cre male mice exhibited a marked decreased survival compared to control counterparts (Supplementary Fig. 3b). Of note, immunostaining with K5 and TRF1 demonstrated that the majority of K5-positive cells stained negative for TRF1 (96%) in the lungs of Trf1Δ/Δ male mice (Supplementary Fig. 3c, d). In accordance with previous results (Fig. 10o–s), airway collagen (Sirius Red) and Vimentin stained areas, as well as airway SM thickness (SMA) were found significantly incremented in Trf1Δ/Δ male mice (Supplementary Fig. 3c, e, f, g).
Discussion
In this study we have analyzed the lung pathological consequences of telomere dysfunction in fibroblasts, club and basal cells. We show that a short-term telomere dysfunction was efficiently induced in lung fibroblasts to study the early lung phenotypes, without affecting alveolar collagen deposition and fibroblast abundance, as previously shown in a similar mouse model41. Additionally, we show that upon telomere dysfunction COL1A2+ fibroblasts exhibited increased telomeric damage, cell cycle arrest, apoptosis and proliferation, as well as increased presence of inflammatory cells in BALF, but we did not observe decreased mouse survival unlike the study of Naikawadi et al. where telomere dysfunction in fibroblasts drastically decreased mouse survival 1 week after tamoxifen administration. These discrepancies could be possibly due to differences in the Col1a2 Cre driver, as well as to the dosing and timing of tamoxifen administration.
We also demonstrate that telomere dysfunction in fibroblasts in the context of a bleomycin (BLM)-induced fibrosis model, exacerbates classical features observed in patients with IPF including increased collagen deposition, fibroblast abundance and EMT, as well as reduced dynamic compliance42,43,44,45. Specifically, we previously demonstrated that a low BLM dose synergizes with short telomeres to trigger pulmonary fibrosis in telomerase-deficient mice15. Interestingly, after the BLM challenge, the majority of COL1A2+ cells stained positive for TRF1 in Trf1Δ/Δ mice, which could be due to the fact that fibroblasts that escaped the deletion of Trf1 are those proliferating. Alternatively, an induction of EMT, indicated by the loss of E-cadherin46, could be leading to a higher number of fibroblasts. Telomere dysfunction in lung fibroblasts exacerbated Th1 and Th2 inflammation upon BLM challenge. In this sense, M1 macrophages contribute to tissue injury after induction of Th1 inflammation and M2 macrophages lead to the resolution of inflammation and tissue repair upon activation of Th2 inflammation47. Specifically, M2 macrophages were reported to promote myofibroblast differentiation and are associated with pulmonary fibrogenesis48.
In our study we also report that a telomere dysfunction was efficiently induced in club cells. We show that deletion of Trf1 in club cells leads to a reduction in the number of these cells. As previously reported, Trf1 deletion not only in club cells but also in lung fibroblasts and basal cells did not affect telomere length10,11. Unlike our study, telomere length was reported to be reduced in mice with dysfunctional telomeres in club cells49. Furthermore, dysfunctional telomeres in club cells increased airway collagen content, inflammation and lung resistance (LR), in accordance to a previous work49. It should be noted that unlike our study, Naikawadi et al. did not assess fibroblast abundance, as well as telomeric damage, cell cycle arrest, proliferation and differentiation in SCGB1A1+ club cells. Noteworthy, telomere dysfunction in club cells did not increase alveolar collagen content, as we have reported in mice upon deletion of Trf1 in ATII cells, which are at the origin of pulmonary fibrosis15. Specifically, we show increased differentiation of club cells upon deletion of Trf1 in this cell type. Interestingly, we have recently reported in mice a significant increase of differentiating SOX2+ club cells with aging, which was anticipated in telomerase-deficient mice50. SOX2 has been previously shown a marker of differentiation of club cells51. Concerning mouse survival, Naikawadi et al. reported that Trf1Δ/Δ mice started to die from 8 months upon TMX administration, unlike our study in which we show that Trf1Δ/Δ mice started to die from week 11, thus, we decided to study the early lung phenotypes. This discrepancy could be due to differences in the Scgb1a1 Cre driver, as well as to the dosing and timing of tamoxifen administration. Interestingly, telomere dysfunction in club cells increased the number of p63+ basal cells specifically in distal airways. In steady state, basal cells are quiescent and only present in the trachea and proximal intrapulmonary airways. Nevertheless, in response to lung injury, airway basal cells are activated to operate as progenitor cells capable of self-renewal and differentiation into Club cells33,35,36,52,53.
Telomere dysfunction in p63+ basal cells caused increased telomeric damage and cell cycle arrest, generating abnormal pathologies in the skin, tongue, esophagus and non-glandular stomach as we reported after deletion of Trf1 in K5+ basal cells10, supporting the reduced mouse survival observed upon telomere dysfunction in both p63+ and K5+ basal cells. Thus, we had to study the early lung phenotypes since Trf1Δ/Δ mice started to die from week 3 upon TMX administration. Trf1 deletion in p63+ lung basal cells decreased the number of these cells but did not alter the number of club cells, which have been shown to be a stem cell population with an important role in lung repair31,32. We also show by the first time that dysfunctional telomeres in lung basal cells does not lead to fibrotic pathologies in the lung parenchyma but did increase lung inflammation, as well as airway collagen deposition, fibroblast abundance and lung resistance (LR). On this basis, we think that increased mortality of Trf1Δ/Δ mice is mainly due to intestinal defects originated by esophagus lumen reduction and stomach pathologies rather than lung dysfunction.
In summary, only male mice with dysfunctional telomeres in club and basal cells exhibited increased lung resistance (LR), airway collagen content and fibroblast abundance. This finding goes in line with the fact that androgens exacerbate lung function impairment and airway collagen deposition in bleomycin-challenged male versus female mice54. Indeed, IPF predominantly affects males55. Similarly, male mice are more prone than females to develop lung fibrosis56. Furthermore, telomere length, a risk factor for IPF19, was described to be shorter in males than in females, since estrogen activates telomerase57,58.
Specifically, dysfunctional telomeres in club and basal cells increased airway remodeling in male mice, which is a critical feature of several chronic bronchial diseases characterized by aberrant repair of the epithelium and accumulation of fibroblasts, which contribute to extracellular matrix deposition that involves the expression of mesenchymal proteins such as α-SMA and vimentin59. Airway remodeling is present in several bronchial diseases including asthma, chronic obstructive pulmonary disease (COPD), bronchiolitis obliterans (BO), bronchopulmonary dysplasia (BPD), cystic fibrosis and bronchiectasis39,60,61,62,63,64. Specifically, airway remodeling upon deletion of Trf1 in club and basal cells is similar to that observed in asthmatic patients, mainly characterized by increased airway smooth muscle mass and sub-epithelial fibrosis39. Of note short telomeres were reported in patients with bronchial diseases including asthma, COPD, BO, BPD65,66,67,68. In addition, dysfunctional telomeres were also observed in patients with COPD69. Our results do not ultimately probe that telomere dysfunction per se in club and basal cells lead to airway fibrosis since airway fibrosis could also be mediated by the inflammatory cascade triggered by TRF1 depletion.
Remarkably, the observed lung phenotypes in club and basal cells are induced by Trf1-dependent telomere dysfunction independently of telomere length. In particular, Trf1 deletion induces telomere uncapping and the activation of a persistent DNA damage response (DDR) at chromosome ends10,11.
On that basis we can conclude that TRF1 could have an important role in lung tissue homeostasis, supported by previous findings in which we demonstrated its importance in tissue regeneration and homeostasis upon conditional deletion of Trf1 from specific cell types10,13,14,15. In summary, here we show the pathological consequences of telomere dysfunction in lung fibroblasts, club and basal cells. Specifically, telomere dysfunction in club and basal cells from male mice increased lung damage, inflammation and airway remodeling. Noteworthy, depletion of TRF1 in fibroblasts, Club and basal cells did not lead to interstitial lung fibrosis, underscoring ATII cells as the relevant cell type for the origin of interstitial fibrosis (Fig. 10t). Our findings contribute to a better understanding of the importance of TRF1 in lung tissue homeostasis.
Methods
Ethical statement
All experiments and animal procedures were approved by our Institutional Animal Care and Use Committee (IACUC) (IACUC.011-2018, CBA_20_2018), by the Ethics Committee for Research and Animal Welfare (CEIyBA) (CBA 20-2018) from the Instituto de Salud Carlos III, and by Consejería de Medio Ambiente, Administración Local y Ordenación del Territorio (Comunidad de Madrid) (PROEX 163/18). All experiments and animal procedures were performed in accordance with the guidelines stated in the International Guiding Principles for Biomedical Research Involving Animals, developed by the Council for International Organizations of Medical Sciences (CIOMS). The animals were bred and maintained under specific pathogen-free (SPF) conditions in laminar flow caging at the CNIO animal facility in accordance with the recommendations of the Federation of European Laboratory Animal Science Associations (FELASA).
Generation of mutant mouse lines
Trf1lox/lox mice were generated as previously described10. To conditionally delete Trf1 in fibroblasts, club and basal cells, homozygous Trf1lox/lox mice were crossed with transgenic mice expressing CreERT2 under the control of the Col1a2, Scgb1a1 and p63 promoters31,37,40 as well as with transgenic mice harboring the Katushka fluorescent protein (KFP) encoding gene that contains a stop cassette flanked by lox sequences, the KFPCAG-lox-STOP-lox allele70 (Figs. 1a, 5a and 8a). Tamoxifen (TMX) (Sigma Aldrich, San Luis, MO) was intraperitoneally (i.p.) injected to eight-to 10-week-old male Trf1+/+ KFP+/t Cre+/t and Trf1lox/lox KFP+/t Cre+/t mice daily for five consecutive days during the first week and then once a week until the sacrifice and sample collection (Figs. 1b, 5b and 8b). Fluorescence intensity of KFP was measured with an IVIS Lumina Series III (Perkin Elmer, Waltham, MA) in vivo imaging system. Additionally, we have generated the Trf1Δ/Δ p53−/− K5-Cre mouse model, in which a Trf1 deletion was induced from embryonic development onwards(Supplementary Fig. 3a)10.
Bleomycin mouse model
TMX (Sigma Aldrich) was i.p. injected to eight-to 10-week-old male Trf1+/+ KFP+/t Cre+/t (Col1a2-Cre; Trf1+/+) and Trf1lox/lox KFP+/t Cre+/t (Col1a2-Cre; Trf1lox/lox) mice for five consecutive days during the first week and then once a week until week (W) 7 to induce the deletion of Trf1 in Col1a2+ fibroblasts (Fig. 3a). Then, at W7, animals were intra-tracheally instilled with either a single dose of 0.8 mg/kg of bleomycin (BLM) (Sigma Aldrich, San Luis, MO) or saline (controls) under a ketamine-medetomidine anesthetic combination (10 μl/g). Sacrifice and sample collection was performed at W10. (Fig. 3a).
In vivo measurement of lung function
The mice were anesthetized using 10 μl/g of ketamine-medetomidine and intubated with a 24-gauge catheter (BD biosciences, Franklin lakes, NJ, USA). Then, lung function was assessed in a plethysmograph (SCIREQ, Montreal, Canada) for the determination of LR (lung resistance) and Cdyn (dynamic compliance)71,72.
Sample collection and processing
Animals were euthanized using 10 μl/g of ketamine-xylazine. Blood was collected by cardiac puncture and lungs were lavaged with 1 ml of cold PBS 1X. Right lung lobes were dissected and snap-frozen in liquid nitrogen for qPCR and ELISA analyses, and the left lung lobe and remaining organs were fixed in 10% buffered formalin, embedded in paraffin and cut into 3 μm sections for histopathological evaluation, immunohistochemistry (IHC) or immunofluorescence (IFC).
Histopathological analyses, IHC and IFC
Hematoxylin and eosin (H&E) staining was performed for histopathological evaluation, and Sirius Red (Sigma-Aldrich) staining served to evaluate collagen deposition. IFC was performed using the following antibodies: COL1A2 (Clone E-6 1:400, Santa Cruz Biotechnology, Dallas, TX) and TRF1 (CNIO Monoclonal Antibodies Core Unit, Madrid, Spain). IHC was performed using the following antibodies: Turbo-RFP (KFP) (1:3000, EVROGEN, Moscow, Russia), COL1A2 (Clone E-6 1:100, Santa Cruz Biotechnology), H2AX (Ser139, Clone JBW301 1:200, EMD Millipore, Burlington, MA), p16 (Clone 33B 1:30, CNIO Monoclonal Antibodies Core Unit), p21 (Clone 291H/B5 1:10, CNIO Monoclonal Antibodies Core Unit), C3 (Asp175 1:300, Cell Signaling Technology, Danvers, MA), Ki-67 (Clone D3B5 1:50, Cell Signaling Technology), SCGB1A1/CC10 (Clone T-18 1:1000, Santa Cruz Biotechnology), TRF1 (Clone 572C 1:50, Abcam, Cambridge, UK), p63 (Clone 4A4, Roche, Basel, Switzerland), SOX2 (Clone C70B1 1:75, Cell Signaling Technology), Myeloperoxidase (1:1250, DAKO, Jena, Germany), CD4 (Clone D7D2Z 1:50, Cell Signaling Technology), CD8 (Clone 94A 1:200, CNIO Monoclonal Antibodies Core Unit), F4/80 (Clone A3-1 MCA497 1:20, AbD Serotec/Bio-Rad, Hercules, CA), Collagen I (1:600, EMD Millipore), Vimentin (Clone D21H3 1:50, Cell Signaling Technology), E-cadherin (Clone 36 1:1000, BD Biosciences) and SMA (Clone 1A4 1:4, DAKO, Agilent technologies, Santa Clara, CA).
Telomere Q-FISH analyses
After deparaffinization and rehydration, tissues underwent antigen retrieval in 10 mM sodium citrate buffer and permeabilization was performed in PBS 0.5% Triton X-100 for 3 h. Next, tissues were washed 3 × 5 min in PBS 1X, fixed in 4% formaldehyde for 5 min, washed 3 × 5 min in PBS and dehydrated in a 70%–90%–100% ethanol series (5 min each). Then, the immuno-telomere-Q-FISH with COL1A2 (Clone E-6 1:400, Santa Cruz Biotechnology), SCGB1A1/CC10 (Clone E-11 1:100, Santa Cruz Biotechnology) and p63 (Clone 4A4, Roche) antibodies was performed and analyzed as previously described50,71. Following the same protocol, an immuno-telomere-Q-FISH with the DNA damage marker 53BP1 (1:500, Novus Biologicals, Centennial, CO) was performed to identify telomeric induced foci (TIF) in COL1A2 (Clone E-6 1:400, Santa Cruz Biotechnology), SCGB1A1 (Clone E-11 AF488 1:100, Santa Cruz Biotechnology) and p63 (Clone 4A4, Roche, Basel, Switzerland) positive cells as previously described71,73.
RNA isolation, reverse transcription, qPCR and ELISAS
Inferior right lung lobes were homogenized in TRIzol reagent (Invitrogen), and RNA was isolated using a RNeasy Mini Kit (Qiagen, Hilden, Germany) and reverse-transcribed to cDNA using SuperScript II First-Strand Synthesis System (Invitrogen). qPCR was performed as previously describe50,74. Primer sets used for qPCR are included within the supplementary information (Supplementary Table 1). TGFB1 levels were assessed in homogenized lung tissue lysates using a TGFB1 Quantikine ELISA Kit (R&D systems, Minneapolis, MN).
Statistics
According to the sample distribution (Shapiro–Wilk normality test), either a Mann–Whitney or umpaired t tests were used to compare differences between 2 independent groups. Following a Shapiro–Wilk normality test, either a one-way ANOVA test or a Kruskal–Wallis test were used and then, the post hoc Dunn–Sidak multiple test was carried out for multiple comparisons between experimental groups. Animal survival was assessed by the Kaplan–Meier analysis, using the log Rank (Mantel–Cox) test.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The authors declare that data supporting the findings of this study are available within the paper (and its supplementary information files). Source data are provided with this paper.
References
Blackburn, E. H. Switching and signaling at the telomere. Cell 106, 661–673 (2001).
De Lange, T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 19, 2100–2110 (2005).
Palm, W. & De Lange, T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 42, 301–334 (2008).
Olovnikov, A. M. A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J. Theor. Biol. 41, 181–190 (1973).
Harley, C. B., Futcher, A. B. & Greider, C. W. Telomeres shorten during ageing of human fibroblasts. Nature 345, 458–460 (1990).
Greider, C. W. & Blackburn, E. H. Identification of a specific telomere terminal transferase activity in tetrahymena extracts. Cell 43, 405–413 (1985).
Bianchi, A. et al. TRF1 binds a bipartite telomeric site with extreme spatial flexibility. EMBO J. 18, 5735–5744 (1999).
Xin, H., Liu, D. & Songyang, Z. The telosome/shelterin complex and its functions. Genome Biol. 9, 232 (2008).
Diotti, R. & Loayza, D. Shelterin complex and associated factors at human telomeres. Nucleus 2, 119–135 (2011).
Martínez, P. et al. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 23, 2060–2075 (2009).
Sfeir, A. et al. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138, 90–103 (2009).
Boué, S., Paramonov, I., Barrero, M. J. & Belmonte, J. C. I. Analysis of human and mouse reprogramming of somatic cells to induced pluripotent stem cells. What is in the plate? PLoS One 5, e12664 (2010).
Schneider, R. P. et al. TRF1 is a stem cell marker and is essential for the generation of induced pluripotent stem cells. Nat. Commun. 4, 1946 (2013).
Beier, F., Foronda, M., Martinez, P. & Blasco, M. A. Conditional TRF1 knockout in the hematopoietic compartment leads to bone marrow failure and recapitulates clinical features of dyskeratosis congenita. Blood 120, 2990–3000 (2012).
Povedano, J. M., Martinez, P., Flores, J. M., Mulero, F. & Blasco, M. A. Mice with pulmonary fibrosis driven by telomere dysfunction. Cell Rep. 12, 286–299 (2015).
Martinez, F. J. et al. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Prim. 3, 17074 (2017).
Richeldi, L., Collard, H. R. & Jones, M. G. Idiopathic pulmonary fibrosis. Lancet 389, 1941–1952 (2017).
Raghu, G. et al. Diagnosis of idiopathic pulmonary fibrosis an official ATS/ERS/JRS/ALAT clinical practice guideline. Am. J. Respir. Crit. Care Med. 198, e44–e68 (2018).
Alder, J. K. et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl Acad. Sci. USA 105, 13051–13056 (2008).
Armanios, M. Y. et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 356, 1317–1326 (2007).
Tsakiri, K. D. et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl Acad. Sci. USA 104, 7552–7557 (2007).
Fingerlin, T. E. et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet. 45, 613–620 (2013).
Cronkhite, J. T. et al. Telomere shortening in familial and sporadic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 178, 729–737 (2008).
Chibbar, R. et al. Familial interstitial pulmonary fibrosis: a large family with atypical clinical features. Can. Respir. J. 17, 269–274 (2010).
Stanley, S. E. et al. Telomerase mutations in smokers with severe emphysema. J. Clin. Invest. 125, 563–570 (2015).
Wijsenbeek, M. & Cottin, V. Spectrum of fibrotic lung diseases. N. Engl. J. Med. 383, 958–968 (2020).
Ushakumary, M. G., Riccetti, M. & Perl, A. K. T. Resident interstitial lung fibroblasts and their role in alveolar stem cell niche development, homeostasis, injury, and regeneration. Stem Cells Transl. Med. 10, 1021–1032 (2021).
Desai, T. J., Brownfield, D. G. & Krasnow, M. A. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 507, 190–194 (2014).
Rawlins, E. L. Emergency back-up for lung repair Stellar clocks. Nature 517, 6–7 (2015).
Olajuyin, A. M., Zhang, X. & Ji, H. L. Alveolar type 2 progenitor cells for lung injury repair. Cell Death Discov. 5, 63 (2019).
Rawlins, E. L. et al. The role of Scgb1a1+ clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem Cell 4, 525–534 (2009).
Hogan, B. L. M. et al. Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell 15, 123–138 (2014).
Dean, C. H. & Snelgrove, R. J. New rules for club development: new insights into human small airway epithelial club cell ontogeny and function. Am. J. Respir. Crit. Care Med. 198, 1355–1356 (2018).
Liu, Z. et al. An obligatory role for club cells in preventing obliterative bronchiolitis in lung transplants. JCI Insight 4, e124732 (2019).
Shaykhiev, R. Multitasking basal cells: combining stem cell and innate immune duties. Eur. Respir. J. 46, 894–897 (2015).
Morrisey, E. E. Basal cells in lung development and repair. Dev. Cell 44, 653–654 (2018).
Zheng, B., Zhang, Z., Black, C. M., de Crombrugghe, B. & Denton, C. P. Ligand-dependent genetic recombination in fibroblasts. Am. J. Pathol. 160, 1609–1617 (2002).
Lee, T.-H. et al. Fibroblast-enriched endoplasmic reticulum protein TXNDC5 promotes pulmonary fibrosis by augmenting TGFβ signaling through TGFBR1 stabilization. Nat. Commun. 11, 4254 (2020).
Hough, K. P. et al. Airway remodeling in asthma. Front. Med. 7, 191 (2020).
Lee, D. K., Liu, Y., Liao, L., Wang, F. & Xu, J. The prostate basal cell (BC) heterogeneity and the p63-positive BC differentiation spectrum in mice. Int. J. Biol. Sci. 10, 1007–1017 (2014).
Naikawadi, R. P. et al. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight 1, e86704 (2016).
Lomas, N. J., Watts, K. L., Akram, K. M., Forsyth, N. R. & Spiteri, M. A. Idiopathic pulmonary fibrosis: immunohistochemical analysis provides fresh insights into lung tissue remodelling with implications for novel prognostic markers. Int. J. Clin. Exp. Pathol. 5, 58–71 (2012).
Plantier, L. et al. Physiology of the lung in idiopathic pulmonary fibrosis. Eur. Respir. Rev. 27, 170062 (2018).
Surolia, R. et al. Vimentin intermediate filament assembly regulates fibroblast invasion in fibrogenic lung injury. JCI Insight 4, e123253 (2019).
Herrera, J. et al. Registration of the extracellular matrix components constituting the fibroblastic focus in idiopathic pulmonary fibrosis. JCI Insight 4, e125185 (2019).
Di Gregorio, J. et al. The epithelial-to-mesenchymal transition as a possible therapeutic target in fibrotic disorders. Front. Cell Dev. Biol. 8, 607483 (2020).
Zhang, L. et al. Macrophages: friend or foe in idiopathic pulmonary fibrosis? Respir. Res. 19, 170 (2018).
Hou, J. et al. M2 macrophages promote myofibroblast differentiation of LR-MSCs and are associated with pulmonary fibrogenesis. Cell Commun. Signal. 16, 89 (2018).
Naikawadi, R. P. et al. Airway epithelial telomere dysfunction drives remodeling similar to chronic lung allograft dysfunction. Am. J. Respir. Cell Mol. Biol. 63, 490–501 (2020).
Piñeiro-Hermida, S. et al. Telomerase treatment prevents lung profibrotic pathologies associated with physiological aging. J. Cell Biol. 219, e202002120 (2020).
Tompkins, D. H. et al. Sox2 is required for maintenance and differentiation of bronchiolar Clara, ciliated, and goblet cells. PLoS One 4, e8248 (2009).
Zuo, W. et al. P63 + Krt5 + distal airway stem cells are essential for lung regeneration. Nature 517, 616–620 (2015).
Yang, Y. et al. Spatial-temporal lineage restrictions of embryonic p63+ progenitors establish distinct stem cell pools in adult airways. Dev. Cell 44, 752–761.e4 (2018).
Voltz, J. W. et al. Male sex hormones exacerbate lung function impairment after bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 39, 45–52 (2008).
Somayaji, R. & Chalmers, J. D. Just breathe: a review of sex and gender in chronic lung disease. Eur. Respir. Rev. 31, 210111 (2022).
Kawano-Dourado, L., Glassberg, M. K., Assayag, D., Borie, R. & Johannson, K. A. Sex and gender in interstitial lung diseases. Eur. Respir. Rev. 30, 210105 (2021).
Bayne, S. et al. Estrogen deficiency leads to telomerase inhibition, telomere shortening and reduced cell proliferation in the adrenal gland of mice. Cell Res. 18, 1141–1150 (2008).
Gardner, M. et al. Gender and telomere length: systematic review and meta-analysis. Exp. Gerontol. 51, 15–27 (2014).
Pain, M. et al. Tissue remodelling in chronic bronchial diseases: from the epithelial to mesenchymal phenotype. Eur. Respir. Rev. 23, 118–130 (2014).
Jonigk, D. et al. Obliterative airway remodeling: molecular evidence for shared pathways in transplanted and native lungs. Am. J. Pathol. 178, 599–608 (2011).
Regamey, N., Jeffery, P. K., Alton, E. W. F. W., Bush, A. & Davies, J. C. Airway remodelling and its relationship to inflammation in cystic fibrosis. Thorax 66, 624–629 (2011).
Popova, A. P. Mechanisms of bronchopulmonary dysplasia. J. Cell Commun. Signal. 7, 119–127 (2013).
Polverino, E. et al. The overlap between bronchiectasis and chronic airway diseases: state of the art and future directions. Eur. Respir. J. 52, e8248 (2018).
Jones, R. L., Noble, P. B., Elliot, J. G. & James, A. L. Airway remodelling in COPD: it’s not asthma! Respirology 21, 1347–1356 (2016).
Córdoba-Lanús, E. et al. Telomere shortening and accelerated aging in COPD: findings from the BODE cohort. Respir. Res. 18, 59 (2017).
Everaerts, S. et al. The aging lung: tissue telomere shortening in health and disease. Respir. Res. 19, 95 (2018).
Barbé-Tuana, F. M. et al. Shorter telomeres in children with severe asthma, an indicative of accelerated aging. Aging 13, 1686–1691 (2021).
Henckel, E. et al. Children with bronchopulmonary dysplasia. Children 8, 80 (2021).
Birch, J. et al. DNA damage response at telomeres contributes to lung aging and chronic obstructive pulmonary disease. Am. J. Physiol. 309, L1124–L1137 (2015).
Diéguez-Hurtado, R. et al. A Cre-reporter transgenic mouse expressing the far-red fluorescent protein Katushka. Genesis 49, 36–45 (2011).
Piñeiro-Hermida, S., Martínez, P. & Blasco, M. A. Short and dysfunctional telomeres protect from allergen-induced airway inflammation. Aging Cell 20, e13352 (2021).
Alfaro-Arnedo, E. et al. IGF1R as a potential pharmacological target in allergic asthma. Biomedicines 9, 912 (2021).
Bosso, G. et al. Early differential responses elicited by BRAF V600E in adult mouse models. Cell Death Dis. 13, 142 (2022).
Alfaro-Arnedo, E. et al. IGF1R acts as a cancer-promoting factor in the tumor microenvironment facilitating lung metastasis implantation and progression. Oncogene 41, 3625–3639 (2022).
Acknowledgements
We are grateful to Dr. J. Xu from the Baylor College of Medicine for providing p63-CreERT2 mouse sperm for the generation of the p63 mutant mouse line. Research in the Blasco Lab is funded by AstraZeneca; Fundación Botín and Banco Santander (Spain); Agencia Estatal de Investigación (AEI/MCI/10.13039/501100011033) with the project RETOS SAF2017-82623-R, cofunded by European Regional Development Fund (ERDF), “A way of making Europe”; Comunidad de Madrid with the Synergy Project COVIDPREclinicalMODels-CM and the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement No 882385) through the project ERC-AvG SHELTERINS. The CNIO, certified since 2011 as Severo Ochoa Centre of Excellence by AEI/MCI/10.13039/501100011033, is supported by the Spanish Government through the Instituto de Salud Carlos III (ISCIII).
Author information
Authors and Affiliations
Contributions
M.A.B. had the original idea and secured funding. M.A.B., P.M., J.C. and R.L. supervised research. M.A.B., P.M. and S.P.-H. wrote the paper. S.P.-H., G.B., J.M.F. and S.S. performed experiments. M.A.B., S.P.-H., G.B. and J.M.F. analyzed the data.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Peter Bitterman and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Piñeiro-Hermida, S., Martínez, P., Bosso, G. et al. Consequences of telomere dysfunction in fibroblasts, club and basal cells for lung fibrosis development. Nat Commun 13, 5656 (2022). https://doi.org/10.1038/s41467-022-32771-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-32771-6
This article is cited by
-
Differential contribution for ERK1 and ERK2 kinases in BRAFV600E-triggered phenotypes in adult mouse models
Cell Death & Differentiation (2024)
-
Co-amplification of CBX3 with EGFR or RAC1 in human cancers corroborated by a conserved genetic interaction among the genes
Cell Death Discovery (2023)
-
Telomerase deficiency and dysfunctional telomeres in the lung tumor microenvironment impair tumor progression in NSCLC mouse models and patient-derived xenografts
Cell Death & Differentiation (2023)
-
Molecular Mechanisms Underlying Systemic Sclerosis–Associated Interstitial Lung Disease and Idiopathic Pulmonary Fibrosis: an Update
Current Treatment Options in Rheumatology (2023)
-
Adverse effects of pristine and aged polystyrene microplastics in mice and their Nrf2-mediated defense mechanisms with tissue specificity
Environmental Science and Pollution Research (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
